

Abstract
While human cells express potent antiviral proteins as part of the host defense repertoire, viruses have evolved their own arsenal of proteins to antagonize them. BST2 was identified as an inhibitory cellular protein of HIV-1 replication, which tethers virions to the cell surface to prevent their release. On the other hand, the HIV-1 accessory protein, Vpu, has the ability to downregulate and counteract BST2. Vpu also possesses the ability to downmodulate cellular CD4 and SLAMF6 molecules expressed on infected cells. However, the role of Vpu in HIV-1 infection in vivo remains unclear. Here, using a human hematopoietic stem cell-transplanted humanized mouse model, we demonstrate that Vpu contributes to the efficient spread of HIV-1 in vivo during the acute phase of infection. Although Vpu did not affect viral cytopathicity, target cell preference, and the level of viral protein expression, the amount of cell-free virions in vpu-deficient HIV-1-infected mice was profoundly lower than that in wild-type HIV-1-infected mice. We provide a novel insight suggesting that Vpu concomitantly downregulates BST2 and CD4, but not SLAMF6, from the surface of infected cells. Furthermore, we show evidence suggesting that BST2 and CD4 impair the production of cell-free infectious virions but do not associate with the efficiency of cell-to-cell HIV-1 transmission. Taken together, our findings suggest that Vpu downmodulates BST2 and CD4 in infected cells and augments the initial burst of HIV-1 replication in vivo. This is the first report demonstrating the role of Vpu in HIV-1 infection in an in vivo model.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1), the causative agent of AIDS, encodes four accessory proteins in its viral genome: negative factor (Nef), virus infectivity factor (Vif), vial protein R (Vpr), and viral protein U (Vpu) (10). Vpu is a 16-kDa integral membrane protein and is expressed from a bicistronic mRNA together with envelope protein (Env) during the late stage of the viral life cycle (10, 65). Vpu was originally acquired by the ancestor of certain simian immunodeficiency viruses (SIVs; SIVmon, SIVmus, and SIVgsn) and was later transferred to SIVcpz and HIV-1 by recombination and cross-species transmission events. On the other hand, Vpu is noticeably absent in human immunodeficiency virus type 2 (HIV-2) and the other SIVs including SIVsmm/mac (7, 26).
Vpu has been found to act on several cellular proteins by affecting their surface expression using two common mechanisms: (i) targeting them for ubiquitin-mediated degradation and/or (ii) trafficking them from the plasma membrane to intracellular compartments. It is widely known that Vpu can recruit β-transducin repeat-containing protein 1 (BTRC; also called β-TrCP1), an E3 ubiquitin ligase, and degrades CD4 molecules by the ubiquitin/proteasome pathway (31, 35, 60, 72). In addition, a recent report demonstrated that signaling lymphocyte activation molecule family member 6 (SLAMF6; also called NTB-A), a transmembrane protein that induces natural killer cell-mediated killing, can be downregulated from the plasma membrane by Vpu (64).
A third molecule influenced by Vpu was discovered after observations that different cell types displayed various degrees of viral replication kinetics after HIV-1 infection with or without Vpu. Virus yields in the supernatant of vpu-deficient HIV-1-infected/transfected cells were reported to be lower than those of vpu-proficient (i.e., wild-type [WT]) HIV-1-infected/transfected cells in certain CD4+ T cell lines (41, 61), primary CD4+ T cells (41, 61–63), monocyte-derived macrophages (59, 62, 63, 66), in vitro tonsil histocultures (59), and HeLa cells (12). However, Vpu was dispensable for HIV-1 virion production in cell lines such as 293T cells and HT1080 cells. Varthakavi et al. later demonstrated that HeLa cells express an inhibitory factor for HIV-1 particle release, which can be counteracted by Vpu (68). In 2008, Neil et al. (42) and Van Damme et al. (67) identified this factor as tetherin and bone marrow stromal cell antigen 2 (BST2; also known as CD317 or HM1.24), respectively. BST2 is a glycosylphosphatidylinositol-anchored transmembrane protein and is endogenously expressed in human CD4+ T cells and macrophages (9). Van Damme et al. (67) and other groups, including ours (56), have reported that BST2 expressed on the surface of HIV-1-infected cells is severely downregulated by Vpu. Moreover, some reports have revealed that Vpu-mediated BST2 downregulation is dependent on BTRC, similar to the manner by which CD4 is downregulated (7, 8, 11, 37).
