

Abstract
High-risk human papillomavirus type 16 (HPV16) is the primary causative agent of cervical cancer and therefore is responsible for significant morbidity and mortality worldwide. Cellular transformation is mediated directly by the expression of viral oncogenes, the least characterized of which, E5, subverts cellular proliferation and immune recognition processes. Despite a growing catalogue of E5-specific host interactions, little is understood regarding the molecular basis of its function. Here we describe a novel function for HPV16 E5 as an oligomeric channel-forming protein, placing it within the virus-encoded “viroporin” family. The development of a novel recombinant E5 expression system showed that E5 formed oligomeric assemblies of a defined luminal diameter and stoichiometry in membranous environments and that such channels mediated fluorescent dye release from liposomes. Hexameric E5 channel stoichiometry was suggested by native PAGE studies. In lieu of high-resolution structural information, established de novo molecular modeling and design methods permitted the development of the first specific small-molecule E5 inhibitor, capable of both abrogating channel activity in vitro and reducing E5-mediated effects on cell signaling pathways. The identification of channel activity should enhance the future understanding of the physiological function of E5 and could represent an important target for antiviral intervention.
INTRODUCTION
Human papillomaviruses (HPVs) are small, double-stranded DNA viruses that infect squamous epithelial cells and produce a range of clinical lesions, including common warts, genital warts, and cancers of the anogenital tract and oropharynx. A subset of HPVs are carcinogenic, and among these high-risk types, HPV16 is detected in approximately 60% of all cervical cancer cases worldwide (4). The virus encodes three oncoproteins: E5, E6, and E7. The roles of E6 and E7 in cervical carcinogenesis have been extensively studied, and the contributions of both proteins to HPV pathogenesis are well accepted. The least characterized of the three oncoproteins is the highly hydrophobic, 83-amino-acid E5 protein, which associates with internal membranes, most notably those of the endoplasmic reticulum, Golgi apparatus, and perinuclear region (24).
HPV16 E5 is classified as an oncoprotein due to its ability to induce anchorage-independent growth in murine fibroblasts and human keratinocytes (34). Transgenic mouse model systems demonstrate that high levels of E5 expression in the skin induce epithelial hyperproliferation, resulting in spontaneous tumor formation (15, 30). These mice also display increased dysplastic disease in the cervical epithelium (29). E5 mRNA is highly abundant in HPV lesions (37), and the protein is expressed in the early stages of malignant transformation, where the episomal viral genome is present (2). Therefore, E5 represents a target for early-stage intervention, prior to the progression of premalignant lesions to cervical cancer.
E5 hyperactivates ligand-dependent epidermal growth factor (EGF) receptor (EGFR) signaling pathways, resulting in enhanced extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) activity (38). This is accomplished by impaired endosome maturation (5, 40), resulting in delayed EGFR degradation. The expression of E5 is also associated with reduced apoptosis (32) and immune evasion (1). However, the mechanisms by which E5 achieves these effects are poorly understood due to inadequate mammalian and bacterial expression systems (41) as well as a lack of immunological reagents. Given its cellular distribution, the production of E5, in a form compatible with analyses of its interaction with membranes, is highly desirable.
E5 shares characteristics reminiscent of a group of viral membrane proteins termed viroporins. These short proteins (50 to 120 residues) contain a hydrophobic domain that contains at least one amphipathic alpha helix. The insertion of these domains into membranes and subsequent homo-oligomerization create a hydrophilic pore formed by the hydrophilic faces of the amphipathic helices and the hydrophobic residues interacting with the phospholipid bilayer. The passage of small molecules and ions through these channels allows the virus to modulate ion homeostasis. Other domains identified in some viroporins include an additional hydrophobic region, a stretch of basic residues that can act as a membrane insertion motif, and a domain rich in aromatic amino acids that is usually inserted into the interfacial phase of the phospholipid bilayer (35). Viroporin-mediated membrane permeability occurs at various stages during infection, often controlling viral entry and release. Although not usually essential for viral genome replication, viroporins are often essential for the production of infectious virions or during virus entry (19). Other cellular functions altered by these proteins include vesicle trafficking and glycoprotein homeostasis. The inhibition of viroporin function is an ideal target for antiviral drug development (17), as exemplified by the clinical use of adamantane drugs to block the M2 proton channel of influenza A virus (IAV) (22).
Here we describe the development of the first system to express and purify recombinant HPV16 E5, which functions as a viroporin in vitro. Furthermore, the rational identification of a specific small-molecule E5 inhibitor provides both a novel tool with which to investigate channel function as well as a basis for exploiting E5 as a target for future drug treatments.
MATERIALS AND METHODS
Construction of plasmid vectors.
For the expression of E5 in mammalian cells, HPV16 E5 (isolate Qv17722E) with an in-frame amino-terminal FLAG epitope was subcloned into the EcoRI and BamHI sites of pSG5 (Stratagene) using forward primer 5′-CCGGAATTCGCCACCATGGACTACAAGGACGATGACGATAAGACAAATCTTGATACT-3′ and reverse primer 5′-GCGGGATCCTTATGTAATTAAAAAGCG-3′. For bacterial expression, FLAG-E5 was subcloned into the EcoRI and NotI sites of pGEX-6P1 (Amersham) and pET-28b-GFP-His10 using forward primer 5′-ATATATGAATTCGCGGCCATGGATTACAAGGATGACGACGATAAGACAAATCTTGATACTGCA-3′ and reverse primer 5′-ATATATACTGCAGGCGGCCGCTTATGTAATTAAAAAGCG-3′. To generate Cherry-FLAG-E5, FLAG-E5 was subcloned into the EcoRI and BamHI sites of pmCherry-C2 (Clontech) using forward primer 5′-ATA TAT GAA TTC GCG GCC ATG GAT TAC AAG GAT GAC GAC GAT AAG ATA ACA AAT CTG GAT ACT-3′ and reverse primer 5′-AAT TGG ATC CTT ATG TAA TCA GAA AGC GTG CAT GTG TAT GGA T-3′. All constructs were verified by sequencing (GATC). Plasmids expressing green fluorescent protein (GFP) fusions of HPV16 oncoproteins were provided by M. Hibma (University of Otago). pGEX-FLAG-p7(J4) was previously described (3).
Cell culture.
HaCaT, C33A, and HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS) as described previously (31). XBHK-21 cells were maintained in minimum essential medium Eagle (Sigma) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM l-glutamine, and 10% FBS.
Expression of FLAG-E5 using vaccinia virus VTF7-3.
BHK-21 cells were infected with 5 PFU per cell of VTF7-3 in phosphate-buffered saline (PBS) and incubated for 50 min with gentle shaking every 10 min. Subsequently, cells were transfected with 4 μg pSG5-FLAG-E5 and mock (pSG5) using Lipofectin transfection agent (Invitrogen) according to the manufacturer's protocol. Cells were lysed at 24 h posttransfection and analyzed by Western blot analysis.
Transfections, growth factor stimulations, and luciferase reporter assays.
Cells were transfected by using polyethyleneimine (PEI) reagent as described previously (28). For the analysis of ERK phosphorylation, cells were maintained in low-serum medium (0.5%) prior to stimulation with recombinant EGF (Sigma) at a working concentration of 100 ng/ml. Where appropriate, cells were treated with rimantadine or MV006 (100 μM) 1 h prior to growth factor stimulation. Dual-luciferase reporter assays were carried out as described previously (12, 27). Briefly, C33A cells were transfected with plasmids expressing HPV16 E5 in combination with a reporter plasmid in which tandem AP-1 promoter elements drive the expression of firefly luciferase. A constitutively expressing Renilla luciferase plasmid was used to assess transfection efficiency. Transfected cells were mock treated or stimulated with 100 ng/ml EGF (Sigma) for 8 h and then lysed and assayed for luciferase activities by using Dual-Luciferase Stop and Glo reagent (Promega) and a luminometer (EG&G Berthold). All assays were performed in triplicate. Promoter activity was calculated by dividing the relative luciferase activity of stimulated cells by that of mock-treated cells.
