

Abstract
The nuclear export of the influenza A virus ribonucleoprotein (vRNP) is crucial for virus replication. As a major component of the vRNP, nucleoprotein (NP) alone can also be shuttled out of the nucleus by interacting with chromosome region maintenance 1 (CRM1) and is therefore hypothesized to promote the nuclear export of the vRNP. In the present study, three novel nuclear export signals (NESs) of the NP—NES1, NES2, and NES3—were identified as being responsible for mediating its nuclear export. The nuclear export of NES3 was CRM1 dependent, whereas that of NES1 or NES2 was CRM1 independent. Inactivation of these NESs led to an overall nuclear accumulation of NP. Mutation of all three NP-NESs significantly impaired viral replication. Based on structures of influenza virus NP oligomers, these three hydrophobic NESs are found present on the surface of oligomeric NPs. Functional studies indicated that oligomerization is also required for nuclear export of NP. Together, these results suggest that the nuclear export of NP is important for virus replication and relies on its NESs and oligomerization.
INTRODUCTION
The influenza A virus genome consists of eight negative-sense single-stranded RNA segments (vRNA) (17). Each vRNA segment is associated with multiple copies of the viral nucleoprotein (NP) and three polymerase subunits (PA, PB1, and PB2), forming the viral ribonucleoprotein (vRNP) complex. During an early stage of infection, the vRNPs are released into the cytoplasm from virions following fusion of the viral membrane and endosomal membrane (41). Subsequently, the incoming vRNPs are transported into the nucleus, where viral genome replication and transcription occur (41).
One of the determinants for vRNP nuclear import is NP (8, 28, 44, 49, 52, 53), the major protein in the vRNP structure. Thus far, two nuclear localization signals (NLSs) and a nuclear accumulation signal (NAS) have been identified in NP. The stronger NLS is an unconventional signal located in the N-terminal basic region (between residues 3 and 13) of NP (28, 44), and the weaker signal, a classical bipartite NLS, is located between residues 198 and 216 (49). The NAS-spanning residues (327 to 345) were identified through analyses of mutants that lacked both of the NLSs but still exhibited partial nuclear distribution (9).
At a late stage of virus infection, the vRNPs exit the nucleus to assemble and bud from the apical plasma membrane of polarized cells (3). The trafficking of the vRNPs into and out of the nucleus is a tightly regulated process (7). The nuclear export of progeny vRNPs is mediated by the CRM1 cellular export receptor (12, 23, 29, 48). Nuclear export protein (NEP; formerly referred to as the NS2 protein), which possesses a nuclear export signal (NES), and the matrix protein (M1) of influenza A virus are deemed responsible for directing export of the vRNPs (29, 31). This process is regulated by the Raf/MEK/ERK pathway (32), which is stimulated by the membrane association of influenza virus hemagglutinin (HA) (25). Interestingly, exogenously expressed NP can also shuttle between the cytoplasm and nucleus (28, 51), and treatment of NP-expressing cells with leptomycin B (LMB) promotes NP toward a more nuclear distribution (12). These data suggest that NP is a candidate for mediating vRNP shuttling out of the nucleus. In the previous study, the N-terminal 38 amino acids of NP have been found capable of shuttling between the nucleus and cytoplasm (28). However, other NP-NESs maybe exist, and the role of NP nuclear export in virus replication is not clear.
In the present study, we identified that NP contained a CRM1-dependent NES and two CRM1-independent NESs. The NESs were all functional in full-length NP, and recombinant virus carrying all deficient NESs could not be successfully rescued, probably due to impairment of viral protein expression. Finally, analyses of point mutations demonstrated that the oligomerization of NP is crucial for its nuclear export.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
Madin-Darby canine kidney (MDCK) cells, human embryonic kidney 293T (HEK293T) cells, human hepatoma Huh7 cells, and mouse NIH 3T3 cells were grown in Dulbecco modified Eagle medium (DMEM) (Gibco-BRL, Inc., Gaithersburg, MD) supplemented with 10% fetal bovine serum (PAA Laboratories, Linz, Austria) at 37°C with 5% CO2. The influenza A virus A/WSN/33 (H1N1) used in the present study was rescued from cDNAs (30). Rabbit polyclonal antibody against NP was prepared as described previously (47). For mouse anti-NP polyclonal antibody and rabbit anti-M1 polyclonal antibody, purified hexahistidine-tagged NP (His-NP) and His-M1 were provided to company (Beijing Cowin Biotech Co., Ltd., Beijing, China) for immunization. Anti-β-actin polyclonal antibody and anti-c-Myc (9E10) antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse anti-FLAG (M2) antibody was purchased from Sigma-Aldrich (St. Louis, MO). The goat anti-rabbit IgG conjugated to TRITC (tetramethyl rhodamine isothiocyanate) and fluorescein isothiocyanate and goat anti-mouse IgG conjugated to TRITC secondary antibodies were from Zhongshan Golden Bridge Biotechnology (Beijing, China).
Plasmid constructs.
The NP gene was amplified from influenza A virus A/WSN/33 (H1N1) cDNA by PCR and then inserted between the KpnI and XhoI sites of the pcDNA4/TO vector (Invitrogen, Carlsbad, CA). The full-length NP mutants, including F39A/I41A (NP-NES1m), M189A/M191A (NP-NES2m), L266A (NP-NES3m), F39A/I41A/M189A/M191A (NP-NES1/2m), F39A/I41A/L266A (NP-NES1/3m), M189A/M191A/L266A (NP-NES2/3m), Y39A/I41A/M189A/M191A/L266A (NP-NES1/2/3m), E339A, R416A, and F479A, were generated by using QuikChange II site-directed mutagenesis (Stratagene, La Jolla, CA) and a Newpep site-directed mutagenesis kit (Beijing Newpep Biotech Co., Ltd., Beijing, China). All plasmids expressing putative NESs in Table 1 and NES mutants in Fig. 3A were constructed by annealing double-stranded oligonucleotides into the pEGFP-C1 expression vector (Clontech Laboratories, Mountain View, CA). The full-length M1 and NP genes from A/WSN/33 virus were subcloned into the bacterial expression vector pET30a(+) for protein purification and antibody preparation. For coimmunoprecipitation, the full-length NP gene was cloned into the pcDNA3-FLAG and pCMV-Myc vectors, respectively. The expression plasmids for the PA, PB1, and PB2 genes from A/WSN/33 virus were generated by cloning into the pcDNA3-FLAG vector as described previously (47). pHH21-cNS-Luc was generated by cloning the cRNA promoter of NS with luciferase into pHH21 as described previously (18). All constructs were verified by DNA sequencing. The oligonucleotide sequences and detailed protocols used in cloning and site-directed mutagenesis are available upon request.