The restriction conferred by BST2 is not limited to retroviruses (9, 23, 26) but also various enveloped viruses belonging to Filoviridae (Ebola virus and Marburg virus) (23, 24, 49), Arenaviridae (Lassafever virus) (49), Herpesviridae (Kaposi's sarcoma-associated herpesvirus) (34), Rhabdoviridae (vesicular stomatitis virus) (71), Orthomyxoviridae (influenza A virus) (69), and Paramyxoviridae (Nipah virus) (47). However, some of these viruses possess their own antagonizing BST2 counterparts instead of Vpu. For instance, HIV-2 counteracts BST2 with its envelope glycoprotein (Env) (8, 13, 15, 30), while SIVsmm/mac impairs the tethering function of simian BST2 with its accessory protein, Nef (15, 58, 73). In addition, it was reported that Ebola virus glycoprotein (24) and the K5 protein of Kaposi's sarcoma-associated herpesvirus (34, 46) can counteract BST2. Given that various diverse viruses have evolved methods to overcome the restriction by BST2, it would appear that its tethering ability is critical for the host defense.
Lines of investigations in cell culture systems have revealed the molecular mechanisms of virion tethering by BST2 and the ability of Vpu to antagonize BST2, which have shed light on host-virus interactions. Moreover, accumulating evidence suggests that BST2 is a potent suppressor of HIV-1 infection. However, the role of Vpu in HIV-1 expansion in vivo, particularly its antagonism of BST2 in vivo, remains unresolved.
In order to elucidate the dynamics of human-specific pathogens such as HIV-1 in vivo, we have constructed a humanized mouse model by xenotransplanting human CD34+ hematopoietic stem cells into an immunodeficient NOD/SCID Il2rg−/− (NOG) mouse (44, 52, 55). In this system, human leukocytes including human CD4+ T cells are differentiated de novo and are stably and longitudinally maintained. By using these humanized mice, we have established novel animal models for HIV-1 and Epstein-Barr virus infections and related diseases (44, 52, 54, 55). Particularly noteworthy is that our humanized mice, named NOG-hCD34 mice, are able to recapitulate the characteristics of HIV-1 pathogenesis, such as the depletion of CD4+ T cells in peripheral blood (PB) and the preferential infection of effector memory CD4+ T cells (44, 53).
In this study, by utilizing our humanized mouse model, we show that Vpu positively associates with efficient HIV-1 replication in vivo during the initial phase of infection. In addition, we investigated the dynamics of three Vpu-associated cellular proteins, BST2, CD4, and SLAMF6, in vivo and found that BST2 and CD4 but not SLAMF6 were severely downregulated on the surface of infected cells in a Vpu-dependent manner. Moreover, immunostaining analyses revealed frequent cell-to-cell interaction of HIV-1-infected cells in the spleen of infected mice, suggesting the occurrence of an efficient cell-to-cell virus transmission in vivo. Furthermore, in vitro transfection experiments demonstrated that BST2 and CD4 distinctively and synergistically suppressed the production of infectious virions while modestly affecting the efficiency of cell-to-cell infection. This is the first report focusing on the fundamental role of Vpu in HIV-1 infection in vivo. Our findings suggest that Vpu enhances the kinetics of cell-free virus propagation, especially during the initial phase of infection, and downregulates BST2 and CD4 in infected cells, which lead to a rapid systemic HIV-1 dissemination in vivo.
MATERIALS AND METHODS
Ethics statement.
All protocols involving human subjects were reviewed and approved by the Kyoto University institutional review board. Informed written consent from the human subjects was obtained in this study.
Humanized mice.
NOD.Cg-Prkdcscid Il2rgtm1Sug/Jic (NOD/SCID Il2rg−/−) mice (14) were obtained from the Central Institute for Experimental Animals (Kanagawa, Japan). The mice were maintained under specific-pathogen-free conditions and were handled in accordance with the Regulation on Animal Experimentation at Kyoto University. Human CD34+ hematopoietic stem cells were isolated from human fetal liver as described previously (1). The humanized mouse (NOG-hCD34 mouse) was constructed as previously described (44, 52, 54, 55). Briefly, 70 newborn (aged 0 to 2 days) NOG mice from 15 litters were irradiated with X-ray (10 cGy per mouse) by an RX-650 X-ray cabinet system (Faxitron X-ray Corporation) and were then intrahepatically injected with the obtained human CD34+ cells (12 × 104 to 35 × 104 cells). A list of the humanized mice used in this study is summarized in Table 1.
Table 1.