Coimmunoprecipitations, Western blotting, and native PAGE.
Coimmunoprecipitations from HEK293 lysates at 48 h posttransfection were performed as described previously (28). Cell lysates or precipitates were resolved by SDS-PAGE (15% Tris-glycine) and probed with antibodies specific for GFP (Clontech), the FLAG epitope (Sigma), or the phosphorylated form of ERK1/2 or with a phosphorylation status-independent ERK1/2 antibody (both from Cell Signaling Technologies). Immunoblots were visualized by using an ECL system (Amersham Biosciences). Native PAGE analysis was performed as described previously (10).
FRET.
Sensitized fluorescence resonance energy transfer (FRET) was performed by using the Delta Vision wide-field system on live cells. Images were captured with a 60×/1.4-numerical-aperture (NA) PlanApo objective on a Roper CoolSnap HQ charge-coupled-device (CCD) camera with a binning of 2 by 2 (pixel size, 0.22 μm2). The filter combinations were as follows: a 480- to 500-nm excitation wavelength (λex) and 581- to 653-nm emission wavelength (λem) for the FRET channel, a 480- to 500-nm excitation and 509- to 547-nm emission for GFP, and a 541- to 569-nm excitation and 581- to 653-nm emission for mCherry. A single image for each channel was collected in a three-layer stack, and stacks were then analyzed by using the PixFRET plug-in in ImageJ. Images were converted to an 8-bit depth before analysis and processed as described previously (9), using the normalization FRET/SqRt(Donor × Acceptor) method (9). Normalized FRET (NFRET) images were then measured for mean FRET signals and analyzed with Excel 2007. Normalized FRET takes into account, and corrects for, differences in expression levels when FRET is performed with transfected cells and controls for spectral bleed-through from the donor (GFP) and acceptor (mCherry) channels.
Expression of recombinant HPV16 FLAG-E5.
FLAG-E5 and FLAG-p7 were expressed as a glutathione S-transferase (GST) fusion in Escherichia coli cells and then cleaved and purified as described previously (3). Purified FLAG-E5 was resuspended in dimethyl sulfoxide (DMSO) to a final concentration of 0.25 μg/μl. The expression of GFP-10×His-FLAG-E5 in Rosetta-2 cells was achieved by autoinduction as described previously (39). The pellet was resuspended in 20 mM HEPES–250 mM NaCl and lysed by sonication (10 s on and 20 s off for 10 cycles). The lysate was centrifuged at 20,000 × g for 30 min at 4°C (Beckman Avanti J-26 XP), and the soluble fraction was filtered and then loaded onto a His-Trap chelating column in the Ni2+ form (GE Healthcare), according to the manufacturer's specifications. GFP-His10-FLAG-E5 was eluted by using a step gradient of imidazole.
Preparation of unilamellar liposomes.
Lipids (Avanti polar lipids), including l-α-phosphatidic acid (egg monosodium salt) (PA) (0.5 mg) and l-α-phosphatidylcholine acid (egg) (PC) (0.5 mg), with a final concentration of 0.5% (wt/wt) l-α-phosphatidylethanolamine with lissamine rhodamine B-labeled head groups (egg), were dried under a stream of argon and then placed under a vacuum for 2 h at room temperature. Lipids were rehydrated (2 mg/ml) with vigorous shaking overnight in a self-quenching concentration of carboxyfluorescein (CF) (Sigma-Aldrich) buffer (50 mM CF in HEPES-buffered saline [HBS] [10 mM HEPES-NaOH {pH 7.4}, 107 mM NaCl]).
Alternatively, buffers containing fluorescein isothiocyanate (FITC)-conjugated dextrans (FDs) of increasing molecular masses (4 kDa [FD-4], 10 kDa [FD-10], and 70 kDa [FD-70] [Sigma]) were used for experiments estimating the E5 pore size (50 mg/ml FITC-dextran, 20 mM HEPES, and 107 mM NaCl [pH 7.4]). Unilamellar liposomes were extruded through a 0.4-μm Nuclepore Track-Etch membrane filter (Whatman), using an Avanti miniextruder with Hamilton glass syringes at 37°C. Liposomes were washed with HBS and purified three times by centrifugation at 100,000 × g for 15 min in a Sorvall S55S ultracentrifuge rotor. Liposomes were finally resuspended in HBS to a stock concentration of 1.0 mM.
Liposome permeability assays.
The real-time release of liposomal contents into the surrounding buffer was monitored by fluorimetry and exploited the self-quenching property of CF. The CF release mediated by FLAG-E5 was assessed by incubating protein (typically 1 μg, dissolved in DMSO) with 50 μM liposomes (determined by the rhodamine absorbance at 570 nm). The total reaction mixture volume was made up to 100 μl with HBS, giving a final E5 concentration of ∼1 μM. Fluorescence measurements were performed with a FLUOstar Optima microplate reader (BMG Technologies) with excitation and emission filters set to 485 and 520 nm, respectively. The real-time measurement of CF release was performed by using 1 μM melittin (Sigma) as a positive control. Fluorescence readings were acquired every 30 s for 30 min at 37°C. All samples were repeated in triplicate, and data were averaged. Initial rates were calculated from the initial linear dye release kinetics (ΔFU s−1), where FU are fluorescence units.
Experiments to determine the effect of pH on FLAG-E5-mediated CF release or assays utilizing FDs could not be performed in real time. This was due to the quenching effect of the acidic pH on CF fluorescence and the lower concentrations of FDs used, respectively. Instead, samples were centrifuged at 160,000 × g for 2 h in an S100-AT3 rotor (Sorvall), and the liposome-free supernatant was transferred onto a 96-well microtiter plate prior to fluorimetry. For FD assays, baseline fluorescence corresponded to spontaneous leakage from vesicle-only controls (liposomes only and 5% DMSO), and maximum CF release was taken as the fluorescence value obtained after the addition of 0.5% (vol/vol) Triton X-100 to liposomes prior to centrifugation. Experiments with all samples were repeated in triplicate, and data were averaged.
For experiments assessing the effect of pH on CF release, liposomes containing CF were resuspended in citrate-phosphate buffers (citric acid and sodium phosphate, dibasic [12H2O], mixed appropriately to give pH 7.4, 6.8, 6.2, and 5.6). Liposome-free supernatants were readjusted to pH 7.4 by the addition of buffering amounts of 1 M Tris-HCl (pH 8.0), determined by the restoration of 100% fluorescence in Triton X-100 controls of the same volume.
In silico modeling of HPV16 E5, rational drug design, and binding studies.
The secondary structure of E5 was predicted by using PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred) and MEMSAT3. Models of the E5 monomer were constructed by using Maestro (Schrodinger Inc.) as previously described (10). Monomers were built amino acid by amino acid with energy minimization (Merck molecular force field [MM/FF] in a simulated water dielectric). Monomers were docked into an oligomer and subjected to further energy minimization. Images of resultant structures were obtained by using either Maestro or Pymol v0.9 (Delano Scientific). Surface representations of the channel complex model revealed cavities within the channel lumen. The Maestro draw function was utilized to design molecules that would fit within the cavities. Molecules were subjected to free-energy minimization, and stable, bound conformations were used as templates for the rapid overlay of chemical structures (ROCS), which created a small subset of analogues available from commercial libraries (Maybridge). Compound-binding studies against E5 complexes employed the program Glide (Schrodinger Inc.). Ligands were prepared by using the LigPrep module of Maestro in the Schrodinger docking program Glide (11). The LigPrep-treated and energy-minimized ligand was docked into the prepared receptor grid, and the binding affinity was evaluated with the Glide score (GScore) parameter (7). The best-docked pose was selected as the one with the lowest GScore, the highest negative value.
Transmission electron microscopy (TEM).
GFP-His10-FLAG-E5, incubated overnight in buffer (20 mM HEPES [pH 7], 100 mM NaCl, 50 mM 1,2-diheptanoyl-sn-glycero-3-phosphocholine [DHPC]), was adsorbed onto thin carbon films. Samples were washed with successive droplets of water to remove detergent, stained by using 2% (wt/vol) phosphotungstic acid, and subsequently adhered to 300-mesh copper grids. Samples were observed by using a Phillips CM10 electron microscope, and micrographs were recorded at ×52,000 magnifications on Kodak SO-163 film.
