

Abstract
Vaccinia virus (VACV) encodes a multifunctional protein, E3L, that is necessary for interferon (IFN) resistance in cells in culture. Interferon resistance has been mapped to the well-characterized carboxy terminus of E3L, which contains a conserved double-stranded RNA binding domain. The amino terminus of E3L has a Z-form nucleic acid binding domain, which has been shown to be dispensable for replication and IFN resistance in HeLa and RK13 cells; however, a virus expressing E3L deleted of the amino terminus has reduced pathogenicity in an animal model. In this study, we demonstrate that the pathogenicity of a virus expressing E3L deleted of the amino terminus was fully rescued in type I IFN receptor knockout (IFN-α/βR−/−) mice. Furthermore, this virus was IFN sensitive in primary mouse embryo fibroblasts (MEFs). This virus induced the phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α) in MEFs in an IFN-dependent manner. The depletion of double-stranded RNA-dependent protein kinase (PKR) from these MEFs restored the IFN resistance of this virus. Furthermore, the virus expressing E3L deleted of the amino terminus was also IFN resistant in PKR−/− MEFs. Thus, our data demonstrate that the amino terminus of E3L is necessary to inhibit the type I IFN response both in mice and in MEFs and that in MEFs, the amino terminus of E3L functions to inhibit the PKR pathway.
INTRODUCTION
Type I interferons (IFNs) (IFN-α/β) are potent cytokines known primarily to inhibit virus replication. In most cells, the induction of IFN synthesis can occur within hours of virus infection unless protein synthesis is inhibited (1). IFNs exert their actions by binding to specific receptors (IFN-α/β receptor [IFN-α/βR]) located on the surfaces of most cells. Binding results in a signaling cascade mediated through the JAK/STAT pathway, which involves the phosphorylation of latent transcription factors, leading to the upregulation of IFN-stimulated genes (ISGs) (7, 13, 14). Hundreds of ISGs are upregulated in response to IFN, some of which function to prime the cells into an antiviral state. Among the antiviral effectors upregulated by IFN is the double-stranded RNA (dsRNA)-dependent protein kinase, PKR. PKR acts as a sentinel to virus infection by recognizing dsRNA, a common by-product of virus infection and a potent activator of the IFN pathway. The activation of PKR occurs by binding to dsRNA, followed by autophosphorylation (12). PKR phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF2α), resulting in the inhibition of protein synthesis (27, 30). Since PKR is a key regulator of translation, viruses employ painstaking measures to circumvent its activation.
Vaccinia virus (VACV) is an IFN-resistant double-stranded DNA (dsDNA) virus, which replicates in the cytoplasm of its infected host. A major contributor to the VACV IFN-resistant phenotype is the double-stranded RNA binding protein E3L. VACV deleted of E3L (VACVΔE3L) is IFN sensitive in rabbit kidney RK13 cells and human Huh7 cells (2, 3, 11). IFN sensitivity seems to be mediated primarily through the functions of PKR, as previous studies showed that RNA interference against PKR in Huh7 cells reversed the IFN-sensitive phenotype of VACVΔE3L (2). E3L deletion studies have shown that the primary inhibition of PKR maps to the carboxy terminus, which encodes a double-stranded-RNA binding domain (dsRBD) (11). Previous studies demonstrated that dsRNA binding of E3L is necessary for the inhibition of PKR and that this effect is reversed upon the addition of excess dsRNA (36). In addition to IFN resistance, E3L is also necessary for the broad-host-range phenotype of VACV, as VACVΔE3L is unable to replicate in HeLa cells (4, 36). Recent reports also showed that E3L inhibits the RNA polymerase III dsDNA-sensing pathway (38). Both functions have also been mapped to the carboxy terminus. The knockdown of PKR expression demonstrated that the host range phenotype of E3L in HeLa cells worked primarily through its ability to inhibit PKR through its dsRBD (40). Although much of the evidence suggests that the inhibition of PKR by E3L is mediated through the sequestration of activator dsRNA molecules, there has been some evidence that PKR is also being inhibited through direct protein-protein interactions with E3L (32, 35).
The function of the amino terminus of E3L is less understood. In cells in culture, this domain is completely dispensable for IFN resistance and replication (11, 36) but is necessary for full pathogenesis in mice (8, 9). Previous animal studies showed that this domain is necessary to spread from nose to brain in intranasal infections (9) and is also necessary for neurovirulence, as the deletion of this domain results in a loss of pathogenicity in intracranial infections (8). Highly conserved among poxviruses, the amino terminus of E3L shares sequence homology to the family of Z-DNA binding proteins and has been shown to bind to Z-DNA in solution (24). Neurovirulence studies of E3L have shown that the replacement of this domain of E3L with a Z-DNA binding domain (ZBD) of distantly related Z-DNA binding proteins, ADAR-1 (adenoisine deaminase acting on RNA-1) or tumor stroma and activated macrophage protein, DLM-1, fully complements pathogenesis. complements pathogenesis. Point mutations that decrease the affinity of Z-DNA binding also correlate with a decrease in neurovirulence in mice (24). Taken together, these results suggest that a functional Z-DNA binding domain of E3L is necessary for full pathogenesis in mice.
Previous studies have suggested a role for the amino terminus of E3L in the PKR pathway. In HeLa cells infected with VACV, this domain is necessary to fully inhibit the actions of PKR (26). In addition, previously reported yeast two-hybrid and λ repressor dimerization studies have shown that the amino terminus of E3L interacts with PKR (32, 35). Although there is mounting evidence that suggests that the amino terminus inhibits PKR, the characterization of this domain at the molecular level has been difficult, as established cell lines have shown that the amino terminus is not necessary for replication or IFN resistance.
In this study, it is demonstrated that the amino terminus of E3 plays a role in inhibiting the IFN response specifically through the actions of PKR. The highly attenuated phenotype associated with VACV expressing E3 deleted of the amino terminus, which includes the Z-DNA binding domain (VACVE3LΔ83N), is completely reversed in mice deleted of the type I IFN receptors (IFN-α/βR−/− mice). Furthermore, this reversal is accompanied by systemic spread and replication in various tissues comparable to those of wild-type VACV (wtVACV). We found that VACVE3LΔ83N is IFN sensitive in primary mouse embryo fibroblasts (MEFs) in culture, thereby granting us a system to characterize its role in the IFN pathway at the molecular level. We demonstrate that the sensitivity is due to a cessation in viral protein synthesis accompanied by the phosphorylation of the translation initiation factor eIF2α. In addition, both PKR−/− MEFs from the same parental strain of mouse and the targeted small interfering RNA (siRNA) knockdown of PKR in MEFs reversed the IFN-sensitive phenotype of VACVE3LΔ83N. Taken together, these data show that the amino terminus of E3L plays a role in inhibiting the IFN response by inhibiting the potent cellular antiviral enzyme PKR.