Humanized mice used in this studya
| Mouse no.b | Recipient mouse |
Transplanted hHSCse |
Inoculated virus | Dose (×103 TCID50) | Inoculation age (no. of wks) | ||
|---|---|---|---|---|---|---|---|
| Lot no.c | Sexa | Donor lotd | No. of cells | ||||
| 1 | 104 | M | A | 170,000 | HIV-1Δvpu | 300 | 15 |
| 2 | 105 | F | B | 140,000 | 14 | ||
| 3 | 105 | M | B | 140,000 | HIV-1Δvpu | 300 | 14 |
| 4 | 105 | M | B | 140,000 | 14 | ||
| 5 | 105 | M | B | 140,000 | HIV-1 | 300 | 14 |
| 6 | 105 | M | B | 140,000 | HIV-1 | 300 | 14 |
| 7 | 106 | F | C | 120,000 | HIV-1Δvpu | 300 | 14 |
| 8 | 106 | F | C | 120,000 | 14 | ||
| 9 | 106 | M | C | 120,000 | HIV-1 | 300 | 14 |
| 10 | 106 | M | C | 120,000 | 14 | ||
| 11 | 106 | M | C | 120,000 | HIV-1Δvpu | 300 | 14 |
| 12 | 108 | F | D | 240,000 | HIV-1 | 300 | 13 |
| 13 | 108 | F | D | 240,000 | HIV-1Δvpu | 300 | 13 |
| 14 | 108 | F | D | 240,000 | HIV-1 | 300 | 13 |
| 15 | 108 | M | D | 240,000 | 13 | ||
| 16 | 108 | M | D | 240,000 | HIV-1 | 300 | 13 |
| 17 | 108 | M | D | 240,000 | 13 | ||
| 18 | 108 | M | D | 240,000 | HIV-1Δvpu | 300 | 13 |
| 19 | 109 | F | E | 350,000 | HIV-1Δvpu | 300 | 12 |
| 20 | 109 | F | E | 350,000 | 12 | ||
| 21 | 109 | M | E | 350,000 | HIV-1 | 300 | 12 |
| 22 | 110 | M | F | 300,000 | HIV-1 | 300 | 15 |
| 23 | 110 | M | F | 300,000 | HIV-1Δvpu | 300 | 15 |
| 24 | 110 | M | F | 300,000 | HIV-1 | 300 | 15 |
| 25 | 113 | F | E | 300,000 | HIV-1 | 300 | 15 |
| 26 | 113 | F | E | 300,000 | HIV-1Δvpu | 300 | 15 |
| 27 | 113 | M | E | 200,000 | HIV-1Δvpu | 300 | 14 |
| 28 | 113 | M | E | 200,000 | HIV-1 | 300 | 14 |
| 29 | 113 | M | E | 200,000 | HIV-1Δvpu | 300 | 14 |
| 30 | 114 | F | D | 180,000 | HIV-1 | 300 | 17 |
| 31 | 114 | M | D | 180,000 | HIV-1Δvpu | 300 | 17 |
| 32 | 116 | F | F | 250,000 | HIV-1 | 300 | 15 |
| 33 | 116 | F | F | 250,000 | HIV-1Δvpu | 300 | 15 |
| 34 | 116 | M | F | 250,000 | HIV-1Δvpu | 300 | 15 |
| 35 | 116 | M | F | 250,000 | HIV-1 | 300 | 15 |
| 36 | 117 | F | C | 170,000 | HIV-1Δvpu | 300 | 15 |
| 37 | 117 | F | C | 170,000 | HIV-1Δvpu | 300 | 15 |
| 38 | 117 | F | C | 170,000 | HIV-1 | 300 | 15 |
| 39 | 117 | F | C | 170,000 | HIV-1 | 300 | 15 |
| 40 | 117 | M | C | 170,000 | HIV-1Δvpu | 300 | 15 |
| 41 | 117 | M | C | 170,000 | HIV-1 | 300 | 15 |
| 42 | 117 | M | C | 170,000 | HIV-1Δvpu | 300 | 15 |
| 43 | 117 | M | C | 170,000 | HIV-1 | 300 | 15 |
| 44 | 130 | F | D | 130,000 | HIV-1 | 3 | 14 |
| 45 | 130 | F | D | 130,000 | HIV-1Δvpu | 3 | 14 |
| 46 | 126 | F | G | 130,000 | HIV-1Δvpu | 3 | 14 |
| 47 | 126 | F | G | 130,000 | HIV-1Δvpu | 3 | 14 |
| 48 | 126 | F | G | 130,000 | HIV-1 | 3 | 14 |
| 49 | 126 | F | G | 130,000 | HIV-1Δvpu | 3 | 14 |
| 50 | 126 | M | G | 130,000 | HIV-1 | 30 | 14 |
| 51 | 126 | M | G | 130,000 | HIV-1 | 30 | 14 |
| 52 | 126 | M | G | 130,000 | HIV-1 | 300 | 14 |
| 53 | 126 | M | G | 130,000 | HIV-1Δvpu | 30 | 14 |
| 54 | 127 | F | G | 130,000 | HIV-1 | 300 | 14 |
| 55 | 127 | F | G | 130,000 | HIV-1 | 3 | 14 |
| 56 | 127 | F | G | 130,000 | HIV-1 | 3 | 14 |
| 57 | 127 | F | G | 130,000 | HIV-1Δvpu | 30 | 14 |
| 58 | 127 | M | G | 130,000 | HIV-1 | 300 | 14 |
| 59 | 127 | M | G | 130,000 | HIV-1Δvpu | 300 | 14 |
| 60 | 127 | M | G | 130,000 | HIV-1Δvpu | 300 | 14 |
| 61 | 128 | F | H | 130,000 | HIV-1 | 3 | 14 |
| 62 | 128 | M | H | 130,000 | HIV-1 | 30 | 14 |
| 63 | 128 | M | H | 130,000 | HIV-1Δvpu | 3 | 14 |
| 64 | 128 | M | H | 130,000 | HIV-1Δvpu | 3 | 14 |
| 65 | 128 | M | H | 130,000 | HIV-1 | 3 | 14 |
| 66 | 129 | F | H | 130,000 | HIV-1 | 30 | 14 |
| 67 | 129 | M | H | 130,000 | HIV-1 | 30 | 14 |
| 68 | 129 | M | H | 130,000 | HIV-1Δvpu | 30 | 14 |
| 69 | 129 | M | H | 130,000 | HIV-1Δvpu | 30 | 14 |
| 70 | 129 | M | H | 130,000 | HIV-1Δvpu | 30 | 14 |
M, male; F, female.
