

Abstract
Herpes simplex virus 1 (HSV-1) protein VP22, encoded by the UL49 gene, is a major virion tegument protein. In the present study, we showed that VP22 was required for efficient redistribution of viral proteins VP16, VP26, ICP0, ICP4, and ICP27 and of cellular protein Hsc-70 to the cytoplasm of infected cells. We found that two dileucine motifs in VP22, at amino acids 235 and 236 and amino acids 251 and 252, were necessary for VP22 regulation of the proper cytoplasmic localization of these viral and cellular proteins. The dileucine motifs were also required for proper cytoplasmic localization of VP22 itself and for optimal expression of viral proteins VP16, VP22, ICP0, UL41, and glycoprotein B. Interestingly, a recombinant mutant virus with alanines substituted for the dileucines at amino acids 251 and 252 had a 50% lethal dose (LD50) for neurovirulence in mice following intracerebral inoculation about 103-fold lower than the LD50 of the repaired virus. Furthermore, the replication and spread of this mutant virus in the brains of mice following intracerebral inoculation were significantly impaired relative to those of the repaired virus. The ability of VP22 to regulate the localization and expression of various viral and cellular proteins, as shown in this study, was correlated with an increase in viral replication and neurovirulence in the experimental murine model. Thus, HSV-1 VP22 is a significant neurovirulence factor in vivo.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) virions, like those of other herpesviruses, consist of three morphologically distinct structures: the nucleocapsid, containing the linear double-stranded DNA viral genome, which encodes at least 84 viral proteins, in an icosahedral capsid; the tegument, a proteinaceous layer surrounding the nucleocapsid; and the envelope, a host cell-derived lipid bilayer with viral glycoproteins enclosing the nucleocapsid and tegument (55). The tegument consists of a large number of proteins, including at least 26 viral proteins (41). Tegument proteins are released into the cytoplasm upon the fusion of the virion envelope with the host cell membrane and therefore may function to establish an environment for effective initiation of very early infection. However, the roles of only a few tegument proteins, including the immediate-early gene transactivator VP16, encoded by the UL48 gene, and the virion host shutoff protein VHS, encoded by the UL41 gene (55), in very early infection have been established. Large amounts of newly synthesized tegument proteins have also been shown to modulate the host cellular environment and to regulate various viral replication processes later in infection (28).
VP22, encoded by the UL49 gene, is one of the major HSV-1 tegument proteins, and its amino acid sequence is conserved in the subfamily Alphaherpesvirinae. It appears that the functional requirements of VP22 homologues in the viral life cycle differ among alphaherpesviruses. For example, the VP22 homologues of Marek's disease virus serotype 1 and varicella-zoster virus are considered to be essential for viral replication in cell cultures, while those of bovine herpesvirus 1 (BHV-1), pseudorabies virus (PRV), and HSV-1 are not (6, 9, 12, 38, 52). Also, deletion of the UL49 gene impaired BHV-1 pathogenicity in a natural host and impaired HSV-1 pathogenicity in an animal model (13, 37) but had no apparent effect on the replication of PRV in cell cultures or on its pathogenicity in an animal model (9).
In infected cells, HSV-1 VP22 forms multimers and is nucleotidylylated, possibly by casein kinase II (CKII) (3, 45, 47). VP22 is also phosphorylated in infected cells, possibly by CKII and a viral protein kinase encoded by the UL13 gene (1, 20). It has been suggested that phosphorylation of VP22 is involved in the regulation of its incorporation into virions and in its dissociation from the tegument upon the entry of the virus into the cell (3, 20, 46). Since VP22 localizes in the cytoplasm or nucleus at different times postinfection (19, 53, 61), it appears to be dynamically trafficked during viral infection. VP22 has been considered to play a critical role in HSV-1 replication and pathogenicity, based on studies showing that recombinant VP22-null mutant viruses have impaired viral growth and spread in cell cultures, as well as impaired viral spread and pathogenesis in infected mouse corneas (13, 15, 52, 58). Although the precise mechanism(s) by which VP22 acts in viral replication and pathogenesis remains largely unknown at present, a number of functions for VP22 have been reported. These include (i) association with and reorganization of microtubules (18, 34); (ii) interaction with chromatin, histones H1 and H4, and template-activating factor 1 (TAF-1), a chromatin-remodeling factor, and impairment of histone acetylation and TAF-1-dependent nucleosome assembly (17, 43, 54, 66); (iii) neutralization of the activity of VHS (62); (iv) binding to a viral RNA and transport of the RNA from cell to cell for expression (59); (v) association with cellular membranes and localization in acidic compartments, including the trans-Golgi network, where HSV-1 nucleocapsids may acquire their final envelope (4, 61); (vi) interaction with various viral tegument and envelope proteins, including VP16, ICP0, Us9, UL46, UL47, UL56, glycoprotein E (gE), gD, and gM (7, 16, 21, 22, 24, 25, 35, 42, 50); (vii) promotion of the synthesis of various viral proteins in infected cells and incorporation of a subset of virion-assembled proteins (13–15); and (viii) regulation of the compartmentalization of VP16 and ICP0 (15, 16). Although many VP22 functions have been clarified, as described above, and the domains responsible for some VP22 functions have been mapped, none of these functions have been explicitly linked to HSV-1 replication and pathogenesis. Since the UL49-null mutation has only a modest effect on viral replication in cell cultures (13, 15, 52, 58), the phenotypes of recombinant viruses, each carrying a mutation(s) that abolishes a specific VP22 function, need to be examined in a mouse model in order to clarify the role of each VP22 function in viral replication and pathogenesis. Such studies have not yet been reported.
In the present study, we have described a novel function of VP22. The generation and characterization of an HSV-1 UL49-null mutant virus showed that VP22 was required for efficient redistribution of viral proteins ICP0, ICP4, ICP27, VP16, and VP26 and cellular protein Hsc-70 to the cytoplasm. We have also shown that two dileucine motifs in VP22, at amino acids 235 and 236 and amino acids 251 and 252, that are well conserved among VP22 homologues in alphaherpesviruses were necessary for this novel VP22 function. In addition, we studied a recombinant virus with amino acid substitutions in the VP22 dileucine motif at amino acids 251 and 252, and we found that this mutation significantly impaired HSV-1 neurovirulence in a murine model.
MATERIALS AND METHODS
Cells and viruses.
Vero and rabbit skin cells have been described previously (64), as has the HSV-1 wild-type strain HSV-1(F) (64).
Generation of recombinant viruses.
A UL49-null mutant virus, YK451 (ΔVP22), in which the UL49 gene was disrupted by the insertion of a foreign gene cassette carrying a stop codon (TGA) just downstream of the UL49 start codon, an I-SceI site, a kanamycin resistance gene, and 60 bp of a sequence upstream of the second codon of the UL49 gene (Fig. 1), was generated by the Red-mediated mutagenesis procedure as described previously (29, 30), except that the primers listed in Table 1 were used. Recombinant virus YK452, in which the foreign gene cassette inserted into the UL49 locus of YK451 was excised (ΔVP22-repair) (Fig. 1), was generated by the Red-mediated mutagenesis procedure as described above using Escherichia coli containing the YK451 genome (29). An additional repaired virus, YK457 (ΔVP22-repair-2), was constructed by the same procedure as YK452 (ΔVP22-repair), but with E. coli GS1783 harboring YK451 circular viral DNA isolated from Vero cells infected with YK451. E. coli GS1783 (27) harboring YK451 circular viral DNA isolated from YK451-infected Vero cells by the Hirt method was generated as described previously (64). The UL41-null mutant virus YK476 (ΔUL41) (Fig. 1) was generated by the same procedure as YK451 (ΔVP22), but with the primers listed in Table 1. Recombinant viruses YK453 and YK455 (Fig. 1), encoding VP22 with alanine substitutions for the dileucine motifs at amino acids 235 and 236 (Di-Leu-235-236) (VP22LL235AA) and amino acids 251 and 252 (Di-Leu-251-252) (VP22LL251AA), respectively, were generated by the two-step Red-mediated mutagenesis procedure using E. coli GS1783 containing pYEbac102 and the primers listed in Table 1 as described previously (30). Recombinant viruses YK454 and YK456, in which the VP22LL235AA and VP22LL251AA mutations in YK453 and YK455, respectively, were repaired, were generated with the primers listed in Table 1 as described previously (30). Additional repaired viruses YK458 (VP22LL235AA-repair-2) and YK459 (VP22LL251AA-repair-2) were constructed by the same procedures as YK454 (VP22LL235AA-repair) and YK456 (VP22LL251AA-repair) but with E. coli GS1783 harboring YK453 and YK455 circular viral DNA isolated from Vero cells infected with YK453 and YK455, respectively. Wild-type HSV-1(F) and the recombinant viruses used in this study were propagated on Vero cells.
Fig 1.