Table 1.
PutativeNES motifs in NP of influenza A virus
| Putative NESa | Sequenceb | Function of NES |
|---|---|---|
| Consensus NES | ΦX2-3ΦX2-3ΦXΦ | |
| NP-pNES[24-49]* (NES1) | EIRASVGKMIDGIGRFYIQMCTELKL | Yes |
| NP-pNES[56-68] | LIQNSLTIERMVL | No |
| NP-pNES[183-197]* (NES2) | VKGVGTMVMELIRMI | Yes |
| NP-pNES[248-274] (NES3) | PGNAEFEDLIFLARSALILRGSVAHKS | Yes |
| NP-pNES[289-314] | YDFEREGYSLVGIDPFRLLQNSQVYS | No |
| NP-pNES[371-381] | METMESSTLEL | No |
| NP-pNES[406-420]* | ISIQPTFSVQRNLPF | No |
| NP-pNES[14-38] | ETDGERQNATEIRASVGKMIDGIGR(28) | No |
| NEP-NES | ILMRMSKMQL(31) | Yes |
*, The single fragment contained two putative NESs.
Φ, A large hydrophobic residue, such as leucine, isoleucine, valine, or methionine; X, any amino acid. Italicized letters indicate the cores of the pNESs, and boldface letters indicate the cores of the second pNESs in the single fragment. Underlining indicates residues critical to NES activity.
Fig 3.
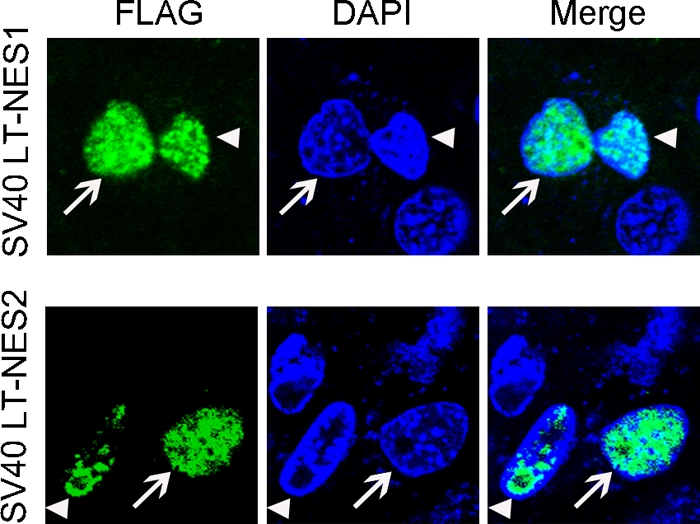
Heterokaryon analysis demonstrates that both NES1 and NES2 of NP are NESs. Huh7 cells transfected with SV40 LT-NES1 or SV40 LT-NES2 chimera were fused with NIH 3T3 cells using PEG 8000. SV40 LT is a nuclear protein which contains an NLS, but without any NES. Upon fusion with NES1 or NES2 of NP, the chimera of SV40 LT-NES can be transported from transfected human to untransfected mouse nuclei. This result suggested that both NES1 and NES2 of NP are NESs. The SV40 LT was detected with mouse anti-FLAG antibody. Arrowhead, human Huh7 cells transfected with SV40 LT-NES1 or SV40 LT-NES2 (green); arrow, untransfected mouse NIH 3T3 cells with mouse characteristic DAPI blue staining.
Construction of simian virus 40 (SV40) LT-NESs. SV40 large T antigen (LT) carrying a linker (Thr-Thr-Thr-Thr-Gly-Ser) was amplified by PCR. The SV40 LT DNA fragment without a stop codon was then subcloned into the pcDNA3-FLAG expression plasmid. NES1 or NES2 of the NP fragment and BamHI/XhoI sites was amplified by PCR. The DNA fragment was cloned to the C terminus of SV40 LT.
Generation of NP-expressing inducible cell lines.
The T-REx system (Invitrogen) was used to generate inducible cell lines for NP expression. A 293T cell line stably expressing the tetracycline (Tet) repressor from the pcDNA6/TR plasmid (293T-6TR) was used for transfection with the pcDNA4/TO or pcDNA4/TO-NP plasmid. Clones were selected using 100 μg of zeocin (Invitrogen)/ml. NP expression was induced by adding 1 μg of Tet/ml to the culture medium and analyzed by immunofluorescence assays and Western blotting with a rabbit anti-NP polyclonal antibody.
Transfection and fluorescence microscopy.
293T cells were grown on coverslips in 24-well plates, followed by transfection with the corresponding plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. At the indicated time points, the cells were rinsed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 30 min, and permeabilized with 0.5% Triton X-100 in PBS for 10 min at room temperature. To detect NP or M1, the cells were incubated for 2 h with anti-NP or anti-M1 antibodies. After washing with PBS, the cells were incubated for 1 h with the appropriate secondary antibodies. Fluorescence images were obtained using a Leica confocal laser scanning microscope.
LMB treatment.
293T cells were transfected with pEGFP-C1-NES1, pEGFP-C1-NES2, or pEGFP-C1-NES3. LMB (Tocris Bioscience, Avonmouth, United Kingdom) was added to the desired wells at a final concentration of 11 nM at 3 h posttransfection. The cells were harvested 3 h later and fixed for 30 min with 4% paraformaldehyde in PBS. For cells stably expressing NP, at 8 h after induction with 1 μg of Tet/ml, the cells were washed with PBS and treated with either 11 nM LMB or vehicle for 3 h.
Generation of infectious influenza virus.
Infectious virus was generated as described by Neumann et al. (30), except that the pcDNA4/TO plasmid was used for expression of NP. At 48 h posttransfection, the virus was harvested to be titrated in MDCK cells.
Heterokaryon assays.
Heterokaryon assays were performed as described previously (19). Briefly, at 24 h posttransfection, human Huh7 cells were treated with trypsin, and 106 Huh7 cells were mixed with 2 × 106 untransfected mouse NIH 3T3 cells, before being seeded on glass coverslips in a 6-cm dish. The cells were incubated at 37°C for 18 h, followed by treatment with 50 μg of cycloheximide (Sigma-Aldrich)/ml for 30 min. Fusion was induced by addition of 50% (wt/vol) polyethylene glycol (PEG) 8,000 (Sigma-Aldrich), followed by incubation for 2 min at 37°C. The cells were washed with PBS, incubated at 37°C for 2 h in culture media plus cycloheximide, fixed, and analyzed by microscopy.
Fractionation of nucleus and cytoplasm.