Mouse numbers 11, 25, 26, 28, and 29 correspond with those shown in Fig. 3A.
Fifteen lots of newborn NOG mice were used for the recipient.
NOG-hCD34 mice were reconstituted with one of eight donors.
hHSCs, human CD34+ hematopoietic stem cells.
Cell culture.
293T cells, HeLa cells, HEK293 cells, and TZM-bl cells (obtained through the NIH AIDS Research and Reference Reagent Program) (70) were maintained in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin as previously described (27, 51, 52, 56). Primary human CD4+ T cells were isolated from the peripheral blood of a healthy donor by using a human CD4+ T cell isolation kit (Miltenyi) according to the manufacturer's protocol. The isolated cells were stimulated with Dynabeads Human T-Activator CD3/CD28 for cell expansion and activation (Invitrogen) for 3 days. The activated human CD4+ T cells were maintained in RPMI 1640 medium containing 10% fetal calf serum, 100 U/ml interleukin-2, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Transfection, Western blotting, and TZM-bl assay.
For in vitro transfection experiments shown in Fig. 1, 1 μg of pAD8+ (a molecular clone of HIV-1 strain AD8 [HIV-1AD8]) (66), pAD8-UDEL2, which is a derivative of AD8 carrying an 81-bp deletion and an 8-bp irrelevant insertion in the vpu region (62, 63), or pUC19 (for mock control [TaKaRa]; shown as “vector” on the figures) was transfected into 293T and HeLa cells by using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. At 48 h posttransfection, the culture supernatant was harvested, centrifuged, and then filtered through a 0.45-μm-pore-size filter (Millipore) to produce virus solutions. To detect viral and cellular proteins, Western blotting was performed as previously described (27, 51, 52, 56). For Western blotting, the following antibodies were used: goat anti-p24 polyclonal antibody (Virostat), rabbit anti-Vpu polyclonal antibody (U2-3; kindly provided by Klaus Strebel) (33), and mouse anti-α-tubulin (TUBA) monoclonal antibody (DM1A; Sigma). To quantify the infectivities of the obtained viruses, a TZM-bl assay was performed as previously described (56). For in vitro transfection experiments shown in Fig. 9, a BST2 expression plasmid (pBST2) and a CD4 expression plasmid (pCD4), which are based on pCMV-SPORT6 (Invitrogen), were prepared. To construct pBST2, a BST2 cDNA was obtained from pKGC-BST2 (27), and the obtained fragment was inserted into pCMV-SPORT6. To construct a CD4 expression plasmid (pCD4), pBCMGSNeo-human CD4 (kindly donated by Masayuki Miyasaka) was digested with XhoI, blunted, and then further digested with NotI. The resulting fragment was inserted into the EcoRV-XhoI site of pCMV-SPORT6. Sequences of these plasmid constructs were confirmed with an ABI Prism 3130xl genetic analyzer (Applied Biosystems). A total of 800 ng of pAD8+, pAD8-UDEL2, or pUC19 (for mock control; termed vector on the figures) was cotransfected with either 10 nanograms of pBST2 or pCD4 or both into HEK293 cells by using Lipofectamine 2000. A total of 100 ng of a green fluorescent protein (GFP)-expressing plasmid, peGFP-C1 (Clontech), was also cotransfected to monitor transfection efficiency, and DNA content in each well was adjusted to 1 μg by using pCMV-SPORT6. At 48 h posttransfection, the culture supernatant was harvested, centrifuged, and then filtered through a 0.45-μm-pore-size filter to produce virus solutions. The transfected cells were washed and suspended with DMEM, and 1/10 of the cell suspension was cocultured with TZM-bl cells. The TZM-bl assay was performed as described above.
Fig 1.