RESULTS
HPV16 E5 exists as an oligomer in cells.
Previous work using transient expression systems showed that E5 is able to oligomerize (16); however, it has not been established whether E5 oligomers form in cell membranes. HPV16 E5 expressed as a GFP fusion protein (GFP-E5) in HEK293 cells formed dimers and hexamers observed by Western blot analysis of anti-GFP immunoprecipitates separated by SDS-PAGE. Notably, GFP remained monomeric under the same denaturing conditions (Fig. 1A). Levels of both GFP and GFP-E5 were increased by treatment with a proteasome inhibitor, although this did not affect the formation of higher-order species, suggesting that oligomerization is not dependent on the abundance of the E5 protein present in the cell lysates (Fig. 1A). Oligomerization was not dependent on the GFP moiety, as FLAG-tagged E5 (FLAG-E5), expressed from a vaccinia virus system in BHK cells, also formed predominantly dimers and oligomers when analyzed by SDS-PAGE (Fig. 1B). To confirm the E5-specific oligomerization, differentially tagged (FLAG or GFP) E5 proteins were coexpressed and assessed for interactions in anti-FLAG immunoprecipitates. FLAG-E5 did not interact with GFP, GFP-E6, or GFP-E7 controls but bound efficiently to GFP-E5 (Fig. 1C). Furthermore, intermolecular fluorescence resonance energy transfer (FRET) studies with the human cervical C33A cell line using E5 fused with either GFP (GFP-E5) or mCherry (mCherry-FLAG-E5) at the amino terminus confirmed that E5 formed close-range contacts in living cells (Fig. 1D). The functionality of E5 fusion proteins was confirmed by assessing the enhanced ligand-dependent activation of EGFR signaling, a known physiological effect of E5 expression (Fig. 1E). Taken together, these data demonstrate that E5 self-associates and that this association occurs independently of epitope tags.
Fig 1.

Oligomerization of HPV16 E5. (A) Anti-GFP Western blot analysis of GFP and GFP-E5 immunoprecipitates from HEK293 cell lysates. A single monomeric species of GFP was observed at low levels of expression or in cells treated with the proteasome inhibitor MG132 to enhance levels of expression. Multiple bands of GFP-E5 were present, likely indicating a monomer (Mon), a dimer (Dim), and a higher-order oligomer (Olig). Note that the distribution of oligomer species did not change in cells treated with MG132. (B) Anti-FLAG Western blot analysis of FLAG precipitates from BHK cell lysates. Multiple bands of FLAG-E5 were present, including a dimer and tetramer. Note that no E5 monomer was observed in these samples. Asterisks indicate antibody heavy and light chains. Hex, hexamer. (C) FLAG immunoprecipitates (IP) from cells expressing FLAG-E5 and GFP, GFP-E5, GFP-E6, or GFP-E7. Precipitates were probed with anti-FLAG and anti-GFP antibodies to demonstrate protein interactions and equal precipitations of FLAG-E5. Analysis of lysates revealed the expression of all GFP fusion proteins. WB, Western blot. (D) FRET analysis of human cervical C33A cells coexpressing either GFP-E5 and mCherry-FLAG-E5 (i), GFP and mCherry empty vector control plasmids (ii), or GFP-E5 and mCherry empty vector control plasmids (III), pseudocolored to highlight the fluorescence intensity within each image. Images show pixel intensities following the normalization of the signal in channels, as described in Materials and Methods. (E) Hyperactivation of EGFR signaling by the E5 protein. C33A cells expressing an AP-1-responsive luciferase reporter plasmid plus untagged or GFP- or mCherry-fused E5 were mock treated or stimulated with EGF (100 ng/ml) and lysed 6 h later. Levels of luciferase were detected by luminometry and are expressed as a fold increase over mock-treated control cells. Results are the averages of data from 3 independent experiments. Error bars represent standard deviations of the means. RLU, relative light units.
Generation of high-purity recombinant HPV16 E5 from bacteria.
The characterization of E5 has been hampered by the lack of an efficient system to purify recombinant proteins. Here we have adapted a method used previously to purify the hepatitis C virus (HCV) p7 protein (3), where FLAG-tagged E5 is fused to the carboxyl terminus of cleavable glutathione S-transferase (GST) (Fig. 2A). A significant proportion of this fusion protein is targeted to inclusion bodies, reducing toxicity and therefore allowing purification. 3C protease cleavage liberates FLAG-E5, which can be solubilized by using the detergent N-lauryl sarcosine and purified to homogeneity by reverse-phase high-performance liquid chromatography (HPLC) (rpHPLC). Figure 2B shows a Western blot analysis of the 38-kDa GST-FLAG-E5 fusion protein following isopropyl-β-d-thiogalactopyranoside (IPTG) induction (Fig. 2B, lane 2) and the efficient cleavage of GST from recombinant FLAG-E5 (Fig. 2B, lane 4). Detergent-solubilized FLAG-E5 migrated predominantly as a dimer upon SDS-PAGE (Fig. 2B, lane 5), whereas protein purified by rpHPLC and resuspended in DMSO was monomeric (Fig. 2B, lane 6).
Fig 2.
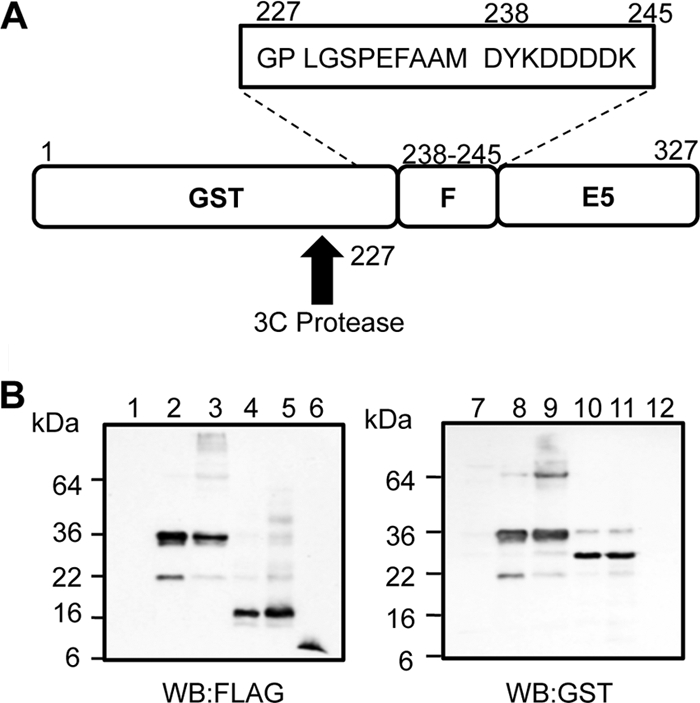
Purification and analysis of recombinant HPV16 FLAG-E5. (A) Schematic of GST-FLAG-E5 designed to express the E5 protein amino-terminally fused to GST and FLAG and containing a 3C cleavage site. The FLAG tag sequence and additional residues fused to the E5 protein postcleavage are boxed. (B) SDS-PAGE and Western blot analysis of expressed E5 fusion proteins during purification using anti-FLAG and anti-GST antibodies. Lanes 1 and 7, pre-IPTG induction; lanes 2 and 8, post-IPTG induction; lanes 3 and 9, precleavage; lanes 4 and 10, postcleavage; lanes 5 and 11, solubilized pre-HPLC sample; lanes 6 and 12, post-HPLC purified FLAG-E5.
Recombinant HPV16 E5 protein associates with liposomes.
