

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV), a human tumor virus, encodes two homologous membrane-associated E3 ubiquitin ligases, modulator of immune recognition 1 (MIR1) and MIR2, to evade host immunity. Both MIR1 and MIR2 downregulate the surface expression of major histocompatibility complex class I (MHC I) molecules through ubiquitin-mediated endocytosis followed by lysosomal degradation. Since MIR2 additionally downregulates a costimulatory molecule (B7-2) and an integrin ligand (intercellular adhesion molecule 1 [ICAM-1]), MIR2 is thought to be a more important molecule for immune evasion than MIR1; however, the molecular basis of the MIR2 substrate specificity remains unclear. To address this issue, we determined which regions of B7-2 and MIR2 are required for MIR2-mediated B7-2 downregulation. Experiments with chimeras made by swapping domains between human B7-2 and CD8α, a non-MIR2 substrate, and between MIR1 and MIR2 demonstrated a significant contribution of the juxtamembrane (JM) region of B7-2 and the intertransmembrane (ITM) region of MIR2 to MIR2-mediated downregulation. Structure prediction and mutagenesis analyses indicate that Phe119 and Ser120 in the MIR2 ITM region and Asp244 in the B7-2 JM region contribute to the recognition of B7-2 by MIR2. This finding provides new insight into the molecular basis of substrate recognition by MIR family members.
INTRODUCTION
Host immune defense against viral infections is mediated by antigen-specific cytotoxic T lymphocytes (CTLs), which are activated by engagement of the T cell receptor (TCR) with viral peptide-major histocompatibility complex class I (MHC I) molecules expressed on the surface of infected cells. MHC I is a heterodimeric glycoprotein that consists of a transmembrane (TM)-type heavy chain and a small light chain (β2-microglobulin) (2). The pathogen-derived peptides are presented within a groove composed of the α1 and α2 domains of the MHC I heavy chain, thereby initiating CTL immunity (36).
Viruses have evolved various mechanisms to evade host immunity, including inhibition of MHC I expression or function through viral immune evasion molecules (28, 29). However, inhibition of MHC I expression renders the infected cells susceptible to natural killer (NK) cell-mediated killing. The activation of NK cells is regulated by the balance between inhibitory and stimulatory signals. MHC I provides an inhibitory signal through killer immunoglobulin (Ig)-like receptors (30), and many immunoreceptors, including intercellular adhesion molecule 1 (ICAM-1) and B7-2, provide stimulatory signals (1, 5, 18, 40). Therefore, to suppress NK-mediated cytotoxicity, viruses have evolved additional evasion strategies (20, 27).
Kaposi's sarcoma-associated herpesvirus (KSHV) is associated with several human tumors: Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease (39). KSHV encodes many types of immune evasion molecules in its genome to establish latent infection (8, 22), among them, modulator of immune recognition 1 and 2 (MIR1 and -2), which are expressed during the early lytic cycle and localize in the endoplasmic reticulum (ER). MIR1 and -2 are homologous E3 ubiquitin ligases and induce a dramatic degradation of human MHC I by ubiquitin-mediated endocytosis (3, 10, 12). Since MHC I is such a critical immune receptor for protection from viral infection, these ubiquitin E3 ligases are considered important molecules for immune evasion by KSHV (8, 26). Intriguingly, MIR2 can downregulate other important immune receptors in addition to MHC I, e.g., B7-2 and ICAM-1 (8, 9, 18). This additional function suggests that MIR2 is a E3 ubiquitin ligase critical for establishing full immune evasion by KSHV, because these additional MIR2 substrates are also involved in the activation of T cells. Thus, it is important to understand the molecular mechanism of MIR2-mediated downregulation; however, it remains unclear how MIR2 recognizes its substrates.
Both MIR E3 ligases are type III membrane proteins and have similar secondary structures, including a variant type of RING domain (RINGv) at the amino terminus located in the cytoplasm, two transmembrane (TM) regions connected by a short intertransmembrane (ITM) region located in the ER lumen, and a long cytoplasmic (CYT) tail at the carboxyl terminus (26, 32). MIR1 and MIR2 have 40% amino acid identity (25, 31), and the only well-conserved region is the RINGv domain, which plays a role in recruiting E2 ubiquitin-conjugating enzymes to catalyze ubiquitination of lysine residues located in the cytoplasmic tail of substrates (26). Regarding substrate recognition, it has been reported that the region, including the two TMs and ITM of MIR2, is sufficient for recognition of B7-2 (32), and that report suggested that MIR2 recognizes B7-2 through the interaction of the TMs of MIR2 and B7-2; however, a precise examination of how MIR2 recognizes B7-2 is still lacking.
In this study, we systematically examined the important regions of MIR2 and B7-2 that are required for MIR2-mediated B7-2 downregulation. By using substrate chimeras constructed by swapping regions between human B7-2 and human CD8α chain (CD8α), which is not an MIR2 substrate (19), we found that the B7-2 juxtamembrane (JM) region is important for its downregulation. The analysis performed using MIR chimeras made by swapping ITMs between MIR1 and MIR2 revealed the importance of the ITM region of MIR2. In addition, structure prediction and mutagenesis analysis results suggest that anchoring the aromatic ring of Phe119 in the plasma membrane supports the suitable positioning of the hydroxyl group of Ser120, which forms an initial interaction core with Asp244 of the B7-2 JM. Thus, this is the first report describing the involvement of the MIR2 ITM in substrate selection. This finding provides new insight into the molecular basis of substrate recognition by MIR family members.
MATERIALS AND METHODS
Plasmids.