293T cells (106) were harvested into 200 μl of CLB buffer (10 mM HEPES, 10 mM NaCl, 1 mM KH2PO4, 5 mM NaHCO3, 1 mM CaCl2, 0.5 mM MgCl2, 5 mM EDTA, Complete protease inhibitor cocktail [Roche Diagnostics, Indianapolis, IN]). The cells were allowed to swell for 5 min on ice and Dounce homogenized 50 times. After centrifugation at 7,500 rpm for 5 min at 4°C, the pellet is nuclei plus debris, and the supernatant is cytosol plus plasma membrane. The nucleus-debris pellet was then resuspended in 1 ml of TSE buffer (10 mM Tris [pH 7.5], 300 mM sucrose, 1 mM EDTA, 0.1% NP-40, Complete protease inhibitor cocktail) and Dounce homogenized 30 times, followed by centrifugation at 5,000 rpm for 5 min. The pellet was resuspended and washed twice to obtain the final nucleus pellet. The final pellet was resuspended in 50 μl of TSE buffer.
Plaque assay.
Plaque assay was performed as described previously (22). Briefly, MDCK cell monolayers (0.4 × 106 cells at 100% confluence in 12-well tissue culture plates) were washed with PBS and infected with different dilutions of virus for 1 h at 37°C. The virus inoculum was then removed by washing with PBS. Next, cell monolayers were overlaid with agar overlay medium (DMEM supplemented with 0.6% low-melting-point agarose and 2 μg of TPCK-treated trypsin/ml) and incubated at 37°C. Visible plaques were counted at 4 days postinfection, and the virus titers were determined. All data are expressed as the means of three independent experiments.
Luciferase assay.
The plasmids for expression of the PA, PB1, PB2, and NP (wild type [WT] or mutant) were simultaneously transfected into 293T cells with a luciferase reporter plasmid (pHH21-cNS-Luc) and pcDNA-β-gal, as described by Li et al. (20), with some modification. As a negative control, 293T cells were transfected with the same plasmids, with the exception of the NP expression plasmid. After transfection, the cells were incubated at 37°C for 48 h, and then the amount of luciferase activity in transfectants was measured and normalized to the amount of β-galactosidase activity, as measured by standard kits (Promega, Madison, WI).
Coimmunoprecipitation.
Cells were lysed in immunoprecipitation buffer, containing 0.5% NP-40, 150 mM NaCl, 20 mM HEPES (pH 7.4), 10% glycerol, and 1 mM EDTA with Complete protease inhibitor cocktail. After centrifugation, the supernatant was incubated with anti-FLAG M2 affinity gel (Sigma-Aldrich) for at least 2 h. After five washes in immunoprecipitation buffer, the bound proteins were eluted by boiling for 10 min in sodium dodecyl sulfate (SDS) protein loading buffer and analyzed by Western blotting.
Western blot analysis.
Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane. Blots were blocked overnight at 4°C in blocking solution (5% skimmed milk powder 0.5% Tween 20 in PBS), and proteins were detected using appropriate antibodies, followed by the addition of anti-rabbit or anti-mouse secondary antibody coupled to horseradish peroxidase. The interest proteins were visualized by enhanced chemiluminescence detection (Amersham, Arlington Heights, IL) as described by the manufacturer.
Nuclear and cytoplasmic mRNA quantification by real-time reverse transcription-PCR (RT-PCR).
293T cells were transfected as described for the luciferase assay. At 6 h posttransfection, nuclear and cytoplasmic RNA was fractionated from cells as described previously (46). Briefly, transfected cells were lysed in RSB (10 mM Tris [pH 7.4], 10 mM NaCl, 3 mM MgCl2) containing 0.5% Nonidet P-40, 10% glycerol, and 100 U of RNasin (Promega)/ml. Nuclei were further washed with 1% Tween 40 and 0.5% sodium deoxycholate, and RNA from both cytoplasmic and nuclear fractions was purified using TRIzol (Invitrogen). The resulting RNA preparations were treated with RNase-free DNase I (Takara, Tokyo, Japan) according to the manufacturer's protocol. One microgram of total RNA was used for cDNA synthesis with the oligo(dT)18 primer, followed by real-time PCR quantification. Real time quantitative PCR was performed with SYBR Premix Ex Taq (TaKaRa, Japan) on a Corbett 6200 real-time detection system (Corbett, Australia) to investigate the expression level of luciferase in luciferase assay (54). The PCR conditions were 95°C for 30 s, followed by 40 cycles of 94°C for 5 s and 60°C for 30 s. The oligonucleotide primers used were as follows for luciferase: forward, CAACTGCATAAGGCTATGAAGAGA; and reverse, ATTTGTATTCAGCCCATATCGTTT (24).
RESULTS
NP possesses three novel NESs.
Previous studies have demonstrated that NP is a nucleocytoplasmic shuttling protein (5, 12, 28), and the CRM1 pathway can mediate nuclear export of NP (12). To locate the NES in the NP, the NP sequence was analyzed by computer-assisted visual inspection. We identified 10 potential NES motifs with striking similarity to leucine-rich NES motifs (Table 1). To verify their function, synthetic oligonucleotides encoding the putative NP-NESs were synthesized and fused to enhanced green fluorescence protein (EGFP). For convenience, two putative NESs (pNESs) located in close proximity were tested in a single segment. As shown in Fig. 1, EGFP alone was localized in both the nucleus and the cytoplasm, which is consistent with the observation in previous reports (15, 26, 39), since its molecular mass (26 kDa) is below the size exclusion limit (∼40 kDa) for passive diffusion through the nuclear pores. However, of the seven sequences tested, three (NP-pNES[24-49], NP-pNES[183-197], and NP-pNES[248-274]) shifted the EGFP to the cytoplasm, suggesting that these NP-NESs are sufficient to direct the fusion protein from the nucleus to cytoplasm. Here, we refer to NP-pNES[24-49], NP-pNES[183-197], and NP-pNES[248-274] as NES1, NES2, and NES3, respectively (Table 1). In addition, previous research has demonstrated that the N-terminal 38 amino acids of NP have the ability to shuttle between the nucleus and cytoplasm (28). Because an NLS was identified in NP at amino acids 3 to 13, residues 14 to 38 were tested for NES activity in the present study. Our results demonstrated that NP-pNES[14-38] cannot direct the EGFP to the cytoplasm. Meanwhile, the NEP-NES was cloned as a positive control. As shown in Fig. 1, the NEP-NES was capable of targeting EGFP into the cytoplasm, confirming the rationality of this method. In this experiment, cycloheximide was used to eliminate the possibility of detecting newly translated proteins in the cytoplasm. Finally, NP-NES sequences were compared among various influenza A virus isolates. The results indicated that each of the three NES motifs is highly conserved among the influenza A viruses (data not shown).
Fig 1.