To determine whether recombinant E5 associated with lipid membranes, protein-liposome suspensions were subjected to ultracentrifugation, resulting in flotation through a discontinuous Ficoll gradient, as described previously (36). Gradient fractions were analyzed by Western blotting to detect FLAG-E5, and the distribution of liposomes was assessed by rhodamine fluorescence. This confirmed that liposomes had migrated to the 10% Ficoll–aqueous buffer interface (Fig. 3A, top) and that a significant portion of FLAG-E5 associated with the liposomes (Fig. 3A, top blot). Exposure to a high pH did not disrupt this association, confirming that FLAG-E5 was integrated within bilayers and not trapped within vesicles or peripherally associated (Fig. 3A, middle blot). Treatment with detergent (Triton X-100) disrupted the liposome–FLAG-E5 interaction (Fig. 3A, bottom blot), indicating that the migration of the protein to the Ficoll-aqueous buffer interface was liposome dependent.
Fig 3.

HPV16 FLAG-E5 associates with liposomes and displays dose-dependent channel activity. (A) Recombinant FLAG-E5 was assessed for membrane associations in the presence or absence of a high pH (100 mM Na2CO3 [pH 11.4]). Anti-FLAG Western blots from a fractionated, discontinuous Ficoll gradient are shown from fractions 1 to 9. The top panel shows the rhodamine fluorescence of gradient fractions with liposomes floating to the 10% Ficoll–aqueous buffer interface. Control reaction mixtures treated with the detergent Triton X-100 resulted in FLAG-E5 remaining at the bottom of the gradient. (B) Liposome assay controls. Data from a real-time analyses of the effects of liposomes only, the solvent control (DMSO), GST, FLAG-E5, melittin, and Triton X-100 on the kinetics of CF release are shown. (C) Initial rates calculated from the linear part of the real-time curve highlight the negligible impact of solvent alone (DMSO [D]) or GST (G) on CF release in comparison to FLAG-E5 (E5). Error bars represent standard deviations of the means, and the statistical significance of CF release was assessed by a one-way analysis of variance (ANOVA) comparing GST alone to FLAG-E5 (*, P < 0.05). (D and E) Increasing concentrations of recombinant FLAG-E5 ranging from 0.1 to 1 mM were incubated with CF-containing liposomes, and dye release was measured in real time (D) and by initial rates calculated from the linear part of the real-time curve (E). L, liposome only; D, DMSO solvent control; M, melittin. Error bars represent standard deviations of the means, and the statistical significance of CF release was assessed by a one-way ANOVA compared to solvent controls and E5 (**, P < 0.01).
Recombinant HPV16 E5 displays membrane channel activity in liposomes.
To address the functional implications of the FLAG-E5 membrane insertion, we employed a convenient liposome-based fluorescent dye release assay used previously to investigate HCV p7 function and inhibitor sensitivity (36). Increasing amounts of FLAG-E5 were incubated with liposomes containing the fluorescent dye carboxyfluorescein (CF) at self-quenching concentrations. The release of this dye resulted in the recovery of the fluorescent signal, monitored in real time by fluorimetry. Melittin, the pore-forming protein of bee venom, was used as a positive control, and Triton X-100 treatment yielded maximal fluorescence. Baselines were calculated from solvent controls (10% DMSO and liposomes alone). GST served as a negative control and induced baseline levels of fluorescence over time (Fig. 3B and C). The addition of increasing concentrations of FLAG-E5 promoted a rapid, dose-dependent release of CF from liposomes (Fig. 3D and E).
Direct visualization of recombinant HPV16 E5 oligomers by transmission electron microscopy.
Attempts to visualize recombinant FLAG-E5 by transmission electron microscopy (TEM) proved challenging due to its small size. Unfortunately, the larger GST-FLAG-E5 fusion protein had an rpHPLC retention time similar to that of N-lauryl sarcosine, precluding its use in TEM studies. It was therefore necessary to create an alternative E5 fusion protein that could be efficiently purified in the absence of detergent. The GFP-His10-FLAG-E5 fusion protein, expressed in E. coli, formed oligomers by SDS-PAGE analyses of bacterial lysates (Fig. 4A), making this an ideal candidate for TEM studies. Accordingly, phosphotungstic acid negative staining of GFP-His10-FLAG-E5 solubilized in the membrane-mimetic compound 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC) revealed the presence of ringlike structures that were absent with GFP-His10 alone (Fig. 4B). The structures contained an electron-dense center of approximately 1 to 2 nm in diameter, indicative of the presence of a pore, as observed previously for p7 protein complexes (3, 26). These findings not only confirm the presence of E5 oligomeric complexes but also suggest the existence of a membrane-integrated channel structure reminiscent of other viroporins.
Fig 4.

Characterization of the HPV16 E5 channel. (A) Oligomerization of the GFP-His10-FLAG-E5 fusion protein. GFP-His10-FLAG-E5, expressed in E. coli, was analyzed by SDS-PAGE and Western blotting with antibodies against GFP or FLAG. In addition to monomeric GFP-His10-FLAG-E5, higher-molecular-weight forms were evident, representing oligomers of E5 formed in the presence of bacterial lipids. In comparison, the GFP-His10 fusion partner was present as a monomer. (B) Transmission electron microscopy of the GFP-His10-FLAG-E5 channel. This typical view from a raw image of GFP-His10-FLAG-E5 oligomers negatively stained with phosphotungstic acid (PTA) at a ×52,000 magnification (left) demonstrates the presence of “ringlike” structures only in the presence of E5. A selected set of individual GFP-His10-FLAG-E5 oligomers (middle) details the ringlike arrangement of the protein. Shown is a view of a raw TEM image of GFP-His10 negatively stained with PTA. (C) FLAG-E5 forms a channel with a restrictive luminal diameter. Liposomes containing CF or FITC-dextrans (FDs) of various molecular masses (0.38 to 70 kDa) and Stokes' radii (0.6 to 6.0 nm) were incubated with FLAG-E5 (E5) (1 μM) and melittin (M) (1 μM). Liposome-free supernatants were assessed by using fluorimetry at an λex of 485 nm and an λem 520 nm. Liposome-only and solvent controls corresponded to baseline fluorescence, and Triton X-100 controls (T) established maximum fluorescence. Baseline controls were subtracted from the fluorescence readings generated by the E5- and melittin-mediated fluorophore release. Melittin allowed the efficient release of CF and FD-4, whereas E5 would permit only the release of CF. Error bars represent standard deviations of the means. The statistical significance of E5-mediated CF release compared to 4-kDa dextran release and melittin-mediated 4-kDa and 10-kDa dextran release was assessed by a one-way ANOVA (***, P < 0.001). (D) FLAG-E5-mediated CF release from liposomes is enhanced at acidic pH. FLAG-E5 and melittin were added to CF liposomes resuspended in citrate-phosphate buffers of various pHs (5.6 to 7.4). Liposome-free supernatants were assessed by fluorimetry, and the pH of each supernatant was readjusted to pH 7.4 by the addition of buffering amounts of 1 M Tris-HCl (pH 8.0) (amounts determined by the fluorescence recovery of Triton X-100 controls). Triton X-100-lysed controls were used to determine maximum fluorescence, and baseline fluorescence was determined with liposome-alone (L) and solvent (DMSO [D]) controls. Error bars represent standard deviations of the means. The statistical significances of differences in E5-mediated CF release between pH 6.8 to 7.4 and pH 5.6 to 7.4 were assessed by a one-way ANOVA (**, P < 0.01; *, P < 0.05).
Recombinant HPV16 E5 forms a channel with a defined luminal diameter and stoichiometry.
