

Abstract
Mature vaccinia virus (vaccinia MV) infects a broad range of animals in vivo and cell cultures in vitro; however, the cellular receptors that determine vaccinia MV tropism and entry pathways are poorly characterized. Here, we performed quantitative proteomic analyses of lipid raft-associated proteins upon vaccinia MV entry into HeLa cells. We found that a type II membrane glycoprotein, CD98, is enriched in lipid rafts upon vaccinia MV infection compared to mock-infected HeLa cells. The knockdown of CD98 expression in HeLa cells significantly reduced vaccinia MV entry. Furthermore, CD98 knockout (KO) mouse embryonic fibroblasts (MEFs) also exhibited reduced vaccinia MV infectivity without affecting MV attachment to cells, suggesting a role for CD98 in the postbinding step of virus entry. Further characterization with inhibitors and dominant negative proteins that block different endocytic pathways revealed that vaccinia MV entry into MEFs occurs through a clathrin-independent, caveolin-independent, dynamin-dependent, fluid-phase endocytic pathway, implying that CD98 plays a specific role in the vaccinia MV endocytic pathway. Infections of wild-type and CD98 KO MEF cells with different strains of vaccinia MV provided further evidence that CD98 plays a specific role in MV endocytosis but not in plasma membrane fusion. Finally, different CD98-C69 chimeric proteins were expressed in CD98 KO MEFs, but none were able to reconstitute MV infectivity, suggesting that the overall structure of the CD98 protein is required for vaccinia MV endocytosis.
INTRODUCTION
Vaccinia virus (VV) is the prototype of the orthopoxvirus genus of the Poxviridae, which includes variola virus, the causative agent of smallpox disease. Vaccinia virus is a large, enveloped, double-stranded DNA virus and replicates in the cytoplasm of the host cell. It has a wide host range and infects many cell cultures in vitro and animal species in vivo (39). Virus-infected cells produce multiple forms of the infectious particles, of which the mature virus (MV) constitutes the majority. While most MV particles are released from the cells upon lysis, a proportion of MV acquires additional membranes from the Golgi apparatus and is exocytosed to form the extracellular virus (EV) (15).
Mature vaccinia virus (vaccinia MV) entry pathways and virus-induced signaling are complex in nature and dependent on viral strains (4, 36) and host cell types (16, 36, 50, 62). MV binds to cell surface glycosaminoglycans (GAGs) (13, 25, 35) and the extracellular matrix protein laminin (12) and subsequently enters cells through either endocytosis (24, 54) or plasma membrane fusion (2, 7, 8, 17, 36, 55). Analyses of mutant vaccinia viruses revealed that two envelope proteins, A25 and A26, control virus entry pathway specificity; i.e., MVs containing A25/A26 proteins are endocytosed into HeLa cells, whereas MV particles with no A25/A26 proteins enter cells through plasma membrane fusion (9). The MV endocytic route in HeLa cells was reported previously as dynamin-independent macropinocytosis (38) and as dynamin- and vaccinia virus penetration factor (VPEF)-dependent fluid-phase endocytosis (26). So far, however, the cell surface membrane proteins necessary for virus binding and penetration have not been reported.
The type II membrane glycoprotein CD98, also known as 4F2 or SLC3A2, is expressed in all cell types, with the exception of platelets (49, 60). CD98 constitutes the heavy chain of the family of the heterodimeric amino acid transporters (HATs) (10, 59). So far, six light subunits have been shown to associate with CD98; these subunits include LAT1, LAT2, y+LAT1, y+LAT2, asc1, and xCT, and they confer specific amino acid transport activity to the heteromeric complex (58). In addition, CD98 has been demonstrated to interact with certain integrin β-subunits to promote integrin-dependent signaling, leading to the activation of focal adhesion kinase (FAK), phosphatidylinositol 3-kinase (PI3K), Akt, Rac, and the adhesion molecule p130Cas (20, 22, 63).
Our previous study showed that vaccinia MV clustered at the lipid rafts of the plasma membrane prior to cell entry and that the interruption of lipid raft integrity significantly inhibited MV infection in HeLa cells (14). To gain more insight into the molecular mechanisms of MV entry, we aimed to identify cellular proteins that are enriched in lipid rafts upon vaccinia virus infection by quantitative proteomic analyses. Here, we show that one of the lipid raft-associated proteins, CD98, is important for vaccinia MV infection of host cells. Our results demonstrate that CD98 mediates MV endocytosis in both mouse embryonic fibroblast (MEF) and HeLa cells and that the entire structure of the CD98 protein is required to preserve its functions for mediating MV entry.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Sigma). MEFs derived from CD98 conditional knockout (KO) embryonic stem (ES) cells were described previously (48). MEFs were cultured in DMEM supplemented with 10% fetal bovine serum (HyClone), 0.1 mM nonessential amino acids (Invitrogen), 2 mM l-glutamine (Invitrogen), 1% penicillin-streptomycin (Sigma), 1% HEPES (Invitrogen), and 0.2% 2-mercaptoethanol. The vaccinia virus Western Reserve (WR) strain was used in this study. The IHD-J strain of vaccinia virus and the IA27L virus were obtained from G. L. Smith. The IA27L virus contains an A27L open reading frame (ORF) under the regulation of isopropyl-β-d-thiogalactopyranoside (IPTG); therefore, it was grown in culture medium containing 5 mM IPTG (51). The truncated A26L ORF of the IA27L virus was subsequently replaced by a full-length A26L (WR) ORF to generate a recombinant virus, IA27L-A26LWR, which was also grown in the presence of IPTG, as described previously (12). The IHD-W strain of vaccinia virus was purchased from the ATCC (ATCC 1441-VR). The WR A4-mCherry virus was prepared as described previously (26). WR, IA27L-WRA26, IHD-J, and IHD-W viruses expressing the dual-expression cassette luc-lacZ (inserted at the tk locus) were subsequently generated with the luciferase (luc) gene driven by a viral early promoter and the lacZ gene driven by the p11k late promoter. WRΔA25L and WRΔA26L, expressing a dual luc-gpt cassette containing the luciferase gene driven by a viral early promoter and the gpt gene driven by the p7.5k promoter, were used as described previously (9). The purification of vaccinia MV was performed as previously described (29).
Antibodies, reagents, and plasmids.
Mouse anti-human CD98 antibody (Ab) derived from the hybridoma cell line 4F2(C13) (anti-CD98) was purchased from the ATCC (HB-22). The CD98 antibody was purified from hybridoma culture medium by protein A affinity chromatography. Rabbit anti-CD98 polyclonal antibody (H300), goat anti-cyclophilin B (CypB) polyclonal antibody (C15), and mouse anti-human CD69 monoclonal antibody (MAb) (HP-4B3) were purchased from Santa Cruz Biotechnology. Mouse anti-β-actin antibody was purchased from Sigma (A5441). Mouse anti-green fluorescent protein (GFP) monoclonal antibody (JL8) was obtained from Clontech. Mouse anti-human CD69 antibody (F509), rat anti-integrin β1 antibody (9EG7) (detects integrin β1 of mouse and human origins), rat anti-mouse CD98 antibody, and fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD98 antibody (clone H202-141) were purchased from BD Pharmingen. Mouse monoclonal anti-hemagglutinin (HA) antibody (16B12) was obtained from Covance. Mouse anti-human CD71 (MEM-75) and rabbit anti-Rab5 (GTX13253) antibodies were purchased from Genetex. Rabbit anti-glutathione S-transferase (GST) antibody (G7781) was purchased from Sigma. Rabbit anti-A4 antiserum was raised against a recombinant viral A4 protein purified from bacteria. Rabbit anti-vaccinia virus antiserum was raised against MVs. The mouse monoclonal antibody (clone 2D5) recognizing the L1 protein was obtained from Y. Ichihashi (27). Secondary antibodies (donkey or goat anti-rabbit, anti-rat, or anti-mouse IgG conjugated to FITC, Texas Red, or cyanine 5 [Cy5]) were purchased from Invitrogen Inc. Horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-mouse and anti-rabbit) were purchased from Jackson ImmunoResearch Inc. HRP-conjugated bovine anti-goat antibody was purchased from Santa Cruz Biotechnology. Alexa Fluor 594-, Texas Red-, or FITC-conjugated cholera toxin B (CTB); FITC-conjugated transferrin; and Texas Red- or FITC-conjugated dextran (molecular weight [MW] of 10,000) were purchased from Invitrogen Inc.
The YFP-pReceiver-M15 vector was purchased from Genecopoeia Inc. Plasmids encoding human CD98, C69T98E98, C98T98E69, C69T98E69, and CD69 were provided by Mark H. Ginsberg (University of California, San Diego). These constructs were cloned into the pReceiver-M15 vector to make the above-mentioned chimera ORFs fused with the yellow fluorescent protein (YFP) ORF. The C98T69E98 chimera was constructed in the pReceiver-M15 vector by using the following primers: 5′-GGCTGGGTACGCACCCGCGTTCCTGTCCTGTGTGCTGTAATG-3′ (forward) and 5′-GCGACAACGCGGCGCTCGGCCCACTGATAAGGCAATGAGAG-3′ (reverse) for PCR of the CD69 transmembrane region, 5′-CGTTCGAACCATGAGCCAGGACACCGAGGTG-3′ (forward) and 5′-GCGACAACGCGGCGCTCGGCCCACTGATAAGGCAATGAGAG-3′ (reverse) to add the CD98 cytoplasmic domain, and 5′-CGTTCGAACCATGAGCCAGGACACCGAGGTG-3′ (forward) and 5′-CCCTCGAGTCAGGCCGCGTAGGGGAAGCGG-3′ (reverse) to add the CD98 extracellular domain. The pEGFP-C1 vector was purchased from Clontech Inc. Eps15D95/295 (dominant negative Eps15 [DN-Eps15]) was kindly provided by Alice Dautry-Varsat (Pasteur Institute, Paris, France). Plasmids encoding wild-type dynamin 1 (WT-Dyn1), dynamin 1 with a K44A mutation (DN-Dyn1 [Dyn1K44A]), WT-Dyn2, and DN-Dyn2 (Dyn2K44A) were kindly provided by Sandra Schmid (Scripps Research Institute, CA). Plasmids encoding constitutively active (G12V)Cdc42 [CA(G12V)Cdc42], DN(T17)NCdc42, CA(G12V)Rac1, and DN(T17)NRac1 were obtained from the University of Missouri—Rolla cDNA Resource Center. GFP–DN-caveolin 1 (Cav1D1-81) was constructed as described previously (26). Dynasore was a gift from Tomas Kirchhausen (Harvard Medical School and CBR Institute for Biomedical Research Inc., MA). Chlorpromazine (Cpz) hydrochloride, amiloride, and blebbistatin were purchased from Sigma Inc. Jasplakinolide was purchased from Invitrogen Inc. [14C]l-leucine was purchased from Perkin-Elmer Inc.