MATERIALS AND METHODS
Cells and viruses.
Rabbit kidney (RK13) (ATCC) cells were maintained in minimum essential medium (MEM) supplemented with 5% fetal bovine serum (FBS) (HyClone) and 50 μg/ml gentamicin sulfate (Mediatech). IFN-α/βR+/+ (129/SV wild-type) and IFN-α/βR−/− (129/SV background) MEFs were generated from embryos at 13 to 16 days of gestation and maintained in Dulbecco's modified minimum essential medium (D-MEM) supplemented with 10% FBS with 50 μg/ml gentamicin (16). PKR−/− MEFs (129/SV background) were generously provided by Lynda Morrison (16) from Saint Louis University and were maintained in D-MEM with 10% FBS and 50 μg/ml gentamicin. The Western Reserve (WR) strain of wtVACV and virus mutants deleted of the full E3L gene (VACVΔE3L), the N-terminal 83 amino acids of E3L (VACVE3LΔ83N), or the 26 C-terminal amino acids of E3L (VACVE3LΔ26C) were generated as previously described (8, 9). The VACVE3LΔ83N revertant (VACVE3LΔ83N REV) was generated by replacing the mutant E3L gene, E3LΔ83N, with a full-length E3L gene. All virus stocks were generated in BHK cells. Crude lysates were purified by pelleting through a 36% sucrose pad, as previously described (38a). For IFN treatment, cells were treated with 1,000 U/ml of recombinant mouse IFN-α (Calbiochem) 18 h prior to infection, as previously described (26).
Mice.
Wild-type 129/SV (IFN-α/βR+/+) mice were purchased from Charles River Laboratories (Wilmington, MA). IFN-α/βR−/− mice (129/SV background) were also kindly provided by Lynda Morrison (16, 29). PKR−/− mice (C57BL/6 and 129/SV background) were kindly provided by Adolfo Garcia-Sastre (6a) and were housed at the Mount Sinai School of Medicine, New York, NY. IFN-α/βR+/+ and IFN-α/βR−/− mice were bred and housed by the Arizona State University Department of Animal Care and Technologies (DACT) according to university IACUC regulations.
Intranasal and intracranial infections of mice.
Anesthesia and infections were carried out as previously described (9). Briefly, wild-type 129/SV and IFN-α/βR−/− mice were anesthetized with a ketamine cocktail containing 37.5 mg/ml ketamine (Lloyd Laboratories, Shenandoah, IA), 7.5 mg/ml xylazine (Vedco, St. Joseph, MO), and 2.5 mg/ml acepromazine maleate (Fort Dodge Laboratories, Fort Dodge, IA) at the age of 4 to 8 weeks. Intramuscular injections of anesthesia were given at a dose of 1 μl per gram of body weight. Anesthetized mice were infected with 5 μl of virus intranasally by the use of a Pipetman instrument loaded with a protein-loading tip or intracranially with 10 μl of virus delivered by a 27-gauge hypodermic needle and a 1-ml syringe. Mice were monitored and weighed every other day during infection to obtain the 50% lethal dose (LD50) (9, 31). Mice dropping below 30% of their initial weight were euthanized by asphyxiation using CO2.
Tissue distribution.
Tissues were harvested from infected wild-type IFN-α/βR+/+ and IFN-α/βR−/− mice euthanized by asphyxiation with CO2 at 6 days postinfection. Tissues were placed into a cryovial and then quick-frozen in liquid N2. Tissues were brought to a l0% (wt/vol) suspension with MEM containing 2% FBS and supplemented with 50 μg/ml gentamicin sulfate (Mediatech). The samples were homogenized on ice by a mechanical hand homogenizer. Virus was released by three cycles of freeze-thaws, which consisted of freezing samples at −80°C and then slowly thawing the samples on ice, followed by a quick thaw at 37°C and sonication. Virus titers were determined by a standard plaque assay using RK13 monolayers.
Single-step growth curves of MEF cultures.
The indicated cells were infected at a multiplicity of infection (MOI) of 5 PFU/cell. After 1 h, the inoculum was removed, and the cells were rinsed 3 times with prewarmed phosphate-buffered saline (PBS) and then overlaid with D-MEM containing 10% FBS. Triplicate cultures were harvested by scraping them into medium at 1 and 30 h postinfection (hpi). Virus was released by three cycles of freeze-thaws, followed by sonication. Replication was quantitated by a standard plaque assay on RK13 monolayers.
In vivo protein labeling.
Wild-type IFN-α/βR+/+ MEFs infected with virus at an MOI of 5 PFU/cell were washed twice with prewarmed PBS to remove all traces of media and then starved with medium lacking methionine (D-MEM) for 30 min at 37°C. Newly synthesized proteins were radiolabeled with 50 μCi [35S]methionine/ml (800 Ci/mmol) (Perkin-Elmer) for 30 min at 37°C. After labeling, the cells were washed twice with PBS and pelleted by centrifugation at 500 × g. The cells were then lysed with 2.5 volumes of ice-cold NP-40 lysis buffer (20 mM HEPES [pH 7.5], 120 mM KCl, 5 mM Mg acetate, 1 mM dithiothreitol, 10% glycerol, 0.5% NP-40, and 1% protease inhibitor cocktail [Sigma]). The samples were incubated on ice for 15 min, followed by centrifugation at 10,000 × g for 10 min at 4°C to remove the cell debris. The supernatant was transferred into a new tube, and 2× SDS loading dye was added in equal amounts. The samples were boiled for 5 min, and equal protein amounts were separated by denaturing 12% SDS-PAGE. Radiolabeled proteins were visualized by autoradiography.
Western immunoblot analysis.