To coexpress recombinant MIR proteins with green fluorescent protein (GFP), the coding regions of KSHV MIR1 and MIR2 were obtained by PCR using the following primers: 5′-GCGAATTCGCCGCCATGGAAGATGAGGATGTT-3′ (with an EcoRI site and a Kozak sequence at its 5′ end) and 5′-CCTCTAGAATGAAACATAAGGGCAGACG-3′ (with an XbaI site at its 5′ end) for MIR1 and 5′-GCGAATTCGCCGCCATGGCGTCTAAGGACGTA-3′ (with an EcoRI site and a Kozak sequence at its 5′ end) and 5′-CCTCTAGAACCGTTGTTTTTTGGATG-3′ (with an XbaI site at its 5′ end) for MIR2. After digestion with EcoRI and XbaI, the PCR products were ligated into the EcoRI/XbaI-digested pTracer-EF vector (Invitrogen). To generate ITM region-swapping chimeras, each of the ITM sequences (residues 109 to 118 of MIR1 and residues 113 to 122 of MIR2) was exchanged between MIR1 and MIR2 by an overlapping PCR method (14). Alanine (or glycine for endogenous alanine residues) scanning and site-saturation mutagenesis of MIR2 were performed by site-directed mutagenesis based on pTracer-EF MIR2.
To examine the surface expression level of substrates, 3×FLAG-tagged substrates were constructed. The coding regions of human CD8α and B7-2 were obtained by PCR using their cDNAs (19) as the templates. The primer sequences were 5′-TGCCCAAGCTTAGCCAGTTCCGGGTGTCGCCGCTG-3′ (with an HindIII site at its 5′ end) and 5′-ATCGCTCTAGATTAGACGTATCTCGCCGAAAGGCT-3′ (with an XbaI site at its 5′ end) for CD8α and 5′-TGCGGAAGCTTGCTTATTTCAATGAGACTGCAGAC-3′ (with an HindIII site at its 5′ end) and 5′-ATCGCTCTAGATTAAAAACATGTATCACTTTTGTC-3′ (with an XbaI site at its 5′ end) for B7-2. After digestion with HindIII and XbaI, the PCR products were ligated into the HindIII/XbaI-digested p3×FLAG cytomegalovirus 9 (CMV-9) vector (Sigma-Aldrich). To generate the chimeras constructed by swapping regions between human CD8α and human B7-2, the CD8α and B7-2 protein sequences were analyzed by the use of SMART software (21, 33) and could be separated into four regions: the extracellular (EX) region (residues 22 to 181 of CD8α and residues 30 to 245 of B7-2), the juxtamembrane (JM) region (residues 172 to 181 of CD8α and residues 236 to 245 of B7-2), the transmembrane (TM) region (residues 182 to 206 of CD8α and residues 246 to 270 of B7-2), and the cytoplasmic (CYT) region (residues 207 to 235 of CD8α and residues 271 to 329 of B7-2). Each region of CD8α and B7-2 was exchanged by an overlapping PCR method (14) and ligated into the p3×FLAG CMV-9 vector. Alanine mutants and site-saturated mutants were generated by site-directed mutagenesis based on the B7-2JM-CYT chimera.
Downregulation assay.
The human Burkitt lymphoma-like (BJAB) cell line was maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 μg/ml), and streptomycin (100 U/ml) at 37°C under 5% CO2. For recombinant protein expression, 1 × 107 cells were transfected with 10 μg of p3×FLAG CMV-9-based substrate-expressing plasmids and 10 μg of pTracer-EF-based plasmids by electroporation at 260 V and 975 μF using a Gene Pulser II system (Bio-Rad). At 24 h after electroporation, cells were stained with 10,000×-diluted phycoerythrin (PE)-conjugated anti-DDDDK tag antibody (clone M2; Abcam) in 100 μl of phosphate-buffered saline (PBS) (pH 7.4)-supplemented 2% FBS at 4°C for 15 min. Stained cells were washed with PBS (pH 7.4)-supplemented 2% FBS and analyzed by FACSCalibur (BD). Data were analyzed with CellQuestPro software (BD).
Calculation of relative sensitivities of substrates and activity of MIRs.
For statistical evaluation of the extent of MIR2-mediated downregulation, the relative sensitivities of substrates and relative activities of MIRs were calculated. In each experiment, the percentage of PE-positive cells (i.e., substrate-expressing cells) was measured in the population of GFP-positive cells, and this percentage was used as the value indicated in the following formula. The relative sensitivity of each mutant type of substrate to MIR2-mediated downregulation was calculated as percent relative sensitivity = (Nx − X)/(Nw − W) × 100, where Nx represents the value obtained from the experiment in which the mutant substrate (i.e., each sample) and GFP were expressed, Nw represents the value obtained from the experiment in which the wild-type (WT) substrate (i.e., the wild type for each sample) and GFP were expressed, X represents the value obtained from the experiment in which the mutant substrate, GFP, and MIR2 were expressed, and W represents the value obtained from the experiment in which the wild-type substrate, GFP, and MIR2 were expressed. The relative activity of each mutant type of MIR was calculated as percent relative activity = (N − X)/(N − W) × 100, where N represents the value obtained from the experiment in which GFP and the indicated substrate were expressed, X represents the value obtained from the experiment in which GFP, each mutant MIR2, and the indicated substrate were expressed, and W represents the value obtained from the experiment in which GFP, wild-type MIR2, and the indicated substrate were expressed. The relative sensitivities and activities are represented as means and standard deviations (SD) calculated from the results of three individual experiments. The statistical significance of the data was calculated by Student's t test.
Detection of MIR-mediated ubiquitination.