Dynamics of WT and vpu-deficient HIV-1 infection in cultured cells. (A to C) HIV-1 release and downregulation of surface BST2. One microgram of pAD8+ (WT HIV-1-producing plasmid) or pAD8-UDEL2 (vpu-deficient HIV-1-producing plasmid) was transfected into 293T or HeLa cells, respectively. The expression levels of Gag (Pr55Gag) and Vpu, the amount of virions retained in the cells (p24CA in the cells), and the amount of released virion (p24CA in the supernatant) in the transfected 293T cells (A) and HeLa cells (B) were determined by Western blotting (left panels). The input of cell lysate was standardized to α-tubulin (TUBA), and representative results are shown. In TZM-bl assays titers of the viruses released from the transfected 293T cells and HeLa cells were determined. The infectivity is shown as the percentage of the value of WT HIV-1. (C) The surface expression level of BST2 on the transfected HeLa cells at 48 h posttransfection was assessed by flow cytometry. The HeLa cells transfected with pAD8+, pAD8-UDEL2, and pUC19 (parental plasmid; Vector) were classified into virus-producing (Gag-positive) or non-virus-producing (Gag-negative) populations by using an antibody against HIV-1 p24 antigen. Representative histograms (left), the percentage of BST2-positive cells (middle), and the MFI of BST2 (right) are shown. In the left panel, the numbers on each histogram indicate the MFI values, and the vertical broken lines indicate the thresholds for the gating of positive cells based on the result from the isotype control. The assay was performed in quadruplicate. (D to F) Primary human CD4+ T cells were prepared as described in Materials and Methods and infected with WT and vpu-deficient HIV-1 at an MOI of 0.1. At 7 dpi, the cells were harvested and used for flow cytometry. The cells were classified into infected (Gag-positive) or uninfected (Gag-negative) populations by using an anti-p24 antibody. Representative histograms (left), the positive percentages (middle), and the MFIs (right) of CD4, BST2, and SLAMF6 in each population are shown. In the left panels, the numbers on each histogram indicate the MFI values, and the vertical broken lines indicate the thresholds for the gating of positive cells based on the result from isotype controls. The assay was performed in triplicate. The statistic difference is determined by a Student's t test. Statistically significant differences are shown as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SEMs. NS, no statistical significance.
Fig 9.

Different potentials of CD4 and BST2 in the production of infectious cell-free virions and cell-to-cell HIV-1 transmission. A total of 800 ng of pAD8+ (WT HIV-1-producing plasmid), pAD8-UDEL2 (vpu-deficient HIV-1-producing plasmid), or pUC19 (parental plasmid; vector) was cotransfected with either 10 ng of CD4 expression plasmid (pCD4) or BST2 expression plasmid (pBST2) or both into HEK293 cells. peGFP-C1 (100 ng) was also cotransfected to monitor transfection efficiency. The assay was performed in quadruplicate. (A to C) Downregulation of surface CD4 and BST2. The surface expression levels of CD4 and BST2 on the transfected HEK293 cells (i.e., GFP-positive cells) at 48 h posttransfection were assessed by flow cytometry. The positive percentage (left) and the MFI (right) are shown. The percentage of the cells negative for both CD4 and BST2 in the cells cotransfected with both pCD4 and pBST2 is also shown (C). (D and E) Infectious potentials of cell-free virions in culture supernatant and transfected cells. The infectivities of the viruses in the supernatant and the transfected cells (coculture) were titrated by TZM-bl assay and were normalized to the values per culture. Statistically significant differences (determined by Student's t test) are shown as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SEMs.
Virus preparation and infection.
To prepare the virus solutions of WT HIV-1AD8 and vpu-deficient HIV-1AD8 (HIV-1Δvpu) for the experiments using humanized mouse, 30 μg of either pAD8+ or pAD8-UDEL2 was transfected into 293T cells by the calcium-phosphate method. At 48 h posttransfection, the culture supernatant was harvested, centrifuged, and then filtered through a 0.45-μm-pore-size filter to produce virus solution. To titrate virus infectivity, the virus solution obtained was serially diluted and then inoculated onto phytohemagglutinin-stimulated human peripheral blood mononuclear cells in a 96-well plate in triplicate. At 14 days postinfection, the endpoint was determined by using an HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) kit (ZetroMetrix), and virus infectivity was calculated as the 50% tissue culture infectious doses (TCID50) according to the Reed-Muench method. The TCID50 of WT and vpu-deficient HIV-1 used in this study were 7.74 × 106/ml and 7.72 × 106/ml, respectively. A virus solution of 3,000, 30,000, or 300,000 TCID50 was intraperitoneally inoculated into NOG-hCD34 mice aged between 12 to 17 weeks old (Table 1). RPMI 1640 medium was used in mock infections.
PB collection and isolation of mononuclear cells from organs.
PB and plasma were routinely collected as previously described (44). The spleen was crushed, rubbed, and suspended, and the bone marrow (BM) was obtained from the dissected thighbones by flushing the interior as previously described (44). The human mononuclear cells (MNCs) in the spleen and the BM were isolated as previously described (44).