Schematic diagrams of the genome structures of wild-type YK304 and the relevant domains of the recombinant viruses used in this study. Diagram 1, the YK304 genome carrying a bacmid (BAC) in the intergenic region between UL3 and UL4. Diagram 2, domains carrying the UL48 to UL50 open reading frames. Diagram 3, the UL49 gene, encoding VP22. Diagrams 4 to 12, recombinant viruses with mutations in the VP22 gene. Diagram 13, domains carrying the UL40 to UL42 open reading frames. Diagram 14, the UL41 gene, encoding VHS. Diagram 15, recombinant virus with a mutation in the UL41 gene.
Table 1.
Primer sequences used for the construction of recombinant viruses
| Mutation | Sequence |
|---|---|
| ΔVP22 | 5′-CCGTCCGAACCCAGGCCTAATTGTCCGCGCATCCGACCCTAGCGTGTT CGTGGAACCATGTAAAGGATGACGACGATAAGTAGGG-3′ |
| 5′-TTCACGGAGCGGCGAGAGGTCATGGTTCCACGAACACGCTAGGGTCG GATGCGCGGACAACAACCAATTAACCAATTCTGATTAG-3′ | |
| VP22LL235AA | 5′-GGACATGTCGCGTCCGCGCACAGACGAAGACCTCAACGAAGCCGCTG GCATCACCACCATCCGCGTAGGATGACGACGATAAGTAGGG-3′ |
| 5′-TTTTGCCCTCGCAGACCGTCACGCGGATGGTGGTGATGCCAGCGG CTTCGTTGAGGTCTTCGTCTGCAACCAATTAACCAATTCTGATTAG-3′ | |
| VP22LL235AA-repair | 5′-GGACATGTCGCGTCCGCGCACAGACGAAGACCTCAACGAACTCCTTG GCATCACCACCATCCGCGTAGGATGACGACGATAAGTAGGG-3′ |
| 5′-TTTTGCCCTCGCAGACCGTCACGCGGATGGTGGTGATGCCAAGGAGT TCGTTGAGGTCTTCGTCTGCAACCAATTAACCAATTCTGATTAG-3′ | |
| VP22LL251AA | 5′-CATCACCACCATCCGCGTGACGGTCTGCGAGGGCAAAAACGCGGCTC AGCGCGCCAACGAGTTGGTAGGATGACGACGATAAGTAGGG-3′ |
| 5′-CCTGCACCACGTCTGGATTCACCAACTCGTTGGCGCGCTGAGCCGCG TTTTTGCCCTCGCAGACCGCAACCAATTAACCAATTCTGATTAG-3′ | |
| VP22LL251AA-repair | 5′-CATCACCACCATCCGCGTGACGGTCTGCGAGGGCAAAAACCTGCTTC AGCGCGCCAACGAGTTGGTAGGATGACGACGATAAGTAGGG-3′ |
| 5′-CCTGCACCACGTCTGGATTCACCAACTCGTTGGCGCGCTGAAGCAGGT TTTTGCCCTCGCAGACCGCAACCAATTAACCAATTCTGATTAG-3′ | |
| ΔUL41 | 5′-GACCGTCGCACCAACCACGCCTTAGTTAGGCCGATCCGCAGTTACAAT TGACCTGACATGTAGAGGATGACGACGATAAGTAGGG-3′ |
| 5′-TTCATCATCCCGAACAAACCCATGTCAGGTCAATTGTAACTGCGGAT CGGCCTAACTAAGCAACCAATTAACCAATTCTGATTAG-3′ |
Antibodies.
To generate rabbit polyclonal antibodies to UL41, two rabbits were immunized with a domain of UL41 encoding amino acids 239 to 489 fused to maltose-binding protein (MBP), which was expressed in E. coli and purified as described previously (32, 56) by the standard protocol at Medical & Biological Laboratories Co., Ltd. Rabbit polyclonal antibodies to VP22 and VP26 have been described previously (48, 61). Mouse monoclonal antibodies to gB (H1817) and ICP0 (5H7) were purchased from East Coast Bio. Mouse monoclonal antibodies to ICP4 (10F1), ICP8 (11E2), and ICP27 (8.F.137B) were purchased from Abcam. A goat polyclonal antibody to VP16 was purchased from Santa Cruz Biotechnology. A rabbit polyclonal antibody to apoptosis-inducing factor (AIF) was purchased from Cell Signaling Technology. A rat monoclonal antibody to Hsc70 was purchased from Stressgen.
Immunoblotting, immunoprecipitation, immunofluorescence, and determination of plaque size.
Immunoblotting, immunoprecipitation, immunofluorescence, and determination of plaque size were performed as described previously (33, 65).
Animal studies.
Female ICR mice were purchased from Charles River. Three-week-old mice were infected intracerebrally with 102 PFU of YK455 (VP22LL251AA) or YK456 (VP22LL251AA-repair) as described previously (64). Mice were monitored daily, and mortality from 1 to 14 days postinfection was attributed to the inoculated virus. To determine viral titers in brains, mice were inoculated intracerebrally with 102 PFU of the YK455 (VP22LL251AA) or YK456 (VP22LL251AA-repair) as described above. At 3 days postinfection, the mice were sacrificed, and whole brains were removed, sonicated in 1 ml Medium 199 containing 1% fetal calf serum (FCS) and antibiotics, and frozen at −80°C. Frozen samples were later thawed and centrifuged, and viral titers in the supernatants were determined by standard plaque assays on Vero cells. All animal studies were carried out with the approval of the Ethics Committee for Animal Experimentation of the University of Tokyo.
Histopathology and immunohistochemistry.
Mice were inoculated intracerebrally with 102 PFU of YK455 (VP22LL251AA or YK456 (VP22LL251AA-repair) as described above. At 5 days postinfection, infected mice were anesthetized and perfused with 20 ml 20% phosphate-buffered formalin. Fixed brains were routinely embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Immunohistochemical detection of HSV-1 antigens was performed on paraffin-embedded sections, as follows. After deparaffinization with xylene, sections were rehydrated in ethanol and were immersed in phosphate-buffered saline (PBS). Antigens were retrieved by hydrolytic autoclaving for 20 min at 121°C in 10 mM sodium citrate-sodium chloride buffer (pH 6.0). After cooling, the sections were immersed in PBS. Endogenous peroxidase was blocked by incubation in 1% hydrogen peroxide in methanol for 30 min. After washing in PBS, the sections were incubated with normal goat serum for 5 min and were then reacted with a rabbit polyclonal antibody against HSV-1 (B0114; DakoCytomation) overnight at 4°C. The sections were incubated with the Dako EnVision+ System, containing a horseradish peroxidase (HRP)-labeled polymer conjugated with anti-rabbit antibodies (K4003; Dako), for 30 min at room temperature. Peroxidase activity was detected by development with diaminobenzidine containing hydrogen peroxide. Nuclei were counterstained with hematoxylin.
RESULTS
Characterization of a UL49-null mutant virus.
To clarify the role(s) of VP22 during HSV-1 infection, we generated UL49-null mutant virus YK451 (ΔVP22) and its repaired virus YK452 (ΔVP22-repair) (Fig. 1). As expected, Vero cells infected with wild-type HSV-1(F) or YK452 (ΔVP22-repair) expressed VP22, whereas those infected with YK451 (ΔVP22) did not (Fig. 2). A previous report has shown that expression of various HSV-1 proteins (i.e., ICP0, VP16, VHS, glycoprotein D [gD], and gE) was impaired in cells infected with the UL49-null mutant virus relative to that in cells infected with the wild-type virus, but the UL49-null mutation had no effect on the expression of ICP4 and ICP27 (14). In agreement with the previous report, the levels of VP16, ICP0, and UL41 expression in Vero cells infected with YK451 (ΔVP22) at a multiplicity of infection (MOI) of 1 for 20 h appeared to be lower than those in cells infected with wild-type HSV-1(F) or the YK452 (ΔVP22-repair) virus, but the UL49-null mutation (ΔVP22) had no effect on ICP4 and ICP27 expression (Fig. 2). We noted that Vero cells infected with YK451 (ΔVP22) produced much less gB than cells infected with wild-type HSV-1(F) or YK452 (ΔVP22-repair) (Fig. 2), indicating that gB expression, like that of the viral proteins listed above, was regulated by VP22.
Fig 2.

Immunoblots of electrophoretically separated lysates of Vero cells that were either mock infected (lane 4) or infected with wild-type HSV-1(F) (lane 1), YK451 (ΔVP22) (lane 2), or YK452 (ΔVP22-repair) (lane 3) at an MOI of 1. Infected Vero cells were harvested at 20 h postinfection and were immunoblotted with an antibody to VP22, VP16, gB, ICP0, ICP4, UL41, ICP27, or α-tubulin.