Cells of the HeLa human cervical cancer cell line were maintained in modified Eagle's medium (MEM) supplemented with 10% FBS, penicillin (100 μg/ml), and streptomycin (100 U/ml) at 37°C under 5% CO2. HeLa cells (70% confluent) were transfected using 4 μg of plasmids and 6 μl of 25 kDa of branched polyethylenimine Max (Polysciences) (1 mg/ml; molecular weight [MW], 40,000). Eighteen hours after transfection, cells were lysed in lysis buffer (1% Nonidet P-40, 0.1% sodium dodecyl sulfate [SDS], 0.1% sodium deoxycholate, 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, Complete EDTA-free protease inhibitor [Roche]) on ice for 30 min and were centrifuged at 10,000 × g at 4°C for 10 min. A 30-μl volume of anti-FLAG M2 affinity gel (Sigma-Aldrich) was added to the lysates and mixed at 4°C with gentle rotation. One hour later, the affinity gel was washed three times with lysis buffer, and then bound proteins were eluted with 100 μl of 3×FLAG peptide (150 μg/ml in lysis buffer). Whole-cell lysates and precipitated proteins were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using 8 or 15% gels. For immunoblotting analysis, separated proteins were transferred to polyvinylidene difluoride membranes (Millipore) by the use of a semidry blotter (Bio-Rad) at 15 V for 45 min. The blotted membranes were incubated with 3% skim milk–PBS-Tween (PBST) (PBS [pH 7.4], 0.05% Tween 20) at room temperature for 60 min and then incubated with 1,000×-diluted anti-6×His tag antibody (polyclonal; MBL), anti-ubiquitin antibody (clone FK2; Enzo Life Sciences), or horseradish peroxidase (HRP)-conjugated anti-FLAG M2 antibody (clone M2; Sigma-Aldrich) in PBST at room temperature for 60 min. After three washes with PBST were performed, the membranes were incubated with 2,000×-diluted HRP-conjugated anti-mouse or -rabbit IgG antibodies at room temperature for 30 min. After three washes with PBST were performed, the membranes were visualized with Immobilon Western chemiluminescent HRP substrate (Millipore).
Analysis of expression of MIR2 mutants.
Cells of the HEK293T human embryonic kidney cell line were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, penicillin (100 μg/ml), and streptomycin (100 U/ml) at 37°C under 5% CO2. For recombinant protein expression, 90% confluent 293T cells in 6-well culture dishes were transfected by lipofection with 4 μg of plasmid and 6 μl of branched polyethylenimine (Sigma-Aldrich) (1 mg/ml; MW, 25,000). After 48 h, cells were lysed in lysis buffer as described above and centrifuged at 10,000 × g at 4°C for 10 min. Cleared lysates were subjected to 15% SDS-PAGE and examined with 1/1,000-diluted anti-6×His tag polyclonal antibody (code PM032; MBL) or anti-alpha tubulin antibody (clone B-5-1-2; Zymed Laboratory) or anti-FLAG antibody (clone M2, Sigma-Aldrich) as the primary antibody.
Structure prediction analysis.
The structures of MIR2 (regions TM1 plus ITM plus TM2), B7-2 (regions JM plus TM), and MIR2 (regions TM1 plus ITM plus TM2)–B7-2 (regions JM plus TM) complex embedded in the membrane were predicted by performing replica-exchange molecular dynamic (REMD) simulations in which a larger conformational space is sampled by swapping temperatures between neighboring replicas based on the metropolis criterion (35). The CHARMM program and all-atom PARM22 force field with dihedral cross-term corrections (CMAP) for proteins were used for the simulation (4, 23). The effect of solvent and membrane was introduced by using the GBSW (17) (Generalized Born model with a simple Switching function) implicit membrane/solvent model, in which the membrane is approximated as an infinite planar low-dielectric slab. The model has been used for predicting transmembrane helix oligomers, e.g., the dimeric structure of glycophorin A (6). We used 0.04 kcal/mol/Å2 for surface tension, 5 Å for each membrane/solvent interface, and 27.5 Å for the thickness of membrane hydrophobic region to mimic a POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) membrane bilayer. The temperature range for the REMD simulations was between 300°K and 600°K. The Langevin thermostat was used to control the temperature. The SHAKE method was applied to bonds that involved hydrogen atoms, and the time step was set to 2 fs. The replica exchanges were performed every 2 ps. A 16-Å cutoff distance for nonbonding interactions was applied. The MMTSB toolset (11) was used to manage the REMD simulation, loop modeling, and analysis. A RMSD K-cluster algorithm was carried out for the structures obtained at 300°K, and the cluster radius was set to 2.5 Å. The illustrations of proteins were prepared by using VMD (15).
In the structural prediction of MIR2, we first prepared two MIR2 transmembrane helices as separated fragments: fragment 1, including TM1 and the partial EX region (E86LFLLMSVVVAGLVGVALCTWTLLVILTAP115), and fragment 2, including the partial EX region and TM2 (A116GTFSPGAVLGFLCFFGFYQIFIVFAFGGICRVSG150). The residues underlined are MIR2 TM regions predicted to be ideal helices by the SOSUI server (13). Then, we placed the fragments at the membrane center. The initial orientations of these fragments were parallel to the normal membrane orientation. The distance between the centers of mass of the two fragments was then set to 20 Å. Each helix was rotated every 45 degrees to generate 64 different initial configurations. A cylindrical wall boundary with a radius of 30 Å was added around the two MIR2 fragments by the use of the MMFP module of CHARMM to prevent the fragments from moving far away from each other. A 64-replica REMD simulation was carried out for 20 ns for enhanced sampling of the TM1-TM2 contact interfaces. The simulated structures were clustered, and the ITM region (residues 112 to 124) of the largest cluster centroid structure was rebuilt by using the LOOPMODEL module in the MMTSB toolset. Another REMD simulation was carried out for 20 ns by taking these helical hairpin structures as the initial structures, and the REMD-simulated structures were clustered. The positions of side chains of selected residues above the membrane center were determined.
In the structural modeling of B7-2, residues 239 to 271 (JM-plus-TM fragment) of B7-2 were built as an ideal helix, and the TM region (residues 249 to 271) was placed at the membrane center and parallel to the normal membrane orientation. REMD simulation was performed for 20 ns, and then simulated structures obtained at 300°K were clustered. In the modeling of the MIR2–B7-2 complex, the predicted MIR2 and B7-2 fragments were placed in the membrane and the distance between the centers of the masses of the two fragments was then set to 35 Å. MIR2 was rotated every 45 degrees and B7-2 was rotated every 90 degrees to generate 32 different initial configurations. A cylindrical wall boundary with a radius of 30 Å was added around the two fragments to prevent the fragments from moving far away from each other. A 32-replica REMD simulation was carried out for 30 ns, and the REMD-simulated structures at 300 K were clustered. The positions of the side chains of selected MIR2–B7-2 residues above the membrane center were determined. The percentages of the clusters and the shortest contact distances between MIR2 and B7-2 in the centroid structure for each cluster were examined, and the cluster structures were determined.