Quantification of HIV-1 RNA and antigen.
The amount of HIV-1 RNA in 50 μl of plasma was quantified by Bio Medical Laboratories, Inc. The detection limit of HIV-1 RNA is 1,600 copies/ml. To quantify the amount of cell-free virions in the supernatant of spleen and the BM fluid, the splenic cell suspension and the BM fluid obtained by flushing were centrifuged; the supernatants were filtered through a 0.45-μm-pore-size filter and then ultracentrifuged at 30,000 rpm for 1 h at 4°C by using a TL-100 instrument (Beckman). The pellets were resuspended with phosphate-buffered saline, and the amount of HIV-1 Gag proteins was quantified using an HIV-1 p24 antigen ELISA kit (ZetroMetrix).
Flow cytometry, hematocytometry, and immunostaining.
Flow cytometry was performed with a FACSCanto II and a FACSCalibur (BD Biosciences) as previously described (44, 52, 54, 55), and the obtained data were analyzed with CellQuest software (BD Biosciences) and FlowJo software (Tree Star, Inc.). For flow cytometry analysis, the following antibodies were used: fluorescein isothiocyanate (FITC)-conjugated rat anti-HIV-1 p24 monoclonal antibody (clone 2C2, kindly provided by Yuetsu Tanaka) (45), phycoerythrin (PE)-conjugated anti-CD45 antibody (HI30; Biolegend) and anti-SLAMF6 antibody (clone 292811; R&D Systems), allophycocyanin (APC)-conjugated anti-BST2 antibody (RS38E; Biolegend) and anti-CD4 antibody (RPA-T4; Biolegend), PE-Cy7-conjugated anti-CD4 antibody (RPA-T4; Biolegend), APC-Cy7-conjugated anti-CD3 antibody (HIT3a; Biolegend) and anti-CD45 antibody (HI30; Biolegend), peridinin chlorophyll protein (PerCP)-Cy5.5-conjugated anti-CD45RA antibody (HI100; Biolegend), and AmCyan-conjugated anti-CD8 antibody (DK25; Dako). For the costaining of CD4, BST2, SLAMF6, and Gag, surface CD4, BST2, and SLAMF6 were stained prior to fixation. Then, the cells were fixed and permeabilized using a Cytofix/Cytoperm solution (BD biosciences), and intracellular Gag proteins were subsequently stained. Hematocytometry was performed with a Celltac a MEK-6450 (Nihon Kohden, Co.) as previously described (54). Immunostaining was performed with a Leica TCS SP2 AOBS confocal laser microscope (Leica Microsystems) as previously described (44, 54). The obtained pictures were assessed by using ImageJ software (http://rsbweb.nih.gov/ij/). For immunostaining analysis, mouse anti-HIV-1 p24 monoclonal antibody (Kal-1; Dako) and Alexa Fluor 594-conjugated goat anti-mouse IgG antibody (Molecular probes) were used. Nuclei were stained with Hoechst 33342 (Molecular Probes).
PCR analyses.
DNA was extracted from the splenic MNCs at 21 days postinfection (dpi) by a urea-lysis method as previously described (25). PCR was performed by using rTaq DNA polymerase (TaKaRa) according to the manufacturer's protocol, and the following primers were used: gag forward (bases 1484 to 1503), 5′-GGG GAA GTG ACA TAG CAG GA-3′; gag reverse (bases 1746 to 1765), 5′-CAT TTT GGA CCA GCA AGG TT-3′; vpu forward 1st (bases 5712 to 5731), 5′-GAT ACT TGG GCA GGA GTG GA-3′; vpu reverse 1st (bases 6222 to 6241), 5′-CCT GAT CCC CTT CAC TTT CA-3′; vpu forward 2nd (bases 5926 to 5945), 5′-TGC CAA GTT TGC TTC ACA AG-3′; vpu reverse 2nd (bases 6205 to 6224), 5′-TCA TTG CCA CTG TCT TCT GC-3′; ACTB forward, 5′-TCA CCC ACA CTG TGC CCA TCT ACG A-3′; ACTB reverse, 5′-CAG CGG AAC CGC TCA TTG CCA ATG G-3′.
Statistical and mathematical analyses.
Data were expressed as averages with standard errors of the means (SEMs). Statistical differences were determined by Student's t test, Welch's t test, or the log rank test. To determine a statistically significant correlation (see Fig. 4C), the Pearson correlation coefficient (r) was applied. To estimate the 50% infectious dose (ID50) for NOG-hCD34 mice, nonlinear regression analyses (39, 43) were performed by using Mathematica, version 8, software (Wolfram Research).
Fig 4.