A previous report (58) showed that a UL49-null mutant virus derived from an HSV-1(F) bacterial artificial chromosome (BAC) clone, which was different from the HSV-1(F) BAC clone used in this study, accumulated deletion and frameshift mutations in the UL41 coding region when propagated on Vero cells. Therefore, we sequenced the UL41 coding region from the wild-type HSV-1(F) virus and the YK451 (ΔVP22) virus but did not detect any DNA sequence differences between these coding regions (data not shown).
Localization of VP16 in VP22-null mutant virus-infected cells.
It has been shown that coexpression of VP16 and VP22 in transient-transfection assays resulted in redistribution of the two viral proteins from their normal locations to a perinuclear cytoplasmic domain, suggesting that each protein regulated the localization of the other (16). To investigate whether VP22 regulates the localization of VP16 in HSV-1-infected cells, Vero cells were infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), or YK476 (ΔUL41) at an MOI of 1. At 8, 15, and 20 h postinfection, VP16 localization was analyzed by immunofluorescence with confocal microscopy. Previous reports showed that VP16 was detected in both the cytoplasm and nuclei of wild-type HSV-1-infected Vero cells early in infection and that cytoplasmic localization of VP16 became predominant at later times postinfection (16). Similar observations were reported for HSV-2 and PRV (23, 49). In agreement with these previous reports, VP16 was detected in both the cytoplasm and nuclei of wild-type HSV-1(F)-infected Vero cells at 8 h postinfection but was localized predominantly in the cytoplasm at later times (15 and 20 h) postinfection (Fig. 3). In contrast, VP16 was detected predominantly in the nuclei of Vero cells infected with YK451 (ΔVP22) throughout infection (Fig. 3). Wild-type localization of VP16 was restored in Vero cells infected with YK452 (ΔVP22-repair), in which the UL49-null mutation was repaired (Fig. 3). In addition, the UL41-null mutation had no effect on the subcellular distribution of VP16 in infected cells (Fig. 3). These results indicated that VP22 is required for proper redistribution of VP16 to the cytoplasm of infected cells late in infection.
Fig 3.

Digital confocal microscope images showing the localization of VP16. Vero cells were infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), or YK476 (ΔUL41) at an MOI of 1. Infected Vero cells were fixed at the indicated times postinfection, permeabilized, stained with an anti-VP16 antibody, and examined by confocal microscopy.
Effects of amino acid substitutions in the two VP22 dileucine motifs on the localization of VP16 in infected cells.
O'Regan et al. (51) previously reported that alanine substitutions of the leucines in both VP22 dileucine motifs (Di-Leu-235-236 and Di-Leu-251-252), which are conserved among VP22 homologues in alphaherpesviruses, abolished the interaction of VP22 with VP16 in cells transiently transfected with a VP22 mutant tagged with green fluorescent protein (GFP) (GFP-VP22), followed by wild-type HSV-1 infection. To examine whether both dileucine motifs were required for VP22 regulation of VP16 localization in infected cells, we generated recombinant viruses YK453 (VP22LL235AA) and YK455 (VP22LL251AA), in which Di-Leu-235-236 and Di-Leu-251-252 were replaced with alanines, and recombinant viruses YK454 (VP22LL235AA-repair) and YK456 (VP22LL251AA-repair), in which these mutations were repaired (Fig. 1). We then used immunofluorescence with confocal microscopy to examine the localization of VP16 in Vero cells infected with either wild-type HSV-1(F), YK451 (ΔVP22), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) at an MOI of 1 for 15 h. As was found with YK451 (ΔVP22) (Fig. 3), VP16 was detected predominantly in the nuclei of cells infected with YK453 (VP22LL235AA) or YK455 (VP22LL251AA) (Fig. 4). These results indicated that both dileucine motifs are necessary for VP22 regulation of the proper cytoplasmic localization of VP16 in infected cells. We carried out DNA sequence analysis of the UL41 coding regions in HSV-1(F), YK453, and YK455 to confirm that the procedure used to produce alanine substitutions in the two VP22 dileucine motifs did not also introduce mutations into the UL41 coding regions in YK453 (VP22LL235AA) and YK455 (VP22LL251AA) (data not shown).
Fig 4.

Digital confocal microscope images showing the localization of VP16 in Vero cells infected with wild-type HSV-1(F), YK451 (ΔVP22), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) at an MOI of 1 for 15 h. Infected Vero cells were fixed, permeabilized, stained with an anti-VP16 antibody, and examined by confocal microscopy.
Effects of amino acid substitutions in the VP22 dileucine motifs on viral replication in cell cultures.
To further characterize the recombinant viruses with alanine substitutions in the two VP22 dileucine motifs, we first analyzed viral growth in cell cultures by infecting Vero cells with wild-type HSV-1(F), YK451 (ΔVP22), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) at an MOI of 1 or 0.01. Total virus was harvested from the infected cells and cell culture supernatants at 24 h postinfection and was assayed for PFU on Vero cells. As shown in Fig. 5, the progeny virus titers in cells infected at both MOIs with recombinant viruses with mutations in VP22 were lower than those in cells infected with wild-type HSV-1(F) or one of the repaired viruses. In addition, we analyzed plaque sizes in Vero cells infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) under the conditions used for the plaque assay. As shown in Fig. 6, YK451 (ΔVP22), YK453 (VP22LL235AA), and YK455 (VP22LL251AA) produced significantly smaller plaques than wild-type HSV-1(F) and their repaired viruses (YK452 [ΔVP22-repair], YK454 [VP22LL235AA-repair], and YK456 [VP22LL251AA-repair]). These results are in agreement with a previous report that a VP22-null mutant virus exhibited reduced virus growth and produced plaques of reduced sizes on Vero cells (13, 52).
Fig 5.
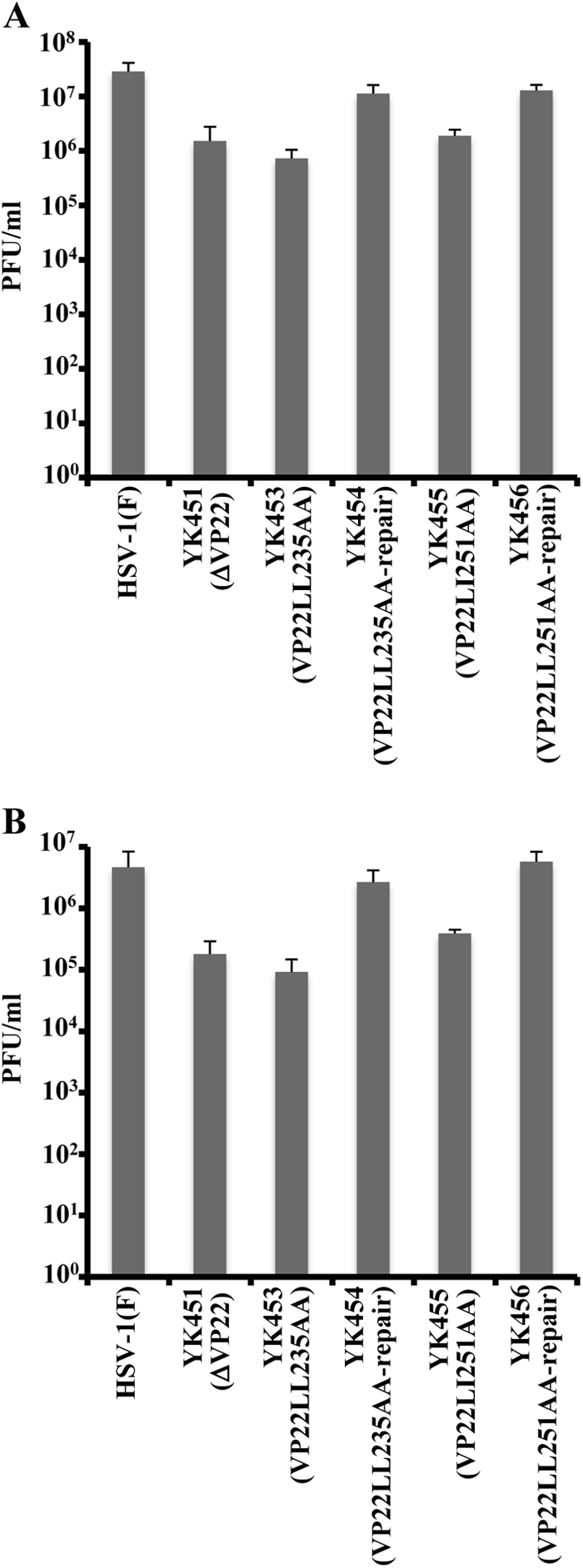
Effects of the ΔVP22, VP22LL235AA, and VP22LL251AA mutations on the production of infectious virus. Vero cells were infected with wild-type HSV-1(F), YK451 (ΔVP22), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) at an MOI of 1 (A) or 0.01 (B). Total virus from the cell culture supernatants and infected cells was harvested at 24 h postinfection and was assayed on Vero cells. Data are means and standard errors for three independent experiments.