Cell extracts were obtained by radioimmunoprecipitation assay (RIPA) cell lysis as previously described (26). Briefly, the indicated cells were collected by scraping into medium and pelleted by centrifugation at 500 × g for 10 min at 4°C. The supernatant was removed, and the cells were washed twice with ice-cold PBS. The cells were pelleted by centrifugation at 500 × g for 10 min at 4°C. The cells were lysed with 2.5 volumes of RIPA lysis buffer (1× PBS, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 100 mM NaF, 2 mM Na3VO4, and 1% protease inhibitor cocktail [Sigma]) and placed on ice for 10 min. Samples were centrifuged at 10,000 × g for 10 min at 4°C. SDS protein loading dye (2×) was added in equal amounts to the supernatants, and the samples were then boiled for 5 min. Samples were loaded and separated on a denaturing 12% SDS-PAGE gel and then transferred onto a polyvinylidene difluoride (PVDF) membrane. Following transfer, the membrane was incubated in blocking buffer (140 mM NaCl, 3 mM KCl, 20 mM Tris [pH 7.8], 0.05% Tween 20, 3% nonfat milk) for 1 h at room temperature. Rabbit polyclonal antibodies were used to detect phospho-eIF2α (Cell Signaling Technology), total eIF2α (Cell Signaling Technology), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam), and total mouse PKR (Santa Cruz Biotechnology Inc.). Antibodies conjugated with horseradish peroxidase directed against rabbit immunoglobulin G (Sigma) were used at a 1:15,000 dilution. Immunoreactive bands were visualized by chemiluminescent development using SuperSignal West Dura Duration substrate (Pierce Biotechnology).
Transient siRNA knockdown.
PKR-specific and nontargeting siRNAs were purchased from Dharmacon (Lafayette, CO). The knockdown of PKR expression was targeted by using four siRNAs (On-Target Plus Smart Pool, catalog number L04080700) directed against mouse PKR according to the manufacturer's protocol, whereas nontargeting siRNA (On-Target Plus Non-Targeting siRNA, catalog number D0018101005) served as the negative control. Briefly, IFN-α/βR+/+ MEFs were seeded in triplicate onto 60-mm dishes at 25% confluence in D-MEM–10% FBS without antibiotics 24 h before transfection. siRNA diluted in DharmaFECT 4 reagent was added to the cells, and cells were allowed to incubate for 24 h. After 24 h, the cells were mock treated or treated with 1,000 U/ml of recombinant mouse IFN-α for 18 h, followed by infection.
Statistical analysis.
The statistical significance of the data was determined by using Student's t test or the Mann-Whitney rank sum test using SigmaStat 3.5 statistical software.
RESULTS
Pathogenesis of VACVE3LΔ83N is restored to wtVACV levels in IFN-α/βR−/− mice.
The VACV E3 protein is necessary for pathogenesis in mice. The deletion of this gene results in a highly attenuated virus in mouse studies. The infection of mice with a high dose of VACVΔE3L by either intranasal or intracranial infection led to no mortality and no detectable morbidity in C57BL/6 mice. Additionally, the amino terminus of E3 is necessary for full pathogenesis in mice (9). Intranasal infections with VACV expressing E3 deleted of the amino terminus (VACVE3LΔ83N) results in a virus that is approximately 1,000 times more attenuated in C57BL/6 mice than a wtVACV infection and is unable to spread and replicate in tissues during infection (9). Since E3L is a major contributor to the IFN-resistant phenotype of VACV in cells in culture, we asked if a function for the amino terminus in the context of the IFN pathway could be determined with animal models. Mice genetically deficient for the IFN-α/β receptor (IFN-α/βR−/−), thereby dismantling the signaling and priming aspect of the IFN system, were used to determine a phenotype associated with mutations in E3L. Since the IFN-α/βR−/− mice were of a 129/SV background, we used wild-type 129/SV (IFN-α/βR+/+) mice as an isogenic control. IFN-α/βR+/+ and IFN-α/βR−/− mice were infected intranasally with increasing doses of wtVACV, VACVΔE3L, and VACVE3LΔ83N, and the morbidities and mortalities of the viruses were monitored and compared. In the IFN-α/βR+/+ mice, wtVACV infection resulted in symptoms of disease as early as 4 days postinfection and exhibited a dose-dependent response. Through the progression of the disease, mice exhibited ruffled unkempt fur, lethargy, labored breathing, and weight loss, as seen in previous studies (9). The LD50 for wtVACV infection of IFN-α/βR+/+ mice was determined to be approximately 3 × 104 PFU (Fig. 1A) (31). Both VACVE3LΔ83N and VACVΔE3L were apathogenic in the IFN-α/βR+/+ mice, consistent with previous pathogenesis studies using C57BL/6 mice (9). All mice in these groups survived and showed little to no signs of disease even at doses as high as 1 × 107 and 1 × 108 PFU (Fig. 1A, triangles and open circles, respectively). To confirm that the apathogenic phenotype of VACVE3LΔ83N was mediated through the loss of the amino terminus of E3 and not through second-site mutations elsewhere in the virus genome, a revertant virus (VACVE3LΔ83N REV) was constructed. The construction of this revertant virus consisted of the swapping of the truncated E3 protein for the full-length E3 protein in the VACVE3LΔ83N backbone. This virus regained pathogenesis, as shown in Fig. 1A (closed circles).
Fig 1.

Comparison of pathogenicity between IFN-α/βR+/+ and IFN-α/βR−/− mice infected intranasally (A and C) or intracranially (B and D) with VACV expressing E3L mutants. Four- to six-week old mice were infected intranasally or intracranially with increasing doses of wtVACV (squares), VACVE3LΔ83N (circles), or VACVΔE3L (triangles). Mice were weighed every other day, and the progression of disease was monitored. Mice dropping 30% of their initial weight were euthanized and considered dead (n = 5). WT, wild type.
In the IFN-α/βR−/− mice, wtVACV was approximately 30 times more pathogenic than in the IFN-α/βR+/+ mice (Fig. 1A and C). wtVACV-infected IFN-α/βR−/− mice exhibited symptoms similar to those exhibited by the IFN-α/βR+/+ mice, including weight loss, hunching of the back, and the development of ruffled and unkempt fur. In addition, mice showed more signs of respiratory distress in the form of labored breathing. VACVΔE3L was apathogenic in these mice, with the exception of a mouse that was euthanized after a dose of 1 × 107 PFU for sudden severe weight loss (Fig. 1C, triangles). With the exception of a minor initial weight loss, VACVΔE3L infection did not cause any symptoms of disease. Surprisingly, VACVE3LΔ83N was as pathogenic as wtVACV in the IFN-α/βR−/− mice, with an LD50 of about 2.5 × 103 PFU (Fig. 1C, compare squares and circles). The symptoms were nearly identical to those seen with wtVACV infection, and both viruses showed dose-dependent responses. IFN-α/βR−/− mice infected with wtVACV and VACVE3LΔ83N showed symptoms of disease which included ruffled, unkempt fur; hunching; labored breathing; weight loss; and, in some cases, abdominal swelling. The loss of pathogenicity associated with the deletion of the amino terminus of E3 was regained when the IFN system was incapacitated.