RESULTS
The B7-2 JM region is involved in MIR2-mediated downregulation.
To determine which region of B7-2 is essential for the substrate recognition by MIR2, we constructed chimeras by swapping regions between CD8α and B7-2. Both immunoreceptors are type-I transmembrane proteins and could be separated into three corresponding regions: EX, TM, and CYT. The chimeras depicted in Fig. 1A were cotransfected with a pTracer-EF MIR2, which expresses both MIR2 and GFP, into human BJAB cells. At 24 h after cotransfection, cells were analyzed by flow cytometry using anti-DDDDK antibody to measure the cell surface expression levels of the chimeras. Since GFP expression denoted cells that had been successfully transfected, we examined chimera expression levels on GFP-positive cells (see Fig. S1 in the supplemental material), and the relative sensitivity of each chimera was calculated as described in Materials and Methods.
Fig 1.

The B7-2 JM region is important for MIR2-mediated downregulation. (A) Schematic representation of the chimeras made by swapping regions between human B7-2 and human CD8α. The B7-2 and CD8α proteins can be separated into three distinct regions: extracellular (EX; residues 22 to 181 of CD8α and residues 30 to 245 of B7-2), transmembrane (TM; residues 182 to 206 of CD8α and residues 246 to 270 of B7-2), and cytoplasmic (CYT; residues 207 to 235 of CD8α and residues 271 to 329 of B7-2). Residues 236 to 245 of B7-2 and 172 to 181 of CD8 were designated the juxtamembrane (JM) region. (B) Downregulation of chimeras by MIR2. The FLAG-tagged chimeras were cotransfected with control GFP (mock) or GFP-MIR2 vectors into BJAB cells and the cell surface expression level of the chimeras was analyzed by flow cytometry. The relative sensitivity of each chimera is shown. Error bars represent SD (n = 3). The asterisk indicates significant difference (P < 0.05). (C) Downregulation analysis of additional chimeras by MIR2. The relative sensitivity of each is shown. Error bars represent SD (n = 3). The asterisk indicates significant difference (P < 0.05).
As shown in Fig. 1B, expression of the B7-2 wild type from the surface of BJAB cells was downregulated by coexpression of MIR2, while the cell surface expression levels of CD8α and the B7-2CYT chimera were unaffected. The lysine residues in the B7-2 CYT are known to be substrates for MIR2-mediated ubiquitination (10), and the positioning of the lysines is essential (7); however, this region by itself was not sufficient for downregulation by MIR2. Consistent with a previous report (32), the B7-2TM-CYT chimera, which contains the TM region of B7-2, was downregulated by MIR2, demonstrating the contribution of this region to MIR2-mediated downregulation. However, the B7-2EX-CYT chimera, which consists of the B7-2 EX and CYT and the CD8 TM regions, was also significantly downregulated by MIR2 and the extent of downregulation was statistically greater than that of the B7-2TM-CYT chimera (Fig. 1B). This finding suggested that the B7-2 EX plays a more important role in the MIR2-mediated downregulation than the TM region. Therefore, we performed extensive experiments to understand how the B7-2 EX region contributes to MIR2-mediated recognition.
B7-2 has two immunoglobulin (Ig)-like domains in its EX region, and the second Ig-like domain (IgC type) (16) is joined to the TM region by a short JM region consisting of nine amino acids (i.e., EDPQPPPDH). To reveal which region is involved in the B7-2 EX-mediated downregulation, we constructed additional chimeras, B7-2JM-CYT and B7-2EXΔJM-CYT, which contained the B7-2 and CD8α JM regions, respectively (Fig. 1A). The downregulation assay demonstrated that the B7-2JM-CYT chimera was more efficiently downregulated than the B7-2EXΔJM-CYT chimera (Fig. 1C), a result clearly demonstrating that the B7-2 JM region plays a major role in the B7-2 EX-mediated downregulation by MIR2. Because the B7-2JM-CYT chimera contains the minimum region for EX-mediated downregulation, we used it as a model substrate for a JM-mediated downregulation assay in the following experiments.
The MIR2 ITM region participates in JM-mediated B7-2 downregulation.
Our finding that B7-2 JM is the important region for EX-mediated downregulation by MIR2 suggested the potential importance of the MIR2 ITM in recognition, since this region is exposed to the extracellular (or ER lumenal) space. Therefore, to address this issue, we constructed ITM chimeras by swapping regions between MIR2 (i.e., TAPAGTFSPG) and MIR1 (i.e., GGHDPEIDHV) as shown in Fig. 2A and performed the downregulation assay using the B7-2JM-CYT chimera as a substrate (Fig. 2B). Consistent with previous reports (9, 18), MIR1 did not downregulate the B7-2JM-CYT chimera (Fig. 2B); however, when its ITM region was replaced by the MIR2 ITM region (MIR1ΔITM MIR2ITM), the chimera acquired downregulation activity. In contrast, the MIR2 chimera with the MIR1 ITM region (MIR2ΔITM MIR1ITM) completely lost activity (Fig. 2B). These results demonstrate that the MIR2 ITM region is required for the B7-2 JM-mediated downregulation and, taken together with the data in Fig. 1, strongly suggest that the MIR2 ITM region directly recognizes the B7-2 JM region.
Fig 2.

The MIR2 ITM region contributes to B7-2 downregulation by ubiquitination. (A) Schematic representation of the intertransmembrane (ITM) chimeras made by swapping regions between KSHV MIR1 and MIR2. MIR1 and MIR2 have short ITM regions (residues 109 to 118 of MIR1 and 113 to 122 of MIR2), and these regions were swapped. (B) Downregulation analysis of B7-2JM-CYT by use of the MIR chimeras. The FLAG-tagged B7-2JM-CYT was cotransfected with the control GFP (mock) or GFP-MIR chimera vector into BJAB cells, and the cell surface expression level of B7-2JM-CYT was analyzed by flow cytometry. The relative downregulation activity of each MIR chimera is shown. Error bars represent SD (n = 3). Double asterisks indicate significant difference (P < 0.01). (C) The detection of JM-mediated ubiquitination. HeLa cells were cotransfected with a B7-2JM-CYT chimera and MIRs. Two days later, the substrates were immunoprecipitated (IP) with anti-FLAG antibody and the precipitated proteins were subjected to immunoblotting (IB) analysis using antiubiquitin antibodies (bottom). Whole-cell extract (WCE) was examined using anti-6×His tag (top).