Fig 6.
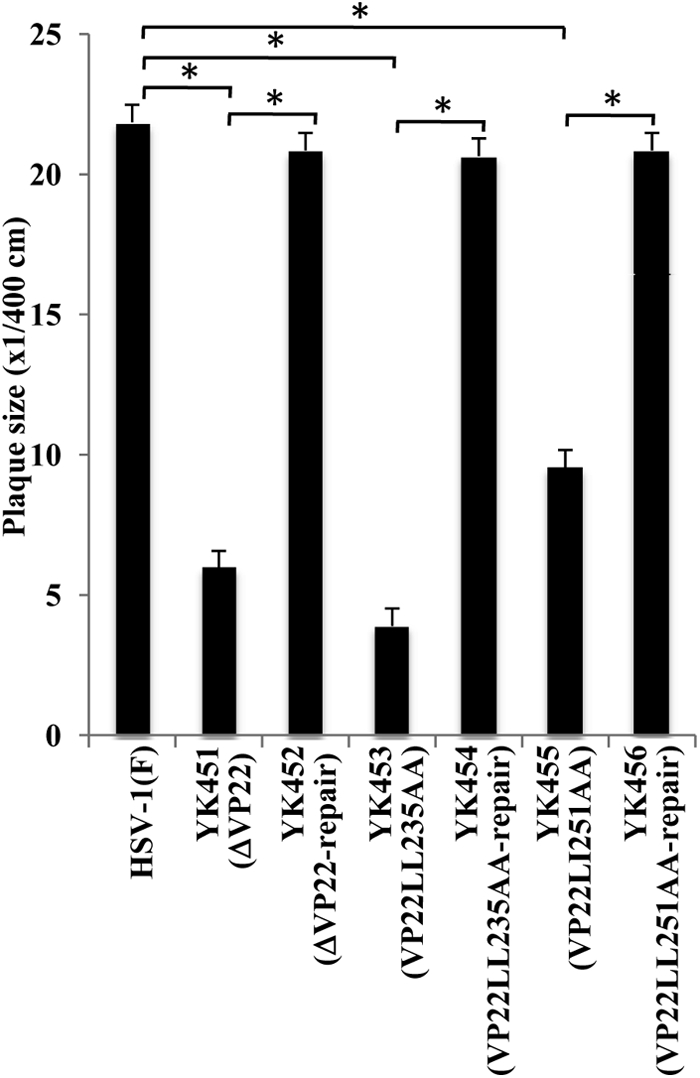
Effects of the ΔVP22, VP22LL235AA, and VP22LL251AA mutations on the production of plaques. Vero cells were infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) at an MOI of 0.0001 under plaque assay conditions. The diameters of 20 single plaques for each of the recombinant viruses were determined 2 days after infection. The data shown are means and standard errors. Asterisks indicate significant differences (*, P < 1 × 10−8) by a two-tailed Student t test.
In the second series of experiments, we used immunofluorescence with confocal microscopy to examine the localization of mutant VP22 in Vero cells infected with each of the recombinant viruses at an MOI of 1 for 8 or 15 h. It has been reported that VP22 is localized predominantly in the cytoplasm of HSV-1(F)-infected Vero cells early in infection and begins to translocate from the cytoplasm to the nucleus at later times (9 to 16 h postinfection) (53, 61). As shown in Fig. 7, at 8 h postinfection, wild-type VP22 was localized predominantly in the cytoplasm of Vero cells infected with wild-type HSV-1(F). In agreement with previous reports (53, 61), some HSV-1(F)-infected cells showed nuclear localization of VP22 at 15 h postinfection. In contrast, as observed for VP16 in cells infected with YK453 (VP22LL235AA) or YK455 (VP22LL251AA), VP22LL235AA and VP22LL251AA were detected predominantly in the nuclei of cells infected with YK453 or YK455, respectively, at 8 and 15 h postinfection (Fig. 7). These results indicated that the two VP22 dileucine motifs are also required for the proper cytoplasmic localization of VP22 itself.
Fig 7.

Digital confocal microscope images showing the localization of VP22. Vero cells were infected with wild-type HSV-1(F), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) at an MOI of 1 for 15 h. Infected Vero cells were fixed at the indicated times postinfection, permeabilized, stained with an anti-VP22 antibody, and examined by confocal microscopy. The left and right columns at each time point show VP22 fluorescence and the simultaneous acquisition of VP22 fluorescence and digital interference contrast, respectively.
In the third series of experiments, we examined the expression of viral proteins VP16, gB, VP22, ICP0, ICP4, ICP27, and UL41 in Vero cells that were either mock infected or infected with wild-type HSV-1(F), YK451 (ΔVP22), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), or YK456 (VP22LL251AA-repair) at an MOI of 1 for 20 h. In agreement with the results obtained for YK451 (ΔVP22) (Fig. 2), the expression of VP16, VP22, gB, ICP0, and UL41 in cells infected with YK453 (VP22LL235AA) or YK455 (VP22LL251AA) decreased compared to that of these proteins in cells infected with wild-type HSV-1(F) or the repaired virus YK454 (VP22LL235AA-repair) or YK456 (VP22LL251AA-repair) (Fig. 8). In contrast, the levels of expression of ICP4 and ICP27 in cells infected with YK453 (VP22LL235AA) or YK455 (VP22LL251AA) were similar to those in cells infected with HSV-1(F), YK454 (VP22LL235AA-repair), or YK456 (VP22LL251AA-repair) (Fig. 8). Furthermore, the level of expression of VP22LL235AA or VP22LL251AA in cells infected with YK453 (VP22LL235AA) or YK455 (VP22LL251AA), respectively, was lower than that of VP22 in cells infected with wild-type HSV-1(F), YK454 (VP22LL235AA-repair), or YK456 (VP22LL251AA-repair). These results indicated that the VP22 dileucine motifs are required for VP22 to regulate proper localization of VP16 and VP22 in infected cells and for efficient expression of VP16, VP22, gB, ICP0, and UL41 in infected cells.
Fig 8.

Immunoblots of electrophoretically separated lysates of Vero cells that were either mock infected (lane 7) or infected with wild-type HSV-1(F) (lane 1), YK451 (ΔVP22) (lane 2), YK453 (VP22LL235AA) (lane 3), YK454 (VP22LL235-repair) (lane 4), YK455 (VP22LL251AA) (lane 5), or YK456 (VP22LL251AA-repair) (lane 6) at an MOI of 1. Infected Vero cells were harvested 20 h postinfection and were immunoblotted with an antibody to VP16, gB, VP22, ICP0, ICP4, ICP27, UL41, or α-tubulin.
O'Regan et al. (51) also reported that when cells were first transfected with the expression vector for GFP-VP22 and then infected with wild-type HSV-1, GFP-VP22 was incorporated into progeny virions. Therefore, they examined the effects of the VP22LL235AA and VP22LL251AA mutations on the incorporation of VP22 into virions and found that the VP22LL235AA mutation abrogated incorporation of the mutant GFP-VP22 into virions but that GFP-VP22 with the VP22LL251AA mutation was incorporated into virions at a level comparable to that of wild-type GFP-VP22. To confirm this observation, in the fourth series of experiments, we purified total intracellular and extracellular virions from Vero cells infected with wild-type HSV-1(F), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), YK456 (VP22LL251AA-repair), or YK476 (ΔUL41) and analyzed them by immunoblotting with an anti-VP22, anti-VP5, or anti-VP16 antibody. In agreement with the report by O'Regan et al. (51), incorporation of VP22LL235AA protein into YK453 (VP22LL235A) virions was barely detectable, whereas VP22LL251AA protein was incorporated into YK455 (VP22LL251AA) virions at a level comparable to those of the other viruses, except YK453 (Fig. 9A). In contrast, the VP22 LL235AA and LL251AA mutations had no detectable effect on the incorporation of VP16 (Fig. 9B). These results indicated that the VP22LL235AA mutation has a defect in the incorporation of VP22 into progeny virions but that the VP22LL251AA mutation does not.
Fig 9.

(A) Immunoblots of electrophoretically separated virions of wild-type HSV-1(F) (lane 1), YK453 (VP22LL235AA) (lane 2), YK454 (VP22LL235AA-repair) (lane 3), YK456 (VP22LL251AA-repair) (lane 4), YK455 (VP22LL251AA) (lane 5), and YK476 (ΔUL41) (lane 6) purified by sucrose gradient centrifugation and blotted with an antibody to VP22. (B) Immunoblots of electrophoretically separated virions of wild-type HSV-1(F) (lane 1), YK453 (VP22LL235AA) (lane 2), YK454 (VP22LL235AA-repair) (lane 3), YK455 (VP22LL251AA) (lane 4), YK456 (VP22LL251AA-repair) (lane 5), and YK476 (ΔUL41) (lane 6) purified by sucrose gradient centrifugation and blotted with an antibody to VP5 or VP16.