VACVE3LΔ83N is neurovirulent in IFN-α/βR−/− mice.
The WR strain of VACV is a mouse-adapted neurovirulence strain (20, 28). The amino terminus of E3 is necessary for this phenotype in C57BL/6 mice, as the deletion of this domain results in the inability of the virus to replicate in the brain to titers comparable to those of wtVACV, and it has a 1,000-times-higher LD50 (8). To determine if the loss of neurovirulence of VACVE3LΔ83N was related to an inability to inhibit the IFN system, IFN-α/βR+/+ and IFN-α/βR−/− mice were infected intracranially with increasing doses of wtVACV, VACVΔE3L, and VACVE3LΔ83N, and the pathogenesis of the viruses was monitored. In IFN-α/βR+/+ mice, wtVACV was highly pathogenic and had an LD50 of about 5 PFU (Fig. 1B), consistent with what was seen previously (8). These mice also exhibited significant weight loss and neurological disorders, including seizures, piloerection, and hind-limb paralysis. VACVΔE3L, on the other hand, was apathogenic, with mice surviving all doses tested (Fig. 1B). VACVE3LΔ83N was 400 times less pathogenic than wtVACV, with an LD50 of 2 × 103 PFU (Fig. 1B, open circles). For IFN-α/βR−/− mice, a pattern similar to that of the intranasal infection was observed, with wtVACV being 10 times more pathogenic than in the IFN-α/βR+/+ mice, with an LD50 of 0.5 PFU (Fig. 1D, squares). The pathogenicity of VACVE3LΔ83N was restored to the level of wtVACV, with an LD50 of 2 PFU (Fig. 1D, circles). The pathogenicity of VACVΔE3L also increased, with an LD50 of 7 × 102 PFU, when the IFN system was debilitated. However, the level of pathogenesis was 2 logs lower than that of wtVACV or VACVE3LΔ83N. These data demonstrate not only that the amino terminus was necessary for neurovirulence but also that the phenotype was dependent on the amino terminus inhibiting the IFN pathway.
VACVE3LΔ83N is able to spread and replicate to high titers similarly to wtVACV in IFN-α/βR−/− mice.
The data obtained from the intranasal (Fig. 1A and C) and intracranial (Fig. 1B and D) infections demonstrated that the amino terminus of E3 was playing a role in inhibiting the IFN pathway. It is likely that the apathogenic phenotype of VACVE3LΔ83N seen in this study in IFN-α/βR+/+ mice infected intranasally is due primarily to the inability of the virus to disseminate systemically because of the IFN system. A previous study demonstrated that VACVE3LΔ83N was unable to spread from the nose to various tissues in C57BL/6 mice (8). To determine if the stark difference in pathogenesis between VACVE3LΔ83N and wtVACV is due in part to the IFN system preventing spread from the site of inoculation, virus spread and replication were compared in the two systems. IFN-α/βR+/+ and IFN-α/βR−/− mice were infected intranasally with 10 times the LD50 of wtVACV and VACVE3LΔ83N (1 × 105 PFU and 7 × 107 PFU, respectively, in IFN-α/βR+/+ mice and 1 × 104 PFU for both viruses in IFN-α/βR−/− mice). Each cohort of mice was sacrificed at 6 days postinfection, and the lungs, brain, spleen, and nasal turbinates were harvested. wtVACV grew to high titers in the sinus in both strains of mice, whereas VACVE3LΔ83N grew to comparable levels only in IFN-α/βR−/− mice (Fig. 2). In IFN-α/βR+/+ mice, titers of VACVE3LΔ83N in nasal tissue were reduced 30-fold. With the exception of the sinus, VACVE3LΔ83N replicated to low titers in the tissues of IFN-α/βR+/+ mice, which was surprising considering that the amount of input virus was almost 3 logs higher than the input amount for wtVACV, suggesting that the virus was unable to spread from the initial site of infection. Furthermore, all the mice survived and recovered from VACVE3LΔ83N infection by day 8 (data not shown). VACVE3LΔ83N replicated to titers similar to those of wtVACV in IFN-α/βR−/− mice in all tissues, with the exception of the lungs. Surprisingly, the removal of the IFN system resulted in a loss of titers in the lungs for wtVACV. However, VACVE3LΔ83N replicated to titers approximately 1 log higher than those of wtVACV. What can account for this discrepancy is not known; however, the titers in this group of mice were consistent. Taken together, these findings demonstrate that the spread of virus deleted for the amino terminus of E3 to distal sites and replication in these distal sites were restored by disabling the IFN system.
Fig 2.

Virus replication and spread in IFN-α/βR+/+ or IFN-α/βR−/− mice. Mice were infected intranasally with wtVACV or VACVE3LΔ83N at a dose 10 times the LD50. At 6 days postinfection, the indicated tissues were harvested, and the amount of virus was determined. Error bars represent standard errors of the means within each group (n = 5), and asterisks (**, P < 0.01; *, P < 0.05) indicate samples that are significant, as determined by Student's t tests.
Deletion of the amino terminus of E3 results in an IFN-sensitive phenotype in wild-type MEFs.
The E3 protein has been shown to be necessary for the VACV IFN-resistant phenotype in cell lines such as RK13 cells (11), mouse L cells (3), and Huh7 cells (2). The amino terminus is dispensable for IFN sensitivity, as it was demonstrated previously that a functional dsRNA binding domain is sufficient and necessary for IFN resistance in established cell lines (3, 11). Since the amino terminus of E3 was necessary to evade the IFN system in 129 IFN-α/βR+/+ mice, we asked if we could recapitulate this system in primary murine embryonic fibroblast (MEFs) harvested from these mice. To determine if the amino terminus of E3 was necessary to circumvent the effects of IFN, single-step growth curves for wtVACV or VACVE3LΔ83N were performed with primary IFN-α/βR+/+ and IFN-α/βR−/− MEFs mock treated or pretreated with IFN. As shown in Fig. 3, both wtVACV and VACVE3LΔ83N replicated to comparable levels in the IFN-α/βR+/+ MEFs 24 h after infection. However, in IFN-α/βR+/+ MEFs primed with IFN, the ability of VACVE3LΔ83N to replicate significantly diminished compared to that of wtVACV. As expected, both wtVACV and VACVE3LΔ83N were able to grow undeterred in IFN-α/βR−/− MEFs either left untreated or pretreated with IFN (Fig. 3). This demonstrates a role for the amino terminus of E3 in inhibiting the effects of the type I IFN system in MEFs in culture.
Fig 3.

