

Abstract
Herpes simplex virus 1 (HSV-1) mutants that lack the γ134.5 gene are unable to replicate in the central nervous system but maintain replication competence in dividing cell populations, such as those found in brain tumors. We have previously demonstrated that a γ134.5-deleted HSV-1 expressing murine interleukin-12 (IL-12; M002) prolonged survival of immunocompetent mice in intracranial models of brain tumors. We hypothesized that M002 would be suitable for use in clinical trials for patients with malignant glioma. To test this hypothesis, we (i) compared the efficacy of M002 to three other HSV-1 mutants, R3659, R8306, and G207, in murine models of brain tumors, (ii) examined the safety and biodistribution of M002 in the HSV-1-sensitive primate Aotus nancymae following intracerebral inoculation, and (iii) determined whether murine IL-12 produced by M002 was capable of activating primate lymphocytes. Results are summarized as follows: (i) M002 demonstrated superior antitumor activity in two different murine brain tumor models compared to three other genetically engineered HSV-1 mutants; (ii) no significant clinical or magnetic resonance imaging evidence of toxicity was observed following direct inoculation of M002 into the right frontal lobes of A. nancymae; (iii) there was no histopathologic evidence of disease in A. nancymae 1 month or 5.5 years following direct inoculation; and (iv) murine IL-12 produced by M002 activates A. nancymae lymphocytes in vitro. We conclude that the safety and preclinical efficacy of M002 warrants the advancement of a Δγ134.5 virus expressing IL-12 to phase I clinical trials for patients with recurrent malignant glioma.
INTRODUCTION
Glioblastoma multiforme (GBM) is both the most common and the most lethal of primary brain tumors. Current treatment consists of surgical resection followed by radiotherapy and chemotherapy (40). However, even with recent advances, the median survival of patients with these tumors is 15 months, with a 2-year survival rate of 26% (49, 50). Thus, there remains a need for new, more potent therapies for GBM and other intracranial malignancies. One such treatment strategy is the use of oncolytic viruses, namely, viruses that exhibit tumor-selective replication (38, 47). Among these, herpes simplex virus 1 (HSV-1) has been studied as a potential therapy for glioma (4, 28). HSV is well suited for this purpose, as it infects and lyses a variety of cell types and has a well-characterized genome. Moreover, the availability of in vivo models allows the study of HSV tumor treatment in a variety of immunocompetent settings. However, because wild-type HSV causes potentially life-threatening encephalitis, attenuation is a prerequisite of oncolytic virus development. To this end, mutations within one or more of the following viral genes have been described: thymidine kinase (31), ribonucleotide reductase (UL39) (22, 34), UTPase (43), or γ134.5 (7, 27). The protein kinase encoded by US3 can also be deleted (42) to generate oncolytic HSV (25). In particular, deletion of the diploid γ134.5 gene ablates neurovirulence of HSV (8). The infected cell protein 34.5 (ICP 34.5) is critical for efficient viral replication in normal cells. In infected nonneoplastic cells, protein kinase R (PKR) is stimulated by the production of double-stranded viral RNAs and phosphorylates eukaryotic initiation factor 2 alpha (eIF-2α) to block protein synthesis. Whereas wild-type HSV is able to reactivate eIF-2α and allow viral replication to proceed, γ134.5-deleted HSVs are unable to replicate efficiently in normal cells. In contrast, tumor cells with ras overexpression or other deficiencies in the PKR response support the replication of γ134.5-deleted HSV.
The γ134.5-deleted HSV G207, which also contains an inactivating insertion of lacZ within the UL39 gene encoding ICP6 (ribonucleotide reductase heavy chain), has demonstrated efficacy in vivo against brain tumors in a number of syngeneic and xenogeneic models of GBM (1, 2, 15, 16, 34), neuroblastoma (52, 53, 55), and meningioma (59). The similar virus HSV1716 is also deleted for γ134.5 but retains the UL39 gene (26, 32) and has demonstrated efficacy in two different brain tumor models (19, 24). Both G207 (29) and HSV1716 (36, 44) have been employed in clinical trials and have been demonstrated to be safe following inoculation into tumor tissue as well as the adjacent tissue of the resection cavity (12, 30). The results of these trials demonstrated that while promising responses occurred in some patients, the majority suffered fatal tumor recurrence. Because of tumor heterogeneity and the aggressive growth properties of GBMs, it is likely that tumor eradication will require a multifaceted treatment approach. Also, intratumoral injection of an oncolytic virus is unlikely to result in 100% tumor cell transduction (35), particularly in the case of highly infiltrative neoplasms such as GBM. This fact, and the presence of intratumoral barriers to viral spread, highlights the need for oncolytic HSVs that have a greater antitumor effect.
We and others have been investigating the use of oncolytic γ134.5-deleted HSVs as vectors for intratumoral delivery of foreign transgenes (56). Although a variety of transgenes have been shown to increase the efficacy of oncolytic HSVs, our efforts have focused chiefly on cytokine-based gene therapy. We hypothesize that cytokine-expressing oncolytic HSV will, through the induction of an antitumor immune response, mediate enhanced efficacy over noncytokine HSV and lead to durable antitumor immunity. To this end, we have previously described an oncolytic HSV that expresses murine interleukin 12 (IL-12) under the control of the murine early growth response-1 (EGR-1) promoter (37). The proinflammatory cytokine IL-12 activates NK cells, mediates Th1 responses, and has additional antiangiogenic properties, and it has been shown to have antitumor effects in vivo (9, 10, 51). This IL-12-expressing HSV, designated M002, lacks both copies of γ134.5 but retains UL39. M002 has been shown to improve survival more effectively than G207 in a murine model of glioma (13). We have also demonstrated that M002 inhibits tumor growth and improves survival more effectively than its noncytokine parent virus R3659 in syngeneic models of neuroblastoma (37) and glioma (13) and that these effects are mediated by increased infiltration of immune cells.
In the current report, we have determined whether an IL-12-expressing Δγ134.5 HSV-1 such as M002 would be suitable for examination in clinical trials for patients with malignant glioma. As such, M002 was assessed for in vivo efficacy in experimental murine brain tumor models compared to other oncolytic HSVs. The safety of M002 following intracranial inoculation was also evaluated in a nonhuman primate model. To summarize our results, we have demonstrated the following: (i) M002 retains sensitivity to acyclovir; (ii) M002 replicates more efficiently than G207 in glioma cells; (iii) M002 significantly enhances inhibition of tumor growth over time compared to the other HSV-1 mutants tested in two different models of murine brain tumors; (iv) inoculation of purified M002 in the right frontal lobes of Aotus nancymae did not produce any significant clinical or magnetic resonance imaging (MRI) evidence of toxicity; and (v) murine IL-12 produced by M002 activates A. nancymae lymphocytes in an in vitro assay. These data provide additional support for the use of an IL-12-expressing Δγ134.5 HSV-1 in a clinical trial for patients with malignant glioma.
MATERIALS AND METHODS
Cells.