Isolation of low-density detergent-insoluble membrane fractions on flotation gradients for differential IMID-H4/IMID-D4 labeling and high-performance liquid chromatography/tandem mass spectrometry (LC/MS/MS) analyses.
Low-density detergent-insoluble membrane microdomains were isolated as described previously, with some modifications described below (14, 47). HeLa cells (108) were mock infected with serum-free medium or infected with VV in serum-free medium at a multiplicity of infection (MOI) of 20 PFU per cell for 30 min at 37°C, washed twice with ice-cold phosphate-buffered saline (PBS) to remove unbound virions, and lysed with 5.0 ml of ice-cold TNE buffer (25 mM Tris [pH 7.5], 150 mM NaCl, 5 mM EDTA) containing 1% Triton X-100 (Merck), 1 mM NaF, and a cocktail of protease inhibitors (Roche). The cells were further incubated at 4°C for 30 min with gentle agitation, and the cell lysates were then centrifuged for 10 min at 3,000 rpm at 4°C in an Eppendorf 5415C centrifuge to remove nuclei and insoluble materials. The precleared supernatants were mixed with equal volumes of 80% (wt/vol) sucrose in TNE buffer and then placed into the bottoms of ultracentrifuge tubes. A discontinuous sucrose gradient was formed by overlaying the homogenates sequentially with 3.9 ml of 30% (wt/vol) sucrose and 3.5 ml of 5% (wt/vol) sucrose in TNE buffer. These mixtures were centrifuged for 18 h at 166,000 × g (38,000 rpm) at 4°C in an SW41 rotor (Beckman). After centrifugation, the Triton X-100-insoluble, low-density material containing lipid rafts was visible as an opaque band migrating at the boundary between the 5 and 30% sucrose solutions. The gradient fractions were collected from the top, and a total of 11 fractions (1.0 ml/fraction) were collected and stored at −80°C. For the detection of the ganglioside GM1, dot blot analyses were performed as described previously (3, 14). The resulting GM1-positive fractions were mixed, diluted in 4 volumes of cold TNE, and centrifuged for an additional 2 h at 166,000 × g at 4°C to pellet the detergent-resistant material.
The pellets from the second centrifugation were solubilized in 90 μl of formic acid (98%; Fluka). Two hundred milligrams of solubilized pellets was incubated with 10 μl CNBr (Fluka) at 25°C overnight. Formic acid was then neutralized with ammonia bicarbonate (ABC; Sigma). Peptides were reduced with 20 mM dithiothreitol (DTT) (Pharmacia) in 6 M urea (Sigma) and 2 M thiourea (Riedel-de Haen) for 1 h at 37°C and alkylated with iodoacetamide (IAA; Sigma) for 1 h at 37°C. Seven volumes of 50 mM ABC were added, followed by digestion with 4 mg trypsin (1:50) (Promega) at 37°C overnight. The reduced tryptic peptides were desalted by C18 reverse-phase traps (Opti-Lynx; Optimize Technologies Inc.), dried, and resolubilized in 200 ml of 2.535 M NH3 (Aldrich) in 50% methanol. The pH was adjusted to 11 with 2.535 M NH3. The lysine residues in mock-infected and infected tryptic peptides were labeled with 12.5 mg of 2-methylthio-2-imidazoline (IMID-H4) (catalog no. 15844-4; Aldrich) and deuterium-labeled IMID-D4 at 40°C overnight, respectively, as previously described (45). The reaction was terminated by the addition 10 ml of 10% trifluoroacetic acid (TFA; Riedel-de Haen) to the mixture. One hundred milligrams of labeled peptides from the light and heavy fractions was mixed and desalted again. The labeled peptides were fractionated by strong cation-exchange (SCX) chromatography (1-mm by 15-cm column). The peptide mixture from 200 μg of detergent-resistant material was fractionated by two-dimensional SCX chromatography–reverse-phase liquid chromatography. The first dimension, SCX, was eluted with a linear gradient of 0 to 300 mM KCl (Riedel-de Haen) in 5 mM ammonium formate (Aldrich) at pH 3.0. The peptides eluted from the SCX column were trapped in the second-dimension reverse phase with two C18 reverse-phase traps operating alternately; the bound peptides were desalted and eluted at 2-min intervals and collected on a fraction collector; a total of 23 fractions were collected. The amount of peptide in each fraction was estimated from the UV absorption at 214 nm.
Mass spectrometric analysis was performed on a nanoscale LC-tandem mass spectrometry (MS) instrument (quadrupole time-of-flight mass spectrometer) (QStar XL; Applied Biosystems). The instrument setup was as follows. The flow (150 μl/min) from the binary pump (Agilent 1100 instrument with solvent A [100% deionized water] and solvent B [90% acetonitrile] [J. T. Baker], with both solvents containing 0.1% formic acid) was split with two T-shaped connectors connected to a self-packed precolumn (C18; 2 cm long with a 150-μm internal diameter) with an appropriate flow restrictor to give column flow rates of 10 μl/min for sample loading and 200 to 300 nl/min for sample elution from the analytical column. A sample of 2 to 5 μg was injected onto the precolumn (15 mm long with a 150-mm internal diameter; C18) via a 20-μl sample loop. The analytical column (C18; 15 cm long with a 75-mm internal diameter) was connected to a 15-mm electrospray emitter (10-mm tip opening) by a 1-cm Teflon sleeve. Chromatographic separation was performed with a 120-min gradient profile as follows: 2% solvent B (0 to 4.5 min) and a linear gradient of 2 to 10% solvent B (4.5 to 5 min), 10 to 40% solvent B (5 to 80 min), 40 to 50% solvent B (80 to 100 min), 50 to 80% solvent B (100 to 105 min), 80 to 2% solvent B (105 to 106 min), and 2% solvent B (106 to 120 min). The mass spectra of the eluted peptides were acquired in the data-dependent mode by first acquiring a full MS scan from m/z 400 to 1,900 for 1 s to determine the three most intense peptide peaks with charge states above 2, and three MS/MS scans between m/z 100 and 2,000 (1.5 s each) were then performed for the MS-scanned parent ions, with a threshold above 20 counts. Once sampled, each MS/MS precursor mass was excluded from further tandem experiments for 2 min. The data files completed from the LC/MS runs were converted into Mascot generic-format files using the Mascot.dll script supplied with the Analyst QS 1.1 software. The Mascot software package (Matrix Science) was used for database searching and protein identification using the Swiss-Prot Human database. The peptide mass tolerance and fragment mass tolerance were set at 100 ppm and 0.25 Da, respectively, for the initial search. An alternative calibration algorithm based on Mascot protein identifications was applied to the raw data file to give mass accuracies within 20 ppm. The ion score cutoff was 25. Quantitation methods were performed manually and by using MSquant.
Immunofluorescence microscopy. (i) Patching analyses.
The patching of cell surface proteins was performed as previously described (26, 52). HeLa cells were seeded onto coverslips in 12-well plates and infected on the next day with vaccinia MV at an MOI of 50 PFU/cell in DMEM for 1 h at 4°C. Cells were washed with PBS, incubated at 37°C for different times, and subsequently incubated for 1 h at 12°C with primary antibodies. Rabbit anti-vaccinia virus (1:500), mouse anti-human CD98 (1:2,000), mouse anti human CD71 (1:250), and Alexa Fluor 594-CTB (10 μg/ml) (Molecular Probes, Invitrogen) antibodies were used. Cells were washed at 4°C and incubated for 1 h at 12°C with secondary antibodies, Cy5-conjugated goat anti-rabbit antibody and FITC-conjugated goat anti-mouse antibody. DNA was visualized by staining with 0.5 μg/ml of DAPI (4′,6-diamidino-2-phenylindole dihydrochloride; Molecular Probes). Cells were washed with PBS at 4°C and fixed for 30 min on ice with 3.7% paraformaldehyde in PBS. Images were collected with an LSM510 Meta confocal laser scanning microscope (Carl Zeiss, Germany) using a 63× objective and confocal microscopy software (Zen 2009; Carl Zeiss). Colocalization analysis was performed by using confocal microscopy software (Zen 2009; Carl Zeiss). The z sections of about 50 cells were analyzed, and the percentage of virion-positive pixels showing colocalization with CD98-positive pixels was quantified.
(ii) Virion binding and uncoating assays.