To verify whether the MIR2 ITM-mediated downregulation depends on ubiquitination, we performed a ubiquitination assay using these MIR chimeras. After coexpression of the B7-2JM-CYT and MIR chimeras in HeLa cells, the B7-2JM-CYT chimera was immunoprecipitated with anti-FLAG and bound proteins were analyzed by immunoblotting (Fig. 2C). The B7-2JM-CYT chimera was found to be ubiquitinated by MIR2 and the MIR1ΔITM MIR2ITM chimera but not by MIR1 or the MIR2ΔITM MIR1ITM chimera (Fig. 2C).
MIR2 Phe119 and Ser120 are important residues for ITM-mediated downregulation.
To further explore the molecular basis of MIR2 ITM-mediated downregulation, we defined the important amino acid residues in both the MIR2 ITM and B7-2 JM regions. First, we performed alanine-scanning mutagenesis of the MIR2 ITM region and then, after confirming that the mutants were expressed at equivalent levels in BJAB cells (see Fig. S2A in the supplemental material), measured their activity using the B7-2JM-CYT chimera as a target molecule (Fig. 3A). This analysis demonstrated a significant loss of activity in the F119A and S120A mutants and a moderate loss of activity in the P115A mutant (Fig. 3A).
Fig 3.

Mutational analysis identifies amino acids important for ITM-mediated B7-2 downregulation. (A) Analysis of the MIR2 ITM region by alanine substitution mutagenesis. FLAG-tagged B7-2JM-CYT was cotransfected with control GFP (mock) or the indicated GFP mutant MIR2 vector, and the cell surface expression level of B7-2JM-CYT was analyzed by flow cytometry. The relative activity of each mutant MIR2 is shown. Error bars represent SD (n = 3). Double asterisks indicate significant difference (P < 0.01). (B) Analysis of the B7-2 JM region by alanine substitution mutagenesis. FLAG-tagged B7-2JM-CYT or the indicated mutant FLAG-tagged B7-2JM-CYT was cotransfected with the control GFP or GFP-MIR2 vector, and the cell surface expression level of each mutant was analyzed by flow cytometry. The relative sensitivity of each mutant B7-2JM-CYT is shown. Error bars represent SD (n = 3). The asterisk indicates significant difference (P < 0.05).
Next, we also performed alanine-scanning mutagenesis on the B7-2 JM by using the B7-2JM-CYT chimera. Compared to the B7-2JM-CYT chimera wild type, the extent of downregulation seen with D244A was significantly reduced (Fig. 3B). However, the level of inhibition with the D244A mutation was incomplete, and all the other mutants except that with the H245A mutation showed a trend of slight insensitivity to MIR2-mediated downregulation, suggesting that not only Asp244 but also other residues in the JM region are involved in this process.
Structure prediction analysis suggests the importance of side chains of Phe119 and Ser120 of MIR2.
To examine how particular residues could contribute to substrate recognition, we used the REMD method to perform a simulation of B7-2 (regions JM plus TM) and MIR2 (regions TM1 plus ITM plus TM2) complex formation in a membrane environment (35). The simulated MIR2–B7-2 complex structures were clustered, and the top 6 clusters are shown in Fig. S3A in the supplemental material. Among those clusters, the majority of B7-2 binding to MIR2 took place from the Ser120 side (in clusters A, B, D, E, and F) (see Fig. S3B in the supplemental material); however, clusters A, B, and D were interpreted as artifacts resulting from the use of a short B7-2 fragment (regions JM plus TM) in the simulation because the residue at the tip of the examined B7-2 (i.e., Pro239) is located at the shortest contact interface with MIR2 (see Table S1 in the supplemental material). Of the remaining clusters (i.e., clusters E and F), we chose cluster E as the representative MIR2–B7-2 complex model, since that cluster has the shortest residue contact pairing between MIR2 and B7-2 (see Table S1 in the supplemental material).
A representative MIR2–B7-2 cluster E complex model is shown in Fig. 4A. Asp244 of the B7-2 JM region was located in the vicinity of the surface of the membrane. REMD simulation of MIR2 showed that it formed a TM helix-ITM loop-TM helix structure as expected. In the MIR2 ITM region, the structure and the side chain of Phe119 were buried in the membrane hydrophobic core by its hydrophobic side chain (Fig. 4). The side chain of Ser120 of the MIR2 ITM region was inserted into the membrane/solvent interface (Fig. 4B); however, the hydroxyl group of Ser120 was located near the extracellular space (Fig. 4A). The side chain of Ser120 of the MIR2 ITM and that of Asp244 of the B7-2 JM were predicted to be located similar distances above the membrane surface (Fig. 4B), and the distance between these residues is sufficiently small to allow the formation of a hydrogen bond (see Table S1 in the supplemental material). Thus, the hydroxyl group of Ser120 in the MIR2 ITM might directly interact with the side chain of Asp244 in the B7-2 JM through a hydrogen bond. With these results taken together, we hypothesized that Phe119 of the MIR2 ITM might generate a suitable loop structure to promote the functional association between the MIR2 ITM and the B7-2 JM. It is worth noting that Thr118 was not observed to form a hydrogen bond with Asp244 in the simulation, even though its position and side chain are similar to those Ser120.
Fig 4.

Simulated three-dimensional structures of B7-2 and MIR2. (A) The overall structure of the simulated B7-2 (regions JM plus TM) and MIR2 (regions TM1 plus ITM plus TM2) in the membrane environment. Main chains are depicted as a cartoon model; orange and gray show B7-2 and MIR2, respectively. Amino acids involved in MIR2-ITM-medicated B7-2 downregulation are represented by spheres; cyan, red, and white show carbon, oxygen, and hydrogen, respectively. (B) The position distribution of the selected residue side chain based on the membrane center for the MIR2–B7-2 complex. The position of a residue side chain was determined by the centers of mass of heavy atoms in a residue side chain. The green lines indicated the membrane/solvent boundary.