Identification of three functional NESs in NP. 293T cells were transiently transfected with plasmids expressing putative NP-NESs which were fused with EGFP. EGFP was used as a negative control; the NEP NES was used as a positive control. At 24 h posttransfection, the cells were treated with 100 ng of cycloheximide/μl for 3 h before fixation with 4% paraformaldehyde. The cells were then mounted with DAPI-containing mounting medium and visualized with a Leica confocal laser scanning microscope. Images of cells that are representative of the entire population are shown.
NES3 is CRM1 dependent, whereas NES1 and NES2 are CRM1 independent.
Because leucine-rich NESs are typically mediated by CRM1 (14, 40), we next investigated whether NP-NES activity was dependent on the CRM1 pathway. LMB blocks CRM1-dependent nuclear export and has been extensively used to probe this process (14). 293T cells were transfected with the pEGFP-C1-NES1, pEGFP-C1-NES2, or pEGFP-C1-NES3 constructs and treated with LMB or ethanol for 3 h at 3 h posttransfection. In the presence of LMB, EGFP-NES3 strikingly accumulated in the nucleus, suggesting that the CRM1 pathway is involved in the shuttling of the fusion protein (Fig. 2E and F). Unexpectedly, incubation of the EGFP-NES1- or EGFP-NES2-transfected cells with LMB had no effect on the localization of the fusion proteins (Fig. 2A, B, C, and D), indicating that NES1 and NES2 are CRM1 independent. Consistent with these results, the nuclear export of NP was still observed in some cells in the presence of LMB (Fig. 2G), implying that NP possesses NESs which are CRM1 independent. These results suggested that, aside from CRM1, other export receptors are also involved in the nuclear export of NP via recognition of NES1 or NES2.
Fig 2.

Effect of LMB on the localization of the three NESs and full-length NP. (A, C, and E) 293T cells were transfected with plasmids expressing EGFP-NES1 (A), EGFP-NES2 (C), or EGFP-NES3 (E). At 3 h posttransfection, transfected cells were treated with 11 nM LMB or ethanol for another 3 h. Cells were then washed, fixed, mounted with DAPI-containing medium, and visualized with a Leica confocal laser scanning microscope. Images of cells that are representative of the entire population are shown. (B, D, and F) Graphs showing the mean values ± the standard deviation of experiments (A, C, and E, respectively). At least 100 cells from each transfection were scored as predominantly nuclear (N), nuclear and cytoplasmic (N+C), or predominantly cytoplasmic (C). (G) The cytoplasmic localization of NP was not completely inhibited by LMB. Stable expression cell lines were induced to express NP for 8 h and then treated with LMB for 3 h. Subsequently, the cells were fixed, and NP was detected with the anti-NP polyclonal antibody.
Heterokaryon analyses confirm that NES1 and NES2 of NP possess nuclear export activity.
Despite the NES1 and NES2 could target EGFP to the cytoplasm, they are not sensitive to LMB. Examining steady-state protein localization in fixed cells could not distinguish a putative NES from a cytoplasmic retention signal (CRS). To differentiate an NES from a CRS, heterokaryon assays were performed with NES1 and NES2. To this end, SV40 LT was fused to the N terminus of the NES1 or NES2 to promote nuclear predominance. The human Huh 7 donor cells expressing SV40 LT-NES1 or SV40 LT-NES2 were then fused to mouse NIH 3T3 recipient cells using PEG 8000. Fusion of cells will expose recipient nuclei to a common cytoplasmic milieu, including any protein exported from the original nucleus. If nuclear export occurs (if an NES exists), the shuttling protein would be able to enter the cytoplasm of heterokaryon and be imported into the NIH 3T3 recipient nuclei. DAPI (4′,6′-diamidino-2-phenylindole) DNA staining was used to distinguish the mouse NIH 3T3 cell nuclei from the human Huh 7 cell nuclei. As shown in Fig. 3, we detected localization of SV40 LT (green) in the nuclei of NIH 3T3 cells, suggesting the transport of SV40 LT-NES1 or SV40 LT-NES2 chimeric protein from one donor nucleus into another recipient nucleus. The results confirmed that NES1 and NES2 are NESs.
Mutation of NP-NESs abrogates the ability of NP to shuttle between the nucleus and cytoplasm.
Although the present results demonstrated that the NP-NESs were active in the context of the EGFP-NES chimeric proteins, it was necessary to examine the function of the NESs within the context of the full-length NP. For this purpose, NP-NESs were inactivated by point mutations, and then the subcellular localization of NP mutants was evaluated by immunofluorescence analysis (IFA). We first introduced point mutations into each NES in the context of the EGFP-NES fusion protein to destroy its function. Because the last two hydrophobic residues of the consensus NES sequences are generally conserved and critical for export activity (16, 50), these residues were substituted by alanine in the NESs to abrogate their nuclear export activities. As expected, the mutations (F39A/I41A in NES1, M189A/M191A in NES2, or L266A in NES3) absolutely impaired the exclusive cytoplasmic distribution of the corresponding NESs, and no mutated EGFP-NESs were detected distributed exclusively in the cytoplasm (Fig. 4A). The NESs were then inactivated alone or in combination in the context of the full-length NP. As shown in Fig. 4B, only the NP mutant with all three deficient NESs was exclusively restricted to the nucleus, indicating that all of the NESs were functional in the full-length NP. The IFA result of nuclear retention of NP with NES1/2/3 mutation was confirmed by Western blot analysis (Fig. 4C). Of the three NESs, NES2 was the strongest in NP, because in the NP mutants containing two deficient NESs, the ones with NES2m (NES1/2m and NES2/3m) were more prone to nuclear localization (Fig. 4B). Moreover, the NES2 was CRM1 independent, suggesting other export pathways are involved in NP nuclear export.
Fig 4.

Effect of NES-inactivating mutations on the localization of NP. (A) Mutations and inactivation of NP-NESs which were fused with EGFP. The last two hydrophobic residues from the WT sequence (underlined), which were also the first two hydrophobic residues from the next overlapping NES, were substituted with alanines, and the export activity was monitored. Images of cells that are representative of the entire population are shown. (B) Effect of NES inactivation on NP localization. Subcellular distribution of NPs possessing singly, doubly, and triply deficient NESs was analyzed in 293T cells. The cells were transfected for 24 h with constructs encoding WT NP and NP mutants. Cells were counterstained with DAPI to show the nuclei. (C) The influence of NESs mutation on the nuclear export of NP was confirmed by fractionation and Western blot analysis with anti-NP polyclonal antibody.
NP-NESs play significant roles in virus replication.