HeLa cells and MEFs were seeded onto coverslips for 24 h and 48 h, respectively, before the experiment. A virion binding assay measuring the amount of cell surface-bound virions was performed as described previously (56). A virion penetration assay measuring the intracellular uncoated cores, which can be stained only with anti-A4 core Ab after virus entry, was also performed as described previously (55). Cells were infected for 1 h at 4°C with vaccinia MV at MOIs of 20 PFU/cell for the binding assay and 40 PFU/cell for the uncoating assay, washed three times with PBS, and either fixed immediately (for the virion binding assay) or incubated for 2 h at 37°C in the presence of cycloheximide (30 μg/ml) and then fixed (for the virus penetration assay). The cells were permeabilized in PBS containing 0.2% saponin and stained with rabbit anti-A4L antibody or mouse anti-L1R monoclonal antibody, followed by Texas Red-conjugated goat anti-rabbit antibody or FITC-conjugated goat anti-mouse antibody, respectively. DNA was visualized by staining with 0.5 μg/ml of DAPI. Images were collected with an LSM510 Meta confocal laser scanning microscope (Carl Zeiss, Germany) using a 63× objective and confocal microscopy software (Zen 2009; Carl Zeiss). Fluorescent particles from multiple images were counted, and the average numbers of surface-bound virions and uncoated cores per cell were determined as described previously (26).
(iii) Ligand and antibody internalization assays.
HeLa and MEF cells were seeded onto coverslips for 24 h and 48 h, respectively, before the experiment. For antibody internalization assays, cells were incubated for 1 h at 4°C in PBS-AM (PBS, 0.05% bovine serum albumin, 10 mM MgCl2) in the presence of 2.5 μg/ml FITC-conjugated rat anti-mouse CD98 antibody and WR-mCherry MV at an MOI of 20 PFU/cell. Cells were then shifted to 37°C to allow the endocytosis of the antibody and the virus. After internalization, surface antibody was removed by low-pH, acidic washing (0.2 M acetic acid, 0.5 M NaCl [pH 2.5]) prior to fixation. The staining of intracellular phosphatidylinositol-3-phosphate (PI3P) was performed based on an established protocol described previously (23). In brief, after the acid washes, the cells were further incubated for 10 min with 20 μg/ml GST (as a negative control) or GST-FYVE recombinant protein in 0.2% saponin (Sigma) diluted in 1× PBS, washed, and then fixed. Images were collected with an LSM510 Meta confocal laser scanning microscope (Carl Zeiss, Germany) using a 63× objective and confocal microscopy software (Zen 2009; Carl Zeiss). For transferrin, dextran, and CTB uptake analyses, cells were starved in serum-free DMEM for 1 h at 37°C and then incubated for 30 min at 37°C in complete medium containing 25 μg/ml Texas Red-conjugated transferrin (Invitrogen Inc.) as previously described (26). For CTB and dextran uptake assays, cells were incubated for 40 min at 37°C in complete medium containing 5 μg/ml of Alexa Fluor 594-conjugated CTB or 5 mg/ml of FITC-conjugated dextran (MW, 10,000; Invitrogen). Subsequently, cells were washed extensively to remove extracellular-bound ligands and fixed as previously described (26).
Inhibitor experiments and virus entry assays.
For inhibitor experiments, cells were pretreated with each of the inhibitors prior to virus infections as described below. For bafilomycin A (BFLA) blocking experiments, cells were pretreated in complete DMEM containing dimethyl sulfoxide (DMSO) or BFLA (0.5, 1, 5, 10, 25, or 50 nM) for 30 min. For dynasore, chlorpromazine (Cpz), and amiloride blocking experiments, cells were preincubated with DMSO, dynasore (40 μM or 80 μM), Cpz (2.5 μg/ml or 5 μg/ml), or amiloride (0.5 mM, 1 mM, 2.5 mM, or 5 mM) in serum-free DMEM for 1 h (for dynasore, 30 min of incubation) at 37°C. For jasplakinolide and blebbistatin blocking experiments, cells were pretreated in complete DMEM containing 250 nM jasplakinolide, blebbistatin (10 μM or 25 μM), or 250 nM jasplakinolide and blebbistatin (10 μM or 25 μM). These drug-treated cells were subsequently infected with vaccinia MV at an MOI of 5 PFU/cell for early gene luciferase expression assays (54) or cooled to 4°C and infected at an MOI of 40 PFU/cell for 1 h at 4°C for virus core-uncoating assays (55). After washing with PBS, cells were incubated in growth medium for 2 h at 37°C and harvested for luciferase assays (54). Alternatively, cells were incubated in growth medium for 2 h at 37°C in the presence of cycloheximide (30 μg/ml) and then fixed for virus-uncoating assays (55). The drugs were present in the medium throughout the experiments. For cellular ligand uptake experiments, cells pretreated with dynasore and Cpz as described above were subsequently pulsed with fluorescence-conjugated transferrin for 30 min, washed with PBS, and then fixed as previously described (26).
siRNA knockdown.
The control CypB small interfering RNA (siRNA) duplex (Accell SMARTpool PPIB) and the two CD98 siRNA duplexes (Accell SMARTPool SLC3A2) were purchased from Dharmacon Inc. HeLa cells were transfected with siRNA duplexes (50 pM) using Lipofectamine 2000 (Invitrogen), the process was repeated, and the cells were then used for virus binding and penetration assays as previously described (26, 55, 56).
Flow cytometry.
Immunolabeled cells were analyzed with a flow cytometer (FACSCalibur; BD) using CellQuest software (BD). CD98 expression levels on MEFs were measured with rat anti-mouse CD98 (BD Pharmingen) and mouse anti-human CD98 antibodies. Mouse and human integrin β1 was measured with a rat anti-integrin β1 antibody (9EG7; BD Pharmingen).
Cell fusion assays (fusion from without).
Cell fusion assays were performed as described previously (9). MEFs were seeded into 96-well plates for 48 h, pretreated with 40 μg/ml cordycepin (Sigma) for 60 min, and subsequently infected with WR, WRΔA26L, IA27L, or IHD-W MVs at an MOI of 100 PFU/cell in triplicate. After infection for 60 min at 37°C, cells were incubated in complete DMEM supplemented with 40 μg/ml cordycepin and incubated for 2 h at 37°C. Cells were fixed and stained with DAPI (0.5 μg/ml) and the fluorescent lipid dye PKH26 (Sigma) (11). Cell images were collected with an LSM510 Meta confocal scanning microscope (Carl Zeiss) using a 40× objective and confocal microscopy software (Zen 2009; Carl Zeiss).
Amino acid uptake assays.
Amino acid uptake assays were performed as previously described (32). In brief, cells were washed three times with a standard uptake solution (125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 25 mM HEPES, 1.2 mM KH2PO4, and 5.6 mM glucose [pH 7.4]) and incubated for 1 min in uptake solution containing 1 μCi/ml [14C]l-leucine. After being washed three times with ice-cold uptake solution, the cells were solubilized with 0.1 N NaOH, and the radioactivity was measured by using a liquid scintillation counter.
RESULTS
CD98 is enriched in lipid rafts upon vaccinia MV infection.
Because plasma membrane lipid rafts are required for vaccinia virus entry (14), we investigated whether any specific host factors or receptors are enriched in lipid rafts for virus binding and/or penetration. A lysine residue-specific isotope-labeling scheme was utilized to determine the variation of relative abundance before and after vaccinia virus infection. We therefore isolated low-density, detergent-resistant membrane microdomains (DRMs) before and after vaccinia virus infection of HeLa cells, as previously described (3, 14, 47). Proteins were extracted from DRMs; digested to peptides; differentially labeled with 2-methylthio-2-imidazoline (IMID-light [D0]) and deuterium-labeled IMID ((IMID-heavy [D4]), respectively, using established methods described previously (45); and analyzed by multidimensional high-performance liquid chromatography/tandem MS (LC/MS/MS) (Fig. 1A). A total of 717 cellular proteins were identified, and among those, 570 proteins (79%) were quantified (see Table S1A in the supplemental material). The relative abundances of these quantified proteins fluctuated between 0.57- and 1.97-fold, with an average of 1.06-fold ± 0.11-fold (see Fig. S1B in the supplemental material) before and after virus infection, suggesting that the levels of the majority of DRM-associated proteins were not drastically changed after virus infection. In fact, 97% of all quantified proteins fell within the range of the ratio of 1.0 ± 0.5, i.e., a ratio that was considered not significantly changed. Interestingly, among these DRM-associated proteins, the level of CD98/SLC3A2 was increased 1.75-fold ± 0.06-fold after vaccinia virus infection. The quantification of the lipid raft association of CD98 (4F2_human/SLC3A2) in mock- or virus-infected cells was performed by using four unique peptides, GLVLGPIHK, GRLDYLSSLK, VAEDEAEAAAAAK, and DLLLTSSYLSDSGSTGEHTK.
Fig 1.

Vaccinia MV particles colocalize with cell surface CD98 in lipid rafts upon vaccinia MV infection. (A) Flow chart of differential IMID-H4/D4 labeling and LC/MS/MS analyses of lipid raft-associated proteins isolated from HeLa cells that were either mock infected or infected with WR strain MV. Experimental details are described in Materials and Methods, and a list of identified and quantified raft-associated cellular proteins is included in Table S1A in the supplemental material. (B) CD98 localizes at plasma membrane lipid rafts. HeLa cells were copatched with mouse anti-human CD98 antibody (green) and the lipid raft marker CTB (CTB-Alexa Fluor 594) (red) without permeabilization, as described in Materials and Methods. (C and D) WR MV copatched with CD98 on cell surface lipid rafts. WR MV was allowed to bind to HeLa cells for 1 h at 4°C and washed, and the cells were transferred to 37°C for 5 min before copatching with mouse anti-human CD98 (green) and rabbit anti-vaccinia virus (cyan) antibodies and the lipid raft marker CTB (red) (C) or with mouse anti-human CD98 (red) or mouse anti-human CD71 (red) and rabbit anti-vaccinia virus (green) antibodies without permeabilization (D). (E) Quantification of the WR MV particle colocalization with CD98 shown in panel D. The data were quantified by calculating the fraction of the total number of WR MV-positive pixels colocalizing with CD98- or CD71-positive pixels per cell. A total of ∼50 cells were quantified, and the statistical analyses were performed by using Student's t test. The P value is shown, where *** indicates a P value of <0.0001.