Vero cells were obtained from the American Type Culture Collection (ATCC; Rockville, MD) and were cultured in minimal essential medium (MEM; Cellgro, Mediatech Inc., Herndon, VA) containing 7% heat-inactivated fetal bovine serum (FBS; HyClone, Logan, UT) and 2 mM l-glutamine (Life Technologies, Carlsbad, CA). The murine 4C8 glioma cell line, syngeneic with B6D2F1 mice, was obtained from C. Dyer (E. K. Shriver Center, Waltham, MA). The human D54MG and U251MG glioma cell lines were provided by D. D. Bigner (Duke University, Durham, NC). The human glioma cell line U87MG was obtained from ATCC, and the glioma xenografts GBM6 and GBM10 (6, 46) were provided by C. D. James (University of California San Francisco, San Francisco, CA). Xenografts were maintained by in vivo passaging, as previously described, to obtain cells for in vitro studies (11). The above lines were maintained in a 50:50 mixture of Dulbecco's modified Eagle medium and Ham's nutrient mixture F-12 (DMEM-F12) supplemented with 2 mM l-glutamine and 7% fetal bovine serum (FBS). All cells were maintained in Corning tissue culture plasticware.
Viruses.
HSV-1 “F” strain is a low-passage clinical isolate used as the prototype HSV-1 strain for subsequent virus construction (17, 39). Viruses R3659 (23), G207 (34), and R8306, which expresses murine IL-4 (3), are γ134.5-deleted viruses that have been described previously. Construction of M002, which expresses murine IL-12 under the transcriptional control of the murine EGR-1 promoter, has been detailed elsewhere (37). A good manufacturing practice (GMP)-like lot of M002 was provided by NeuroVir Therapeutics, Inc. (San Diego, CA), and used for neurotoxicity studies.
Acyclovir susceptibility.
Vero cells were seeded in 6-well tissue culture plates at 4 × 105 cells per well in MEM with 7% FBS and incubated overnight. The cells were then rinsed with phosphate-buffered saline and infected with HSV-1 F, G207, or M002 at 25 PFU per well in 0.2 ml MEM + 1% FBS for 2 h at 37°C, 5% CO2. Next, 2 ml of acyclovir (Sigma-Aldrich, St. Louis, MO) diluted in MEM + 2% FBS + 0.5% human IgG (Polygam S/D; Baxter Healthcare Corp., Westlake Village, CA) at 0, 0.03, 0.16, 0.8, 4, 20, and 100 μg/ml was added to each well in triplicate. Plates were incubated for 72 h at 37°C in 5% CO2 and then stained with 0.25% May-Grünwald (Sigma-Aldrich) in methanol (Fisher Scientific, Pittsburgh, PA). The plaques in each well were counted to determine the effective concentration in which 50% (EC50) of the plaques remained versus wells in which no acyclovir was present (41). An EC50 10-fold greater than that of wild-type virus is considered resistant (45).
Viral replication.
Cells from the human U87MG and U251MG glioma lines and GBM6 and GBM10 xenografts were grown to confluence in 6-well plates and then infected with M002 or G207 at a multiplicity of infection (MOI) of 0.1 PFU/cell. The cells were harvested at 24, 48, and 72 h postinfection and then lysed by multiple rounds of freeze-thawing and sonication. The titers of progeny virions in the lysates were determined on monolayers of Vero cells, and the average number of PFU per ml was calculated from quadruplicate wells. Determination of significant differences in titer between M002 and G207 was done by two-tailed, unpaired t tests.
In vivo tumor responses.
All animal experiments were carefully reviewed and approved by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee and adhered to the Public Health Service Policy on the Humane Care and Use of Laboratory Animals, the National Research Council Guide for the Care and Use of Laboratory Animals, and the United States Department of Agriculture (USDA) Animal Welfare Regulations. UAB is licensed as an animal research facility by the USDA and has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare. The animal care and use program at UAB has maintained accreditation with the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) since 1971.
Specific-pathogen-free female SCID and B6D2F1 mice were obtained from Charles River Laboratories and used at approximately 8 weeks of age. For analysis of efficacy in murine brain tumors, 5 × 105 4C8 or D54MG cells in 5 μl serum-free DMEM-F12 with 5% methylcellulose were stereotactically injected into the right caudal nucleus of B6D2F1 or SCID mice, respectively, using methods detailed previously (7, 37). In the D54MG model, tumors were injected 7 days after implantation with 1 × 107 PFU of G207 or M002 in 5 μl sterile saline, or with saline only as a control. Seven days following virus injection, mice that received saline were inoculated with saline. Half the mice in each virus treatment group received an additional 5 μl of the same virus, while the other half received saline only. In the 4C8 model, mice were given intratumoral injections 14 days after tumor implantation of 1 × 107 PFU of R3659, R8306, M002, or saline only. In a second experiment, at 7 days after tumor implantation, mice were given intratumoral injections of 1 × 107 PFU of G207 or M002 or given saline. Seven days after virus injection, mice were reinjected with virus or saline as above. In all experiments, mice were monitored for survival and euthanized when moribund, and necropsies were performed to confirm that tumor growth was the cause of death. Deaths were recorded as described previously (7). Kaplan-Meier survival plots were then constructed and statistical significances in survival between the various cohorts were determined by log rank (Mantel-Cox) pairwise comparisons.
Toxicity in nonhuman primates.
One male and three female adult New World owl monkeys (Aotus nancymae) were purchased through the USDA and housed according to the UAB animal husbandry guidelines for nonhuman primates (NHP), thus allowing social interaction between animals. All intracranial inoculations and magnetic resonance imaging procedures were done under conditions of general anesthesia, in which intramuscular ketamine (4 mg/kg) and acepromazine (0.4 mg/kg) were given as sedatives prior to the administration of isoflurane in oxygen (2.4% induction, 1.5 to 1.8% maintenance). Respiration was assessed by tidal CO2 monitoring and oxygenation by pulse oximetry. The NHPs were placed in a prone position on a water-controlled heating blanket to maintain body temperature. The head was immobilized in a Kopf stereotaxy, the scalp was clipped and shaved, and the animals were covered with a sterile drape. The skin at the surgical site was then sanitized with betadine surgical scrub, and an incision was made to expose the skull. After placement of a burr hole 2 mm anterior to the coronal suture and 7 mm lateral to the midline, virus was injected with a Hamilton syringe attached to the stereotaxy into the right frontal lobe of the brain at a depth of 5 mm. Following injection, the burr hole was closed with bone wax and the incision was sutured.
PCR studies of biodistribution following intracerebral inoculation in A. nancymae.
At both 30 days and 5.5 years postinjection of M002, animals were euthanized by the intravenous administration of 100 mg/kg pentobarbital, and tissue samples were collected aseptically for PCR-based determination of viral biodistribution. Brains were harvested, and individual samples were collected from the injection site as well as different sites of both hemispheres (frontal, temporal, and occipital lobes, cerebellum) and the brainstem. Ocular, nasal, and oral swabs were collected from the 30-day animal. Additionally, the following tissues were harvested: skin (superficial to injection site), heart, liver, pancreas, stomach, lung, spleen, small intestine, large intestine, adrenal glands (left and right), kidneys (left and right), skin (from site remote to the injection), skeletal muscle, and bladder. DNA was extracted from the tissue samples using a Qiagen EZ1 tissue kit and BioRobot EZ1 workstation (Qiagen, Valencia, CA) and then amplified by PCR on an ABI 2400 GeneAmp thermal cycler (Applied Biosystems, Carlsbad, CA) using primers specific for HSV gB and polymerase as previously reported (54).