To ensure that the FLAG-E5-mediated CF release was due to the formation of a channel with a size-selective lumen, rather than nonspecific membrane destabilization, FLAG-E5 (1 μM) was incubated with liposomes containing CF or fluorescein dextrans (FDs) of increasing molecular masses and Stokes' radii, the release of which was assessed by fluorimetry endpoint assays. Melittin was used as a positive control, as it was previously shown to form channels with defined size constraints in similar systems (25, 33, 36). As described above, melittin and FLAG-E5 mediated the efficient release of CF (Stokes' radius of 0.6 nm) from liposomes, validating protein integrity (Fig. 4C). However, FLAG-E5 did not permit the efficient release of FD-4 (Stokes' radius of 1.4 nm), suggesting that this larger molecule could not transit through the E5 channel lumen (Fig. 4C). In contrast, melittin allowed the efficient release of FD-4 from liposomes (Fig. 4C), consistent with previous findings (36). The release of FD-10 (Stokes' radius of 2.3 nm) and FD-70 (Stokes' radius of 6.0 nm) was significantly reduced for both proteins (Fig. 4C). The minimal release of FD-10 and FD-70 by melittin likely reflects either nonspecific escape or the release of FD during liposome fusion. These data suggest that the E5 channel lumen has a diameter equal to or greater than 1.2 nm and less than 2.8 nm. Our data are also consistent with the luminal diameter of melittin (3 nm) (25, 33), as the efficient release of the 4-kDa dextran would require a luminal diameter of at least 2.8 nm. Furthermore, these data agree with the FLAG-E5 lumen size of 1 to 2 nm inferred from our TEM data (Fig. 4B).
Recombinant HPV16 E5-mediated CF release is enhanced at acidic pH.
The gating of some viroporins, including IAV M2 and HCV p7, is influenced by changes in pH, whereby the protonation of ionizable histidine side chains effects altered permeability states of the channel. As E5 also possesses potentially ionizable residues, the effect of reducing the external buffer pH (pHex) relative to that of the liposome interior (pH 7.4) on FLAG-E5 channel activity was investigated. This was assessed by endpoint assays where the pH was rebuffered by the addition of buffering amounts of Tris-HCl (pH 8.0) (assessed by the recovery of Triton X-100 controls) due to the effect of a reduced pH on CF solubility (36). A reduction of the pHex from 7.4 to 6.8 led to a significant increase in FLAG-E5 channel activity, with a further increase between pH 6.8 and pH 5.6 (Fig. 4D). The channel activity exhibited by melittin remained largely stable with decreasing pHs, with liposome and solvent controls validating the integrity of the liposomes in each buffer (Fig. 4D).
Construction of an in silico model of an HPV16 E5 oligomer.
The development of potential E5-inhibitory compounds to study E5 channel function will be dependent on information pertaining to the stoichiometry and structure of channel complexes. Predictions based on Kyte-Doolittle hydrophobicity proposed a three-trans-membrane-domain (TMD) monomer (23). Recent investigations of HPV16 E5 membrane topology support this model, identifying a luminal amino terminus and a cytoplasmic carboxyl terminus (24).
In the absence of high-resolution structural information for the E5 channel complex, we applied a robust molecular modeling approach used previously to generate validated models of heptameric HCV p7 (10). The HPV16 E5 primary sequence was imported into PSIPRED and the MEMSAT3 membrane topology prediction software. This generated a three-TMD prediction model for E5 (Fig. 5A). Models of the channel were constructed by using Maestro (Schrodinger Inc.), and monomers were built amino acid by amino acid with energy minimization. This predicted that the E5 monomer conformed to a triangular wedge-like arrangement, which tessellated at 60° angles, favoring a hexameric stoichiometry. Monomers were therefore docked into a hexameric structure and subjected to further energy minimization (Fig. 5A). Pentameric and/or heptameric channels could potentially form but were predicted to be less stable by the program (data not shown). The hexameric model predicted the channel lumen to be 1.0 nm in diameter at the widest point and to be lined by a mixture of hydrophilic (S37 and S41), hydrophobic (L34, I44, L48, and I51), and potentially ionizable (His75/77) residues (Fig. 5B), which may mediate ion sensing and gating functions, respectively.
Fig 5.

Molecular modeling of full-length HPV16 E5 as a monomer and hexamer. Models of the E5 channel complex were generated by using Maestro as described in Materials and Methods. (A) E5 monomers were modeled with free-energy minimization using the Maestro program and then manually docked into a symmetrical hexameric complex. The top panel shows a side projection (two monomers showing), and the bottom panel shows a top-down projection revealing the channel lumen. (B) Residues from TM2 that are predicted to line the lumen of the channel are highlighted.
HPV16 E5 viroporin activity shows differential sensitivity to adamantane compounds.
Adamantanes specifically inhibit sensitive variants of IAV M2 and HCV p7 channel activity (6, 20). To assess whether these compounds acted similarly against E5, the abilities of amantadine and its methylated derivative rimantadine to block the FLAG-E5-mediated release of CF from liposomes were tested. The incubation of FLAG-E5 with high concentrations (400 μM) of amantadine did not significantly affect the release of CF from liposomes (Fig. 6A). However, the same concentration of rimantadine reduced channel activity by approximately 70% (Fig. 6A), consistent with its increased potency relative to that of amantadine (18). New, E5-specific compounds were therefore required to efficiently block channel function.
Fig 6.
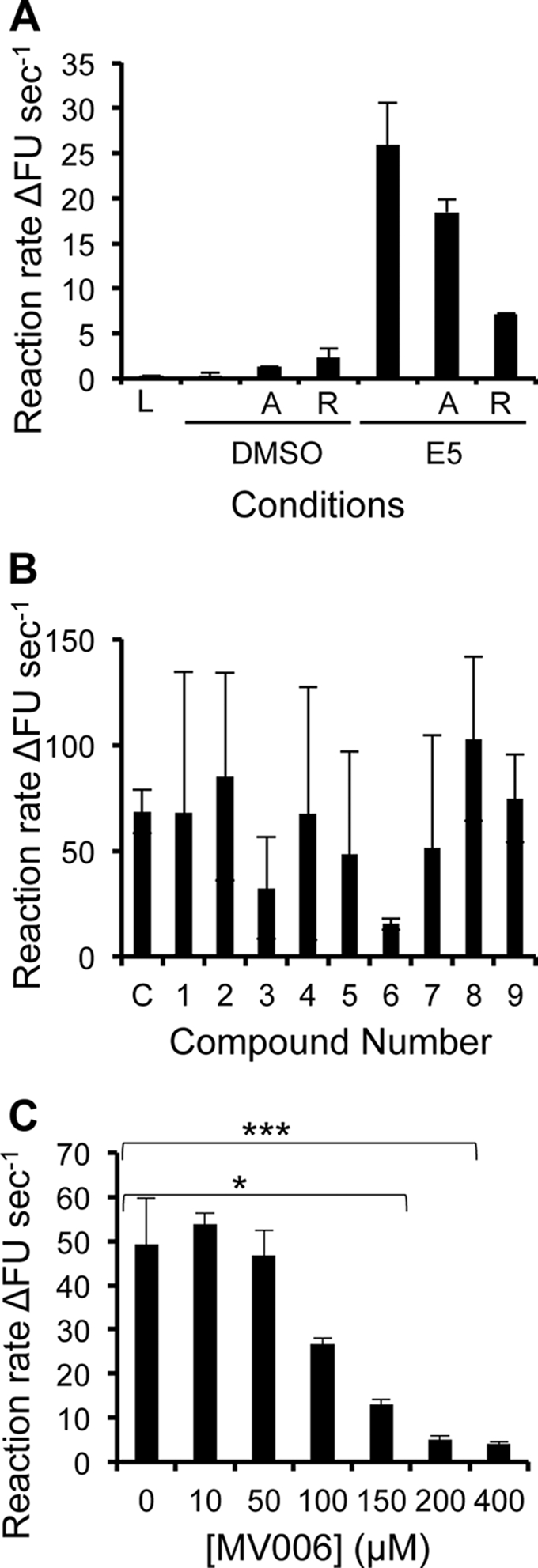
HPV16 FLAG-E5 exhibits mixed sensitivity to adamantane compounds in vitro but can be inhibited, in a dose-dependent manner, by a novel inhibitor. (A) The sensitivity of FLAG-E5 to adamantane drugs was assessed in vitro (1 μM FLAG-E5 displayed resistance to amantadine [denoted A] but was sensitive to rimantadine [denoted R]). DMSO and FLAG-E5 in the absence of compound (denoted C) showed background and maximum activities, respectively. Error bars represent standard deviations of the means, and the statistical significance of CF release was assessed by a one-way ANOVA compared to solvent controls and FLAG-E5 (*, P < 0.05; **, P < 0.01). (B) Effects of candidate E5 inhibitors on FLAG-E5-mediated CF release. The ability of FLAG-E5 to cause the release of CF in the presence of candidate inhibitory compounds (400 μM) was assessed in real time by fluorimetry and is represented as an initial rate. Error bars represent standard deviations of the means. (C) Dose-dependent inhibition of FLAG-E5-mediated CF release by the compound MV006, as depicted by the initial rate. Error bars represent standard deviations of the means, and the statistical significance of CF release was assessed by a one-way ANOVA compared to the control of FLAG-E5 in the absence of compound (*, P < 0.05; ***, P < 0.001).