Following the proteomic analyses, we performed copatching of lipid raft experiments by staining HeLa cell surface CD98 and found a colocalization of CD98 with the lipid raft marker cholera toxin B (CTB) (Fig. 1B), confirming CD98 as a DRM-associated membrane protein. Furthermore, when HeLa cells were infected with vaccinia virus Western Reserve (WR) at 37°C for 5 min, a colocalization of vaccinia MV with CD98 and CTB was detected (Fig. 1C). As a control, we performed a copatching analysis using another receptor, the transferrin receptor (CD71) (Fig. 1D). After quantifying WR MV colocalization with CD98 or CD71, we confirmed that a significant amount of MV colocalized with CD98 compared to CD71 (Fig. 1E). Altogether, these results imply a role for CD98 in vaccinia virus infection.
CD98 depletion in HeLa cells reduces vaccinia MV WR endocytosis.
To investigate whether CD98 is important for MV entry, we knocked down its expression in HeLa cells using siRNA (si-CD98). HeLa cells either mock treated or treated with siRNA knocking down cyclophilin B expression (si-CybB) were used as controls (Fig. 2A). We then tested these knockdown cells for the infectivity of the wild-type WR virus and a recombinant WR virus, IA27L (12, 51), because the former enters HeLa cells via endocytosis, whereas the latter fuses with the plasma membrane (9). HeLa cells were infected at an MOI of 5 PFU/cell and harvested at 2 h postinfection (p.i.) for early gene luciferase expression assays (Fig. 2B). The results suggested that the depletion of CD98, but not CypB, reduced vaccinia virus WR infections to 50%, whereas it had no effect on IA27L infectivity. To investigate whether CD98 is important for the WR virus entry step, we performed MV binding assays, which determine the amount of cell-bound MV particles prior to membrane fusion (56), and virus core-uncoating assays, which measure the number of viral cores after virus membrane fusion (55). As shown in Fig. 2C, the knockdown of CD98 had no effect on WR or IA27L MV binding to HeLa cells (Fig. 2C), demonstrating that CD98 is not involved in the attachment of either virus to HeLa cells. On the other hand, the CD98 knockdown significantly reduced the intracellular viral core numbers of the WR virus but not the IA27L virus (Fig. 2D; quantified in Fig. 2E). Taken together, our results suggest that CD98 is specifically required for the endocytic route of vaccinia MV into HeLa cells.
Fig 2.

CD98 depletion in HeLa cells reduces mature vaccinia virus WR endocytosis. (A) Immunoblots of HeLa cell lysates prepared from mock-treated cells (Mock) or cells transfected with CypB siRNA (si-CypB) or CD98 siRNA (si-CD98) with antibodies against CD98, cyclophilin B, or β-actin (control). (B) Early gene luciferase expression assays in HeLa cells. Mock-, si-CypB-, and si-CD98-treated HeLa cells were infected with WR or IA27L MV at an MOI of 5 PFU/cell for 60 min and harvested at 2 h p.i. for early gene luciferase expression assays. Values for mock-treated HeLa cells were set as 100%. (C) Quantitative results of virion binding assays. Mock-, si-CypB-, and si-CD98-treated HeLa cells were infected for 60 min at 4°C with WR or IA27L MV at an MOI of 20 PFU/cell, washed, and subsequently fixed with 4% paraformaldehyde. The bound particles were stained by using anti-L1R MAb 2D5. Bound virions of 40 cells were counted in each experiment to obtain an average number of bound virions/cell. The number of bound virions obtained from mock-siRNA-treated cells (control) was set as 100%. The experiment was performed three times, and the standard deviations are shown. (D) Immunofluorescence microscopy of virus-uncoating assays with mock-, si-CypB-, and si-CD98-treated HeLa cells infected with WR or IA27L MV. The core was stained by using an anti-A4 antibody (red), and the nuclei were stained with DAPI (blue). (E) Quantitative results of the virus-uncoating assays shown in panel D. Viral core numbers in 40 cells were determined to obtain an average number of viral cores/cell. The viral core number obtained from mock-treated HeLa cells was used as the 100% control. The experiments were performed three times, and the standard deviations are shown.
Vaccinia virus WR enters MEFs via a CD98-dependent fluid-phase endocytic pathway, while the IA27L virus enters MEFs via CD98-independent plasma membrane fusion.
In order to confirm our above-described observations with HeLa cells, we next obtained MEFs derived from CD98 conditional knockout (KO) mouse embryos that expressed either an endogenous level of CD98 (CD98+/+) or no CD98 (CD98−/−) (48). Before we addressed the role of CD98 in vaccinia virus entry, we first characterized vaccinia MV WR and IA27L entry pathways in MEFs to ensure that there was no cell type-specific difference between HeLa and MEF cells. Bafilomycin treatment of MEFs at a range of 5 to 50 nM inhibited WR MV entry but not IA27L virus (Fig. 3A), suggesting that WR MV, but not IA27L MV, enters MEF cells through low-pH-dependent endocytosis. On the contrary, IA27L MV, but not WR, entered MEFs through plasma membrane fusion, resulting in robust cell fusion from without at a neutral pH (Fig. 3B). To further dissect the WR MV endocytic pathway in MEF cells, we used different inhibitors and dominant negative (DN) constructs that specifically block individual endocytic pathways. The results revealed that, similar to HeLa cells, vaccinia MV WR is endocytosed into MEFs through a clathrin-independent, dynamin-dependent (Fig. 4A), and caveolin-independent (Fig. 4B) endocytic pathway. Control experiments showed that transferrin uptake into MEFs was reduced by inhibitors such as chlorpromazine (Cpz), dynasore, DN-dynamins 1 and 2, and DN-Eps15, all of which inhibited clathrin-mediated endocytosis (Fig. 4C), and that CTB uptake through caveolae was blocked by DN-Cav1 (Fig. 4D). Finally, mature vaccinia virus WR endocytosis into MEFs was sensitive to the inhibitors of actin dynamics (Fig. 5A), GDP-bound Rac and CDC42 mutants (Fig. 5B), and the macropinocytosis inhibitor amiloride (Fig. 5C). Overall, the results demonstrated that vaccinia MV WR enters MEFs in a dynamin-dependent fluid-phase endocytic manner, whereas IA27L MV entry was through plasma membrane fusion (results are summarized in Table 1).
Fig 3.
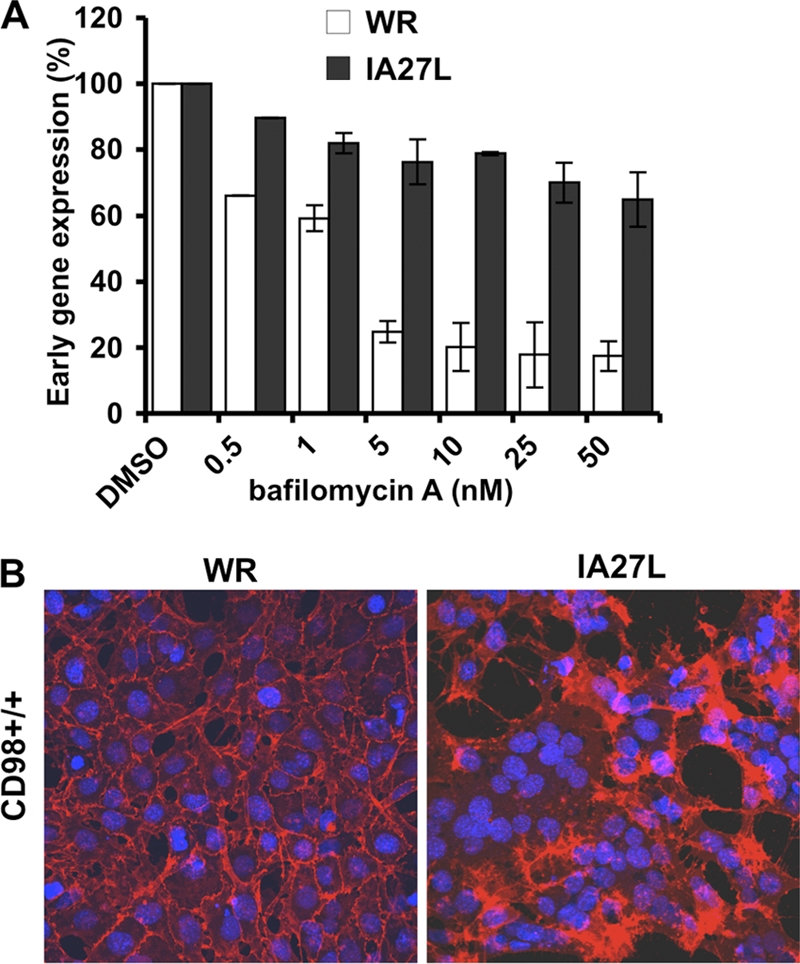
Mature vaccinia virus WR enters CD98+/+ MEFs through endocytosis. (A) Bafilomycin A (BFLA) sensitivity of WR and IA27L MVs in MEFs. CD98+/+ MEFs were pretreated with DMSO or BFLA for 30 min and subsequently infected with WR or IA27L MV at an MOI of 5 PFU/cell for 60 min at 4°C. Cells were harvested and assayed for early luciferase gene expression at 2 h p.i. Luciferase activities obtained from MEFs treated with DMSO were used as the 100% control. (B) IA27L virus but not WR triggers fusion from without at a neutral pH. CD98+/+ MEFs were infected with WR or IA27L virus at an MOI of 100 PFU/cell for 60 min at 37°C at a neutral pH. At 2 h p.i., cells were fixed and stained with DAPI and a fluorescent lipid dye (PKH26).
Fig 4.