Single-cycle growth kinetics of wtVACV and VACVE3LΔ83N in wild-type MEFs mock treated or pretreated with IFN. IFN-α/βR+/+ or IFN-α/βR−/− MEFs treated with or without 1,000 U/ml of IFN-α were infected in triplicate with wtVACV or VACVE3LΔ83N at an MOI of 5 PFU/cell. At 24 h postinfection, cells were harvested, and viral replication was quantitated by a standard plaque assay on RK13 cells. Error bars represent standard errors of the means, and asterisks indicate samples that are significant (**, P < 0.01), as determined by Student's t test.
Viral protein synthesis is inhibited in IFN-pretreated wild-type MEFs infected with VACVE3LΔ83N, and inhibition is mediated by the phosphorylation of eIF2α.
Translational control during virus infection is a basic antiviral mechanism to inhibit virus replication and subsequent spread. To gain further insight into the mechanism of VACVE3LΔ83N sensitivity to IFN, we compared levels of viral protein synthesis in IFN-α/βR+/+ MEFs pretreated with or without IFN and infected with wtVACV, VACVE3LΔ83N, VACVE3LΔ26C, a VACV expressing E3L deleted of the dsRNA binding domain, and VACVΔE3L. Protein synthesis was determined by the radiolabeling of proteins with [35S]methionine at 6 h postinfection. For both wtVACV and VACVE3LΔ83N, protein synthesis clearly shifted from cellular to viral proteins in the mock-treated cells (Fig. 4A, lanes 2 and 4). Both VACVΔE3L and VACVE3LΔ26C infections resulted in the inhibition of total protein synthesis in both cells with and cells without IFN treatment (Fig. 4A, lanes 3, 5, 8, and 10). In MEFs pretreated with IFN, wtVACV showed some inhibition of protein synthesis, which correlated with the slight sensitivity to IFN seen in the growth curves (Fig. 3). VACVE3LΔ83N exhibited a drastic loss of protein synthesis (Fig. 4A, lane 9), clearly demonstrating the need for the amino terminus to maintain viral protein synthesis in IFN-pretreated MEF cells.
Fig 4.

Protein synthesis shutoff of VACV mutants is mediated by the phosphorylation of eIF2α in wild-type MEFs. (A) IFN-α/βR+/+ MEFs were mock treated (lanes 1 to 5) or pretreated with IFN (lanes 6 to 10) for 18 h and then infected with VACV expressing the indicated E3L mutants (lanes 1 to 10) at an MOI of 5 PFU/cell. At 5.5 hpi, cells were starved and then labeled with [35S]methionine. Cells were harvested, proteins from equal cell volumes were separated on 12% SDS-PAGE gels, and radiolabeled proteins were analyzed by autoradiography. (B) IFN-α/βR+/+ MEFs mock treated or pretreated with IFN-α for 18 h were infected with the indicated viruses at an MOI of 5 PFU/cell. At 6 hpi, cells were harvested, and proteins from equal cell volumes were separated on 12% SDS-PAGE gels and subjected to immunoblotting using antibodies specific for phospho-eIF2α or total eIF2α. Asterisks denote viral proteins.
The E3 protein acts as an antagonist of the IFN pathway by binding to and sequestering dsRNA away from PKR, effectively preventing its activation (3, 26). Activated PKR phosphorylates the alpha subunit of the translation initiation factor eIF2, which prevents the initiation of protein synthesis, effectively shutting off protein synthesis and preventing virus replication (15, 17, 25). Since there was a reduction in the level of protein synthesis in VACVE3LΔ83N-infected IFN-α/βR+/+ MEFs pretreated with IFN (Fig. 4A, lane 9), we asked if this response was mediated by the phosphorylation of eIF2α. At 6 h postinfection, in untreated IFN-α/βR+/+ MEF cells, eIF2α phosphorylation occurred only with VACVΔE3L and VACVE3LΔ26C infections (Fig. 4B, lanes 3 and 5). As expected, both wtVACV and VACVE3LΔ83N infections did not lead to eIF2α phosphorylation (Fig. 4B, lanes 2 and 4), concurrent with a shift from cellular to viral protein synthesis. In cells pretreated with IFN, eIF2α phosphorylation occurred with VACVΔE3L, VACVE3LΔ26C, and VACVE3LΔ83N infections (Fig. 4B, lanes 8 to 10), which strongly correlated with the reduction in the level of protein synthesis seen in these infections. wtVACV infection showed a small amount of phosphorylation of eIF2α (Fig. 4B, lane 7), which was not unexpected considering the minor decrease in the amount of protein synthesis along with the slight sensitivity to IFN seen in the growth curves. Since PKR is a downstream effector of the IFN pathway, which regulates translation by phosphorylating eIF2α, we hypothesized that the amino terminus of E3 is inhibiting the actions of PKR.
VACVE3LΔ83N sensitivity to IFN in wild-type MEFs is restored when the expression of PKR is diminished.
We have demonstrated that the amino terminus of E3 was necessary for the inhibition of the IFN pathway. In addition, this domain was necessary to prevent eIF2α phosphorylation and the subsequent inhibition of protein synthesis in IFN-pretreated cells. To confirm that the inhibitory activity of the amino terminus of E3 was acting on the level of PKR, PKR expression was targeted by siRNA to determine if diminished expression levels of PKR would reverse the IFN-sensitive phenotype of VACVE3LΔ83N. IFN-α/βR+/+ MEFs were transfected with siRNA targeting PKR and then treated with IFN 24 h later. Levels of PKR were significantly reduced in IFN-treated cells pretreated with siRNA targeting PKR (Fig. 5A), whereas the nonspecific siRNA did not affect PKR levels (data not shown). The transient knockdown of PKR expression completely restored the VACVE3LΔ83N IFN-resistant phenotype, leading to titers that were similar to those of wtVACV. Transfection with a nonspecific siRNA (Dharmacon) did not restore the replication of VACVE3LΔ83N in IFN-treated MEFs (Fig. 5B). The effect of the diminished levels of PKR on protein synthesis was also measured. Radiolabeled proteins in MEFs that were pretreated with IFN alone or pretreated with IFN and siRNA targeting PKR were analyzed by autoradiography. As expected, in MEFs pretreated with IFN, VACVE3LΔ83N showed a reduction in the level of viral protein synthesis compared to that in untreated MEFs (Fig. 6, lane 6); however, the sensitivity was reversed when PKR expression was targeted with siRNA (Fig. 6, lane 9).
Fig 5.