To investigate potential roles for NP nuclear export in influenza virus replication, we attempted to rescue the recombinant virus with deficient NESs (NES1/2/3m) using the 12-plasmid reverse genetics system (30). Briefly, mutated constructs (pcDNA4/TO-mNP and pHH21-mNP) were used for transfection instead of the WT ones to express the mutated NP and vRNA, respectively. At 48 h posttransfection, cell-free supernatants were titrated in MDCK cells, and the remaining transfected cells were lysed for Western blot analysis. The use of WT NP expression plasmids led to efficient production of infectious recombinant virus. In contrast, when plasmids encoding NP with deficient NESs were transfected, no virus was rescued (Fig. 5A, B, and C). Western blot analysis of the corresponding cell-free supernatants demonstrated the absence of the characteristic NP and M1 (Fig. 5B).
Fig 5.

NP-NESs are crucial for viral replication. The 12-plasmid reverse genetics system was used to rescue recombinant virus with deficient NESs (NES1/2/3m). (A) At 48 h posttransfection, the culture supernatants were harvested and subjected to plaque assays in MDCK cells. Superscript “a”, viruses were generated using an established plasmid-based reverse-genetics system. ND, plaques were not detected. (B) At 48 h posttransfection, cell-free supernatants and the remaining transfected cells were lysed for Western blot analysis. NP and M1 were detected with anti-NP and anti-M1 polyclonal antibodies, respectively. The blots were also probed with an anti-β-actin antibody as a loading control. WT, wild type. (C) Immunofluorescence revealed that viral replication was abolished by mutation of NP-NESs. At 24 h posttransfection, the cells were fixed, and then NP (red) and M1 (green) were detected with mouse anti-NP polyclonal antibody and rabbit anti-M1 polyclonal antibody, respectively. (D) Pre-expression of NP with deficient NESs reduced the WT virus replication. 293T cells were transfected with plasmids expressing WT NP or NP with deficient NESs. At 36 h posttransfection, cells were infected with WT virus (WSN, multiplicity of infection = 0.1). M1 expression level was evaluated by Western blotting at 6 h postinfection in virus-infected cells. The relative protein level of M1 was calculated by quantifying the results. The data are presented as the mean standard deviation from three independent experiments. Significant differences (P < 0.01, Student t test) are indicated by two asterisks. (E) The ability of mutant NP to support virus replication/transcription in luciferase assay was examined in 293T cells as described in Materials and Methods. The expression levels of the WT NP and NP mutant were analyzed by Western blotting. (F) Mutation of NP-NESs only slightly affects the transcriptional activity of the vRNP, as shown by real-time RT-PCR. 293T cells were transfected with plasmids as described in Materials and Methods. At 6 h posttransfection, the total RNA from the nucleus and cytoplasm fractions was isolated, and mRNA from the luciferase reporter gene (luci) was assessed with real-time RT-PCR. The expression values were averaged from three replicates, each of which contained three independent samples. Ratio of N/C luci mRNA, the ratio of nuclear to cytoplasmic luciferase mRNA.
Immunofluorescence assays also support the fact that no recombinant virus was rescued when all of the NP-NESs were inactivated. As shown in Fig. 5C, NP was detected in both producer cells at 24 h posttransfection, although NP was observed only in the nucleus in cells transfected with the NP mutant expression plasmid. However, the M1 protein was barely detectable in the cells transfected with the NP mutants compared to WT NP. These results indicated that mutation of NP-NESs abolishes viral replication.
Exogenously expressed NP-NESs mutant interferes with virus replication in virus-infected cells.
Since the recombinant virus containing all deficient NESs could not be recovered, we examined the effect of NP-NESs mutation on virus replication by infecting NP-NES1/2/3m-expressing cells with WT virus. Western blot analysis showed that the expression of M1 was partially depressed in NP mutant-expressing cells in comparison to that in WT NP expressing or empty plasmid transfected cells at 6 h postinfection (Fig. 5D), suggesting that mutation of NP-NESs reduced virus replication. These results confirmed that NP-NESs play important roles in virus replication.
Mutation of the NP-NESs inhibits the viral replication at the posttranscription level.
It has been reported that NP is a major component of the vRNP (34) and plays important roles in viral transcription and replication (35). Indeed, single point mutations of conserved residues in NP can result in the loss of vRNP polymerase activity (20, 27). To investigate the mechanism by which mutations of NP-NESs abolished virus replication, we examined the effect of these mutations on the vRNP polymerase activity using a previously established luciferase assay (20). Our results showed that inactivation of NES1 and NES3 caused only ca. 22% reduction in luciferase activity, and inactivation of NES2 reduced luciferase activity to 65.11% (data not shown), but when all three NESs were inactivated, the luciferase activity was reduced to the background level (no NP) (Fig. 5E and data not shown). Thus, the NES1/2/3 mutation maybe impaired polymerase activity, viral mRNA nuclear export or translation, and the reduction of luciferase activity caused by NES1/3 or NES2 inactivation maybe attributed to attenuated NP nuclear export.
To further investigate whether loss of luciferase activity was related to NP nuclear retention, some NP mutants displaying nuclear retention were constructed, and then the effects of NP mutants on luciferase activity were evaluated using luciferase assay. The highly hydrophobic amino acids in NP-NES2 were selected for mutagenesis in the pcDNA4/TO-NP-NES1/3m plasmid, since the hydrophobic amino acids are usually critical for NES activity. Among the NP mutants, those devoid of luciferase activity all displayed nuclear retention in the 293T cells (data not shown). In contrast, mutants that did not cause an apparent reduction in luciferase activity could all shuttle out of the nucleus. Together, these data supported the notion that the loss of luciferase activity is likely due to the nuclear retention of NP.
NP is an RNA-binding protein, and thus, we speculated that mutations leading to NP nuclear retention maybe do not affect the transcription of the vRNP but prevent nuclear export of the corresponding mRNA for translation. To determine whether the impairment of luciferase activity was caused by failure of mRNA export, we performed real-time reverse transcription-PCR (RT-PCR) of lysates from nucleus and cytoplasm fractions. At 6 h posttransfection, the total RNA from the nucleus and cytoplasm was isolated, and mRNA from the luciferase reporter gene was assessed with real-time RT-PCR. The results indicated that the NP-NESs mutation only slightly affect the total luciferase mRNA level in cells (Fig. 5F), suggesting that mutation did not impair the transcription of vRNP. However, we found that the ratios of mRNA expression levels of luciferase in the nucleus to that in the cytoplasm showed some difference between NES1/2/3m and WT transfected cells (∼2-fold) (Fig. 5F), indicating there is more mRNA accumulated within the nucleus of NP-NES mutant transfected cells. Thus, mutation of the NP-NESs inhibits the viral replication at the posttranscription level.
All three NESs are presented on the surface of the oligomeric NP.