Mature vaccinia virus WR entry in MEFs is independent of clathrin- and caveolin-mediated endocytosis but relies on dynamin GTPase activity. (A) Vaccinia MV-uncoating assays with HeLa and MEF cells pretreated with inhibitors of clathrin-mediated endocytosis and dynamin GTPase activity. HeLa cells and MEFs were transfected with plasmids for the expression of GFP, dominant negative Eps15 (DN-Eps15), DN-Dyn1, and DN-Dyn2 or pretreated with DMSO, chlorpromazine (Cpz), or dynasore and infected with WR at an MOI of 40 PFU/cell, and virus core numbers in cells were determined as described in Materials and Methods. For each group, viral cores from at least 40 cells were counted to obtain an average number of viral cores/cell; the core numbers obtained from DMSO-treated cells (for drug treatments) and GFP-transfected cells (for transfections with DN constructs) were used as 100% controls. (B). Vaccinia MV uncoating in HeLa and MEF cells transfected with DN-caveolin 1. HeLa cells and MEFs were transfected with plasmids expressing GFP or GFP–DN-caveolin 1 (DN-Cav1), infected with WR MV at an MOI of 40 PFU/cell, and harvested for virus-uncoating assays as described above for panel A. (C) Transferrin uptake in HeLa and MEF cells pretreated with inhibitors or plasmid transfections as described above for panel A. The treated cells were subsequently pulsed with fluorescent transferrin for 30 min, washed, fixed, and analyzed by confocal microscopy to quantify the uptake of fluorescent transferrin ligand. (D) Cholera toxin B (CTB) uptake in transfected HeLa and MEF cells, as described above for panel B. The transfected cells were pulsed with fluorescent CTB for 40 min, washed, fixed, and analyzed by confocal microscopy to quantify the uptake of fluorescent CTB ligand. All the experiments were repeated three times, and the standard deviations are shown.
Fig 5.
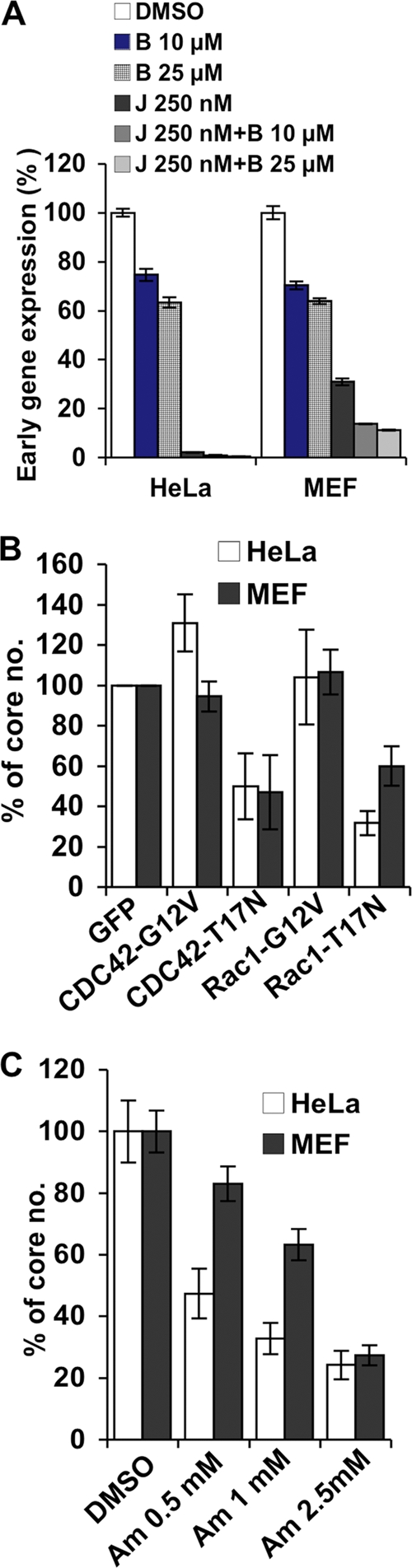
Mature vaccinia virus WR enters MEFs through macropinocytosis. (A) Myosin inhibition and blockage of actin dynamics. HeLa cells and MEFs were pretreated with DMSO, blebbistatin (B), jasplakinolide (J), or blebbistatin along with jasplakinolide (J + B); infected with WR MV at an MOI of 5 PFU/cell for 60 min; and harvested at 2 h p.i. for early gene luciferase expression assays. Luciferase activities obtained from the DMSO-treated cells were used as the 100% control. (B) Small GTPase inhibition of vaccinia virus core uncoating. HeLa cells and MEFs cells were transfected with plasmids for the expression of GFP and the GTP-bound forms (G12V) and GDP-bound forms (T17N) of Rac1 and Cdc42. Cells were infected with WR MV at an MOI of 40 PFU/cell for virus core-uncoating assays. For each group, viral cores from at least 40 cells were counted to obtain an average number of viral cores/cell; viral core numbers obtained from GFP-transfected cells were used as the 100% control. (C) Inhibition of macropinocytosis upon vaccinia virus core uncoating. HeLa cells and MEFs were treated with various concentrations of amiloride (Am) and subsequently infected with WR MV for virus core-uncoating assays as described above for panel B. Viral core numbers obtained from DMSO-treated cells were used as the 100% control. All the experiments were repeated three times, and the standard deviations are shown.
Table 1.
Comparison of WR entry pathways between HeLa cells and MEFs with different inhibitor treatments
| Treatment and inhibitor | Resulta |
|
|---|---|---|
| HeLa cells | MEFs | |
| Actin dynamics | ||
| Jasplakinolide + blebbistatin | S | S |
| Rac1-DN | S | S |
| Cdc42-DN | S | S |
| Dynamin | ||
| Dynasore | S | S |
| DN-dynamin 1 | S | S |
| DN-dynamin 2 | S | S |
| Clathrin-mediated treatment | ||
| Chloropromazine | R | R |
| DN-Eps15 | R | R |
| Fluid phase | ||
| Amiloride | S | S |
| Caveolae | ||
| DN-caveolin 1 | R | R |
S, sensitive; R, resistant.
We next compared MV entry into wild-type (CD98+/+) and CD98 KO (CD98−/−) MEF cells. In addition, plasmid constructs expressing yellow fluorescent protein (YFP) alone or YFP fused with CD98 were individually transfected into CD98−/− MEFs to generate two reconstituted cell lines stably expressing either YFP (CD98−/− YFP) or the YFP-CD98 protein (CD98−/− YFP-CD98) (Fig. 6A). We then performed infections with vaccinia MV WR and IA27L on all four MEF cell lines as described above and harvested cells to monitor virus entry. Binding of mature vaccinia virus WR and IA27L to all four MEF cell lines was comparable (Fig. 6B), whereas viral core uncoating (Fig. 6C) and subsequent early luciferase gene expression levels (Fig. 6D) of WR MV, but not IA27L MV, were reduced by 60% in CD98−/− MEFs. The reconstituted expression of YFP-CD98, but not YFP, in CD98−/− MEFs restored WR MV infectivity (Fig. 6C and D). Imaging analyses consistently showed a reduction in the number of viral cores in CD98−/− MEF cells infected with WR (Fig. 6E) and a specific rescue of WR infectivity by the overexpression of YFP-CD98 (Fig. 6F). Taken together, our results showed that CD98 is important for vaccinia MV WR but not for IA27L entry into MEFs.
Fig 6.

CD98 is important for mature vaccinia virus WR entry into mouse embryonic fibroblasts. (A) CD98 expression in CD98+/+ and CD98−/− MEFs. CD98+/+, CD98−/−, and CD98−/− MEFs stably expressing YFP (CD98−/− YFP) or YFP-CD98 (CD98−/− YFP-CD98) were stained with rat anti-mouse CD98 (for CD98+/+ and CD98−/− cells) or mouse anti-human CD98 (for CD98−/− YFP and CD98−/− YFP-CD98 cells) antibodies and analyzed by FACS analysis. (B) Vaccinia MV binding assays with MEF cells. The above-mentioned MEFs were infected with WR or IA27L MV at an MOI of 20 PFU/cell at 4°C for 60 min, washed, and subsequently fixed with 4% paraformaldehyde. The bound particles were stained by using an anti-L1R antibody (2D5). For each group, viral cores from at least 40 cells were counted to obtain an average number of viral cores/cell; the number of bound virions obtained from CD98+/+ MEFs was used as the 100% control. The experiments were performed three times, and the standard deviations are shown. (C) Vaccinia virus-uncoating assays with MEF cells. The above-mentioned MEFs were infected with WR or IA27L MV at an MOI of 40 PFU/cell, and viral core numbers were determined by staining with an anti-A4 antibody. For each group, viral cores from at least 40 cells were counted to obtain an average number of viral cores/cell; the viral core number obtained from CD98+/+ MEF cells was used as the 100% control. The experiments were performed three times, and the standard deviations are shown. (D) Early gene luciferase expression assays with MEF cells. MEF cells were infected with WR or IA27L virus at an MOI of 5 PFU/cell for 60 min and harvested at 2 h p.i. for early gene luciferase expression assays. Luciferase activities in CD98+/+ MEFs were used as the 100% control. The experiments were performed three times, and the standard deviations are shown. (E) Immunofluorescence microscopy of virus-uncoating assays of CD98+/+ and CD98−/− MEF cells infected with WR or IA27L MV. The core was stained by using an anti-A4 antibody (red). (F) Immunofluorescence microscopy of virus-uncoating assays of CD98−/− MEF cells expressing YFP-CD98 or YFP alone (green). MEF cells were infected with WR MV at an MOI of 40 PFU/cell, and viral cores were stained with an anti-A4 antibody (red) as described above for panel E.
Different strains of vaccinia MV require CD98 during endocytic entry.