Site-saturation mutagenesis supports the hypothesis obtained by structure prediction analysis.
To examine the hypothesis concerning the contribution of Phe119 and Ser120 of the MIR2 ITM and Asp244 of the B7-2 JM region that was obtained by structure prediction analysis, we performed site-saturation mutagenesis analysis of these three residues. First, we performed the downregulation assay using the B7-2JM-CYT chimera and site-saturated mutants of MIR2 Phe119 (F119X mutants). None of the mutants was as effective as the WT; however, the downregulation activity of the F119H, F119R, and F119Y mutants was greater than that of the other mutants (Fig. 5A). Since histidine and arginine both have positively charged side chains, it was expected that F119H and F119R would participate in an artificial charge-charge interaction with the negatively charged amino acid side chains in the B7-2 JM region (i.e., E237, D239, and D244). Indeed, F119H and F119R could not downregulate a D244N mutant of B7-2JM-CYT (data not shown) that was significantly downregulated by wild-type MIR2 (Fig. 5C). Therefore, F119Y was identified as the only mutant with significant activity corresponding to downregulation of B7-2JM-CYT. As discussed below, this result supports the structure prediction hypothesis, since both phenylalanine and tyrosine contain a common hydrophobic six-member ring structure.
Fig 5.

Site-saturation mutagenesis supports the importance of Phe119 and Ser120 of MIR2 ITM and Asp244 of B7-2 JM. (A and B) Downregulation of B7-2JM-CYT by the site-saturated mutants of MIR ITM Phe119 (A) and Ser120 (B). FLAG-tagged B7-2JM-CYT was cotransfected with the control GFP (mock) or the indicated GFP mutant MIR2 vector, and the cell surface expression level of B7-2JM-CYT was analyzed by flow cytometry. The relative activity of each MIR2 mutant is shown. Error bars represent SD (n = 3). The asterisk indicates significant difference (P < 0.05) between the results obtained for the indicated mutant and others after excluding the mutants that were replaced by basic amino acids. (C) Analysis of site-saturated mutants of B7-2JM-CYT Asp244. The site-saturated mutants of FLAG-tagged B7-2JM-CYT were cotransfected with control GFP or GFP-MIR2 vector, and the cell surface expression level of each mutant was analyzed by flow cytometry. The relative sensitivity of each mutant is shown. Error bars represent SD (n = 3).
Next, we performed site-saturation mutagenesis of MIR2 Ser120 (S120X mutants) and a downregulation assay using the B7-2JM-CYT chimera as a substrate. Among the 19 mutants, S120K, S120R, and S120T showed the highest activity (Fig. 5B). However, similar to the situation with F119X, the existence of artificial charge-charge interactions was suspected in the case of S120K and S120R. Indeed, both S120K and S120R were unable to downregulate a B7-2JM-CYT D244N mutant (data not shown), suggesting that a serine or threonine is required at position 120 of the MIR2 ITM region. Since both of these amino acids contain the hydroxyl group, this result also supports our hypothesis. We confirmed the equal expression levels of the two MIR mutants (see Fig. S2B and S2C in the supplemental material).
Finally, we also generated site-saturated mutants of Asp244 of the B7-2 JM region by using the B7-2JM-CYT chimera (D244X mutants) and examined the extent of MIR2-mediated downregulation (Fig. 5C). This experiment revealed that mutants in which the Asp was replaced with hydrophobic residues showed a tendency toward hyposensitivity to MIR2. In particular, D244C, D244I, D244L, and D244W exhibited a significant reduction in their sensitivity to MIR2-mediated downregulation (Fig. 5C). Since hydrophobic amino acids would reduce the probability of interaction with a Ser hydroxyl group, Asp244 might be one of acceptors for Ser120.
DISCUSSION
Here we have shown the first evidence that MIR2 utilizes its ITM region for B7-2 recognition. Based on our results, we propose a model where MIR2 recognizes B7-2 through a divalent recognition mode as depicted in Fig. 6. The extent of B7-2TM-CYT downregulation was less than that of B7-2EX-CYT downregulation (Fig. 1B), indicating an important contribution of the MIR2 ITM. Our result clearly demonstrated that the MIR2 ITM, but not the MIR1 ITM, contributes to the downregulation of B7-2 (Fig. 2B). The amino acid sequence of the MIR1 ITM (i.e., GGHDPEIDHV) is quite different from that of the corresponding MIR2 ITM (i.e., TAPAGTFSPG). Both ITMs contain glycine and proline residues, which are frequently located in loop structures; however, the MIR1 ITM has many charged amino acids, whereas MIR2 ITM does not. Instead, MIR2 has three hydroxyl amino acids (Thr113, Thr118, and Ser120) and a hydrophobic residue (Phe119) in its ITM. Thus, MIR2 might have evolved this additional recognition system to allow a more efficient evasion of KSHV-infected cells from immune targeting.
Fig 6.
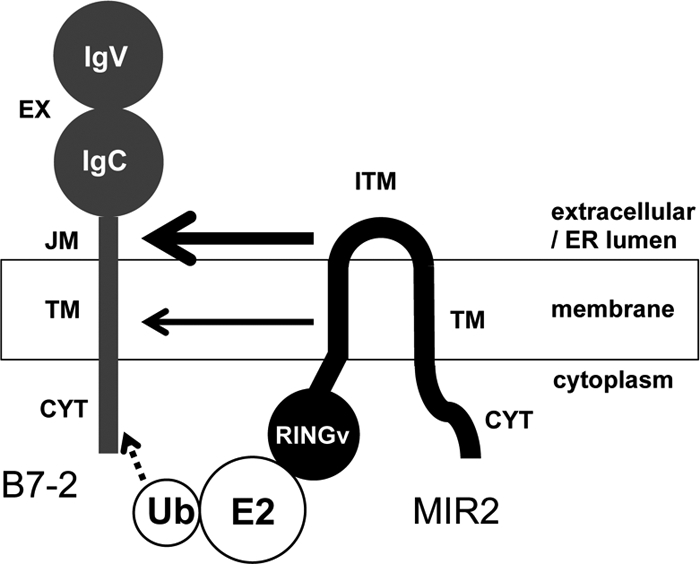
A model of divalent recognition by MIR2. Schematic model of B7-2 recognition by MIR2. MIR2 recognizes B7-2 both through the interaction between the MIR2 ITM and the B7-2 JM regions and through the interaction between the MIR2 TM and the B7-2 TM regions. The contribution of the MIR2 ITM and B7-2 JM regions seems larger than that of the MIR2 TM and B7-2 TM regions. After recognition, the MIR2 N-terminal RINGv domain recruits an E2 ubiquitin-conjugating enzyme with activated ubiquitin.