It has been previously shown by other investigators that NP is a hydrophilic protein (37, 42, 56), whereas its hydrophobicity increases after NP oligomerization (36), which indicates that more hydrophobic residues are exposed on the surface of NP oligomers. Generally, an NES is a short amino acid sequence of four hydrophobic residues that directs a protein from the nucleus to the cytoplasm through the nuclear pore complex. These data raise the possibility that NP oligomerization causes conformational changes in the NP tertiary structure, allowing the hydrophobic NESs to be exhibited on the surface of the protein. To examine this hypothesis, the presentation positions of NESs in oligomeric and monomeric NPs were compared. Because the structure of the biologically active influenza virus ribonucleoprotein complex (PDB ID 2WFS) has been clearly resolved by electron microscopy previously (6), these three NESs were first labeled in the vRNP using PyMOL software (www.pymol.org). Obviously, the NESs are all presented on the surface of the complex (Fig. 6B). Because the structure of the NP monomer has not yet been solved, the positions of the NESs in the NP monomer were predicted by using Protean (DNAStar, Madison, WI), which takes into account hydrophilicity and surface probability. The prediction results implied that the three NP-NESs are all highly hydrophobic and buried in the NP monomer (Fig. 6A). In addition, NES1 and NES2 are presented on the surface of published trimeric NP (PDB ID 2IQH) (55), whereas NES3 is only partially exposed (data not shown). It should be noted that, despite the fact that NESs are exposed on the NP oligomers, Fig. 6B shows that the NES2 was just located in the putative RNA binding cleft which was described in reference 55, suggesting that bound RNA maybe influences accessibility of NES2 to its receptor. These results indicated that oligomerization possibly promotes the exposure of NESs on the surface of NP so that the NESs can be recognized by the nuclear export receptors.
Fig 6.

Location of NESs in the tertiary structure of the influenza virus vRNP. (A) Hydrophobicity and surface possibility of residues in NP predicted using Protean program. The three NESs are localized between every two adjacent vertical lines. (B) Presentation of the three NESs on the surface of biologically active influenza virus ribonucleoprotein complex (PDB ID 2WFS) (6). The positions of the three NESs were labeled in green using PyMOL, red indicates the crucial residues of the NESs, yellow represents the α-helical region at amino acids 438 to 453, pink represents the α-helical region at 422 to 428 from the neighboring NP, and blue indicates the putative RNA binding residues (55).
Oligomerization is critical for the nuclear export of NP.
To confirm the contribution of NP oligomerization to nuclear export, mutational analysis was performed to increase or decrease the ability of NP to oligomerize. It has been demonstrated that alanine substitution of R416 or E339 results in the complete loss of NP oligomerization (13, 55), whereas mutation of F479 to alanine increases self-association 5-fold (13). To investigate the effect of NP oligomerization on the nuclear export of NP, these residues were mutated separately, and then the NP mutants were transfected into 293T cells. At 48 h posttransfection, NP was then detected with anti-NP polyclonal antibody. As shown in the Fig. 7B, monomeric NP mutants (i.e., E339A or R416A) were mostly located in the nucleus. On the contrary, the F479A NP mutant was more abundant in the cytoplasm than WT NP. The IFA result was confirmed by subcellular fractionation and analysis of the resulting protein fractions by Western blotting (Fig. 7C). These results indicated that NP oligomerization is required for the nuclear export of the protein.
Fig 7.

The nuclear export of NP depends on NP oligomerization. (A) Locations of point mutations decreasing (down arrows) or increasing (up arrow) self-association. Blue indicates NP NLSs, and green indicates NP-NESs. Yellow indicates the region that prevents NP oligomerization. CAS, cytoplasmic accumulation signal. (B) Localization of WT NP and NP mutants overexpressed in 293T cells. Cells were transfected with the pcDNA4/TO-NP, pcDNA4/TO-NP-E339A, pcDNA4/TO-NP-R416A, or pcDNA4/TO-NP-F479A constructs. At 48 h posttransfection, the cells were treated with 100 ng of cycloheximide/μl for 3 h and then fixed with 4% paraformaldehyde, and NP was detected with anti-NP polyclonal antibody. (C) Graphs displaying the mean values ± the standard deviations from at least three independent experiments. At least 100 cells from each transfection were scored as predominantly nuclear (N), nuclear and cytoplasmic (N+C), or predominantly cytoplasmic (C). (D) The influence of mutations on the nuclear export of NP was confirmed by fractionation and Western blot analysis with anti-NP polyclonal antibody.
In addition, we also performed coimmunoprecipitation assay to investigate whether mutation of NP-NESs affects NP oligomerization. The results indicated that mutation of NESs did not impair the oligomerization of NP (Fig. 8), suggesting that inactivation of NESs leads to NP nuclear retention not through destroying NP oligomerization.
Fig 8.

Coimmunoprecipitation showed that mutation of NP-NESs does not affect its self-association. pcDNA3-FLAG-NP and pCMV-Myc-NP or pcDNA3-FLAG-NP-NES1/2/3m and pCMV-Myc-NP-NES1/2/3m plasmids were simultaneously transfected into 293T cells. At 48 h posttransfection, the cells were lysed in immunoprecipitation buffer, coimmunoprecipitation was performed with anti-FLAG, and the proteins immunoprecipitated (IP) were assayed with anti-Myc monoclonal antibody. The “Input” results show 1/10 of the total proteins included in each binding reaction.
DISCUSSION
Influenza virus NP undergoes nucleocytoplasmic shuttling during viral replication as a part of the virus assembly pathway. Export of NP from the nucleus depends upon the CRM1 export pathway. In the present study, we identified three novel NESs that were responsible for the nuclear export of NP. NES3 was CRM1 dependent, whereas NES1 and NES2 were CRM1 independent, suggesting that other export pathways are also involved in the NP nuclear export. Nuclear retention of NP was observed when all three NESs were simultaneously inactivated. Furthermore, recombinant virus bearing deficient NP-NESs could not be successfully rescued likely due to failure of viral protein expression. In addition, NP oligomerization was required for NP nuclear export possibly by unmasking its NESs. For the first time, we discussed that NP nuclear export might be relevant to viral protein expression, with implications for understanding influenza virus replication.
In an early stage of influenza virus infection, newly synthesized NP is imported into the nucleus mediated by its NLS likely in the form of monomer (4). It has been reported that NP oligomerizes in vivo during virus life cycle (2, 6, 33, 34, 36), which suggested that NPs form oligomers to participate in virus replication. Because oligomerized NPs associate with cRNA or vRNA in the cRNP or vRNP complex and bound RNA maybe covers the NP-NES2, the nuclear export of NP is consequently attenuated, which facilitates virus replication in early stages of virus infection. When newly synthesized NP is so abundant as to excess the quantity needed for forming cRNP or vRNP complex, it tends to aggregate and assemble into trimer, since NP monomer could not be exported out of the nucleus. Simultaneously, the NESs in NP are possibly exposed on the surface of oligomeric NPs and recognized by nuclear export receptors. It also should be noted that several other factors have been identified to regulate the nuclear export of NP, including NP expression level, cell density, phosphorylation, and interaction with F-actin (5, 10). Thus, NP localization is regulated by multiple factors.