Since vaccinia virus entry was shown to be strain specific, we wanted to corroborate this conclusion by including several vaccinia virus strains and deletion mutants whose entry pathways into HeLa cells have been well characterized (9). For example, wild-type WR, the IHD-J strain, and IA27L-A26WR, a recombinant IA27L virus expressing the full-length A26 protein, use the endocytic entry route, whereas the IA27L strain, IHD-W, and a deletion mutant virus, WRΔA26L, enter HeLa cells through plasma membrane fusion (9). We thus infected CD98+/+ and CD98−/− MEFs with each MV at an MOI of 5 PFU/cell and performed early gene luciferase expression assays at 2 h p.i. As shown in Fig. 7A, all the endocytic MVs (WR, IHD-J, and IA27L-A26WR) had a reduced infectivity in CD98−/− MEFs compared to CD98+/+ MEFs. Furthermore, the reduced infectivity of the above-described viruses, such as WR and IA27L-WRA26, in CD98−/− MEFs was not due to a delay in the kinetics of MV entry, as a longer exposure of viruses with the target cells did not increase virus infectivity in CD98−/− cells (Fig. 7B). In contrast, the MVs targeting the plasma membrane fusion route, such as the IA27L and WRΔA26L viruses, showed comparable infectivities of both CD98+/+ and CD98−/− cells (Fig. 7C). Consistently, the latter viruses triggered robust cell fusion from without at a neutral pH on both CD98+/+ and CD98−/− MEFs (Fig. 7D), demonstrating that CD98 is dispensable for vaccinia virus-induced plasma membrane fusion. These results show that vaccinia MV strains, which enter cells through endocytosis, not plasma membrane fusion, require CD98 for cell entry.
Fig 7.

Vaccinia MV strains that enter cells through endocytosis require CD98. (A) Infectivities of WR, IHD-J, IA27L-A26WR, IA27L, IHD-W, and WRΔA26L in CD98+/+ versus CD98−/− MEFs. Pairwise MEFs were infected with various viruses at an MOI of 5 PFU/cell for 60 min and harvested at 2 h p.i. for early gene luciferase expression assays. Luciferase activities obtained from the CD98+/+ MEFs were used as the 100% control for each virus strain tested. (B) Time courses of WR, IA27L-A26WR, IA27L, and WRΔA26L infectivity in CD98+/+ versus CD98−/− MEFs. Pairwise MEFs were infected with various viruses at an MOI of 10 PFU/cell for 60 min and harvested at 30 min, 60 min, 90 min, and 120 min p.i. for early gene luciferase expression assays. Luciferase activities obtained from the CD98+/+ MEFs were used as the respective 100% controls for each virus strain tested. (C) Time course of IA27L and WRΔA26L virus infectivity in CD98+/+ versus CD98−/− MEFs. Pairwise MEFs were infected with various viruses and harvested for early gene luciferase expression assays as described above for panel B. (D) Fluorescence microscopy of cell fusion from without on CD98+/+ and CD98−/− MEFs triggered by IA27L, IHD-W, and WRΔA26L viruses at a neutral pH. Pairwise MEFs were infected with IA27L, IHD-W, or WRΔA26L virus at an MOI of 100 PFU/cell at 37°C for 60 min at a neutral pH, fixed at 2 h p.i., and stained with DAPI and a fluorescent lipid dye (PKH26).
Vaccinia MV WR localizes in endocytic structures in cells that are positive for CD98.
If CD98 is required for vaccinia virus endocytosis, it is anticipated that vaccinia MV is internalized into CD98-positive intracellular vesicles. One approach to investigate this issue is to use an anti-CD98 MAb to track the internalization of CD98 from the plasma membrane into intracellular vesicles (19). This method has been widely used and shown not to alter the ligand binding and intracellular trafficking of the targeted plasma membrane proteins (19). Accordingly, wild-type MEFs were incubated with an FITC-conjugated anti-CD98 MAb together with fluorescent vaccinia MV WR expressing the viral core protein A4 fused with mCherry for 1 h at 4°C to allow the virus and the antibody to bind the cells. These cells were then incubated at 37°C for 5, 15, 30, or 45 min to allow internalization; washed extensively with acidic buffer to remove any extracellular-bound antibody; and fixed for confocal microscopy analyses. Individual z sections of images of cells at the different time points are shown in Fig. 8. At a time of 0 min, all surface-bound FITC-conjugated anti-CD98 MAbs were not internalized and thus were effectively removed by washing prior to fixation (Fig. 8A). Cells fixed at 5 min and 15 min contained detectable acid-resistant FITC fluorescence, mostly at the cell periphery, suggesting that an initial internalization of surface CD98 occurred (Fig. 8B and C). Furthermore, vaccinia MVs were found within internalized CD98-positive vesicles as soon as 5 min after the temperature shift to 37°C (Fig. 8, arrowheads). Images of cells at 15, 30, and 45 min after the temperature shift showed that vaccinia MV was located within CD98-positive vesicles that already trafficked away from the cell periphery (Fig. 8D and E). The majority of infected cells (≥80%) contained MV particles located within CD98-positive vesicles as early as 5 min after a temperature shift to 37°C (Fig. 8F). Furthermore, 40 to 50% of internalized MV particles in cells were localized inside CD98-positive vesicles at 5 and 15 min (Fig. 8G), demonstrating that CD98 and vaccinia MV WR cointernalization occurred very quickly after the temperature shift.
Fig 8.

Mature vaccinia virus WR localizes within endocytic structures that are positive for CD98 in cells. (A to E) Imaging of WR MV kinetics of internalization into CD98-positive vesicles. CD98+/+ MEFs were incubated for 60 min at 4°C with FITC-conjugated anti-CD98 antibody (green) and WR-A4-mCherry (red) at an MOI of 20 PFU/cell. Cells were shifted to 37°C for 0 min (A), 5 min (B), 15 min (C), 30 min (D), or 45 min (E) and individually washed with acidic buffer to remove surface-bound antibody prior to fixation. The confocal images in each panel are enlarged views of the areas outlined by the white squares. Arrowheads point to the CD98-positive vesicles containing WR MV. (F) Percentages of MEF cells showing WR MV localizing in CD98-positive vesicles. For each time point, z sections of 60 cells were analyzed, and the standard deviations are shown. (G) Percentages of WR MV particles per cell that localize within CD98-positive vesicles. The numbers of WR MV particles localizing in CD98-positive structures per cell and total WR MV particles per cell were quantified. For each time point, z sections of 60 cells were analyzed, and the standard deviations are shown.
To characterize the identity of the above-described CD98-positive vesicles that contain WR MV, we performed antibody internalization assays in the presence of WR MV and immunostained for various cellular endocytic markers after fixation. We observed a colocalization of CD98-positive vesicles containing WR MV with phosphoinositol-3-phosphate (PI3P), a marker for macropinosomes (31) (Fig. 9A, arrowheads), and the early endosome marker Rab5 (Fig. 9B, arrowheads). These results show that WR MV and CD98 are internalized into PI3P-positive macropinosomes, which subsequently fused with early endosomes.
Fig 9.

The CD98-positive endocytic structures are positive for phosphoinositol-3-phosphate (PI3P) and Rab5. (A) Colocalization of vaccinia MV-containing CD98+ vesicles with PI3P. CD98+/+ MEFs were incubated for 60 min at 4°C with FITC-conjugated anti-CD98 antibody (green) and WR-A4-mCherry (red) at an MOI of 20 PFU/cell; subsequently shifted to 37°C for 15 min; and washed with acidic buffer to remove the surface-bound antibody. The cells were fixed and stained for intracellular PI3P-containing macropinosomes (cyan) as described in Materials and Methods. Arrowheads point to the MV-containing CD98+ vesicles that are positive for PI3P. The large insets in each panel are enlarged views of the areas outlined by the small squares. (B) Colocalization of vaccinia MV-containing CD98+ vesicles with an endosome marker, Rab5. CD98+/+ MEFs were treated with an anti-CD98 antibody (green) and WR-A4-mCherry (red) as described above for panel A. The cells were fixed, permeabilized, and stained for an endosome marker, Rab5 (cyan). Arrowheads point to the MV-containing CD98+ vesicles that are positive for Rab5.
Expression of CD98-CD69 chimeras in CD98−/− MEFs reveals that full-length CD98 is required to facilitate WR MV endocytosis.
The above-described results prompted us to investigate whether the cellular function of CD98 is important for vaccinia MV endocytosis. CD98 is known to associate with light chains of amino acid transporters and modulates amino acid transport into cells (5, 53). CD98 also associates with integrin β1 and regulates integrin downstream Akt signaling (20, 22). It was previously shown that the amino acid transporter activity of CD98 depends on its extracellular domain, whereas the integrin signaling activity requires transmembrane and cytoplasmic domains of CD98 (21).
A series of four chimeric constructs was described previously (21), in which domains of CD98 were replaced with portions of another type II transmembrane protein, CD69. These four chimeric CD69-CD98 constructs (Fig. 10A) were fused with YFP and transfected individually into CD98−/− MEFs, and the resulting stable cell lines expressing each of the chimeric YFP fusion proteins at comparable levels were confirmed by both fluorescence-activated cell sorter (FACS) analysis (Fig. 10B) and immunoblotting (Fig. 10C). These cells were subsequently infected with WR MV and harvested at 2 h p.i. for early gene luciferase assays (Fig. 10D) and virus core-uncoating assays (Fig. 10E), as described above. The results showed that while the ectopic expression of full-length YFP-CD98 reconstituted WR MV infectivity to about 80%, all of the CD69-CD98 chimeric proteins did not significantly increase virus infectivity compared to that with control YFP or YFP-CD69 (Fig. 10D and E). We observed only a moderate increase in infectivity with three chimeras (C69T98E98, C98T69E98, and C98T98E69), which did not share a specific domain structure. Thus, the results suggest that the entire CD98 molecule is required to facilitate WR MV endocytosis.
Fig 10.