siRNA targeting of PKR reverses the sensitivity of VACVE3LΔ83N to IFN in wild-type MEFs. (A) Effective and specific knockdown of PKR in IFN-α/βR+/+ MEFs by siRNA. MEFs were mock transfected or transfected with siRNA against PKR. Twenty-four hours later, MEFs were mock treated or treated with IFN-α. Eighteen hours later, whole-cell extracts were assayed for PKR using antiserum against total PKR. GAPDH served as a loading control. (B) MEFs were transfected with siRNA against PKR or a nonspecific sequence. Twenty-four hours later, the cells were treated with or without IFN-α for 18 h. Cells were infected with the indicated viruses, and virus was harvested 24 h later. Virus titers were determined by a standard plaque assay on RK13 cells. Error bars represent standard errors of the means within each group (n = 3), and asterisks (**, P < 0.05) indicate samples that are significant, as determined by Student's t test.
Fig 6.
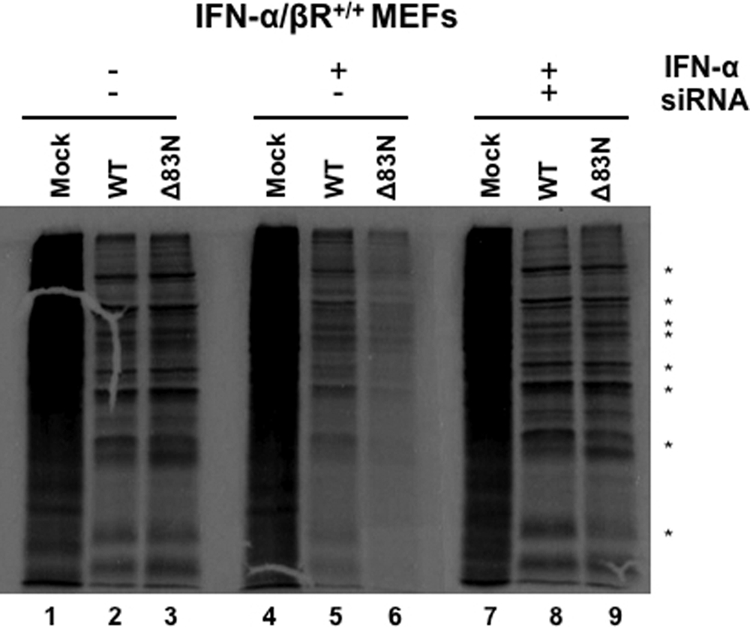
Viral protein synthesis is restored in IFN-pretreated cells infected with VACVE3LΔ83N when PKR expression is diminished. IFN-α/βR+/+ MEFs were mock treated or treated with siRNA against PKR for 24 h. After 24 h, MEFs were mock treated or treated with 1,000 U/ml of IFN-α. Eighteen hours later, the cells were infected with wtVACV or VACVE3LΔ83N at an MOI of 5 PFU/cell, and proteins were radiolabeled after 6 h, as described in the legend of Fig. 4. Extracts were prepared, and proteins were separated on 12% polyacrylamide gels and then visualized by autoradiography.
We also analyzed the replication of VACVE3LΔ83N in a PKR null cell line (PKR−/− MEFs). Similar to the siRNA-mediated PKR knockdown in IFN-α/βR+/+ MEFs, VACVE3LΔ83N replicated to levels indistinguishable from those of wtVACV in IFN-treated PKR−/− MEFs (Fig. 7). Taken together, this strongly suggests that like the carboxy terminus of E3, the amino terminus is necessary to inhibit the antiviral effects of IFN through the actions of PKR.
Fig 7.

The IFN sensitivity of VACVE3LΔ83N is reversed in PKR knockout (PKR−/−) MEFs. IFN-α/βR+/+ and PKR−/− MEFs mock treated or pretreated with IFN were infected at an MOI of 5 PFU/cell with either wtVACV or VACVE3LΔ83N. After 24 h, virus replication was determined by a standard plaque assay on RK13 cells. Error bars represent standard errors of the means within each group (n = 3), and asterisks (**, P < 0.01) indicate samples that are significant, as determined by Student's t tests.
Since the loss of PKR restored the IFN-resistant phenotype of VACVE3LΔ83N in MEFs, we asked if the pathogenesis of VACVE3LΔ83N could be rescued in mice genetically deficient for PKR. PKR−/− mice were infected intranasally with wtVACV and VACVE3LΔ83N. The mice were monitored and weighed over the course of infection. Wild-type VACV infection was mildly pathogenic, with PKR−/− mice displaying signs of illness and with some deaths occurring at the highest dose tested (105 PFU). These mice also exhibited weight loss in a dose-dependent manner (Fig. 8A). VACVE3LΔ83N was not pathogenic in these mice. There was a minor weight loss associated with the highest doses of infection (105 and 106 PFU) (Fig. 8B); however, the mice appeared healthy and remained active during the course of infection.
Fig 8.

VACVE3LΔ83N pathogenicity is not restored in PKR−/− mice. Seven- to eight-week old PKR−/− mice were infected with wtVACV or VACVE3LΔ83N intranasally at various doses. The mice were monitored and weighed every other day. Data show mean changes in body weight after infection (n = 5).
DISCUSSION
In this study, we investigated the role of the amino terminus of E3 in inhibiting the IFN system. Prior to this study, the function of the amino terminus in inhibiting the IFN system has been elusive. Growth kinetic analysis has shown previously that the amino terminus of E3 is dispensable for replication in RK13 cells pretreated with IFN (11). Therefore, the function of this domain in the context of the IFN system had not been established in cells in culture. Pathogenesis studies carried out with mice revealed that the deletion of this domain results in a highly attenuated virus, clearly demonstrating that this domain contributes to the overall function of E3 (8, 9). Since the E3 protein is a major inhibitor of the IFN system, we examined in more detail the function of the amino terminus in an animal model. By using mice devoid of IFN-α/β receptors, we demonstrate that the amino terminus of E3 is necessary to inhibit the IFN response in the mouse model. We show that VACVE3LΔ83N, an otherwise highly attenuated virus, regains its virulence, in both intranasal and intracranial infections, to the same level as that of wtVACV when the IFN system is debilitated (Fig. 1). With the removal of a functional IFN system, VACVE3LΔ83N spreads and replicates to levels comparable to those of wtVACV in all tissues analyzed in mice with an intranasal infection. Conversely, in IFN-α/βR+/+ mice, this virus replicates only to low levels in all the tissues harvested (Fig. 1 and 2). These data suggest that in 129/SV mice, the function of the amino terminus of E3 is to counteract the IFN system.