As for the roles of NP nuclear export in virus replication, our results supported that it might be involved in the viral protein expression at the posttranscription level. NP-NES is possibly involved in the viral mRNA nuclear export or translation. With respect to influenza A virus mRNA nuclear export, the mechanism is not clearly understood. Nevertheless, some studies have demonstrated that viral mRNA nuclear export depends on active RNA polymerase II (1, 11, 45), and NXF1/TAP are required for nuclear export of some viral mRNA (38, 45), but not all transcripts show the same requirement (1, 43). In addition, viral mRNA nuclear export is not sensitive to treatment with LMB (1, 38, 45). Whether viral proteins promote the viral mRNA nuclear export is worth investigating. In the present study, we found that NP-NESs mutant caused a 2-fold increase in the ratio of nuclear to cytoplasmic luciferase mRNA at 6 h after transfection compared to WT NP (Fig. 5F), indicating there is more mRNA accumulated within the nucleus of NP-NES mutants transfected cells. The present data suggested NP could partially contribute to the viral mRNA trafficking.
In addition, despite the fact that the nuclear export of NP was impaired in the present study, we could not demonstrate the role of NP in the nuclear export of the vRNP because other types of viral protein expression were abolished by the mutation of NP-NESs. It is possible that the absence of NEP/NS2 and M1 led to the nuclear retention of the vRNP. However, we could not rule out that NP assists the nuclear export of vRNP mediated by NEP/NS2 and M1.
Interestingly, we also found that some posttranslational modification sites are localized in the NP-NESs. For example, K184 is within the NES2, which has been identified as the ubiquitination site and is critical for virus replication (21). Whether K184 plays important roles in regulating the nuclear export of NP is worth investigating.
The NP-NESs identified in our study are highly conserved among the influenza A virus isolates (data not shown), implying its important roles in virus replication. In addition, we also found consensus NES-like motifs in the influenza B and C virus NPs (data not shown). Whether the NPs of influenza B or C virus are capable of shuttling from the nucleus to cytoplasm should be further studied.
On the whole, we found that NP nuclear export requires the three novel NP-NESs which are critical for virus replication, likely involved in viral protein expression. In addition, oligomerization of NP is also crucial to its nuclear export. The above results provide clues for investigating the roles of NP nuclear export in virus replication.
ACKNOWLEDGMENTS
This study was supported by the National Basic Research Program (no. 973) of China (2011CB504705), the Chinese Academy of Sciences Innovation Projects (KSCX2-YW-R-158), the National Natural Science Foundation of China (30972185 and 30901073), the National Key Technologies Research and Development Program of China (2010BAD04B01), and the Beijing Municipal Natural Science Foundation (6102018). Wenjun Liu, Xin Ye, and George F. Gao are the principal investigators of the Innovative Research Group of the National Natural Science Foundation of China (NSFC grant 81021003).
Footnotes
Published ahead of print 15 February 2012
REFERENCES
- 1. Amorim MJ, Read EK, Dalton RM, Medcalf L, Digard P. 2007. Nuclear export of influenza A virus mRNAs requires ongoing RNA polymerase II activity. Traffic 8:1–11 [DOI] [PubMed] [Google Scholar]
- 2. Becht H, Weiss HP. 1991. The influenza virus-infected host cell, a target for the immune response. Behring Inst. Mitt. 89:1–11 [PubMed] [Google Scholar]
- 3. Boulan ER, Sabatini DD. 1978. Asymmetric budding of viruses in epithelial monolayers: a model system for study of epithelial polarity. Proc. Natl. Acad. Sci. U. S. A. 75:5071–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boulo S, et al. 2011. Human importin alpha and RNA do not compete for binding to influenza A virus nucleoprotein. Virology 409:84–90 [DOI] [PubMed] [Google Scholar]
- 5. Bui M, Myers JE, Whittaker GR. 2002. Nucleocytoplasmic localization of influenza virus nucleoprotein depends on cell density and phosphorylation. Virus Res. 84:37–44 [DOI] [PubMed] [Google Scholar]
- 6. Coloma R, et al. 2009. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 5:e1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cros JF, Palese P. 2003. Trafficking of viral genomic RNA into and out of the nucleus: influenza, Thogoto and Borna disease viruses. Virus Res. 95:3–12 [DOI] [PubMed] [Google Scholar]
- 8. Cros JF, Garcia-Sastre A, Palese P. 2005. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 6:205–213 [DOI] [PubMed] [Google Scholar]
- 9. Davey J, Dimmock NJ, Colman A. 1985. Identification of the sequence responsible for the nuclear accumulation of the influenza virus nucleoprotein in Xenopus oocytes. Cell 40:667–675 [DOI] [PubMed] [Google Scholar]
- 10. Digard P, et al. 1999. Modulation of nuclear localization of the influenza virus nucleoprotein through interaction with actin filaments. J. Virol. 73:2222–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dolan BP, Knowlton JJ, David A, Bennink JR, Yewdell JW. 2010. RNA polymerase II inhibitors dissociate antigenic peptide generation from normal viral protein synthesis: a role for nuclear translation in defective ribosomal product synthesis? J. Immunol. 185:6728–6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elton D, et al. 2001. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J. Virol. 75:408–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elton D, Medcalf E, Bishop K, Digard P. 1999. Oligomerization of the influenza virus nucleoprotein: identification of positive and negative sequence elements. Virology 260:190–200 [DOI] [PubMed] [Google Scholar]
- 14. Fornerod M, Ohno M, Yoshida M, Mattaj IW. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051–1060 [DOI] [PubMed] [Google Scholar]
- 15. Kim FJ, Beeche AA, Hunter JJ, Chin DJ, Hope TJ. 1996. Characterization of the nuclear export signal of human T-cell lymphotropic virus type 1 Rex reveals that nuclear export is mediated by position-variable hydrophobic interactions. Mol. Cell. Biol. 16:5147–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. la Cour T, et al. 2004. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17:527–536 [DOI] [PubMed] [Google Scholar]
- 17. Lamb RA, Krug RM. 2001. Orthomyxoviridae: the viruses and their replication, p 1487–1531 In Knipe DM, Howley PM. (ed), Fields virology. Lippincott/Williams & Wilkins Co, Philadelphia, PA [Google Scholar]