The overall structure of CD98 is important for vaccinia MV endocytosis in MEF cells. (A) Schematic drawings of the chimeric constructs between CD98 (white) and CD69 (blue) and the anticipated antibody recognition patterns. Cytop, cytoplasmic domain; TM, transmembrane domain; Extracell, extracellular domain. (B) Cell surface expression of the CD98-CD69 chimera proteins in CD98 knockout MEFs. CD98−/− MEFs stably expressing each of the CD98-CD69 chimera proteins were stained with mouse anti-human CD98 (CD98, C69T98E98, and C98T69E98) or mouse anti-human CD69 (C69T98E69, C98T98E69, and CD69) antibodies and analyzed by FACS analysis. (C) Immunoblot analysis of the YFP fusion proteins that were fused with CD98-CCD69 chimera constructs and expressed in CD98−/− MEFs. The blot was probed with a mouse anti-GFP antibody to detect YFP expression. Mock represents the CD98−/− MEFs without transfection. The asterisks mark the expected sizes of each chimera construct. (D) Expression of the CD98-CD69 chimera proteins in CD98 knockout MEFs did not increase viral early gene luciferase expression. The above-mentioned cells were infected with WR MV at an MOI of 5 PFU/cell for 60 min and harvested at 2 h p.i. for viral early gene luciferase assays. Luciferase activities in CD98+/+ MEFs were used as the 100% control. The experiments were performed three times, and the standard deviations are shown. (E) Expression of the CD98-CD69 chimera proteins in CD98 knockout MEFs did not increase vaccinia virus core uncoating. The above-mentioned cells were infected with WR MV at an MOI of 40 PFU/cell for viral core-uncoating assays as described above. For each group, viral cores from at least 40 cells were counted to obtain an average number of viral cores/cell; the viral core number obtained from CD98+/+ MEFs was used as the 100% control. The experiments were performed three times, and the standard deviations are shown. (F) Amino acid transport assay with CD98 knockout MEFs expressing CD98-CD69 chimera proteins. The above-mentioned cells were washed, preincubated in amino acid uptake solution, and then pulsed for 1 min with 1 μCi/ml [14C]l-leucine as described in Materials and Methods. Cells were washed and lysed, and the radioactivity was measured by a liquid scintillation counter. The counts per minute (CPM)/mg protein in the lysate are shown. The experiments were performed five times, and the standard deviations are shown.
To analyze the functions associated with the CD98 domains in the chimeric proteins used, we examined which of the constructs could reconstitute amino acid transport in CD98−/− MEFs by performing [14C]l-leucine uptake assays as previously described (32) and quantifying the intracellular radioactivity with a liquid scintillation counter. The level of [14C]l-leucine uptake was very low in CD98−/− MEFs as well as in CD98−/− MEFs expressing YFP, YFP fused with CD69, or a construct containing the extracellular and cytoplasmic domains of CD69 (C69T98E69) (Fig. 10F). The chimeras containing the extracellular domain of CD98 (C69T98E98 and C98T69E98) and the construct comprising the cytoplasmic and transmembrane domains of CD98 but lacking its extracellular domain (C98T98E69) partially reconstituted the amino acid transport activity (Fig. 10F). These results demonstrate that chimeras lacking one of the three domains (i.e., the extracellular, the transmembrane, or the cytoplasmic domain) of CD98 are still able to transport the light chain to the plasma membrane and induce amino acid transport albeit with a reduced efficiency. To reconstitute complete amino acid transport activity, however, all three domains of CD98 were required. Taken together, our results show that amino acid transport activity appears to be necessary but not sufficient to explain the fact that the entire CD98 protein is required for MV endocytosis, suggesting that additional functions provided by full-length CD98 are important for MV entry.
DISCUSSION
The identification of cellular receptors employed by vaccinia virus to enter different types of cells represents an important step in virus research, and a detailed understanding of the cellular targets of vaccinia virus will help us to learn more about virus tropism and pathogenesis. Although several receptors were reported previously to participate in vaccinia virus entry, evidence pertaining to their significance is still lacking (18, 34, 37, 50). Here, we show that CD98 is enriched in the host cell membrane upon vaccinia MV infection and that CD98 and vaccinia MV colocalize in lipid rafts. Moreover, we demonstrate that WR MV can be found within CD98-positive endocytic vesicles. In addition, by using CD98 KO MEFs and YFP-CD98-reconstituted MEFs as well as CD98 knockdown in HeLa cells, we demonstrate here that the type II transmembrane glycoprotein CD98 plays a role in vaccinia MV WR strain entry through endocytosis.
Interestingly, CD98 is widely expressed on multiple cell types (49, 60), which correlates with the broad spectrum of vaccinia virus infectivity. This could explain why vaccinia MV targets this cellular receptor in order to gain entry into many different cell types. CD98 was previously shown to be endocytosed in HeLa cells in a clathrin- and dynamin-independent manner (19). Our study shows that the vaccinia virus WR strain is endocytosed into MEF cells in a non-clathrin- and non-caveola-dependent but dynamin-dependent pathway, which is similar to what was previously observed for HeLa cells (26). Our findings are in agreement with the previously described CD98 endocytic route, with the exception that WR MV endocytosis required dynamin GTPase activity, while CD98 endocytosis itself does not (19). This finding suggests that vaccinia MV first targets CD98 in order to initiate fluid-phase endocytic uptake and then subsequently recruits dynamin to accommodate the membrane fusion event.
Although CD98 was initially identified as a molecule involved in cell-cell fusion (28, 41–44), it is not required for vaccinia MV membrane fusion per se. In our fusion-from-without assays, the CD98 KO MEFs underwent cell-cell fusion more rapidly than did the CD98+/+ cells, suggesting that CD98 plays a minor role in the regulation of vaccinia MV-induced cell-cell fusion. Nevertheless, our study shows clearly that the whole structure of CD98 is essential for WR MV endocytosis.
CD98 accomplishes two main cellular functions: by forming heterodimers with several light chains, it functions as an amino acid transporter (HATs) (10, 59), and it interacts with integrins to modulate integrin-dependent signaling (20, 22, 63). We performed a reverse transcription (RT)-PCR analysis and found that several CD98-associated light chains, i.e., LAT1, LAT2, y+LAT1, y+LAT2, and xCT, were expressed in HeLa and MEF cells (data not shown). While previous studies showed that the extracellular domain of CD98 is required for its association with its light chain and for mediating amino acid transport (21), others have demonstrated that the elimination of the disulfide bridge that forms between CD98 and the light chain or the extracellular domain of CD98 does not completely abolish the expression of the heterodimer at the plasma membrane and the transport of amino acids (6, 40, 46, 61). Using CD98-CD69 chimeric constructs, we found that even though some chimeras could partially reconstitute amino acid transport, none of them reconstituted WR infectivity in MEF cells. Thus, we concluded that the activation of amino acid transport by CD98 is necessary but not sufficient to explain the role of CD98 in WR infectivity. One reason for this could be that the CD98-CD69 chimeric constructs are not efficiently endocytosed or are not endocytosed at the same rate as wild-type CD98. Another function of CD98 is to regulate raft-associated receptor functions, such as integrin β1 (20, 22, 63). In that respect, another study recently conducted in our laboratory revealed that integrin β1 plays an important role in the binding and entry of WR MV into HeLa cells (R. Izmailyan et al., submitted for publication).
Previously, other viruses were shown to target CD98 or amino acid transporters. The cationic amino acid transporter CAT1 was shown to be required for Moloney leukemia virus (MLV) to enter rodent cells (1, 33). The CD98 light chain xCT was also suggested to play a role in the entry (30) and replication (57) of Kaposi's sarcoma-associated herpesvirus (KHSV).
In summary, our study demonstrates that CD98 is specifically required for WR MV endocytic entry into HeLa cells and MEFs. No single CD98 domain is sufficient to provide help for vaccinia MV entry, suggesting that vaccinia MV requires both functions of CD98, i.e., the regulation of amino acid transport as well as integrin β1 signaling. Our results, however, do not exclude the possibility that vaccinia virus simply targets the overall structure of CD98, irrespective of its functions in cells, to gain entry into the cells. Whether such a structural requirement of CD98 is influenced by its associated molecules, such as integrin β1 and light chains of amino acid transporters, is currently unknown. Further studies are needed to better understand how CD98 participates in vaccinia virus endocytosis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mark H. Ginsberg for providing the MEF cells and the CD98 chimeric constructs. We thank Jye-Lin Hsu and Yin-Liang Tang for plasmid constructions and CD98 antibody purification.
This work was supported by grants from the Academia Sinica and the National Science Council of the Republic of China (NSC100-2320-B-001-006).