Effects of the ΔVP22, VP22LL235AA, and VP22LL251AA mutations on the localization of viral proteins in infected cells.
As described above (Fig. 2 and 8) and elsewhere (14, 15), VP22 appeared to regulate the expression of multiple viral proteins in infected cells. These results raised the possibility that VP22 may also regulate the localization of viral proteins other than VP16. To investigate this possibility, Vero cells were infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), YK456 (VP22LL251AA-repair), or YK476 (ΔUL41), and the localization of ICP0 was examined at 4, 8, and 15 h postinfection, while that of ICP4, ICP8, ICP27, and VP26 was examined at 8 and 15 h postinfection. It has been shown that ICP8 localizes in the nucleus throughout HSV-1 infection, while ICP0, ICP4, ICP27, and VP26 localize predominantly in the nucleus at early times postinfection and, at later times postinfection, translocate from the nucleus to the cytoplasm and localize throughout the infected cells or predominantly in the cytoplasm (8, 10, 31, 36, 60, 61). In agreement with these previous reports, ICP0 was detected mainly in the nuclei of wild-type HSV-1(F)-infected cells at 4 h postinfection. At 8 h postinfection, ICP0 was detected throughout the infected cells, and ICP4, ICP27, and VP26 were detected mainly in the nucleus (Fig. 10 to 12). At 15 h postinfection, ICP0, ICP4, ICP27, and VP26 were detected throughout the infected cells, and, in some HSV-1(F)-infected cells, ICP0 was detected predominantly in the cytoplasm (Fig. 10 to 12). Similar results were obtained in cells infected with YK476 (ΔUL41) (Fig. 10 to 12). In contrast, in cells infected with YK451 (ΔVP22), YK455 (VP22LL251AA) (Fig. 10 to 12), or YK453 (VP22LL235AA) (data not shown), ICP0 was localized in the nucleus at 8 and 15 h postinfection and ICP4 and ICP27 were localized in the nucleus at 15 h postinfection (Fig. 10 to 12). Similarly, in cells infected with YK451 (ΔVP22), YK455 (VP22LL251AA) (Fig. 11), or YK453 (VP22LL235AA) (data not shown), VP26 was detected predominantly in the nucleus at 8 and 15 h postinfection, although VP26 was more evident in the cytoplasm than ICP0, ICP4, and ICP27 at 15 h postinfection. Wild-type localization of these viral proteins was restored in cells infected with the repaired virus YK452 (ΔVP22-repair), YK456 (VP22LL251AA-repair) (Fig. 10 to 12), or YK454 (VP22LL235AA-repair) (data not shown). Furthermore, the ΔVP22, VP22LL251AA, and VP22LL235AA mutations had no effect on the localization of ICP8 in infected cells at 8 and 15 h postinfection (Fig. 10; also data not shown). Taken together, these observations indicated that VP22 is required for proper redistribution of ICP0, ICP4, ICP27, and VP26 to the cytoplasm in infected cells and that the two VP22 dileucine motifs are the sites responsible for this VP22 function.
Fig 10.

Digital confocal microscope images showing the localization of ICP4 and ICP8 in Vero cells infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK455 (VP22LL251AA), YK456 (VP22LL251AA-repair), or YK476 (ΔUL41) at an MOI of 1. Infected Vero cells were fixed at the indicated times postinfection, permeabilized, stained with an antibody to ICP4 or ICP8, and examined by confocal microscopy. Left and right columns at each time point show protein fluorescence and simultaneous acquisition of protein fluorescence and digital interference contrast, respectively.
Fig 12.

Digital confocal microscope images showing the localization of ICP0 in Vero cells infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK455 (VP22LL251AA), YK456 (VP22LL251AA-repair), or YK476 (ΔUL41) at an MOI of 1. Infected Vero cells were fixed at the indicated times postinfection, permeabilized, stained with an antibody to ICP0, and examined by confocal microscopy. Left and right columns at each time point show protein fluorescence and simultaneous acquisition of protein fluorescence and digital interference contrast, respectively.
Fig 11.

Digital confocal microscope images showing the localization of ICP27 and VP26 in Vero cells infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK455 (VP22LL251AA), YK456 (VP22LL251AA-repair), or YK476 (ΔUL41) at an MOI of 1. Infected Vero cells were fixed at the indicated times postinfection, permeabilized, stained with an antibody to ICP27 or VP26, and examined by confocal microscopy. Left and right columns at each time point show protein fluorescence and simultaneous acquisition of protein fluorescence and digital interference contrast, respectively.
Effects of the ΔVP22, VP22LL235AA, and VP22LL251AA mutations on the localization of cellular proteins in infected cells.
The cellular chaperon protein Hsc70 is localized throughout uninfected cells (5, 40). It has been reported that this cellular protein is dramatically redistributed upon HSV-1 infection and is dynamically trafficked during HSV-1 infection (5, 39, 40). Thus, Hsc70 localizes predominantly in the nuclei of HSV-1-infected cells at early times postinfection and forms nuclear domains, designated virus-induced chaperon-enriched (VICE) domains, which have been suggested to function as nuclear protein quality control centers in infected cells (5, 39, 40). At later times postinfection, Hsc70 undergoes translocation from the nucleus to the cytoplasm and localizes predominantly in the cytoplasm (2). Since this feature of Hsc70 localization in infected cells was similar to the pattern of localization of viral proteins ICP0, ICP4, ICP27, and VP26, as described above, we investigated whether VP22 also regulated the subcellular localization of Hsc70 in infected cells by determining the localization of Hsc70 at 6 and 15 h postinfection in Vero cells infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK453 (VP22LL235AA), YK454 (VP22LL235AA-repair), YK455 (VP22LL251AA), YK456 (VP22LL251AA-repair), or YK476 (ΔUL41) at an MOI of 1. In agreement with previous reports (2, 5, 39, 40), Hsc70 was detected throughout normal cells and mainly in the nuclei of wild-type HSV-1(F)-infected cells at 6 h postinfection (Fig. 13A and B). At 15 h postinfection, Hsc70 was detected predominantly in the cytoplasm of wild-type HSV-1(F)-infected cells (Fig. 13B). In contrast, in cells infected with YK451 (ΔVP22), YK455 (VP22LL251AA) (Fig. 13B), or YK453 (VP22LL235AA) (data not shown), Hsc70 was detected predominantly in the nucleus at both 8 and 15 h postinfection. Wild-type localization of Hsc70 was restored in cells infected with the repaired virus YK452 (ΔVP22-repair), YK456 (VP22LL251AA-repair) (Fig. 13B), or YK454 (VP22LL235AA-repair) (data not shown), and the UL41-null mutation had no effect on Hsc-70 localization in infected cells (Fig. 13B). Furthermore, the ΔVP22, VP22LL251AA, and VP22LL235AA mutations had no effect on the localization of another cellular protein, mitochondrial apoptosis-inducing factor (AIF), in infected cells at 8 and 15 h postinfection (Fig. 13A and B; also data not shown). These observations indicated that VP22 is required for proper redistribution of Hsc70 to the cytoplasm in infected cells and that the two dileucine motifs in VP22 are the sites responsible for this VP22 function. In a closer examination of VICE domain formation, we noted that VP22 appeared to play no significant role in the formation of VICE domains in infected cells, based on the observation that infection with wild-type HSV-1(F) and with YK451 (ΔVP22) induced similar levels of nuclear VICE domains, as detected by an anti-Hsc70 antibody (data not shown).
Fig 13.

Digital confocal microscope images showing the localization of Hsc70 and AIF in normal Vero cells (A) and in Vero cells infected with wild-type HSV-1(F), YK451 (ΔVP22), YK452 (ΔVP22-repair), YK455 (VP22LL251AA), YK456 (VP22LL251AA-repair), or YK476 (ΔUL41) at an MOI of 1 (B). Infected Vero cells were fixed at the indicated times postinfection, permeabilized, stained with an antibody to Hsc70 or AIF, and examined by confocal microscopy. Left and right columns at each time point show protein fluorescence and simultaneous acquisition of protein fluorescence and digital interference contrast, respectively.
Effect of the VP22LL251AA mutation on viral neurovirulence and replication in murine brains.