MRI.
MRI was performed on anesthetized NHPs at 30 days and 7 months postinoculation with M002 using a 1.5 T Philips (Philips Healthcare, Andover, MA) scanner and knee coil. T1-weighted images were acquired both precontrast and postadministration of gadolinium. Also acquired were T2-weighted and fluid attenuated inversion recovery (FLAIR) images.
Histology.
All tissue samples in which viral DNA was detected by PCR were analyzed by immunohistochemistry (IHC). This included multiple sites from the brains of NHPs euthanized 30 days and 5.5 years postinoculation of M002, as well as a sample of spleen from the 30-day animal. Formalin-fixed paraffin-embedded sections, 10 μm thick, were stained with hematoxylin and eosin. Additional sections, 6 μm thick, were prepared for IHC staining by deparaffinization in xylene, rehydration through graded alcohols, and antigen retrieval with pH 6.0 sodium citrate buffer. Endogenous peroxidases were quenched with hydrogen peroxide, and blocking was done with horse or goat serum (Vector Laboratories, Inc., Burlingame, CA), depending upon the antibody to be used. HSV antigen staining was done using a rabbit polyclonal antibody (BioGenex Laboratories, Inc., Fremont, CA) diluted 1:400. T cells were detected with a mouse anti-CD3 monoclonal antibody (Ventana Medical Systems, Tucson, AZ), which was supplied ready to use. Staining for macrophages was done using a mouse monoclonal antibody diluted 1:100 (MAC387; AbD Serotec, Raleigh, NC) or as provided by the manufacturer (HAM-56; Cell Marque Co., Rocklin, CA). Secondary antibodies specific for rabbit or mouse IgG were supplied as Vectastain ABC kits (Vector Laboratories). Slides were developed using a Vector VIP (Vector Laboratories) peroxidase substrate detection kit and counterstained with methyl green. All slides were examined by a board-certified neuropathologist.
Primate lymphocyte activation.
The responsiveness of A. nancymae lymphocytes to the murine IL-12 expressed by M002 was assessed by a lymphocyte reactivation assay. Briefly, peripheral blood lymphocytes were isolated from pooled blood samples harvested from anesthetized A. nancymae. The cells were cultured for 72 h in RPMI 1640 with 10% (vol/vol) FBS, supplemented with 2.5 μg/ml phytohemagglutinin (PHA), 1,000 U/ml recombinant gamma interferon, and 50 μM β-mercaptoethanol. The cells were then plated in 96-well tissue culture plates at 2.25 × 104 cells/well and incubated in the absence of PHA for 24 h. Next, the cells were cultured for 72 h in RPMI 1640 medium with 10% (vol/vol) FBS and 50 μM β-mercaptoethanol, and replicate wells were stimulated with a range of dilutions of PHA (0.01 to 32 μg/ml) as a control or with dilutions of recombinant murine IL-12 (1.25 to 40 ng/ml) obtained from the concentrated conditioned medium of M002-infected Vero cells. The cells were then pulsed with tritiated thymidine ([3H]Tdr) at 1 μCi/well for 21 h. Lymphocyte activation, as determined by incorporation of [3H]Tdr into the cellular DNA, was assessed by scintillation count and expressed as counts per minute (cpm).
RESULTS
M002 retains sensitivity to acyclovir.
To verify the sensitivity of M002 to acyclovir, a plaque reduction assay was performed. Sensitivity was compared to that of wild-type strain F, the parent virus of M002, and G207, a virus previously used in North American clinical trials (29, 30). M002 exhibited sensitivity to acyclovir at levels similar to that of wild-type virus (Fig. 1). These data translate into an EC50 of <1.0 μg/ml for M002, a value similar to those for wild-type isolates tested in our laboratory (57).
Fig 1.
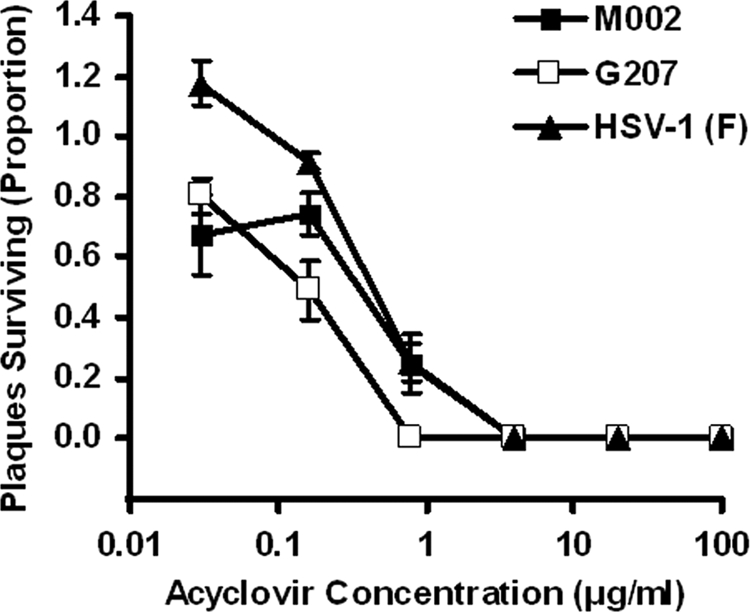
Replication of the IL-12-expressing γ134.5-deleted HSV M002 is inhibited by acyclovir. In a plaque-reduction assay, M002 was found to have an EC50 of <1.0 μg/ml, similar to its wild-type parent F as well as G207, another γ134.5-deleted HSV-1 mutant already used in clinical trials. Shown are the proportions of surviving plaques and standard deviations at various concentrations of acyclovir, relative to infected control cells grown without the drug.
M002 replicates more efficiently than G207 in human glioma cells.
Next, we compared the abilities of M002 and G207 to replicate in glioma cells. Monolayers of the human glioma lines U87MG and U251MG or cells derived from the xenografts GBM6 and GBM10 were infected with M002 or G207 at an MOI of 0.1 PFU/cell. At multiple time points postinfection (24, 48, and 72 h), viral replication was determined. As shown in Fig. 2, M002 replicated to a titer 1 log to 2 logs higher than that of G207 in both the established cell lines and the xenograft-derived cells. This enhanced replication was seen across all three time points.
Fig 2.

M002 replicates more efficiently than G207 in glioma cells. Monolayers of the human glioma cell lines U87MG (A) and U251MG (B), and cells derived from glioma xenografts GBM6 (C) and GBM10 (D), were infected with M002 (filled boxes) or G207 (open boxes) at 0.1 PFU per cell, and viral replication was assayed at the indicated times. No G207 progeny were recovered from GBM10 at 72 h. Shown are the averages and standard deviations of quadruplicate wells at each time point. Significant differences in titer at each time point are indicated: *, P < 0.05; **, P < 0.01; ***, P < 0.0005.