A novel small-molecule inhibitor of HPV16 E5 channel activity.
An important validation of de novo molecular models is their use as accurate templates for the design of functional inhibitory molecules. A surface representation of the E5 hexamer model revealed cavities within the lining of the channel lumen, potentially amenable to small-molecule binding. As described recently (10), the Maestro “draw” function was used to create a small panel of molecules predicted to bind the FLAG-E5 interior with high affinity and so inhibit channel activity through the occlusion of the pore. The effects of the candidate molecules on FLAG-E5-induced CF release were assessed at 400 μM, and the compounds MV003 and MV006 significantly inhibited FLAG-E5 activity, with MV006 showing the highest potency (Fig. 6B). Furthermore, MV006 showed a dose-dependent inhibitory effect on FLAG-E5 activity (Fig. 6C).
MV006 is a more effective inhibitor of FLAG-E5 than the adamantanes in vitro.
The Glide program (Schrodinger) was used to further interrogate binding sites on E5 for the adamantanes and MV006 (Fig. 7A). In each case, the Glide grid was extended to the maximum area, allowing the free docking of each compound to the entire protein surface. The top-binding pose for each ligand, based on the Glide score (GScore), was selected for study. Both adamantanes and MV006 clustered to a highly lipophilic pocket at the interface created by adjacent monomer trans-membrane domains in the pore. A model of the rimantadine-binding site is shown as an example (Fig. 7A). The binding pocket is aligned with lipophilic residues on adjacent trans-membrane domains (Ile44, Leu48, Leu45, Leu23, Leu47, Ile51, Leu48, and Ser41). An inspection of the binding poses reveals that the adamantyl cage of amantadine and rimantadine occupies the lipophilic pocket and that the protonatable amine extends into the lumen of the pore, whereas the head heterocyclic component of MV006 occupies the pocket. A comparison of predicted binding affinities based on the GScore revealed that amantadine bound with a significantly lower avidity than did rimantadine (Fig. 7B), possibly explaining its decreased potency (Fig. 6A). Binding values for MV006 and rimantadine were similar, although MV006 achieved an improved inhibitory effect in vitro, possibly due to the stoichiometry of the E5-inhibitor interaction (Fig. 7C). The alignment between predicted binding affinities and in vitro effects validated our molecular design approach.
Fig 7.

Molecular modeling of HPV16 E5 inhibitor compounds. (A) Molecular model of E5 indicating the position of the rimantadine-binding site. The binding pocket is aligned with lipophilic residues on adjacent trans-membrane domains (Ile44, Leu48, Leu45, Leu23, Leu47, Ile51, Leu48, and Ser41). The left panel shows the top view for rimantadine bound to E5. The right panel indicates a side view of rimantadine bound to E5. For simplicity, only one rimantadine molecule is shown. (B) Predicted ligand binding scores for amantadine, rimantadine, and MV006 against a hexameric E5 molecular model. (C) Comparison of the dose-dependent inhibition of FLAG-E5-mediated CF release by MV006 (black bars) and rimantadine (gray bars) highlighting the differences between the two inhibitors (***, P < 0.001).
MV006 activity is specific for assembled FLAG-E5 complexes.
Controls in which MV006 was incubated with liposomes in the absence of FLAG-E5 showed no adverse effects on liposome stability (data not shown), and their addition to melittin-positive controls had no effect on CF release (Fig. 8A). Together, these data indicate that MV006 does not prevent permeability by nonspecifically coating liposomes or indiscriminately blocking channel complexes. While MV006 was predicted to share a luminal binding mode with rimantadine (Fig. 7A), other prototype viroporin inhibitors, such as alkylated imino-sugars, can prevent channel oligomerization (10). To give a clearer picture of the MV006 mode of action, we sought to demonstrate that its effects were due to the inhibition of assembled E5 channels rather than an effect on oligomerization or membrane insertion. We have previously shown that DHPC and native PAGE revealed the inhibitory effects of imino-sugars on p7 oligomers in DHPC micelles, whereas the detergent lyso-myristoylphosphatidylglycerol (LMPG) does not efficiently promote the formation of channel complexes. As our TEM studies suggested that E5 should behave similarly to p7 (Fig. 4D), we tested whether MV006 could affect FLAG-E5 oligomerization under similar conditions using native PAGE. The reconstitution of FLAG-p7, or FLAG-E5, in DHPC resulted in the formation of a single oligomeric species, indicative of a hexameric E5 stoichiometry (Fig. 8B), which was unaffected by high concentrations of MV006 (4 mM). In addition, FLAG-E5 remained associated with liposome membranes following Ficoll flotation in the presence of MV006 (4 mM) (Fig. 8C). Together, these data support our theory that MV006 inhibits assembled E5 channel complexes, as predicted by molecular models, thereby confirming their relative accuracy in vitro.
Fig 8.

Effects of a novel E5 inhibitor on melittin-mediated CF release, FLAG-E5 oligomerization, and membrane insertion. (A) MV006 does not prevent the melittin-induced release of CF from liposomes, as depicted by an endpoint analysis. Error bars represent standard deviations of the means, and the statistical significance of CF release was assessed by a one-way ANOVA compared to the melittin (M) control in the absence of compound (***, P < 0.001). Initial rates for liposomes alone (L), the DMSO solvent control (D), and Triton X-100 (T) are also shown. (B) Oligomerization of the FLAG-E5 protein. Mild detergents were tested for their abilities to induce the oligomerization of FLAG-p7 and FLAG-E5 using native PAGE. DHPC induced the oligomerization of both FLAG-p7 and FLAG-E5, whereas LMPG did not. The addition of MV006 (4 mM) did not impair FLAG-E5 oligomerization. The a asterisk indicates the detergent front. (C) Membrane association of FLAG-E5 in the presence of MV006. Anti-FLAG Western blots from fractionated discontinuous Ficoll gradients are shown from fractions 1 to 9. The presence of MV006 (4 mM) had no inhibitory effect on membrane insertion.
Inhibitors of viroporin activity reduce ERK phosphorylation in HPV16 E5-expressing cells.
The expression of E5 in mammalian cells inhibits endosome acidification, resulting in increased EGFR activation and phosphorylation of the ERK MAPK (34, 40). We tested the small-molecule E5 inhibitors rimantadine and MV006 to determine whether viroporin activity is responsible for the decreased endosome acidification using phosphorylated ERK-MAPK as an indicator. First, using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, we showed that the addition of high concentrations (up to 4 mM) of MV006 or rimantadine was not directly toxic to HaCaT cells (data not shown). Next, HaCaT cells stably expressing HPV16 E5 or an empty vector were pretreated with rimantadine, MV006, or the DMSO carrier 1 h prior to EGF stimulation. The cells were then incubated for increasing time periods prior to lysis and then probed for phosphorylated ERK by Western blotting (Fig. 9). Similar to previous findings (41), E5-expressing cells exhibited enhanced EGFR signaling, as evident by the increased ERK phosphorylation compared to control cells. Of interest, preincubation with rimantadine or MV006 resulted in a reproducible decrease of ERK phosphorylation in E5-expressing cells, resulting in levels comparable to those of vector controls at 30 min poststimulation, whereas untreated E5-expressing cells retained elevated phosphorylated-ERK levels at this time point. This was not a result of nonspecific inhibitor effects on EGFR signaling, since the levels of ERK phosphorylation were not significantly affected in inhibitor-treated control cells following stimulation. This indicates that the viroporin activity of E5 may be required for enhanced ERK signaling and thereby linked to pro-oncogenic phenotypes.