To examine the significance of VP22-mediated regulation of the localization and expression of the viral and cellular proteins in the studies described above for HSV-1 pathogenesis in vivo, mice were infected intracerebrally with 10-fold dilutions of YK455 (VP22LL251AA) or YK456 (VP22LL251AA-repair), and mortality was monitored. In addition, viral titers and the distribution of HSV-1 antigens in the brains of mice infected with 102 PFU of YK455 (VP22LL251AA) or YK456 (VP22LL251AA-repair) were examined at 3 or 5 days postinfection, respectively. To eliminate the effect of the efficiency of VP22 incorporation into virions on viral pathogenesis in vivo, we did not examine the recombinant virus YK453 (VP22LL235AA) in these experiments, since the VP22LL235AA mutation abrogated VP22 regulation of the localization and expression of the viral and cellular proteins in this study, as well as the incorporation of VP22 into virions, as described above. As shown in Fig. 14A, the 50% lethal dose (LD50) of YK455 (VP22LL251AA) was approximately 103-fold greater than that of YK456 (VP22LL251AA-repair), indicating that the VP22 dileucine motif Di-Leu-251-252 was critical for viral virulence in mice following intracerebral inoculation. In agreement with the effect of the VP22LL251AA mutation on virulence in mice, the titer of YK455 (VP22LL251AA) in the brains of infected mice was significantly (14.3-fold) lower than that of YK456 (VP22LL251AA-repair) (Fig. 14B). Furthermore, viral antigens were detected in many regions of the brains of mice infected with YK456 (VP22LL251AA-repair) but only in a restricted region(s) of the brains of mice infected with YK455 (VP22LL251AA) (Fig. 15). These results indicated that VP22 is required for efficient neurovirulence in mice and for viral replication and spread in the brains of mice following intracerebral inoculation and that one of the sites in VP22 responsible for these functions in mice is Di-Leu-251-252.
Fig 14.
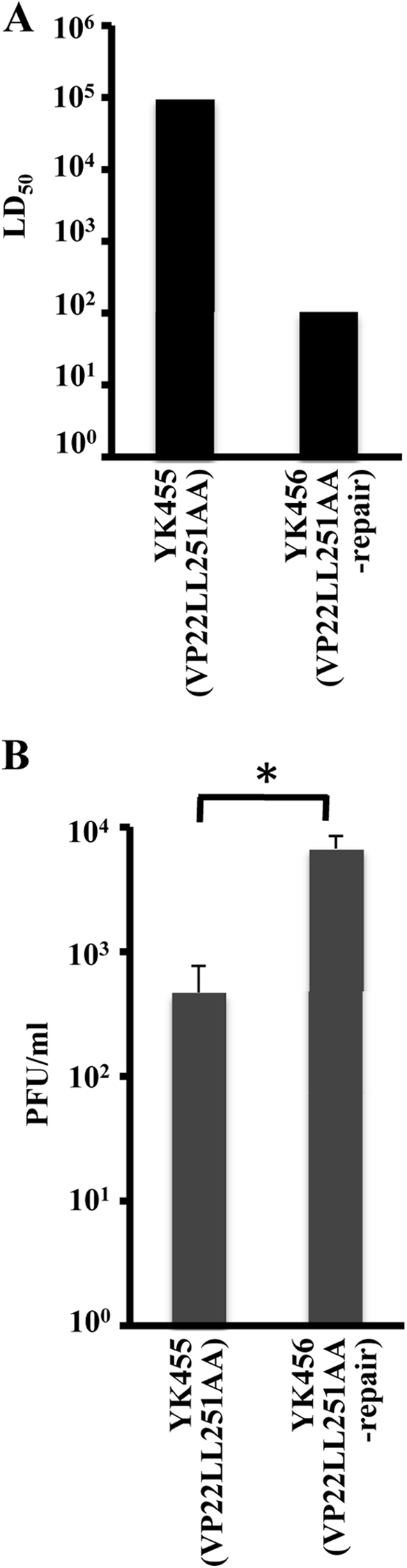
(A) LD50 values of YK455 (VP22LL251AA) and YK456 (VP22LL251AA-repair) in mice following intracerebral inoculation. Three-week-old female mice were inoculated intracerebrally with serial 10-fold dilutions of each virus in groups of 6 per dilution and were monitored for 14 days. LD50 values were determined by the Behrens-Karber method. (B) Virus titers in the brains of mice following intracerebral inoculation. Seventeen 3-week-old female mice were inoculated intracerebrally with 102 PFU of each virus. At 3 days postinfection, the brains of infected mice were harvested, and virus titers were determined by standard plaque assays on Vero cells. The data shown are means and standard errors. A statistically significant difference in virus titer between mice infected with YK455 (VP22LL251AA) and those infected with YK456 (VP22LL251AA-repair) was noted (*, P < 0.005).
Fig 15.

Histopathological features of the brains of mice following intracerebral inoculation. Five 3-week-old female mice were inoculated intracerebrally with 102 PFU of YK455 (VP22LL251AA) (A, B, and C) or YK456 (VP22LL251AA-repair) (D, E, and F). At 5 days postinfection, the brains of infected mice were harvested, sectioned, and stained with hematoxylin and eosin (A and D) or with an antibody to HSV-1 antigens (B, C, E, and F). Panels C and F show magnified images of the regions indicated in panels B and E, respectively. Representative images are shown. Bars, 50 μm.
DISCUSSION
In the present study, we identified a novel function of HSV-1 VP22, namely, regulation of the proper subcellular localization of viral proteins VP16, VP26, ICP0, ICP4, and ICP27 and cellular protein Hsc-70 in infected cells. These VP22-regulated proteins have been reported to be localized in the nucleus early in infection and then to be translocated to the cytoplasm later in infection. We have presented data showing that these viral and cellular proteins were localized in the nuclei of Vero cells at 15 h postinfection when those cells were infected with the UL49-null mutant virus YK451 (ΔVP22) or with recombinant viruses carrying alanine substitutions in one of the dileucine motifs, i.e., YK453 (VP22LL235AA) and YK455 (VP22LL251AA). In contrast, at 15 h postinfection, these VP22-regulated proteins were localized predominantly in the cytoplasm of Vero cells infected with wild-type HSV-1(F) or the repaired virus YK452, YK454, or YK456. From these observations, we concluded that VP22 promotes the redistribution of these viral and cellular proteins to the cytoplasm and that the two VP22 dileucine motifs (Di-Leu-235-236 and Di-Leu-251-252) are required for this VP22 function. Elliott et al. (15, 16) reported previously that transient coexpression of VP22 and VP16 led to the localization of VP22 and VP16 to a perinuclear domain in COS-1 cells and that the absence of VP22 impaired the formation of discrete cytoplasmic ICP0 domains. These observations indicated that VP22 regulated the localization of VP16 and ICP0, in agreement with our conclusions on VP22 functions. However, there were apparent differences between our observations as presented here and the report by Elliott et al. (15) that ICP0 was translocated from the nucleus to the cytoplasm in MDCK, BHK, and Vero cells infected with a UL49-null mutant of HSV-1 strain 17 [HSV-1(17)] but that its localization in discrete cytoplasmic domains was greatly reduced. In contrast, in the studies presented here, we showed that translocation of ICP0 from the nucleus to the cytoplasm was apparently inhibited in Vero cells infected with an HSV-1(F) UL49-null mutant virus, YK451 (ΔVP22). These differences in the effects of VP22 on the localization of ICP0 in infected cells may be due to differences in the HSV-1 strains used in each of the studies, since it has been reported that some properties of VP22 in HSV-1-infected cells appear to depend on the HSV-1 strain used. Thus, in Vero cells infected with wild-type HSV-1(F) or recombinant HSV-1(F) expressing VP22 fused to green fluorescent protein (GFP-VP22), VP22 and GFP-VP22 were localized predominantly in the cytoplasm at early times postinfection and were then translocated from the cytoplasm to the nucleus at later times (53, 61). In contrast, in Vero cells infected with wild-type HSV-1(17) or recombinant HSV-1(17) expressing VP22 fused to a fluorescent protein, VP22 and VP22 fused to the fluorescent protein were localized predominantly in the cytoplasm throughout infection, although VP22 fused to the fluorescent protein translocated from the cytoplasm to the nucleus during cell division (16, 17, 19). Moreover, in Vero cells infected with an HSV-1(F) VP22-null mutant, most viral protein synthesis was at a wild-type level at early times postinfection (3 to 6 h postinfection) but was dramatically impaired relative to that of wild-type HSV-1(F) at later times, beginning at 9 h postinfection (14). However, no significant drop-off in viral protein synthesis was observed at late times (15 to 25 h postinfection), and there was only a delay in viral protein synthesis earlier in infection (5 h postinfection) in Vero cells infected with a VP22-null mutant of HSV-1(17) (15).