- 18. Li G, Zhang J, Tong X, Liu W, Ye X. 2011. Heat shock protein 70 inhibits the activity of Influenza A virus ribonucleoprotein and blocks the replication of virus in vitro and in vivo. PLoS One 6:e16546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li HC, et al. 2010. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 6:e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Z, et al. 2009. Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J. Virol. 83:4153–4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao TL, Wu CY, Su WC, Jeng KS, Lai MM. 2010. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J. 29:3879–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu X, et al. 2009. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell Microbiol. 11:730–741 [DOI] [PubMed] [Google Scholar]
- 23. Ma K, Roy AM, Whittaker GR. 2001. Nuclear export of influenza virus ribonucleoproteins: identification of an export intermediate at the nuclear periphery. Virology 282:215–220 [DOI] [PubMed] [Google Scholar]
- 24. Maekawa T, et al. 2008. ATF-2 controls transcription of Maspin and GADD45 alpha genes independently from p53 to suppress mammary tumors. Oncogene 27:1045–1054 [DOI] [PubMed] [Google Scholar]
- 25. Marjuki H, et al. 2006. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Cα-mediated activation of ERK signaling. J. Biol. Chem. 281:16707–16715 [DOI] [PubMed] [Google Scholar]
- 26. Matthews GD, Gur N, Koopman WJ, Pines O, Vardimon L. 2010. Weak mitochondrial targeting sequence determines tissue-specific subcellular localization of glutamine synthetase in liver and brain cells. J. Cell Sci. 123:351–359 [DOI] [PubMed] [Google Scholar]
- 27. Mena I, et al. 1999. Mutational analysis of influenza A virus nucleoprotein: identification of mutations that affect RNA replication. J. Virol. 73:1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neumann G, Castrucci MR, Kawaoka Y. 1997. Nuclear import and export of influenza virus nucleoprotein. J. Virol. 71:9690–9700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neumann G, Hughes MT, Kawaoka Y. 2000. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 19:6751–6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neumann G, et al. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. U. S. A. 96:9345–9350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Neill RE, Talon J, Palese P. 1998. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 17:288–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pleschka S, et al. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signaling cascade. Nat. Cell Biol. 3:301–305 [DOI] [PubMed] [Google Scholar]
- 33. Pons MW. 1967. Studies on influenza virus ribonucleic acid. Virology 31:523–531 [DOI] [PubMed] [Google Scholar]
- 34. Pons MW, Schulze IT, Hirst GK, Hauser R. 1969. Isolation and characterization of the ribonucleoprotein of influenza virus. Virology 39:250–259 [DOI] [PubMed] [Google Scholar]
- 35. Portela A, Digard P. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 83:723–734 [DOI] [PubMed] [Google Scholar]
- 36. Prokudina-Kantorovich EN, Semenova NP. 1996. Intracellular oligomerization of influenza virus nucleoprotein. Virology 223:51–56 [DOI] [PubMed] [Google Scholar]
- 37. Prokudina EN, Semenova NP. 1991. Localization of the influenza virus nucleoprotein: cell-associated and extracellular non-virion forms. J. Gen. Virol. 72(Pt 7):1699–1702 [DOI] [PubMed] [Google Scholar]
- 38. Read EK, Digard P. 2010. Individual influenza A virus mRNAs show differential dependence on cellular NXF1/TAP for their nuclear export. J. Gen. Virol. 91:1290–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singhal PK, Rajendra Kumar P, Subba Rao MR, Mahalingam S. 2006. Nuclear export of simian immunodeficiency virus Vpx protein. J. Virol. 80:12271–12282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stade K, Ford CS, Guthrie C, Weis K. 1997. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 90:1041–1050 [DOI] [PubMed] [Google Scholar]
- 41. Stegmann T, White JM, Helenius A. 1990. Intermediates in influenza induced membrane fusion. EMBO J. 9:4231–4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stitz L, et al. 1990. Characterization and immunological properties of influenza A virus nucleoprotein (NP): cell-associated NP isolated from infected cells or viral NP expressed by vaccinia recombinant virus do not confer protection. J. Gen. Virol. 71(Pt 5):1169–1179 [DOI] [PubMed] [Google Scholar]
- 43. Vogel U, Kunerl M, Scholtissek C. 1994. Influenza A virus late mRNAs are specifically retained in the nucleus in the presence of a methyltransferase or a protein kinase inhibitor. Virology 198:227–233 [DOI] [PubMed] [Google Scholar]
- 44. Wang P, Palese P, O'Neill RE. 1997. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza A virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 71:1850–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang W, et al. 2008. Imaging and characterizing influenza A virus mRNA transport in living cells. Nucleic Acids Res. 36:4913–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y, Zhu W, Levy DE. 2006. Nuclear and cytoplasmic mRNA quantification by SYBR green based real-time RT-PCR. Methods 39:356–362 [DOI] [PubMed] [Google Scholar]
- 47. Wang Z, et al. 2011. Cyclophilin E functions as a negative regulator to influenza virus replication by impairing the formation of the viral ribonucleoprotein complex. PLoS One 6:e22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watanabe K, et al. 2001. Inhibition of nuclear export of ribonucleoprotein complexes of influenza virus by leptomycin B. Virus Res. 77:31–42 [DOI] [PubMed] [Google Scholar]
- 49. Weber F, Kochs G, Gruber S, Haller O. 1998. A classical bipartite nuclear localization signal on Thogoto and influenza A virus nucleoproteins. Virology 250:9–18 [DOI] [PubMed] [Google Scholar]
- 50. Wen W, Meinkoth JL, Tsien RY, Taylor SS. 1995. Identification of a signal for rapid export of proteins from the nucleus. Cell 82:463–473 [DOI] [PubMed] [Google Scholar]
- 51. Whittaker G, Bui M, Helenius A. 1996. Nuclear trafficking of influenza virus ribonucleoproteins in heterokaryons. J. Virol. 70:2743–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu WW, Pante N. 2009. The directionality of the nuclear transport of the influenza A genome is driven by selective exposure of nuclear localization sequences on nucleoprotein. Virol. J. 6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu WW, Sun YH, Pante N. 2007. Nuclear import of influenza A viral ribonucleoprotein complexes is mediated by two nuclear localization sequences on viral nucleoprotein. Virol. J. 4:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu C, Meng S, Liu X, Sun L, Liu W. 2011. Chicken cyclophilin A is an inhibitory factor to influenza virus replication. Virol. J. 7:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082 [DOI] [PubMed] [Google Scholar]
- 56. Yewdell JW, Frank E, Gerhard W. 1981. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J. Immunol. 126:1814–1819 [PubMed] [Google Scholar]