M002 improves survival more effectively than other cytokine-expressing HSV in a murine brain tumor model.
The antitumor efficacy of M002 was next compared directly with the antitumor efficacy of R8306, a γ134.5-deleted HSV-1 that expresses IL-4. R8306 was previously shown to be an effective antiglioma agent in a preclinical murine brain tumor model (3). To compare these viruses, the murine 4C8 tumor model was employed. These cells form invasive brain tumors in vivo, with histological features typical of human gliomas (13). M002 replicates to similar levels in 4C8 as in human glioma cells and expresses physiological amounts of IL-12 (13). Intracranial tumors were established in B6D2F1 mice and then treated after 14 days with M002 or R8306. Control cohorts of mice were given saline or treated with the noncytokine-expressing virus R3659, an oncolytic HSV with a diploid γ134.5 deletion but lacking the UL39 deletion mutation present in G207. The mice were monitored for survival for 84 days, at which point surviving mice were euthanized. Treatment with M002 resulted in a significantly increased median survival time (MST) (P < 0.05) compared to either R8306 or the control virus R3659 (Fig. 3). Whereas mock-treated mice exhibited an MST of 52 days, mice treated with either R3659 or M002 had MSTs of 69 days and >84 days, respectively. Although mice treated with R8306 had a shorter MST (31 days) than mock-treated mice, the R8306 group had the second greatest proportion (40%) of long-term survivors after the M002 group (80%). In a similar experiment conducted in A/J mice bearing syngeneic Neuro-2A murine neuroblastoma tumors, M002 also improved survival more effectively than M004, a similar HSV that expresses granulocyte-macrophage colony-stimulating factor (unpublished data). Together, these results provide evidence supporting the use of IL-12 as a rational choice among other cytokine-expressing HSVs for clinical use.
Fig 3.

Survival of B6D2F1 mice following intracranial injection of syngeneic 4C8 murine glioma cells. Fourteen days after tumor implantation, the mice were inoculated with saline (open triangles) or 1 × 107 PFU of either IL-12-expressing M002 (filled circles), IL-4-expressing R8306 (open circles), or the control virus R3659 (filled triangles). Median survival was highest in the M002 treated group (>84 days, P < 0.05), followed by the R3659 group (69 days), saline group (52 days), and the R8306 group (31 days). The R8306 group had the second greatest proportion of long-term survivors after M002.
M002 is superior to G207 in xenogeneic and syngeneic models of glioma.
In a syngeneic murine model of glioma, M002 improves survival more effectively than G207 following a single intratumoral injection of virus (13). M002 is also more effective than R3659 in both syngeneic (13) and xenogeneic (48) models of glioma. We next sought to directly compare the antitumor activity of M002 with G207 in human glioma xenografts in vivo. Multiple dosing was also assessed for potential ability to further enhance efficacy.
To compare the effects of treatment with M002 and G207 against human glioma cells in vivo, a survival study was conducted in SCID mice implanted orthotopically with human D54MG xenografts. These cells form relatively well-circumscribed brain tumors in vivo that grow rapidly by expansion. Viral cytotoxicity and IL-12 expression by M002 has previously been confirmed in this line (37). Mice were treated after 7 days with G207 or M002, or given saline only as a control. Seven days after the first injection, half of the mice in each treatment group received a second injection of the same virus, while the other half was given saline only. A single injection of G207 did not significantly improve survival versus mock treatment, with MSTs of 22 days and 18 days, respectively (P = 0.157) (Fig. 4). A second dose of G207 did not further improve survival (MST of 22 days; P = 0.0754 versus mock, 0.429 versus single dose). In contrast, treatment with M002 significantly improved survival over that of mock treatment. A single dose resulted in an MST of 29 days (P = 0.00254), and two doses resulted in an MST of 30 days (P = 0.000473). The differences in survival between two doses (P = 0.0002) of G207 and M002 were also significant.
Fig 4.

Superior efficacy of M002 versus G207 against intracranial human glioma xenografts. SCID mice implanted with D54MG cells were injected 7 days after tumor establishment with 1 × 107 PFU of M002 or G207 or with saline only. Seven days later, mice that received saline were reinjected with saline. Half of the mice that received virus received an additional 5 μl of the same virus, and the other half received 5 μl of saline. Significant differences in median survival times (MST) were as follows: saline/saline versus M002/saline (P = 0.00254), versus M002/M002 (P = 0.000473); M002/saline versus G207/G207 (P = 0.00149); M002/M002 versus G207/G207 (P = 0.0002).
In a similar multiple-dosing study of M002 versus G207 conducted in an immunocompetent intracranial model of glioma, G207 was less effective than M002 against 4C8 tumors (data not shown). Whereas the MST of mock-treated mice was 39.5 days, those treated with G207 exhibited MSTs of 60 days (single dose) and 49 days (two doses). M002-treated mice survived longer when either a single dose (MST = 71.5 days) or two doses (MST = 94 days) were given. Overall, these results indicate that M002 is more effective against human glioma xenografts than G207 and that additional doses may enhance the antitumor effect.
M002 does not produce encephalitis or other significant effects in primate toxicity studies.
Intracerebral inoculation of G207 in Aotus nancymae primates, a New World monkey highly susceptible to HSV-1 replication (18, 33), does not result in virus-associated toxicity (14, 54). However, whether this same safety profile would be exhibited by an IL-12-expressing HSV such as M002 was unclear. Thus, to verify safety in a nonhuman primate model, M002 was directly inoculated into A. nancymae brains. A preparation of M002 that was made according to clinical GMP specifications was inoculated into the monkeys at doses up to 4.8 × 108 PFU. No clinical evidence of toxicity was observed in any of the animals, as assessed by changes in temperature, neurologic performance, feeding or social behavior, or weight. Examination of injected animals by MRI both 1 month and 7 months postinjection demonstrated a lack of encephalitis or any other toxicity (Fig. 5). One female animal, which had been injected in the right frontal lobe with 4.8 × 108 PFU of M002, was euthanized 1 month postinjection for histological evaluation (Fig. 6). Overall, the areas of the brain examined (left and right frontal, temporal, and occipital lobes; brainstem, cerebellum) appeared normal, with a few potentially study-related features noted as follows. The left temporal lobe showed a single perivascular chronic inflammatory cell cuff, deep at the gray-white junction. In the superficial left frontal lobe, a focal encephalitis with active astrogliosis, glial nodules, and neuronphagia was observed. Accordingly, the presence of CD3+ T cells was confirmed by immunostaining. Rare perivascular lymphocytes were observed along the wound tract. No macrophage or HSV antigens were detected by immunostaining in any part of the brain. Five and one-half years after inoculation, one of the injected females, along with a cagemate that had not been treated with virus, was found to demonstrate delayed-type hypersensitivity to purified protein derivative on routine tuberculosis surveillance monitoring. Both animals were euthanized and necropsied. No evidence of mycobacterial infection was found, and no evidence of toxicity related to M002 treatment was seen in the treated animal. Overall, the sections of the brain examined, including the injection site, appeared normal. Reactive changes, which may have been age-related or treatment-related, were observed in some areas of the brain. These changes were deemed clinically insignificant. No HSV or CD3+ cells were detected in any part of the brain. An additional treated female is now more than 10 years status postinjection and remains alive and healthy. One male A. nancymae was injected with a lower dose of M002 (1 × 108 PFU nonpurified standard laboratory preparation). This animal died under anesthesia during follow-up MRI 3 days after injection of virus. No abnormalities were observed by MRI. A postmortem necropsy revealed that subclinical naturally occurring glomerulonephritis and moderate bronchopneumonia contributed to anesthetic complications. The brain was fixed and serially sectioned and then stained both by hematoxylin and eosin as well as by IHC for HSV antigens. Regardless of histopathologic approach, no evidence of HSV encephalitis or virus-related toxicity was found. No hydrocephalus or ventriculomegaly was present in any animal.