Sciortino et al., (58) showed previously that a UL49-null mutant virus accumulated mutations in the UL41 coding region when propagated on Vero cells. Similarly, Mbong et al. reported recently that passage of another UL49-null mutant virus on cells that did not express VP22 led to the introduction of a mutation in a specific codon of the UL41 coding region (44). In the present study, VP22 mutant viruses were propagated on Vero cells. Therefore, one may argue that aberrant subcellular localization of the viral and cellular proteins in cells infected with each of the VP22 mutant viruses resulted from additional mutations in the UL41 coding region and/or in some other genes. However, this is highly unlikely, based on the following observations. (i) We worked with very low passage number VP22 mutant viruses and confirmed that the DNA sequences of the UL41 coding regions in the VP22 mutant viruses grown in Vero cells and used in the experiments of this study were identical to those of the wild-type virus as described above. (ii) Duffy et al., reported that two passages of a UL49-null mutant virus on Vero cells resulted in no sequence changes within the UL41 coding region (14). (iii) As shown in Fig. 3 and 8 to 13, all of the phenotypes observed with the UL41-null mutant virus were apparently different from those observed with the VP22 mutant viruses but were identical to those observed with wild-type HSV-1(F). (iv) As described in Materials and Methods, we generated additional repaired viruses YK457 (ΔVP22-repair-2), YK458 (VP22LL235AA-repair-2), and YK459 (VP22LL251AA-repair-2), based on YK451 (ΔVP22), YK453 (VP22LL235AA), and YK455 (VP22LL251AA), which were grown in Vero cells. We tested them in cell cultures and obtained evidence that the patterns of localization of VP16, VP22, ICP4, ICP27, VP26, ICP0, and Hsc70 in Vero cells infected with YK457 (ΔVP22-repair-2), YK458 (VP22LL235AA-repair-2), or YK459 (VP22LL251AA-repair-2) were apparently different from those in Vero cells infected with YK451 (ΔVP22) but were identical to those in Vero cells infected with wild-type HSV-1(F).
At present we can only speculate about the mechanism(s) by which VP22 acted in promoting the redistribution of VP16, VP26, ICP0, ICP4, ICP27, and Hsc-70 to the cytoplasm in infected cells. It would be interesting to understand the relationship between the function of VP22 in regulating the proper subcellular localization of a subset of viral and cellular proteins and its function in regulating the steady-state levels of certain viral proteins. We have shown that mutations in VP22 resulted in aberrant localization of ICP4 and ICP27 in infected cells, whereas the mutations had no effect on steady-state levels of these viral proteins. These observations suggest that the two functions of VP22 may be unrelated. Since the VP22LL235AA and VP22LL251AA mutations impaired not only the translocation of these viral and cellular proteins but also that of VP22 in infected cells, the mechanism may be that VP22 interacted with these viral and cellular proteins and cotranslocated with them to the cytoplasm, like many other HSV proteins that regulate the localization of a viral or cellular protein by interacting with it (11, 29). In agreement with our observation, Zheng et al. previously identified a leucine-rich motif at amino acids 204 to 216 of BHV-1 VP22, which, interestingly, contains a dileucine motif aligned with Di-Leu-235-236 of HSV-1 VP22, as a nuclear export signal (NES) of the protein, based on the observation that the mutations in the motif resulted in extensive nuclear localization of BHV-1 VP22 (67). Since leucine-rich sequences are a hallmark of CRM-1-dependent NESs, these observations suggested that either Di-Leu-235-236 alone or both Di-Leu-235-236 and Di-Leu-251-252 might be NESs of HSV-1 VP22 and might be involved in exporting a subset of viral and cellular proteins from the nucleus to the cytoplasm.
It has been suggested that some tegument proteins, including VP22, UL11, and VP16, are highly prone to nonspecific binding to other proteins and that this “sticky” nature may be important for tegument assembly (21). Therefore, VP22 may regulate the localization of a variety of viral and cellular proteins in infected cells by interacting and colocalizing with them. Alternatively, VP22 may regulate the localization of various viral and cellular proteins indirectly, via another protein(s). It has been reported that ICP27 promotes the cytoplasmic localization of ICP0 and ICP4 in infected cells (68–70). At present it is not known whether ICP27 also regulates the localization of VP16, VP26, VP22, and Hsc70 in infected cells. However, it has been reported that ICP27 is a multifunctional nucleocytoplasmic shuttling protein that interacts with various cellular and viral proteins and regulates their localization (57). It is of particular interest that ICP27 interacts with VHS (63), which forms a complex with VP16 and VP22 (62). Therefore, VP22 may affect the ability of ICP27 to regulate the localization of viral and cellular proteins. We are currently studying this possibility.
Another significant finding of this study was that HSV-1 VP22 is an important neurovirulence factor in vivo. In experimental animal models of HSV-1 infection, viral pathogenesis in sites peripheral to the initial infection site (pathogenic manifestations in peripheries) and destruction of the central nervous system caused by viral replication (neurovirulence) are semi-independent indicators of viral virulence and can be studied by peripheral and intracerebral inoculation, respectively (26, 29). To our knowledge, a viral protein that is involved in neurovirulence in mice is, in most cases, required for viral pathogenic manifestations in peripheries as well, whereas a viral protein that is involved in pathogenic manifestations in peripheries in mice often has no effect on viral neurovirulence. Although Duffy et al. (13) previously reported that VP22 was required for efficient development of corneal lesions in mice following ocular inoculation, the role of VP22 in neurovirulence in mice following intracerebral inoculation had not been determined. In the present study, we showed that the VP22LL251AA mutation significantly (approximately 103-fold) impaired HSV-1 neurovirulence in mice. In agreement with this observation, the VP22LL251AA mutation also impaired viral replication and spread in the brains of infected mice. The lower levels of replication and spread of the YK455 (VP22LL251AA) mutant than of the repaired mutant YK456 (VP22LL251AA-repair) may account for the lower level of neurovirulence of YK455 in mice. Thus, VP22 is a significant HSV-1 virulence factor in vivo and is critical for both neurovirulence and pathogenic manifestations in peripheries in vivo.
At present, the mechanisms by which VP22 regulates viral replication and neurovirulence in vivo remain unclear. The finding that the VP22LL251AA mutation had no apparent effect on VP22 incorporation into virions suggested that the efficiency of VP22 incorporation into virions was not involved in viral replication and neurovirulence in mice. The finding that the VP22LL251AA mutation, which impaired viral replication and neurovirulence in mice, also attenuated the ability of VP22 to promote the cytoplasmic localization and expression of a number of viral and cellular proteins suggested that these VP22 functions may be critical for viral replication and neurovirulence in vivo. However, the VP22LL251AA mutation may affect a VP22 function(s) other than those identified in this study, since we have not investigated whether Di-Leu-251-252 is required for other VP22 functions, such as functional interaction with microtubules, chromatin, RNA, and cellular membranes (4, 17, 18, 34, 43, 54, 59, 66). Further studies to identify other VP22 functions that require Di-Leu-251-252 are needed and are under way in our laboratories.
ACKNOWLEDGMENTS
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS); a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases and Global COE Program “Center of Education and Research for the Advanced Genome-Based Medicine—For personalized medicine and the control of worldwide infectious diseases” from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan; and grants from the Takeda Science Foundation, the Uehara Memorial Foundation, the Naito Foundation; and the Sumitomo Foundation.