Footnotes
Published ahead of print 15 February 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Albritton LM, Tseng L, Scadden D, Cunningham JM. 1989. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell 57:659–666 [DOI] [PubMed] [Google Scholar]
- 2. Armstrong JA, Metz DH, Young MR. 1973. The mode of entry of vaccinia virus into L cells. J. Gen. Virol. 21:533–537 [DOI] [PubMed] [Google Scholar]
- 3. Bender FC, et al. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542–9552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bengali Z, Townsley AC, Moss B. 2009. Vaccinia virus strain differences in cell attachment and entry. Virology 389:132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bertran J, et al. 1992. Stimulation of system y(+)-like amino acid transport by the heavy chain of human 4F2 surface antigen in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. U. S. A. 89:5606–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Broer A, et al. 2001. Association of 4F2hc with light chains LAT1, LAT2 or y+LAT2 requires different domains. Biochem. J. 355:725–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carter GC, Law M, Hollinshead M, Smith GL. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 86:1279–1290 [DOI] [PubMed] [Google Scholar]
- 8. Chang A, Metz DH. 1976. Further investigations on the mode of entry of vaccinia virus into cells. J. Gen. Virol. 32:275–282 [DOI] [PubMed] [Google Scholar]
- 9. Chang SJ, Chang YX, Izmailyan R, Tang YL, Chang W. 2010. Vaccinia virus A25 and A26 proteins are fusion suppressors for mature virions and determine strain-specific virus entry pathways into HeLa, CHO-K1, and L cells. J. Virol. 84:8422–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chillaron J, Roca R, Valencia A, Zorzano A, Palacin M. 2001. Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am. J. Physiol. 281:F995–F1018 [DOI] [PubMed] [Google Scholar]
- 11. Ching YC, et al. 2009. Disulfide bond formation at the C termini of vaccinia virus A26 and A27 proteins does not require viral redox enzymes and suppresses glycosaminoglycan-mediated cell fusion. J. Virol. 83:6464–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiu WL, Lin CL, Yang MH, Tzou DL, Chang W. 2007. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 81:2149–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72:1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chung CS, Huang CY, Chang W. 2005. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 79:1623–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31–124 [DOI] [PubMed] [Google Scholar]
- 16. de Magalhaes JC, et al. 2001. A mitogenic signal triggered at an early stage of vaccinia virus infection: implication of MEK/ERK and protein kinase A in virus multiplication. J. Biol. Chem. 276:38353–38360 [DOI] [PubMed] [Google Scholar]
- 17. Doms RW, Blumenthal R, Moss B. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64:4884–4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eppstein DA, et al. 1985. Epidermal growth factor receptor occupancy inhibits vaccinia virus infection. Nature 318:663–665 [DOI] [PubMed] [Google Scholar]
- 19. Eyster CA, et al. 2009. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic (Copenhagen) 10:590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fenczik CA, Sethi T, Ramos JW, Hughes PE, Ginsberg MH. 1997. Complementation of dominant suppression implicates CD98 in integrin activation. Nature 390:81–85 [DOI] [PubMed] [Google Scholar]
- 21. Fenczik CA, et al. 2001. Distinct domains of CD98hc regulate integrins and amino acid transport. J. Biol. Chem. 276:8746–8752 [DOI] [PubMed] [Google Scholar]
- 22. Feral CC, et al. 2005. CD98hc (SLC3A2) mediates integrin signaling. Proc. Natl. Acad. Sci. U. S. A. 102:355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gillooly DJ, et al. 2000. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19:4577–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gong SC, Lai CF, Esteban M. 1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology 178:81–91 [DOI] [PubMed] [Google Scholar]
- 25. Hsiao JC, Chung CS, Chang W. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang CY, et al. 2008. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into HeLa cells through fluid phase endocytosis. J. Virol. 82:7988–7999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ichihashi Y, Oie M. 1996. Neutralizing epitope on penetration protein of vaccinia virus. Virology 220:491–494 [DOI] [PubMed] [Google Scholar]
- 28. Ito Y, et al. 1992. Fusion regulation proteins on the cell surface: isolation and characterization of monoclonal antibodies which enhance giant polykaryocyte formation in Newcastle disease virus-infected cell lines of human origin. J. Virol. 66:5999–6007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joklik WK. 1962. The purification of four strains of poxvirus. Virology 18:9–18 [DOI] [PubMed] [Google Scholar]
- 30. Kaleeba JA, Berger EA. 2006. Kaposi's sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science 311:1921–1924 [DOI] [PubMed] [Google Scholar]
- 31. Kerr MC, Teasdale RD. 2009. Defining macropinocytosis. Traffic (Copenhagen) 10:364–371 [DOI] [PubMed] [Google Scholar]
- 32. Kim DK, et al. 2002. Characterization of the system L amino acid transporter in T24 human bladder carcinoma cells. Biochim. Biophys. Acta 1565:112–121 [DOI] [PubMed] [Google Scholar]
- 33. Kim JW, Closs EI, Albritton LM, Cunningham JM. 1991. Transport of cationic amino acids by the mouse ecotropic retrovirus receptor. Nature 352:725–728 [DOI] [PubMed] [Google Scholar]
- 34. Lalani AS, et al. 1999. Use of chemokine receptors by poxviruses. Science 286:1968–1971 [DOI] [PubMed] [Google Scholar]
- 35. Lin CL, Chung CS, Heine HG, Chang W. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74:3353–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Locker JK, et al. 2000. Entry of the two infectious forms of vaccinia virus at the plasma membrane is signaling-dependent for the IMV but not the EEV. Mol. Biol. Cell 11:2497–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marsh YV, Eppstein DA. 1987. Vaccinia virus and the EGF receptor: a portal for infectivity? J. Cell. Biochem. 34:239–245 [DOI] [PubMed] [Google Scholar]
- 38. Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535 [DOI] [PubMed] [Google Scholar]
- 39. Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2945 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 40. Nakamura E, et al. 1999. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J. Biol. Chem. 274:3009–3016 [DOI] [PubMed] [Google Scholar]
- 41. Ohgimoto S, et al. 1995. Molecular characterization of fusion regulatory protein-1 (FRP-1) that induces multinucleated giant cell formation of monocytes and HIV gp160-mediated cell fusion. FRP-1 and 4F2/CD98 are identical molecules. J. Immunol. 155:3585–3592 [PubMed] [Google Scholar]
- 42. Ohgimoto S, et al. 1996. Regulation of human immunodeficiency virus gp160-mediated cell fusion by antibodies against fusion regulatory protein 1. J. Gen. Virol. 77(Pt 11):2747–2756 [DOI] [PubMed] [Google Scholar]
- 43. Okamoto K, et al. 1997. Paramyxovirus-induced syncytium cell formation is suppressed by a dominant negative fusion regulatory protein-1 (FRP-1)/CD98 mutated construct: an important role of FRP-1 in virus-induced cell fusion. J. Gen. Virol. 78(Pt 4):775–783 [DOI] [PubMed] [Google Scholar]
- 44. Okamoto K, et al. 1997. An anti-fusion regulatory protein-1 monoclonal antibody suppresses human parainfluenza virus type 2-induced cell fusion. J. Gen. Virol. 78(Pt 1):83–89 [DOI] [PubMed] [Google Scholar]
- 45. Peters EC, Horn DM, Tully DC, Brock A. 2001. A novel multifunctional labeling reagent for enhanced protein characterization with mass spectrometry. Rapid Commun. Mass Spectrom. 15:2387–2392 [DOI] [PubMed] [Google Scholar]
- 46. Pfeiffer R, et al. 1998. Functional heterodimeric amino acid transporters lacking cysteine residues involved in disulfide bond. FEBS Lett. 439:157–162 [DOI] [PubMed] [Google Scholar]
- 47. Popik W, Alce TM, Au WC. 2002. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J. Virol. 76:4709–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prager GW, Feral CC, Kim C, Han J, Ginsberg MH. 2007. CD98hc (SLC3A2) interaction with the integrin beta subunit cytoplasmic domain mediates adhesive signaling. J. Biol. Chem. 282:24477–24484 [DOI] [PubMed] [Google Scholar]
- 49. Quackenbush E, et al. 1987. Molecular cloning of complementary DNAs encoding the heavy chain of the human 4F2 cell-surface antigen: a type II membrane glycoprotein involved in normal and neoplastic cell growth. Proc. Natl. Acad. Sci. U. S. A. 84:6526–6530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rahbar R, et al. 2006. Vaccinia virus activation of CCR5 invokes tyrosine phosphorylation signaling events that support virus replication. J. Virol. 80:7245–7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rodriguez JF, Smith GL. 1990. IPTG-dependent vaccinia virus: identification of a virus protein enabling virion envelopment by Golgi membrane and egress. Nucleic Acids Res. 18:5347–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Spiegel S, Kassis S, Wilchek M, Fishman PH. 1984. Direct visualization of redistribution and capping of fluorescent gangliosides on lymphocytes. J. Cell Biol. 99:1575–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Torrents D, et al. 1998. Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J. Biol. Chem. 273:32437–32445 [DOI] [PubMed] [Google Scholar]
- 54. Townsley AC, Weisberg AS, Wagenaar TR, Moss B. 2006. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J. Virol. 80:8899–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vanderplasschen A, Hollinshead M, Smith GL. 1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 79(Pt 4):877–887 [DOI] [PubMed] [Google Scholar]
- 56. Vanderplasschen A, Smith GL. 1997. A novel virus binding assay using confocal microscopy: demonstration that the intracellular and extracellular vaccinia virions bind to different cellular receptors. J. Virol. 71:4032–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Veettil MV, et al. 2008. Kaposi's sarcoma-associated herpesvirus forms a multimolecular complex of integrins (alphaVbeta5, alphaVbeta3, and alpha3beta1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J. Virol. 82:12126–12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Verrey F, et al. 2004. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 447:532–542 [DOI] [PubMed] [Google Scholar]
- 59. Verrey F, Jack DL, Paulsen IT, Saier MH, Jr, Pfeiffer R. 1999. New glycoprotein-associated amino acid transporters. J. Membr. Biol. 172:181–192 [DOI] [PubMed] [Google Scholar]
- 60. Verrey F, Meier C, Rossier G, Kuhn LC. 2000. Glycoprotein-associated amino acid exchangers: broadening the range of transport specificity. Pflugers Arch. 440:503–512 [DOI] [PubMed] [Google Scholar]
- 61. Wagner CA, et al. 2000. The heterodimeric amino acid transporter 4F2hc/LAT1 is associated in Xenopus oocytes with a non-selective cation channel that is regulated by the serine/threonine kinase sgk-1. J. Physiol. 526(Pt 1):35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Whitbeck JC, Foo CH, Ponce de Leon M, Eisenberg RJ, Cohen GH. 2009. Vaccinia virus exhibits cell-type-dependent entry characteristics. Virology 385:383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zent R, et al. 2000. Class- and splice variant-specific association of CD98 with integrin beta cytoplasmic domains. J. Biol. Chem. 275:5059–5064 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.