Fig 5.

Shown are MRI images of two different A. nancymae animals after inoculation with M002, (A) Animal 1. One month prior, 1.2 × 108 PFU of M002 was inoculated in the right frontal lobe. Images are in the axial plane. Shown are (i) FLAIR, (ii) T2-weighted, (iii) T1-weighted pregadolinium, and (iv) T1 postgadolinium images. (B) Animal 2. Seven months prior, 4.8 × 108 PFU of M002 was inoculated in the right frontal lobe. Images are in coronal plane and inverted. Shown are (i) FLAIR, (ii) T2-weighted, (iii) T1-weighted postgadolinium, and (iv) additional plane, T1 postgadolinium images. No pathological changes are seen after M002 administration.
Fig 6.

(A and B) Shown are micrographs of A. nancymae brain 30 days after injection with M002, stained with hematoxylin and eosin at low (×40, panel A) and higher (×200, panel B) magnification. Note that while mild inflammatory changes are present, as would be expected from local expression of IL-12, no significant neuronal loss or glial scarring is seen. (C and D) Also shown is a section stained by IHC for CD3-positive cells at two different magnifications (×200, panel C; ×400, panel D).
M002 DNA can be demonstrated in A. nancymae after inoculation in both short- and long-term treated animals.
To examine the persistence and biodistribution of M002 following intracerebral inoculation, it was necessary to confirm that copies of HSV DNA were present in A. nancymae brain after treatment and to assay for HSV DNA in other organs. Multiple sites within the brain, multiple organ sites, and, in the 30-day animal, swabs to evaluate potential shedding sites were all tested for HSV DNA by PCR. Notably, HSV DNA was present in both the 30-day and the 5.5-year animal at multiple sites of the forebrain and brain stem but not the cerebellum (Table 1). In the 30 day animal, HSV DNA was present in the skin at the inoculation site and in the nasal swab but not in the ocular or oral swabs. Systemically, HSV DNA was detected in the spleen but could not be detected in any other major organ. In the 5.5-year animal, no samples outside the brain tested positive for HSV DNA. HSV antigens were not detected by IHC in any sample at either time point. These findings are consistent with previous studies with G207 in patients, in which HSV DNA can be detected by PCR up to 157 days postinoculation, but HSV is not detectable by IHC (29).
Table 1.
Results of PCR testing for HSV DNA in treated A. nancymae at 30 days and 5.5 years postinjection
| Locationa | 30 daysb | 5.5 yrb |
|---|---|---|
| Frontal lobe L | + | + |
| Frontal lobe R | + | + |
| Temporal lobe L | + | + |
| Temporal lobe R | + | + |
| Parietal L | ND | + |
| Parietal R | ND | + |
| Occipital L | + | − |
| Occipital R | + | + |
| Cerebellum L | − | − |
| Cerebellum R | − | − |
| Pons | ND | + |
| Brain stem | + | + |
| Wound injection site | + | + |
| Ocular swab | − | ND |
| Nasal swab | + | ND |
| Oral swab | − | ND |
| Heart | − | ND |
| Liver | − | ND |
| Pancreas | − | − |
| Stomach | − | ND |
| Lung | − | ND |
| Spleen | + | − |
| Small intestine | − | − |
| Large intestine | − | − |
| Adrenal L | − | − |
| Adrenal R | − | − |
| Kidney L | − | − |
| Kidney R | − | − |
| Skin | + | − |
| Skeletal muscle | − | − |
| Bladder | − | − |
L, left; R, right.
ND, not determined.
Murine IL-12 is biologically active in Aotus nancymae.
To confirm that A. nancymae lymphocytes respond to murine IL-12 produced by M002, a lymphocyte activation assay was performed using pooled lymphocytes. Lymphocytes were activated in the presence of PHA, murine recombinant IL-12, or medium alone. As determined by [3H]Tdr incorporation, the lymphocytes showed similar activation to murine recombinant IL-12 as to PHA (Fig. 7). These results confirm that the lack of toxicity seen in M002-injected A. nancymae is not due to a failure of the primate lymphocytes to recognize murine IL-12.
Fig 7.

The ability of A. nancymae peripheral blood lymphocytes to respond to recombinant murine IL-12 was assessed based on incorporation of [3H]Tdr into cellular DNA. Pooled lymphocytes were stimulated with dilutions of concentrated supernatants from Vero cells infected with M002 (IL-12), or with PHA as a control, and then pulsed with [3H]Tdr. Activation was assessed by scintillation count and is expressed as cpm, plotted in comparison to a linear IL-12 response.
DISCUSSION
Oncolytic HSVs are being actively investigated as potential antiglioma therapies. HSVs can be neuroattenuated by a number of strategies, with deletion of the diploid γ134.5 gene being the approach taken for two different HSVs employed in clinical trials. Overall, the results of these trials have underscored the need for HSVs with improved antitumor effects. Our research group has investigated the potential use of oncolytic HSVs as a platform for the delivery of foreign transgenes. Our efforts have focused mainly on cytokine delivery, under the general hypothesis that a cytokine-expressing HSV will activate an antitumor immune response that will lead to the elimination of noninfected tumor cells. The properties of IL-12 warranted its consideration for introduction into an oncolytic HSV and subsequent evaluation as an antitumor agent. IL-12 activates both cytotoxic T lymphocyte and Th1 responses by stimulating gamma interferon production from both NK cells and T cells. Furthermore, by NK cell activation and subsequent cytokine secretion, IL-12 also stimulates an antiangiogenic effect against tumor vasculature. M002 was engineered to express murine IL-12 (37). While murine IL-12 exhibits biological activity on human lymphocytes, human IL-12 does not activate murine lymphocytes. Thus, any construct designed to examine the effects of IL-12 in a murine system must necessarily utilize murine IL-12. As a consequence of its multiple antitumor activities, M002 has been shown to mediate a superior antitumor effect versus other noncytokine HSVs (13, 37, 48).
In the current study, we sought to preclinically evaluate the suitability of M002 for use against glioma in clinical trials. First, it was important to demonstrate that M002 was susceptible to an antiviral drug routinely used in the therapy of HSV infections. In a plaque-reduction assay, M002 replication was inhibited by acyclovir to a similar extent as the parent HSV-1 strain F. These data indicate that neither the expression of IL-12 nor other possible uncharacterized genetic differences from F strain have caused M002 to lose sensitivity to acyclovir. Although the establishment of encephalitis by a γ134.5-deleted HSV has never been documented in humans, the ability to control M002 replication pharmacologically serves as an important backup safety mechanism.