Viral dissemination in the PB and lymphoid organs of humanized mice. (A) Cell-free virions in the PB and lymphoid organs of infected mice. The amounts of cell-free virus in the plasma (left), the supernatant of splenic suspension (middle), and the BM fluid (right) of WT HIV-1-infected mice (300,000 TCID50, 7 and 21 dpi; n = 7) and vpu-deficient HIV-1-infected mice (300,000 TCID50, 7 and 21 dpi; n = 8) were quantified by p24 ELISA. (B) The percentage of HIV-1 Gag-positive cells in the PB (left), the spleen (middle), and the BM (right) of WT HIV-1-infected mice (300,000 TCID50, 7 and 21 dpi; n = 7), vpu-deficient HIV-1-infected mice (300,000 TCID50, 7 and 21 dpi; n = 8), and mock-infected mice (21 dpi; n = 7) were analyzed by flow cytometry. (C) Correlation of the amount of cell-free virions and the percentage of infected cells. The results of the percentage of Gag-positive cells in spleen at 7 dpi (x axis) and the amount of cell-free Gag proteins in splenic fluid at 7 dpi (y axis) from WT HIV-1-infected mice (300,000 TCID50; n = 10) and vpu-deficient HIV-1-infected mice (300,000 TCID50; n = 9) are plotted. The lines represent exponential approximation. The Pearson correlation coefficient (r) was adopted to determine statistically significant correlation between each value, and a P value of <0.05 was considered statistically significance. (D) The MFIs of Gag (Pr55Gag) in Gag-positive cells (right) in the spleen of WT HIV-1-infected mice (300,000 TCID50; n = 7) and vpu-deficient HIV-1-infected mice (300,000 TCID50; n = 7) were analyzed by flow cytometry at 7 dpi. Error bars represent SEMs. ND, not detected; NS, no statistical significance.
The GenBank (http://www.ncbi.nlm.nih.gov/GenBank/index.html) accession numbers for the genes and virus mentioned in the text are as follows: for BST2, NM_004335.2; for CD4, NM_000616.4, for ACTB, NM_001101.3; and for HIV-1 strain AD8, AF004394.1.
RESULTS
Dynamics of vpu-deficient HIV-1 infection in cultured cells.
To assess the impact of HIV-1 Vpu on virus replication, we first confirmed the ability of Vpu, derived from a CCR5-tropic HIV-1 strain AD8, to downregulate BST2 in 293T and HeLa cells. These cell lines were transfected with a plasmid carrying WT HIV-1 strain AD8, pAD8+ (66), or a vpu-deficient derivative of AD8, pAD8-UDEL2 (62, 63). As previously reported (42, 56, 67), in 293T cells, which do not express endogenous BST2, the absence of vpu did not affect the expression level of Gag precursor (Pr55Gag), the amount of produced virions, or the infectivity of the released virions (Fig. 1A). On the other hand, in HeLa cells, which endogenously express BST2, the yield of vpu-deficient virions released was 60-fold lower than that of WT released virions although the expression level of Pr55Gag was comparable (Fig. 1B). We also found that the surface expression of BST2 on WT HIV-1-producing (i.e., positive for HIV-1 Gag protein) but not vpu-deficient HIV-1-producing HeLa cells was significantly and severely downregulated (Fig. 1C). In line with previous reports (42, 56, 67), our results suggest that the Vpu protein encoded by strain AD8 potently downregulates cell surface BST2 and antagonizes its function to enhance the release of nascent virions.
Based on previous reports (8, 26, 31, 64), Vpu is known to downregulate BST2, CD4, and SLAMF6 from the surface of infected cells. To confirm that the Vpu protein encoded by strain AD8 is able to exert these effects, primary human CD4+ T cells were infected with WT and vpu-deficient HIV-1. As shown in Fig. 1D to 1F, we observed that CD4, BST2, and SLAMF6 were significantly downregulated from the surface of WT HIV-1-infected but not of HIV-1Δvpu-infected cells. Because it is well known that CD4 can be downregulated by not only Vpu but also HIV-1 Env and Nef (31), the decrease in CD4 on the surface of HIV-1Δvpu-infected cells is most likely due to the effect of these proteins. Taken together, these findings strongly suggest that the Vpu protein encoded by strain AD8 has the potential to downregulate the reported three molecules in vitro.
Dynamics of vpu-deficient HIV-1 infection in humanized mice.