The alanine (or glycine for endogenous alanine residues)-scanning analysis identified Phe119 and Ser120 of MIR2 as key residues for the ITM-mediated downregulation (Fig. 3A). As might be expected, these amino acids are unique to the MIR2 ITM. The alanine-scanning analysis of the B7-2 JM showed that the downregulation activity of the D244A mutant was significantly decreased; however, the reduction in activity was incomplete (Fig. 3B). Moreover, other mutations around the Asp244 of the B7-2 JM showed slightly reduced sensitivity (Fig. 3B). Thus, these findings suggested that Phe119 and Ser120 of MIR2 are critical and D224 of B7-2 JM is relatively important for MIR2 ITM-mediated B7-2 downregulation.
To help understand how these residues contribute to MIR2 ITM-mediated downregulation of B7-2, we performed structure prediction analysis of the complex and site saturation mutagenesis of these residues. Interestingly, the structure prediction analysis of MIR2 showed that the ITM region of MIR2 forms a loop structure through anchoring of Phe119 in the membrane (Fig. 4). Since aromatic amino acids, including phenylalanine, are frequently utilized for membrane anchoring (24, 34, 37, 38, 41), the hydrophobicity of the six-member ring structure is assumed to be critical for ITM-mediated B7-2 downregulation. This hypothesis was supported by site saturation mutagenesis of Phe119; tyrosine, which also contains a six-member ring structure, could function instead of phenylalanine at position 119. In addition, this analysis suggested that both the size and the hydrophobicity of the six-member ring structure are required at position 119, because tryptophan, which has a more hydrophobic side chain based on the larger indole ring, was not functional. Thus, a suitable conformation of the ITM region, which is mediated through the six-member ring structure of Phe119, is critical for ITM-mediated recognition. The predicted structure also showed that Ser120 of the MIR2 ITM is buried in the membrane/solvent interface but that a hydroxyl group of its side chain is located near the extracellular space (Fig. 4). This finding suggests that the presence of the hydroxyl group might be required for the MIR2 ITM to downregulate B7-2. In fact, the site-saturation mutagenesis demonstrated that threonine could function similarly to serine at position 120 (Fig. 5B). Consistently, the predicted distance between Ser120 of the MIR2 ITM region and Asp244 of the B7-2 JM region was sufficiently small to allow formation of a hydrogen bond (see Table S1 in the supplemental material). Thus, our results suggested a crucial role of Phe119 in forming a suitable conformation and the hydroxyl group in Ser120 of the MIR2 ITM region for recognition of the B7-2 JM region.
In terms of how the JM region of B7-2 is involved in MIR2-mediated downregulation, we focused on Asp244, which was shown by mutational analysis to be the most important residue in the JM region (Fig. 3B). Structure prediction analysis revealed that the B7-2 JM forms an unfolded structure on the membrane surface and that Asp244 is located at the lower position in B7-2 JM (Fig. 4), indicating the possibility that Asp244 interacts with Ser120 of MIR2. Given the importance of the Ser120 hydroxyl group, a hydrogen bond might be formed between the side chains of Asp244 and Ser120. This hypothesis was partially supported by the results of the site-saturation mutagenesis of Asp244; the hydrophobic amino acids were not preferred at this position of the B7-2 JM region (Fig. 5C). In addition to this hypothesis, further consideration is necessary, since the D224A mutant still showed considerable sensitivity to MIR2-mediated downregulation.
Although the results discussed above strongly suggest a direct interaction between B7-2 and MIR2, such a hypothesis is additionally supported by the following findings. In site-saturation mutagenesis of Phe119 or Ser120 of the MIR2 ITM region, replacement with basic amino acids resulted in considerable activity with respect to the downregulation of B7-2JM-CYT (Fig. 5A and B). This result was interpreted as being due to the artificial charge interaction with the acidic Asp244 of the B7-2 JM, since a D244N mutant was not downregulated by such mutants. This observation strongly supports the idea that the distance between the MIR2 ITM region and the B7-2 JM region is small enough to allow this artificial interaction.
In addition, the ITM-mediated downregulation activity of P115A was not completely but was moderately reduced compared with that seen with the wild-type MIR2 (Fig. 3A). We hypothesized that the Pro115 mutation might result in a structure suitable for promotion of the ability to interact with the B7-2 JM region; however, site-saturated mutagenesis of Pro115 demonstrated that proline is not necessary at position 115 of the MIR2 ITM (data not shown). Therefore, at present, the contribution of Pro115 remains unknown.
In summary, we propose the following scheme as the molecular basis for the MIR2 ITM-mediated B7-2 downregulation. (i) The MIR2 ITM recognizes Asp244 of B7-2 JM. (ii) The hydrogen bond between Ser120 of the MIR2 ITM and Asp244 of B7-2 JM is initially required. (iii) The anchoring by the six-member ring structure of Phe119 into the lipid bilayer supports the suitable positioning of the hydroxyl group of Ser120 on MIR2 ITM for interaction with Asp244 of B7-2 JM.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Burrows for critical reading of the manuscript and S. Dohi for secretarial assistance.
This work was supported in part by the Ministry of Education, Culture, Sports, Science and Technology and the Ministry of Health (to M.A.-K., Y.S., and S.I.). Computer time was provided by RICC (RIKEN Integrated Cluster of Clusters).