Next, the antitumor activity of M002 was directly compared in vivo in immunocompetent mice to that of another cytokine-expressing HSV, R8306. In previously published studies, we have shown that M002 stimulates the intratumoral infiltration of immune cells, including CD4, CD8, NK, and macrophages (13, 37) in both the N2A (37) and 4C8 (13) models. Since we have also previously shown that R8306, which expresses IL-4, is effective against glioma in an immunocompetent model (3), we next wanted to compare the efficacy of that virus to M002. Here, we have shown that M002 treatment was superior to that of R8306, leading to a higher proportion of long-term survivors. We speculate that this difference may be due to the differential effects of the respective cytokines. Both R8306 (3) and M002 (37) stimulate an influx of CD4+ and CD8+ T cells and macrophages, but M002 also stimulates NK cell infiltration (13). Furthermore, IL-4 stimulates the differentiation of Th2 cells, whereas IL-12 stimulates a Th1 response. Finally, the antiangiogenic effects of IL-12 may have also contributed to the increased efficacy of M002 versus R8306, as it has been reported that IL-12 expression from an oncolytic HSV reduces tumor angiogenesis (58). While M002 treatment resulted in 80% long-term survival in this model, some of the mice eventually succumbed to tumor growth. This may have been a consequence of incomplete viral spread throughout these infiltrative tumors.
In additional experiments, M002 treatment was also superior to that of G207 in both immunodeficient and immunocompetent models. In the first, M002 was more effective than G207 against human xenografts in SCID mice. Our in vitro replication data indicate that M002 replicates to higher titer in glioma cells than does G207. The superior antitumor effect of M002 over G207 in the immunodeficient model is likely due to its enhanced replicative ability, since adaptive immune responses are not elicited and thus not able to contribute to the antitumor effect. Unlike the immunocompetent model, in which long-term survivors were observed, in the immunodeficient model, all animals eventually died of tumor burden, probably a result of incomplete local virus replication that allowed surviving tumor cells to continue growth. Other factors that may have contributed to the enhanced efficacy of M002 relative to G207 in this model include the antiangiogenic properties of IL-12 and NK cell stimulatory effects. Also, the UL39 gene retained by M002 may confer a replication advantage over G207, which lacks this gene, in this particular cell line. In this model, an additional dose of M002 did not lead to a statistically significant increase in efficacy over a single dose. This may be evidence that in the absence of a potential antitumor effect mediated by the adaptive immune system, innate antiviral responses may offset the potential benefit of additional virus. In an unpublished experiment conducted in immunocompetent mice, we observed that a single injection of M002 was superior to a single injection of G207 and that an additional dose of virus further extended the benefit of M002 treatment but did not extend the benefit of G207 treatment. In this case, it is possible that an antiviral immune response prevented an increase in efficacy for a double dose of G207, but the many antitumor effects of IL-12 expression may have allowed double dosing of M002 to overcome potential antiviral responses to result in an overall increase in efficacy. The potential benefits of multiple dosing with M002 may simply be model dependent and/or require a greater number of mice (more power) to demonstrate, and further clarification of this issue is needed. However, previous murine studies have shown that the increase in immune cell infiltration mediated by M002 may begin to wane 7 days after treatment. In the clinical setting, in which treatment schedules occur over a longer time frame (i.e., months), multiple dosing may still increase efficacy.
All of the efficacy experiments in this report were conducted with the same viral dose (1 × 107 PFU), which was based upon previous work in which we determined the maximum tolerated dose (PFU/50% lethal dose [LD50]) of M002 in the HSV-sensitive A/J mouse strain to be 2 × 107 PFU (37). We therefore routinely use 1 × 107 PFU as the highest dose that can be used without risk of toxicity. Whether smaller doses of virus would also be effective remains to be determined, although given the replicating nature of these constructs, the concept of a minimally effective dose is subject to debate. Regarding the dose to be used in a clinical trial, rather than try to determine starting doses based upon murine data, we will take advantage of prior human data with G207 and HSV 1716. HSV 1716 showed no toxic responses at up to 1 × 105 PFU (44), whereas G207 was used at even higher doses (3 × 109 PFU) (29). Neither study established a maximum tolerated dose. We propose that doses in a clinical trial with an IL-12-expressing HSV begin at 1 × 105 PFU and increase to 1 × 109 PFU in the absence of limiting toxicities. The highest likely clinical dose is therefore 100 times higher than the dose used in the murine studies described here. However, given that the intracranial volume of the human is approximately 2,000-fold greater than that of a mouse, the relative dose to be used in a clinical trial is approximately 20-fold less than what has been used in the mouse experiments.
To elucidate the potential safety profile of M002 in a readily translatable model, we evaluated the toxicity and biodistribution of M002 following intracerebral viral injection in the nonhuman primate Aotus nancymae. Similar studies have previously been conducted with G207 (14, 54). Over the course of this study, no toxicities related to M002 injection were observed, either clinically or histologically. While mild inflammation was present at the site of inoculation in the 30-day posttreatment A. nancymae specimen, other major pathological features were not observed, and no pathological changes were detected by MRI. These observations are similar to those following intracerebral injection of G207, in which mild inflammatory changes occurred at the injection site, with no MRI-based evidence of toxicity (14), although the dose of G207 used (109 PFU) was higher than the highest dose in this study (4.8 × 108 PFU). We observed infiltration of CD3+ T cells but did not observe HSV-positive cells as reported by Hunter et al. (14), although our examination was conducted at 30 days rather than 5 days postinjection. We did not directly assay for IL-12 (either murine or endogenous) in this experiment, but given the lack of HSV staining and the relatively mild immune response, we speculate that presence of IL-12 would have been difficult to detect. Also, no signs of IL-12 toxicity were observed in any animal. In this study, the long-term safety of M002 treatment is suggested by the lack of toxicities seen in the 5.5-year animal and further supported by the continued health of the surviving M002-treated animal, now at 10 years postinjection. Previously, the safety of G207 administration in A. nancymae had been followed out to 41 months (14). Regarding viral persistence and biodistribution, both short-term and long-term animals in this study showed retention of HSV DNA in the brain and at the inoculation sites, consistent with the ability of γ134.5-deleted viruses to undergo latency but not emerge from latency (20). This is not altogether surprising, as tumor resected from malignant glioma patients more than 150 days after treatment with G207 has been positive for HSV DNA (29). It has also been reported that HSV DNA can be detected in autopsy specimens from the brains of normal humans who were seropositive for HSV (5). We observed HSV DNA at multiple sites throughout the brain but not the cerebellum, a finding also noted in the G207 study by Todo et al. (54), who reported the presence of HSV DNA 2 years postinjection. Although G207 was not detected outside the brain (54), HSV DNA was present in the spleen of the 30-day animal in this study. We also detected HSV DNA in a nasal swab at 30 days and in the skin superficial to the injection site, neither of which was assayed in the G207 study. Both of these sites were negative for HSV at 5.5 years, however. Since HSV antigens were not detected by IHC in any of these samples, the presence of DNA may be a product of immune cell clearance or a very low level of retained virus. The significance of this finding remains to be determined. However, it should be noted that in the G207 phase I study mentioned above, viral antigens similarly were not detected by IHC at 5 months postinjection, in spite of PCR-based detection of viral DNA (29). In total, the distribution of M002 after intracerebral injection is not markedly different from that of G207.