Footnotes
Published ahead of print 22 February 2012
REFERENCES
- 1. Asai R, Ohno T, Kato A, Kawaguchi Y. 2007. Identification of proteins directly phosphorylated by UL13 protein kinase from herpes simplex virus 1. Microbes Infect. 9:1434–1438 [DOI] [PubMed] [Google Scholar]
- 2. Bastian TW, Livingston CM, Weller SK, Rice SA. 2010. Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection. J. Virol. 84:2384–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blaho JA, Mitchell C, Roizman B. 1994. An amino acid sequence shared by the herpes simplex virus 1 alpha regulatory proteins 0, 4, 22, and 27 predicts the nucleotidylylation of the UL21, UL31, UL47, and UL49 gene products. J. Biol. Chem. 269:17401–17410 [PubMed] [Google Scholar]
- 4. Brignati MJ, Loomis JS, Wills JW, Courtney RJ. 2003. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J. Virol. 77:4888–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burch AD, Weller SK. 2004. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J. Virol. 78:7175–7185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Che X, et al. 2008. Functions of the ORF9-to-ORF12 gene cluster in varicella-zoster virus replication and in the pathogenesis of skin infection. J. Virol. 82:5825–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chi JH, Harley CA, Mukhopadhyay A, Wilson DW. 2005. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 86:253–261 [DOI] [PubMed] [Google Scholar]
- 8. Corbin-Lickfett KA, Rojas S, Li L, Cocco MJ, Sandri-Goldin RM. 2010. ICP27 phosphorylation site mutants display altered functional interactions with cellular export factors Aly/REF and TAP/NXF1 but are able to bind herpes simplex virus 1 RNA. J. Virol. 84:2212–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. del Rio T, Werner HC, Enquist LW. 2002. The pseudorabies virus VP22 homologue (UL49) is dispensable for virus growth in vitro and has no effect on virulence and neuronal spread in rodents. J. Virol. 76:774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dobrikova E, Shveygert M, Walters R, Gromeier M. 2010. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J. Virol. 84:270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dorange F, Tischer BK, Vautherot JF, Osterrieder N. 2002. Characterization of Marek's disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 76:1959–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duffy C, et al. 2006. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 80:8664–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duffy C, Mbong EF, Baines JD. 2009. VP22 of herpes simplex virus 1 promotes protein synthesis at late times in infection and accumulation of a subset of viral mRNAs at early times in infection. J. Virol. 83:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elliott G, Hafezi W, Whiteley A, Bernard E. 2005. Deletion of the herpes simplex virus VP22-encoding gene (UL49) alters the expression, localization, and virion incorporation of ICP0. J. Virol. 79:9735–9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elliott G, Mouzakitis G, O'Hare P. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69:7932–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elliott G, O'Hare P. 2000. Cytoplasm-to-nucleus translocation of a herpesvirus tegument protein during cell division. J. Virol. 74:2131–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elliott G, O'Hare P. 1998. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 72:6448–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elliott G, O'Hare P. 1999. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J. Virol. 73:4110–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elliott G, O'Reilly D, O'Hare P. 1996. Phosphorylation of the herpes simplex virus type 1 tegument protein VP22. Virology 226:140–145 [DOI] [PubMed] [Google Scholar]
- 21. Farnsworth A, Wisner TW, Johnson DC. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 81:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fossum E, et al. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 5:e1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 76:6729–6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuchs W, et al. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J. Virol. 76:8208–8217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hafezi W, Bernard E, Cook R, Elliott G. 2005. Herpes simplex virus tegument protein VP22 contains an internal VP16 interaction domain and a C-terminal domain that are both required for VP22 assembly into the virus particle. J. Virol. 79:13082–13093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J. Virol. 84:153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jarosinski KW, et al. 2007. Horizontal transmission of Marek's disease virus requires US2, the UL13 protein kinase, and gC. J. Virol. 81:10575–10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 29. Kato A, et al. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 85:9599–9613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kato A, et al. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1δ: ICP0 affects translational machinery. J. Virol. 71:1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kawaguchi Y, et al. 2003. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1δ. J. Virol. 77:2359–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kotsakis A, Pomeranz LE, Blouin A, Blaho JA. 2001. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 75:8697–8711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354 [DOI] [PubMed] [Google Scholar]
- 36. Lengyel J, Strain AK, Perkins KD, Rice SA. 2006. ICP27-dependent resistance of herpes simplex virus type 1 to leptomycin B is associated with enhanced nuclear localization of ICP4 and ICP0. Virology 352:368–379 [DOI] [PubMed] [Google Scholar]
- 37. Liang X, Chow B, Babiuk LA. 1997. Study of immunogenicity and virulence of bovine herpesvirus 1 mutants deficient in the UL49 homolog, UL49.5 homolog and dUTPase genes in cattle. Vaccine 15:1057–1064 [DOI] [PubMed] [Google Scholar]
- 38. Liang X, et al. 1995. Characterization of bovine herpesvirus 1 UL49 homolog gene and product: bovine herpesvirus 1 UL49 homolog is dispensable for virus growth. J. Virol. 69:3863–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Livingston CM, DeLuca NA, Wilkinson DE, Weller SK. 2008. Oligomerization of ICP4 and rearrangement of heat shock proteins may be important for herpes simplex virus type 1 prereplicative site formation. J. Virol. 82:6324–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Livingston CM, Ifrim MF, Cowan AE, Weller SK. 2009. Virus-induced chaperone-enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog. 5:e1000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maringer K, Elliott G. 2010. Recruitment of herpes simplex virus type 1 immediate-early protein ICP0 to the virus particle. J. Virol. 84:4682–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin A, O'Hare P, McLauchlan J, Elliott G. 2002. Herpes simplex virus tegument protein VP22 contains overlapping domains for cytoplasmic localization, microtubule interaction, and chromatin binding. J. Virol. 76:4961–4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mbong M, et al. 2011. Secondary mutations in vhs restore HSV-1 protein synthesis in the absence of VP22 in a cell-type independent manner. 36th Annu. Int. Herpes Virus Workshop, abstr. 2.04 [Google Scholar]
- 45. Mitchell C, Blaho JA, Roizman B. 1994. Casein kinase II specifically nucleotidylylates in vitro the amino acid sequence of the protein encoded by the alpha 22 gene of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 91:11864–11868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morrison EE, Wang YF, Meredith DM. 1998. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J. Virol. 72:7108–7114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mouzakitis G, McLauchlan J, Barreca C, Kueltzo L, O'Hare P. 2005. Characterization of VP22 in herpes simplex virus-infected cells. J. Virol. 79:12185–12198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nozawa N, et al. 2005. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 79:6947–6956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nozawa N, Yamauchi Y, Ohtsuka K, Kawaguchi Y, Nishiyama Y. 2004. Formation of aggresome-like structures in herpes simplex virus type 2-infected cells and a potential role in virus assembly. Exp. Cell Res. 299:486–497 [DOI] [PubMed] [Google Scholar]
- 50. O'Regan KJ, Bucks MA, Murphy MA, Wills JW, Courtney RJ. 2007. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology 358:192–200 [DOI] [PubMed] [Google Scholar]
- 51. O'Regan KJ, Murphy MA, Bucks MA, Wills JW, Courtney RJ. 2007. Incorporation of the herpes simplex virus type 1 tegument protein VP22 into the virus particle is independent of interaction with VP16. Virology 369:263–280 [DOI] [PubMed] [Google Scholar]
- 52. Pomeranz LE, Blaho JA. 2000. Assembly of infectious herpes simplex virus type 1 virions in the absence of full-length VP22. J. Virol. 74:10041–10054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pomeranz LE, Blaho JA. 1999. Modified VP22 localizes to the cell nucleus during synchronized herpes simplex virus type 1 infection. J. Virol. 73:6769–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ren X, Harms JS, Splitter GA. 2001. Bovine herpesvirus 1 tegument protein VP22 interacts with histones, and the carboxyl terminus of VP22 is required for nuclear localization. J. Virol. 75:8251–8258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott-Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 56. Sagou K, Uema M, Kawaguchi Y. 2010. Nucleolin is required for efficient nuclear egress of herpes simplex virus type 1 nucleocapsids. J. Virol. 84:2110–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sandri-Goldin RM. 2008. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Front. Biosci. 13:5241–5256 [DOI] [PubMed] [Google Scholar]
- 58. Sciortino MT, et al. 2007. Replication-competent herpes simplex virus 1 isolates selected from cells transfected with a bacterial artificial chromosome DNA lacking only the UL49 gene vary with respect to the defect in the UL41 gene encoding host shutoff RNase. J. Virol. 81:10924–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sciortino MT, Taddeo B, Poon AP, Mastino A, Roizman B. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. U. S. A. 99:8318–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Simpson-Holley M, Baines J, Roller R, Knipe DM. 2004. Herpes simplex virus 1 UL31 and UL34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 78:5591–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sugimoto K, et al. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Taddeo B, Sciortino MT, Zhang W, Roizman B. 2007. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. U. S. A. 104:12163–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Taddeo B, Zhang W, Roizman B. 2010. Role of herpes simplex virus ICP27 in the degradation of mRNA by virion host shutoff RNase. J. Virol. 84:10182–10190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tanaka M, Nishiyama Y, Sata T, Kawaguchi Y. 2005. The role of protein kinase activity expressed by the UL13 gene of herpes simplex virus 1: the activity is not essential for optimal expression of UL41 and ICP0. Virology 341:301–312 [DOI] [PubMed] [Google Scholar]
- 66. van Leeuwen H, et al. 2003. Herpes simplex virus type 1 tegument protein VP22 interacts with TAF-I proteins and inhibits nucleosome assembly but not regulation of histone acetylation by INHAT. J. Gen. Virol. 84:2501–2510 [DOI] [PubMed] [Google Scholar]
- 67. Zheng C, Brownlie R, Babiuk LA, van Drunen Littel-van den Hurk S. 2005. Characterization of the nuclear localization and nuclear export signals of bovine herpesvirus 1 VP22. J. Virol. 79:11864–11872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhu Z, Cai W, Schaffer PA. 1994. Cooperativity among herpes simplex virus type 1 immediate-early regulatory proteins: ICP4 and ICP27 affect the intracellular localization of ICP0. J. Virol. 68:3027–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhu Z, DeLuca NA, Schaffer PA. 1996. Overexpression of the herpes simplex virus type 1 immediate-early regulatory protein, ICP27, is responsible for the aberrant localization of ICP0 and mutant forms of ICP4 in ICP4 mutant virus-infected cells. J. Virol. 70:5346–5356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhu Z, Schaffer PA. 1995. Intracellular localization of the herpes simplex virus type 1 major transcriptional regulatory protein, ICP4, is affected by ICP27. J. Virol. 69:49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]