Fig 9.

Viroporin inhibitors decrease ERK phosphorylation in E5-expressing cells. (A) HaCaT cells stably expressing HPV16 E5 or an empty plasmid were preincubated with MV006 (100 mM), rimantadine (100 mM), or the DMSO carrier for 1 h prior to stimulation with recombinant EGF (100 ng/ml). Cells were lysed, and equal amounts of protein were analyzed by SDS-PAGE. The levels of phosphorylated ERK (P-ERK) were determined by Western blotting. (B) Quantitative analysis of data from panel A using ImageJ software analysis to compare levels of phosphorylated ERK to total levels of ERK.
DISCUSSION
The precise molecular mechanisms by which the expression of the high-risk E5 protein contributes to cellular transformation and HPV pathogenesis remain elusive, although the disruption of cellular ionic gradients appears to be central to this process. In this study, we provide evidence for a previously undocumented function of E5 as a virus-encoded channel or viroporin. To the best of our knowledge, this is the oncogene possessing channel activity, and future work will be essential to understand how viroporin activity contributes to the physiological function of E5 during the HPV life cycle.
Key to this discovery was the establishment of the first robust system for the expression and purification of a recombinant E5 protein (Fig. 2), which should permit a comprehensive biophysical characterization of the function of E5. Initial studies confirmed that recombinant FLAG-E5 exists as an oligomer in membrane-like environments. As seen previously for HCV p7, SDS acts both as a membrane mimetic and as a denaturant, leading to a laddering effect of E5 oligomeric species by SDS-PAGE, with higher-ordered forms being less abundant than monomers/dimers. Furthermore, the presence of a reducing agent in the SDS sample buffer argues against disulfide linkages being responsible for oligomer formation. Under native DHPC-PAGE conditions, which allow proteins to maintain their native state, a single hexameric FLAG-E5 species with a molecular mass similar to that of the FLAG-p7 heptamer (63 kDa) was detected. This finding suggests that the E5 complex is energetically stable and likely represents the native channel complex in vivo (Fig. 8B). Moreover, the design of the active inhibitor MV006, using a hexameric E5 template, is consistent with these observations (discussed below) and further validates our molecular model as a good approximation of the channel structure.
The efficient release of CF and retention of large FDs within liposomes incubated with FLAG-E5 are further evidence for the formation of FLAG-E5 channels with defined stoichiometries and luminal diameters (Fig. 4A). These data show that the E5 channel will allow the release of molecules with a Stokes' radius of 0.6 nm but not those with a radius of 1.4 nm or larger. These data infer that the luminal diameter of the E5 channel is equal to or greater than 1.2 nm but less than 2.8 nm. These findings are consistent with our TEM data that suggest that E5 forms a pore with a luminal diameter of ∼1 to 2 nm and our in silico model that predicts a lumen diameter of ∼1 nm, consistent with a hexameric stoichiometry. This robust assay has also successfully validated the luminal diameter and pore-forming abilities of HCV p7 (36) and melittin (33, 36). Our data show that the release of CF in our real-time assay is not a result of nonspecific membrane disruption and that any unincorporated FLAG-E5 that might aggregate is not causing membrane destabilization. It was suggested previously that E5 oligomerization is an artifact of overexpression and cell lysis conditions (24). While the paucity of immunological reagents against HPV16 E5 precludes a direct analysis of E5, using physiologically relevant expression levels, we have utilized a variety of techniques to demonstrate E5 oligomerization in cells. Of note, FRET in live cells was apparent for E5 fluorescent protein fusions (Fig. 1D), which can occur only if the donor and acceptor fluorophores are within defined molecular distances of each other, consistent with specific protein-protein interactions.
Viroporins are attractive targets for antiviral therapy, with the adamantanes amantadine and rimantadine providing a clinical precedent (21). Our analysis demonstrates that E5 is resistant to high concentrations of amantadine but can be inhibited by relatively high (50% inhibitory concentration [IC50] of ∼100 μM) concentrations of rimantadine (Fig. 6A). This highlights differences between E5 and the prototypic viroporins M2 and p7, several variants of which can be highly sensitive to both compounds. However, single-amino-acid variations can drastically affect the binding of small molecules to viroporins, as illustrated by circulating adamantane-resistant IAV and our recent investigations into p7 (10).
A complete understanding of the molecular mechanisms of viroporin function requires high-resolution structural information, which is lacking for most viroporins. However, we have previously demonstrated our ability to generate de novo molecular models in the absence of such information, which recapitulate many facets of channel behavior (10, 17). The E5 model predicted a hexameric channel (Fig. 5), in agreement with native PAGE studies of the recombinant protein (Fig. 8B). This model revealed the possible three-dimensional arrangement of conserved E5 residues and will guide future mutagenesis studies to explore the molecular basis of channel activity.
The interaction of small molecules is an important way of validating molecular models through the experimental confirmation of relative binding predictions. Accordingly, the inactive amantadine was predicted to bind to the E5 channel model with a low affinity (Fig. 7B), whereas rimantadine was predicted to bind with a relatively higher affinity within the channel lumen. Modeling suggested that in addition to hydrophobic contacts with the adamantyl cage, rimantadine would also gain additional hydrophobic binding potential through its additional methyl group, which, obviously, amantadine cannot. De novo molecular design was also used to generate novel inhibitors targeting the luminal binding pocket, based on virtual screening algorithms. In agreement with predictions, the compound MV006 exhibited a specific, dose-dependent inhibition of E5 viroporin activity in vitro and, as predicted, was more potent than rimantadine. In addition to our recent studies of p7 (10), this is another convincing example of the use of de novo molecular modeling to produce novel compounds with antiviroporin activity. Further studies are required to determine the mechanisms by which MV006 and rimantadine prevent E5 channel function. Based on our binding studies, it is plausible that the molecules act to inhibit a conformational change to the active form or stabilize an inactive conformation.
Accumulating evidence supports an important role for E5 in the HPV life cycle (8, 14). Expression analysis studies have identified an increasing incidence of E5 expression, both in cervical biopsy specimens and in head and neck cancers, where the HPV genome is not integrated into host chromosomes. Accordingly, an important target of future studies will be to assign a role for viroporin activity in the myriad of E5-associated functions documented in the literature. The absence of E5 has been shown to have deleterious effects on several late events in the HPV life cycle, including cell cycle progression and HPV genome amplification (8, 14). Indeed, many of the interactions attributed to E5 require the modification of host membranes and the subversion of ionic gradients (5, 13, 40), phenotypes often associated with viroporins. In particular, E5 expression can enhance EGFR signaling (Fig. 1E) through a mechanism that may require the deacidification of endosomes (5). While this was initially thought to occur by the binding of E5 to the vacuolar ATPase (v-ATPase), it is now clear that E5 modulates the maturation of endosome compartments independently of this protein interaction (40). The enhanced FLAG-E5-mediated release of CF at an acidic pH (Fig. 4B) suggests that, like IAV M2 and HCV p7, E5 may be gated by changes in pH. No measurement of proton transport was recorded in this study; however, the increased release of CF at lower pHex values is indicative of an increased frequency of channel opening, presumably due to side-chain ionization as a result of the enhanced electrochemical gradient across the membrane. As endosome function is absolutely dependent on the correct establishment of ionic gradients, it is tempting to speculate that one role for the pH-sensitive E5 viroporin may be to subvert ionic gradients to enhance EGFR signaling. Our observation that E5 inhibitors reduce EGF-stimulated ERK phosphorylation in E5-expressing cells suggests that viroporin function is required for the hyperactivation of mitogenic signaling. At this stage, it is unclear whether the viroporin is directly modulating endosome acidification or is required for a secondary effect. A more comprehensive analysis of the role of the viroporin in this pathway will require viroporin-null E5 mutants or the generation of more potent small-molecule inhibitors with improved bioavailability in order to preclude any potential off-target effects.