If murine IL-12 was not functional in A. nancymae, the results of the toxicity study would be less meaningful. Presumably, with function being retained across species from mouse to human, murine IL-12 would be expected to function in the A. nancymae as well as in mice. A study conducted by Kim et al. demonstrated that murine IL-12 expressed from an injected DNA plasmid was biologically active in rhesus macaques, another nonhuman primate species similar to A. nancymae (21). We have confirmed here that murine IL-12 activates A. nancymae lymphocytes, validating the safety profile of M002 in this model.
In conclusion, we have conducted a preclinical determination of whether an IL-12-expressing γ134.5-deleted HSV-1 might be a suitable virus to advance into clinical studies for patients with malignant brain tumors. To summarize our results: (i) M002 mediates efficacy in preclinical brain tumor models superior to that of other mutant HSV-1, including one currently under clinical evaluation; (ii) no significant toxicity is seen after intracerebral inoculation of M002 into mice or the HSV-sensitive primate A. nancymae despite long-term persistence of viral DNA as measured by PCR, and the potential for acyclovir treatment and patient rescue exists should such toxicity occur; and (iii) murine IL-12 expressed by M002 activates A. nancymae lymphocytes, thus confirming the validity of this model for evaluation of safety. Findings consistent with efficacy greater than that of G207, lack of significant neurologic and systemic toxicity by clinical, radiologic, and pathological evaluation despite long-term persistence of HSV DNA, and confirmation of the activity of IL-12 produced by the virus on A. nancymae lymphocytes all support the advancement of an IL-12-expressing γ134.5-deleted HSV-1 into clinical trials for patients suffering from malignant glioma. Although species specificity of human IL-12 prevents its use in the murine model, we have also generated a syngeneic virus that expresses human IL-12. This virus might be appropriate for use in clinical trials, to avoid the possibility of any unforeseen differences in activity between human and murine IL-12, and possible generation of an immune response against a murine protein when used in human patients.
ACKNOWLEDGMENTS
This work was supported by the National Cancer Institute grant P01 CA71933 (J.M.M., J.N.P., G.Y.G., and R.J.W.). We also acknowledge support from the Training Program in Brain Tumor Biology, grant T32NS048039 (J.J.C.).
We thank Deidra Isbell for veterinary assistance and Timothy J. Ness for anesthesia assistance with the primates.
We declare that J.M.M., J.N.P., G.Y.G., and R.J.W. maintain a consulting relationship with Catherex, Inc. We are also stockholders and cofounders.
Footnotes
Published ahead of print 29 February 2012
REFERENCES
- 1. Aghi M, Rabkin S, Martuza RL. 2006. Effect of chemotherapy-induced DNA repair on oncolytic herpes simplex viral replication. J. Natl. Cancer Inst. 98:38–50 [DOI] [PubMed] [Google Scholar]
- 2. Aghi MK, Liu TC, Rabkin S, Martuza RL. 2009. Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol. Ther. 17:51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andreansky S, et al. 1998. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 5:121–130 [DOI] [PubMed] [Google Scholar]
- 4. Andreansky SS, et al. 1996. The application of genetically engineered herpes simplex viruses to the treatment of experimental brain tumors. Proc. Natl. Acad. Sci. U. S. A. 93:11313–11318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baringer JR, Pisani P. 1994. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann. Neurol. 36:823–829 [DOI] [PubMed] [Google Scholar]
- 6. Carlson BL, et al. 2009. Radiosensitizing effects of temozolomide observed in vivo only in a subset of O6-methylguanine-DNA methyltransferase methylated glioblastoma multiforme xenografts. Int. J. Radiat. Oncol. Biol. Phys. 75:212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chambers R, et al. 1995. Comparison of genetically engineered herpes simplex viruses for the treatment of brain tumors in a scid mouse model of human malignant glioma. Proc. Natl. Acad. Sci. U. S. A. 92:1411–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1266 [DOI] [PubMed] [Google Scholar]
- 9. Colombo MP, Trinchieri G. 2002. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 13:155–168 [DOI] [PubMed] [Google Scholar]
- 10. Del Vecchio M, et al. 2007. Interleukin-12: biological properties and clinical application. Clin. Cancer Res. 13:4677–4685 [DOI] [PubMed] [Google Scholar]
- 11. Giannini C, et al. 2005. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 7:164–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harrow S, et al. 2004. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 11:1648–1658 [DOI] [PubMed] [Google Scholar]
- 13. Hellums EK, et al. 2005. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol. 7:213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hunter WD, et al. 1999. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J. Virol. 73:6319–6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huszthy PC, et al. 2008. Oncolytic herpes simplex virus type-1 therapy in a highly infiltrative animal model of human glioblastoma. Clin. Cancer Res. 14:1571–1580 [DOI] [PubMed] [Google Scholar]
- 16. Huszthy PC, et al. 2010. Cellular effects of oncolytic viral therapy on the glioblastoma microenvironment. Gene Ther. 17:202–216 [DOI] [PubMed] [Google Scholar]
- 17. Jenkins FJ, Roizman B. 1986. Herpes simplex virus 1 recombinants with noninverting genomes frozen in different isomeric arrangements are capable of independent replication. J. Virol. 59:494–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katzin DS, Connor JD, Wilson LA, Sexton RS. 1967. Experimental herpes simplex infection in the owl monkey. Proc. Soc. Exp. Biol. Med. 125:391–398 [DOI] [PubMed] [Google Scholar]
- 19. Kesari S, et al. 1995. Therapy of experimental human brain tumors using a neuroattenuated herpes simplex virus mutant. Lab. Invest. 73:636–648 [PubMed] [Google Scholar]
- 20. Kesari S, Lee VM, Brown SM, Trojanowski JQ, Fraser NW. 1996. Selective vulnerability of mouse CNS neurons to latent infection with a neuroattenuated herpes simplex virus-1. J. Neurosci. 16:5644–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim JJ, et al. 1999. Antigen-specific humoral and cellular immune responses can be modulated in rhesus macaques through the use of IFN-gamma, IL-12, or IL-18 gene adjuvants. J. Med. Primatol. 28:214–223 [DOI] [PubMed] [Google Scholar]
- 22. Kramm CM, et al. 1997. Therapeutic efficiency and safety of a second-generation replication-conditional HSV1 vector for brain tumor gene therapy. Hum. Gene Ther. 8:2057–2068 [DOI] [PubMed] [Google Scholar]