Extending the study to cells in culture, primary MEFs taken from mice used in the animal experiments provided a platform to characterize the molecular mechanism by which the amino terminus of E3 inhibits the IFN system. In IFN-α/βR+/+ MEFs, VACVE3LΔ83N is sensitive to the effects of IFN. We routinely measured a 2-log reduction in the ability of VACVE3LΔ83N to replicate under these conditions, while wtVACV was fully IFN resistant. This IFN sensitivity is associated with a reduction in levels of viral protein synthesis and with eIF2α phosphorylation. The IFN sensitivity of VACVE3LΔ83N in MEFs was fully reversed by knocking down the expression of PKR or in PKR null cells (Fig. 5 to 7). This finding suggests that in MEFs, a function of the amino terminus of E3 is to inhibit PKR.
Although the amino terminus of E3 has been difficult to characterize in cells in culture in the past, there has been evidence of PKR inhibition associated with this domain. Studies with heterologous yeast systems have shown that inhibition occurs through the direct interaction of the amino terminus of E3 with the kinase domain of PKR. Interaction was suggested previously to occur through an E3-PKR-dsRNA complex (32, 35). The amino terminus is also necessary for full PKR inhibition in HeLa cells and in C57BL/6 mice. VACVE3LΔ83N infection is able to suppress eIF2α phosphorylation at early time points of infection (3 to 6 hpi), but at later times of infection (9 to 12 hpi), eIF2α phosphorylation occurs at levels similar to those for VACVΔE3L infection of HeLa cells. Interestingly, this phosphorylation event does not lead to an inhibition of viral protein synthesis in these cells, and VACVE3LΔ83N is able to replicate similarly to wtVACV. Thus, the biological significance of the phosphorylation event in this system is unknown (26). Although the mechanism of inhibition is not completely understood, immunofluorescence studies have shown a difference in the subcellular distributions of wild-type E3 and E3 deleted of the amino terminus (11). In addition, the level of dsRNA increases during the course of infection, as seen by immunofluorescence (our unpublished data). As the level of dsRNA increases, the N-terminal domain may be necessary to mask excess dsRNA. The change in localization coupled with the inability to mask the overwhelmingly large amounts of unbound dsRNA could explain the activation of the PKR pathway during a VACVE3LΔ83N infection.
A number of previous studies suggested that Z nucleic acid binding plays a role in the IFN pathway. In mammalian cells, the IFN-inducible proteins ADAR and DAI both have ZBDs (6, 18, 23, 34). Recently, DAI was recognized as a cytosolic sensor which recognizes dsDNA and activates the IFN pathway (37, 39). Also, fish encode a PKR-like protein that has ZBDs instead of dsRBDs (21, 33). This protein has been shown to be upregulated by immunostimulation and can phosphorylate eIF2α, strengthening the notion that Z-form nucleic acid binding has a role in the host response (5). The amino terminus of E3 is highly conserved among orthopoxviruses, and E3 shares sequence homology to a family of Z-DNA binding proteins, such as IFN-inducible ADAR-1 and DLM (19, 22, 24). Domain-swapping studies where the ZBD of E3 was interchanged with ZBDs of the distantly related Z-DNA binding proteins ADAR-1 and DLM showed that the virus retains its lethality in intracranial infections (24). Furthermore, point mutations that diminish Z-DNA binding in either the chimera or wild-type E3 decrease the pathogenicity of the virus (24). Currently, we have not tested whether a disruption of Z DNA binding could affect the ability of E3 to inhibit the IFN system. Furthermore, we do not know the role of Z nucleic acid binding in the inhibition of PKR in cells in culture, but it is under investigation. Since ZBD proteins can bind to both Z-form RNA and dsRNA (10, 33), it could be speculated that during the course of infection, as the accumulation of dsRNA increases, the ZBD of E3 is necessary to mask alternative secondary structures of RNA, which can activate the IFN system. This binding/interaction could account for the subcellular localization differences seen between wild-type E3 and E3Δ83N.
In this study, we show that the amino terminus of E3 is necessary to inhibit PKR in primary MEFs. We have shown previously that the amino terminus of E3 is necessary to prevent eIF2α phosphorylation (26). Surprisingly, the pathogenicity of VACVE3LΔ83N was not restored after intranasal infections of PKR−/− mice (Fig. 8). In these mice, VACVE3LΔ83N was apathogenic, suggesting that other IFN-inducible pathways can inhibit the pathogenicity of VACVE3LΔ83N in the whole-animal model, in addition to PKR. This is consistent with the restoration of the pathogenesis of VACVE3LΔ83N when the IFN system was completely debilitated by the removal of the IFN-α/β receptors in mice.
A recent paper (17a) described the modification of the E3 protein by SUMOylation at lysines 40 and 99 and the presence of a SUMO-interacting motif in E3, at residues 119 to 122. One of the two SUMOylation sites described for the E3 protein will be missing in E3Δ83N (lysine 40), while the SUMO-interacting motif should be intact. Since an E3 protein lacking both SUMOylation sites has a largely wild-type phenotype, we do not believe that the deletion of one of the two SUMOylation sites will have an effect on the phenotype of a virus expressing E3Δ83N.
In conclusion, the amino terminus of E3 is necessary for the full inhibition of the IFN response. We show that the ZBD of E3 is necessary to inhibit the IFN pathway in a whole-animal model. Furthermore, we show that the molecular mechanism of inhibition in primary MEFs occurs through the inhibition of a key antiviral enzyme, PKR. Thus, this study, along with previously published data, demonstrates that the VACV E3 protein in its entirety is necessary for full IFN and PKR inhibition.
ACKNOWLEDGMENTS
This work was supported by grant numbers AI066326 and AI052347 from the National Institutes of Health.
We thank Lynda Morrison and Adolfo Garcia-Sastre for providing IFN-α/βR−/− and PKR−/− mice, respectively.