Footnotes
Published ahead of print 29 February 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Barber DF, Faure M, Long EO. 2004. LFA-1 contributes an early signal for NK cell cytotoxicity. J. Immunol. 173:3653–3659 [DOI] [PubMed] [Google Scholar]
- 2. Bjorkman PJ, Parham P. 1990. Structure, function, and diversity of class I major histocompatibility complex molecules. Annu. Rev. Biochem. 59:253–288 [DOI] [PubMed] [Google Scholar]
- 3. Boname JM, Stevenson PG. 2001. MHC class I ubiquitination by a viral PHD/LAP finger protein. Immunity 15:627–636 [DOI] [PubMed] [Google Scholar]
- 4. Brooks BR, et al. 2009. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30:1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. 2005. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J. Exp. Med. 202:1001–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bu L, Im W, Brooks CL., III 2007. Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys. J. 92:854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cadwell K, Coscoy L. 2008. The specificities of Kaposi's sarcoma-associated herpesvirus-encoded E3 ubiquitin ligases are determined by the positions of lysine or cysteine residues within the intracytoplasmic domains of their targets. J. Virol. 82:4184–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coscoy L. 2007. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Nat. Rev. Immunol. 7:391–401 [DOI] [PubMed] [Google Scholar]
- 9. Coscoy L, Ganem D. 2001. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 107:1599–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coscoy L, Sanchez DJ, Ganem D. 2001. A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 155:1265–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feig M, Karanicolas J, Brooks CL., III 2004. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 22:377–395 [DOI] [PubMed] [Google Scholar]
- 12. Hewitt EW, et al. 2002. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J. 21:2418–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirokawa T, Boon-Chieng S, Mitaku S. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379 [DOI] [PubMed] [Google Scholar]
- 14. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59 [DOI] [PubMed] [Google Scholar]
- 15. Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 16. Ikemizu S, et al. 2000. Structure and dimerization of a soluble form of B7-1. Immunity 12:51–60 [DOI] [PubMed] [Google Scholar]
- 17. Im W, Feig M, Brooks CL., III 2003. An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 85:2900–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishido S, et al. 2000. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 13:365–374 [DOI] [PubMed] [Google Scholar]
- 19. Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74:5300–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jonjić S, Babic M, Polic B, Krmpotic A. 2008. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 20:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Letunic I, Doerks T, Bork P. 2009. SMART 6: recent updates and new developments. Nucleic Acids Res. 37:D229–D232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang C, Lee JS, Jung JU. 2008. Immune evasion in Kaposi's sarcoma-associated herpes virus associated oncogenesis. Semin. Cancer Biol. 18:423–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mackerell AD, Jr, Feig M, Brooks CL., III 2004. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25:1400–1415 [DOI] [PubMed] [Google Scholar]
- 24. Modis Y, Ogata S, Clements D, Harrison SC. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319 [DOI] [PubMed] [Google Scholar]
- 25. Nicholas J, et al. 1997. A single 13-kilobase divergent locus in the Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genome contains nine open reading frames that are homologous to or related to cellular proteins. J. Virol. 71:1963–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohmura-Hoshino M, et al. 2006. A novel family of membrane-bound E3 ubiquitin ligases. J. Biochem. 140:147–154 [DOI] [PubMed] [Google Scholar]
- 27. Orange JS, Fassett MS, Koopman LA, Boyson JE, Strominger JL. 2002. Viral evasion of natural killer cells. Nat. Immunol. 3:1006–1012 [DOI] [PubMed] [Google Scholar]
- 28. Petersen JL, Morris CR, Solheim JC. 2003. Virus evasion of MHC class I molecule presentation. J. Immunol. 171:4473–4478 [DOI] [PubMed] [Google Scholar]
- 29. Ploegh HL. 1998. Viral strategies of immune evasion. Science 280:248–253 [DOI] [PubMed] [Google Scholar]
- 30. Raulet DH. 1996. Recognition events that inhibit and activate natural killer cells. Curr. Opin. Immunol. 8:372–377 [DOI] [PubMed] [Google Scholar]
- 31. Russo JJ, et al. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanchez DJ, Coscoy L, Ganem D. 2002. Functional organization of MIR2, a novel viral regulator of selective endocytosis. J. Biol. Chem. 277:6124–6130 [DOI] [PubMed] [Google Scholar]
- 33. Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. U. S. A. 95:5857–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stopar D, Spruijt RB, Hemminga MA. 2006. Anchoring mechanisms of membrane-associated M13 major coat protein. Chem. Phys. Lipids 141:83–93 [DOI] [PubMed] [Google Scholar]
- 35. Sugita Y, Okamoto Y. 1999. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314:141–151 [Google Scholar]
- 36. Tanaka K, Kasahara M. 1998. The MHC class I ligand-generating system: roles of immunoproteasomes and the interferon-gamma-inducible proteasome activator PA28. Immunol. Rev. 163:161–176 [DOI] [PubMed] [Google Scholar]
- 37. Ulmschneider MB, Sansom MS. 2001. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta 1512:1–14 [DOI] [PubMed] [Google Scholar]
- 38. Victor K, Jacob J, Cafiso DS. 1999. Interactions controlling the membrane binding of basic protein domains: phenylalanine and the attachment of the myristoylated alanine-rich C-kinase substrate protein to interfaces. Biochemistry 38:12527–12536 [DOI] [PubMed] [Google Scholar]
- 39. Whitby D, Boshoff C. 1998. Kaposi's sarcoma herpesvirus as a new paradigm for virus-induced oncogenesis. Curr. Opin. Oncol. 10:405–412 [DOI] [PubMed] [Google Scholar]
- 40. Wilson JL, et al. 1999. NK cell triggering by the human costimulatory molecules CD80 and CD86. J. Immunol. 163:4207–4212 [PubMed] [Google Scholar]
- 41. Winter U, Chen X, Fasshauer D. 2009. A conserved membrane attachment site in alpha-SNAP facilitates N-ethylmaleimide-sensitive factor (NSF)-driven SNARE complex disassembly. J. Biol. Chem. 284:31817–31826 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.