- 23. Lagunoff M, Randall G, Roizman B. 1996. Phenotypic properties of herpes simplex virus 1 containing a derepressed open reading frame P gene. J. Virol. 70:1810–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lasner TM, et al. 1996. Therapy of a murine model of pediatric brain tumors using a herpes simplex virus type-1 ICP34.5 mutant and demonstration of viral replication within the CNS. J. Neuropathol. Exp. Neurol. 55:1259–1269 [DOI] [PubMed] [Google Scholar]
- 25. Liu TC, Wakimoto H, Martuza RL, Rabkin SD. 2007. Herpes simplex virus Us3(-) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt targeting molecular therapeutics. Clin. Cancer Res. 13:5897–5902 [DOI] [PubMed] [Google Scholar]
- 26. MacLean AR, Mul-Fareed Robertson L, Harland J, Brown SM. 1991. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the “a” sequence. J. Gen. Virol. 72:631–639 [DOI] [PubMed] [Google Scholar]
- 27. Markert JM, Malick A, Coen DM, Martuza RL. 1993. Reduction and elimination of encephalitis in an experimental glioma therapy model with attenuated herpes simplex mutants that retain susceptibility to acyclovir. Neurosurgery 32:597–603 [DOI] [PubMed] [Google Scholar]
- 28. Markert JM, Gillespie GY, Weichselbaum RR, Roizman B, Whitley RJ. 2000. Genetically engineered HSV in the treatment of glioma: a review. Rev. Med. Virol. 10:17–30 [DOI] [PubMed] [Google Scholar]
- 29. Markert JM, et al. 2000. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 7:867–874 [DOI] [PubMed] [Google Scholar]
- 30. Markert JM, et al. 2009. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre- and post-tumor resection for recurrent GBM. Mol. Ther. 17:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. 1991. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252:854–856 [DOI] [PubMed] [Google Scholar]
- 32. McKay EM, McVey B, Marsden HS, Brown SM, MacLean AR. 1993. The herpes simplex virus type 1 strain 17 open reading frame RL1 encodes a polypeptide of apparent M(r) 37K equivalent to ICP34.5 of herpes simplex virus type 1 strain F. J. Gen. Virol. 74:2493–2497 [DOI] [PubMed] [Google Scholar]
- 33. Melendez LV, Espana C, Hunt RD, Daniel MD, Garcia FG. 1969. Natural herpes simplex infection in the owl monkey (Aotus trivirgatus). Lab Anim. Care 19:38–45 [PubMed] [Google Scholar]
- 34. Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. 1995. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1:938–943 [DOI] [PubMed] [Google Scholar]
- 35. Mok W, Stylianopoulos T, Boucher Y, Jain RK. 2009. Mathematical modeling of herpes simplex virus distribution in solid tumors: implications for cancer gene therapy. Clin. Cancer Res. 15:2352–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Papanastassiou V, et al. 2002. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 9:398–406 [DOI] [PubMed] [Google Scholar]
- 37. Parker JN, et al. 2000. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. U. S. A. 97:2208–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parker JN, Bauer DF, Cody JJ, Markert JM. 2009. Oncolytic viral therapy of malignant glioma. Neurotherapeutics 6:558–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Post LE, Roizman B. 1981. A generalized technique for deletion of specific genes in large genomes: a gene 22 of herpes simplex virus 1 is not essential for growth. Cell 25:227–232 [DOI] [PubMed] [Google Scholar]
- 40. Preusser M, et al. 2011. Current concepts and management of glioblastoma. Ann. Neurol. 70:9–21 [DOI] [PubMed] [Google Scholar]
- 41. Prichard MN, et al. 1990. A microtiter virus yield reduction assay for the evaluation of antiviral compounds against human cytomegalovirus and herpes simplex virus. J. Virol. Methods 28:101–106 [DOI] [PubMed] [Google Scholar]
- 42. Purves FC, Longnecker RM, Leader DP, Roizman B. 1987. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3, which is not essential for virus growth in cell culture. J. Virol. 61:2896–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pyles RB, Warnick RE, Chalk CL, Szanti BE, Parysek LM. 1997. A novel multiply-mutated HSV-1 strain for the treatment of human brain tumors. Hum. Gene Ther. 8:533–544 [DOI] [PubMed] [Google Scholar]
- 44. Rampling R, et al. 2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 7:859–866 [DOI] [PubMed] [Google Scholar]
- 45. Sarisky RT, et al. 2003. Profiling penciclovir susceptibility and prevalence of resistance of herpes simplex virus isolates across eleven clinical trials. Arch. Virol. 148:1757–1769 [DOI] [PubMed] [Google Scholar]
- 46. Sarkaria JN, et al. 2006. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin. Cancer Res. 12:2264–2271 [DOI] [PubMed] [Google Scholar]
- 47. Shah AC, Benos D, Gillespie GY, Markert JM. 2003. Oncolytic viruses: clinical applications as vectors for the treatment of malignant gliomas. J. Neurooncol. 65:203–226 [DOI] [PubMed] [Google Scholar]
- 48. Shah AC, et al. 2006. Serial passage through human glioma xenografts selects for a Deltagamma134.5 herpes simplex virus type 1 mutant that exhibits decreased neurotoxicity and prolongs survival of mice with experimental brain tumors. J. Virol. 80:7308–7315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stupp R, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352:987–996 [DOI] [PubMed] [Google Scholar]
- 50. Stupp R, et al. 2009. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10:459–466 [DOI] [PubMed] [Google Scholar]
- 51. Tahara H, Lotze MT. 1995. Antitumor effects of interleukin-12 (IL-12): applications for the immunotherapy and gene therapy of cancer. Gene Ther. 2:96–106 [PubMed] [Google Scholar]
- 52. Todo T, Rabkin SD, Chahlavi A, Martuza RL. 1999. Corticosteroid administration does not affect viral oncolytic activity, but inhibits antitumor immunity in replication-competent herpes simplex virus tumor therapy. Hum. Gene Ther. 10:2869–2878 [DOI] [PubMed] [Google Scholar]
- 53. Todo T, et al. 1999. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum. Gene Ther. 10:2741–2755 [DOI] [PubMed] [Google Scholar]
- 54. Todo T, et al. 2000. Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in aotus. Mol. Ther. 2:588–595 [DOI] [PubMed] [Google Scholar]
- 55. Todo T, Rabkin SD, Martuza RL. 2000. Evaluation of ganciclovir-mediated enhancement of the antitumoral effect in oncolytic, multimutated herpes simplex virus type 1 (G207) therapy of brain tumors. Cancer Gene Ther. 7:939–946 [DOI] [PubMed] [Google Scholar]
- 56. Todo T. 2008. “Armed” oncolytic herpes simplex viruses for brain tumor therapy. Cell Adh. Migr. 2:208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Williams-Aziz SL, et al. 2005. Comparative activities of lipid esters of cidofovir and cyclic cidofovir against replication of herpesviruses in vitro. Antimicrob. Agents Chemother. 49:3724–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wong RJ, et al. 2004. Angiogenesis inhibition by an oncolytic herpes virus expressing interleukin 12. Clin. Cancer Res. 10:4509–4516 [DOI] [PubMed] [Google Scholar]
- 59. Yazaki T, Manz HJ, Rabkin SD, Martuza RL. 1995. Treatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1. Cancer Res. 55:4752–4756 [PubMed] [Google Scholar]