Footnotes
Published ahead of print 14 March 2012
REFERENCES
- 1. Alsharifi M, Mullbacher A, Regner M. 2008. Interferon type I responses in primary and secondary infections. Immunol. Cell Biol. 86:239–245 [DOI] [PubMed] [Google Scholar]
- 2. Arsenio J, Deschambault Y, Cao J. 2008. Antagonizing activity of vaccinia virus E3L against human interferons in Huh7 cells. Virology 377:124–132 [DOI] [PubMed] [Google Scholar]
- 3. Beattie E, et al. 1995. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J. Virol. 69:499–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beattie E, et al. 1996. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 12:89–94 [DOI] [PubMed] [Google Scholar]
- 5. Bergan V, Jagus R, Lauksund S, Kileng O, Robertsen B. 2008. The Atlantic salmon Z-DNA binding protein kinase phosphorylates translation initiation factor 2 alpha and constitutes a unique orthologue to the mammalian dsRNA-activated protein kinase R. FEBS J. 275:184–197 [DOI] [PubMed] [Google Scholar]
- 6. Berger I, et al. 1998. Spectroscopic characterization of a DNA-binding domain, Z alpha, from the editing enzyme, dsRNA adenosine deaminase: evidence for left-handed Z-DNA in the Z alpha-DNA complex. Biochemistry 37:13313–13321 [DOI] [PubMed] [Google Scholar]
- 6a. Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T. 2000. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 74:6203–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borden EC, et al. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6:975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brandt T, et al. 2005. The N-terminal domain of the vaccinia virus E3L-protein is required for neurovirulence, but not induction of a protective immune response. Virology 333:263–270 [DOI] [PubMed] [Google Scholar]
- 9. Brandt TA, Jacobs BL. 2001. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 75:850–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown BA, II, Lowenhaupt K, Wilbert CM, Hanlon EB, Rich A. 2000. The zalpha domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc. Natl. Acad. Sci. U. S. A. 97:13532–13536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang HW, Uribe LH, Jacobs BL. 1995. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J. Virol. 69:6605–6608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clemens MJ. 1997. PKR—a protein kinase regulated by double-stranded RNA. Int. J. Biochem. Cell Biol. 29:945–949 [DOI] [PubMed] [Google Scholar]
- 13. Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:15623–15628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Veer MJ, et al. 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69:912–920 [PubMed] [Google Scholar]
- 15. Dey M, et al. 2005. PKR and GCN2 kinases and guanine nucleotide exchange factor eukaryotic translation initiation factor 2B (eIF2B) recognize overlapping surfaces on eIF2alpha. Mol. Cell. Biol. 25:3063–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duerst RJ, Morrison LA. 2004. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virology 322:158–167 [DOI] [PubMed] [Google Scholar]
- 17. Garcia MA, Meurs EF, Esteban M. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811 [DOI] [PubMed] [Google Scholar]
- 17a. Gonzalez-Santamaria J, Campagna M, Garcia MA, Marcos-Villar L, Gonzalez D, Gallego P, Lopitz-Otsoa F, Guerra S, Rodriguez MS, Esteban M, Rivas C. Regulation of vaccinia virus E3 protein by small ubiquitin-like modifier proteins. J. Virol. 85:12890–12900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herbert A, et al. 1997. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc. Natl. Acad. Sci. U. S. A. 94:8421–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kahmann JD, et al. 2004. The solution structure of the N-terminal domain of E3L shows a tyrosine conformation that may explain its reduced affinity to Z-DNA in vitro. Proc. Natl. Acad. Sci. U. S. A. 101:2712–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaplan C. 1989. Vaccinia virus: a suitable vehicle for recombinant vaccines? Arch. Virol. 106:127–139 [DOI] [PubMed] [Google Scholar]
- 21. Kim D, Hwang HY, Kim YG, Kim KK. 2009. Crystallization and preliminary X-ray crystallographic studies of the Z-DNA-binding domain of a PKR-like kinase (PKZ) in complex with Z-DNA. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65:267–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim YG, Lowenhaupt K, Oh DB, Kim KK, Rich A. 2004. Evidence that vaccinia virulence factor E3L binds to Z-DNA in vivo: implications for development of a therapy for poxvirus infection. Proc. Natl. Acad. Sci. U. S. A. 101:1514–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim YG, Lowenhaupt K, Schwartz T, Rich A. 1999. The interaction between Z-DNA and the Zab domain of double-stranded RNA adenosine deaminase characterized using fusion nucleases. J. Biol. Chem. 274:19081–19086 [DOI] [PubMed] [Google Scholar]
- 24. Kim YG, et al. 2003. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 100:6974–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kimball SR, Mellor H, Flowers KM, Jefferson LS. 1996. Role of translation initiation factor eIF-2B in the regulation of protein synthesis in mammalian cells. Prog. Nucleic Acid Res. Mol. Biol. 54:165–196 [DOI] [PubMed] [Google Scholar]
- 26. Langland JO, Jacobs BL. 2004. Inhibition of PKR by vaccinia virus: role of the N- and C-terminal domains of E3L. Virology 324:419–429 [DOI] [PubMed] [Google Scholar]
- 27. Lu J, O'Hara EB, Trieselmann BA, Romano PR, Dever TE. 1999. The interferon-induced double-stranded RNA-activated protein kinase PKR will phosphorylate serine, threonine, or tyrosine at residue 51 in eukaryotic initiation factor 2alpha. J. Biol. Chem. 274:32198–32203 [DOI] [PubMed] [Google Scholar]
- 28. Ludvikova V, Kutinova L, Simonova V, Otavova M. 1994. Evaluation of various virulence tests with low virulence vaccinia virus in mice. Biologicals 22:187–190 [DOI] [PubMed] [Google Scholar]
- 29. Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 77:9337–9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pain VM. 1996. Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem. 236:747–771 [DOI] [PubMed] [Google Scholar]
- 31. Reed LJ, Muench H. 1938. A simple method for determining fifty percent endpoints. Am. J. Hyg. 1938:493–497 [Google Scholar]
- 32. Romano PR, et al. 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol. Cell. Biol. 18:7304–7316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rothenburg S, et al. 2005. A PKR-like eukaryotic initiation factor 2alpha kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc. Natl. Acad. Sci. U. S. A. 102:1602–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A. 2001. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Struct. Biol. 8:761–765 [DOI] [PubMed] [Google Scholar]
- 35. Sharp TV, et al. 1998. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: implications for PKR autoregulation. Virology 250:302–315 [DOI] [PubMed] [Google Scholar]
- 36. Shors T, et al. 1997. Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology 239:269–276 [DOI] [PubMed] [Google Scholar]
- 37. Takaoka A, et al. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505 [DOI] [PubMed] [Google Scholar]
- 38. Valentine R, Smith GL. 2010. Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J. Gen. Virol. 91:2221–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38a. Vijaysri S, Jentarra G, Heck MC, Mercer AA, McInnes CJ, Jacobs BL. 2008. Vaccinia viruses with mutations in the E3L gene as potential replication-competent, attenuated vaccines: intra-nasal vaccination. Vaccine 26:664–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Z, et al. 2008. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. U. S. A. 105:5477–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang P, Jacobs BL, Samuel CE. 2008. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J. Virol. 82:840–848 [DOI] [PMC free article] [PubMed] [Google Scholar]