The role fulfilled by E5 appears to be unique among viroporins, which exert their function predominantly during virus entry and/or release. Establishing how viroporin activity relates to mitogenic signaling could provide a novel anticancer target to be exploited through future rational drug design. The identification of MV006 establishes a proof of principle that continued studies will produce improved, more potent compounds to target the activity of E5 within infected cells. Such inhibitors could drastically reduce the productive population of HPV in individuals at disease stages prior to cellular transformation and could theoretically be used prophylactically. Our description of E5 channel activity therefore provides a foundation for both future hypothesis-led studies and the development of therapeutic strategies for HPV-associated malignancies, driven by the need to treat those for whom HPV vaccination is not appropriate.
ACKNOWLEDGMENTS
We are grateful to Toshana Foster and Elizabeth Atkins (University of Leeds) for advice with protein purification and liposome preparation and Iain Manfield at the Wellcome Trust-funded Centre for Biomolecular Interactions (University of Leeds) for advice in the use of HPLC. We also thank Merilyn Hibma (University of Otago) for providing the GFP-HPV16 plasmids.
L.F.W. is supported by a Medical Research Council (MRC) studentship, K.K.H. and M.V. are funded by Biotechnology and Biological Sciences Research Council (BBSRC) Collaborative Awards in Science and Engineering (CASE) studentships, and M.M. is funded by a Yorkshire Cancer Research studentship. S.G. is the recipient of a University of Leeds Biomedical and Health Research Centre (BHRC) senior translational fellowship. A.M. is a Research Council UK (RCUK) fellow, and research in his laboratory is funded by Yorkshire Cancer Research (grants PP015, LPP041, and L339), Cancer Research UK (grant C43832/A14246), and the Royal Society.
Footnotes
Published ahead of print 22 February 2012
REFERENCES
- 1. Ashrafi GH, Haghshenas MR, Marchetti B, O'Brien PM, Campo MS. 2005. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 113:276–283 [DOI] [PubMed] [Google Scholar]
- 2. Chang JL, et al. 2001. The expression of HPV-16 E5 protein in squamous neoplastic changes in the uterine cervix. J. Biomed. Sci. 8:206–213 [DOI] [PubMed] [Google Scholar]
- 3. Clarke D, et al. 2006. Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J. Biol. Chem. 281:37057–37068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clifford GM, Smith JS, Plummer M, Munoz N, Franceschi S. 2003. Human papillomavirus types in invasive cervical cancer worldwide: a meta-analysis. Br. J. Cancer 88:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Disbrow GL, Hanover JA, Schlegel R. 2005. Endoplasmic reticulum-localized human papillomavirus type 16 E5 protein alters endosomal pH but not trans-Golgi pH. J. Virol. 79:5839–5846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duff KC, Ashley RH. 1992. The transmembrane domain of influenza A M2 protein forms amantadine-sensitive proton channels in planar lipid bilayers. Virology 190:485–489 [DOI] [PubMed] [Google Scholar]
- 7. Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. 1997. Empirical scoring functions. I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 11:425–445 [DOI] [PubMed] [Google Scholar]
- 8. Fehrmann F, Klumpp DJ, Laimins LA. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 77:2819–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feige JN, Sage D, Wahli W, Desvergne B, Gelman L. 2005. PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc. Res. Tech. 68:51–58 [DOI] [PubMed] [Google Scholar]
- 10. Foster TL, et al. 2011. Resistance mutations define specific antiviral effects for inhibitors of the hepatitis C virus (HCV) p7 ion channel. Hepatology 54:79–90 [DOI] [PubMed] [Google Scholar]
- 11. Friesner RA, et al. 2004. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47:1739–1749 [DOI] [PubMed] [Google Scholar]
- 12. Gamlen T, et al. 2010. Expression of the NS3 protease of cytopathogenic bovine viral diarrhea virus results in the induction of apoptosis but does not block activation of the beta interferon promoter. J. Gen. Virol. 91:133–144 [DOI] [PubMed] [Google Scholar]
- 13. Gao P, Zheng J. 2010. High-risk HPV E5-induced cell fusion: a critical initiating event in the early stage of HPV-associated cervical cancer. Virol. J. 7:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Genther SM, et al. 2003. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 77:2832–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Genther Williams SM, et al. 2005. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Res. 65:6534–6542 [DOI] [PubMed] [Google Scholar]
- 16. Gieswein CE, Sharom FJ, Wildeman AG. 2003. Oligomerization of the E5 protein of human papillomavirus type 16 occurs through multiple hydrophobic regions. Virology 313:415–426 [DOI] [PubMed] [Google Scholar]
- 17. Gonzalez ME, Carrasco L. 2003. Viroporins. FEBS Lett. 552:28–34 [DOI] [PubMed] [Google Scholar]
- 18. Griffin S, et al. 2008. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 48:1779–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Griffin SD. 2009. Plugging the holes in hepatitis C virus antiviral therapy. Proc. Natl. Acad. Sci. U. S. A. 106:12567–12568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Griffin SD, et al. 2003. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, amantadine. FEBS Lett. 535:34–38 [DOI] [PubMed] [Google Scholar]
- 21. Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. 1985. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 4:3021–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Intharathep P, et al. 2008. How amantadine and rimantadine inhibit proton transport in the M2 protein channel. J. Mol. Graph. Model. 27:342–348 [DOI] [PubMed] [Google Scholar]
- 23. Kell B, et al. 1994. Detection of E5 oncoprotein in human papillomavirus type 16-positive cervical scrapes using antibodies raised to synthetic peptides. J. Gen. Virol. 75(Pt 9):2451–2456 [DOI] [PubMed] [Google Scholar]
- 24. Krawczyk E, Suprynowicz FA, Sudarshan SR, Schlegel R. 2010. Membrane orientation of the human papillomavirus type 16 E5 oncoprotein. J. Virol. 84:1696–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ladokhin AS, Selsted ME, White SH. 1997. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: pore formation by melittin. Biophys. J. 72:1762–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luik P, et al. 2009. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 106:12712–12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Macdonald A, et al. 2003. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J. Biol. Chem. 278:17775–17784 [DOI] [PubMed] [Google Scholar]
- 28. Mankouri J, et al. 2010. Optineurin negatively regulates the induction of IFNbeta in response to RNA virus infection. PLoS Pathog. 6:e1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maufort JP, Shai A, Pitot HC, Lambert PF. 2010. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 70:2924–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maufort JP, Williams SM, Pitot HC, Lambert PF. 2007. Human papillomavirus 16 E5 oncogene contributes to two stages of skin carcinogenesis. Cancer Res. 67:6106–6112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McFadden N, et al. 2011. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog. 7:e1002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oh JM, et al. 2010. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 31:402–410 [DOI] [PubMed] [Google Scholar]
- 33. Park SC, et al. 2006. Investigation of toroidal pore and oligomerization by melittin using transmission electron microscopy. Biochem. Biophys. Res. Commun. 343:222–228 [DOI] [PubMed] [Google Scholar]
- 34. Pim D, Collins M, Banks L. 1992. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 7:27–32 [PubMed] [Google Scholar]
- 35. Sanz MA, Madan V, Carrasco L, Nieva JL. 2003. Interfacial domains in Sindbis virus 6K protein. Detection and functional characterization. J. Biol. Chem. 278:2051–2057 [DOI] [PubMed] [Google Scholar]
- 36. StGelais C, et al. 2007. Inhibition of hepatitis C virus p7 membrane channels in a liposome-based assay system. Antiviral Res. 76:48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoler MH, et al. 1992. Human papillomavirus type 16 and 18 gene expression in cervical neoplasias. Hum. Pathol. 23:117–128 [DOI] [PubMed] [Google Scholar]
- 38. Straight SW, Hinkle PM, Jewers RJ, McCance DJ. 1993. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 67:4521–4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41:207–234 [DOI] [PubMed] [Google Scholar]
- 40. Suprynowicz FA, et al. 2010. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J. Virol. 84:10619–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang DH, Wildeman AG, Sharom FJ. 2003. Overexpression, purification, and structural analysis of the hydrophobic E5 protein from human papillomavirus type 16. Protein Expr. Purif. 30:1–10 [DOI] [PubMed] [Google Scholar]