In order to investigate the role of Vpu in HIV-1 expansion in vivo, three different doses (3,000, 30,000, and 300,000 TCID50) of WT or vpu-deficient HIV-1 (strain AD8) were intraperitoneally inoculated into humanized mice. Seventy humanized mice, which were reconstituted with CD34+ hematopoietic stem cells from eight individual donors, were used for this study (Table 1). Out of the 70 humanized mice, 32 mice were infected with WT HIV-1 (3,000 TCID50, n = 6; 30,000 TCID50, n = 5; and 300,000 TCID50, n = 21), 31 mice were infected with vpu-deficient HIV-1 (3,000 TCID50, n = 6; 30,000 TCID50, n = 5; and 300,000 TCID50, n = 20), and the remaining 7 mice were used for mock infection. The amounts of viral RNA in plasma were assessed at 3, 7, 14, and 21 dpi. As shown in Fig. 2A, humanized mice were more efficiently infected with WT HIV-1 than vpu-deficient HIV-1 (300,000 TCID50, P = 0.013 by the log rank test). In addition, nonlinear regression analyses revealed that the ID50 in NOG-hCD34 mice for WT HIV-1 was 2.3-fold lower than that for vpu-deficient HIV-1 (for WT HIV-1, 1 ID50 is 1,343 TCID50; r = 0.999, P = 1.6 × 10−7; for HIV-1Δvpu, 1 ID50 is 3,046 TCID50; r = 0.990, P = 0.019). These findings suggest that our humanized mice are more susceptible to WT HIV-1 than vpu-deficient HIV-1. Moreover, the amount of viral RNA in the plasma of WT HIV-1-infected mice increased more rapidly (Fig. 2B) and was 8.5-fold higher at 7 dpi than in HIV-1Δvpu-infected mice (300,000 TCID50; P = 0.049). Taken together, these findings suggest that WT HIV-1 propagates more rapidly and efficiently in humanized mice than vpu-deficient HIV-1 during the initial phase of infection.
Fig 2.

Dynamics of WT and vpu-deficient HIV-1 infection in humanized mice. (A and B) Expansion of WT and vpu-deficient HIV-1 in humanized mice. WT HIV-1 (3,000 TCID50, n = 6; 30,000 TCID50, n = 5; and 300,000 TCID50, n = 21) and vpu-deficient HIV-1 (3,000 TCID50, n = 6; 30,000 TCID50, n = 5, and 300,000 TCID50, n = 20) were inoculated into humanized mice aged between 12 and 17 weeks old, and the amount of viral RNA in plasma was quantified at 3, 7, 14, and 21 dpi. (A) Frequency of infection in mice. The percentage of infected mice in which viral RNA in plasma was detected at each time point is presented as Kaplan-Meier curves. (B) Viral load in infected humanized mice. The horizontal broken line indicates the detection limit of the assay (1,600 copies/ml). (C to E) Cytopathic effect of WT and vpu-deficient HIV-1 in the PB of humanized mice. The numbers of total CD4+ T cells (CD45+ CD3+ CD4+ cells; C), Tn cells (CD45+ CD3+ CD4+ CD45RA+ cells; D), and Tm cells (CD45+ CD3+ CD4+ CD45RA− cells; E) in the PB of WT HIV-1-infected mice (3,000 TCID50, n = 6; 30,000 TCID50, n = 5, and 300,000 TCID50, n = 11), vpu-deficient HIV-1-infected mice (3,000 TCID50, n = 6; 30,000 TCID50, n = 5, and 300,000 TCID50, n = 11), and mock-infected mice (n = 7) were routinely analyzed by flow cytometry and hematocytometry. (F) Cytopathic effect of WT and vpu-deficient HIV-1 in the lymphoid tissues of humanized mice. The percentages of total CD4+ T cells, Tn cells, and Tm cells in the spleen (left) and the BM (right) of WT HIV-1-infected mice (300,000 TCID50, n = 7) and vpu-deficient HIV-1-infected mice (300,000 TCID50, n = 7), and mock-infected mice (21 dpi, n = 6) were analyzed by flow cytometry. In panel A, statistical difference was determined by a log rank test. In panel B, statistical difference between WT HIV-1 versus vpu-deficient HIV-1 was determined by Welch's t test. In panels C to F, statistical difference between mock-infected versus WT HIV-1- or vpu-deficient HIV-1-infected mice was determined by Welch's t test. Statistically significant differences are shown as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SEMs. NS, no statistical significance.
We next analyzed the numbers of total CD4+ T cells, CD45RA+ naive CD4+ T cells, and CD45RA− memory CD4+ T cells in the PB of infected mice. As shown in Fig. 2C to E, virus infection at lower doses (3,000 and 30,000 TCID50) did not affect the numbers of peripheral CD4+ T cells. On the other hand, at a high infection dose (300,000 TCID50) in WT HIV-1-infected mice, the number of total CD4+ T cells, naive T (Tn) cells, and memory T (Tm) cells decreased by 4.0-fold, 2.4-fold, and 4.8-fold, respectively (Fig. 2C to E). Interestingly, this observed decrease in analyzed T cells in WT HIV-1-infected mice at 14 and 21 dpi was indistinguishable from that in HIV-1Δvpu-infected mice (Fig. 2C to E). Similarly, we observed that the percentages of CD4+ T cells, particularly Tm cells, in spleen and BM of both WT and vpu-deficient HIV-1-infected mice at 7 and 21 dpi was significantly lower than those of uninfected mice and that the reduced levels of total CD4+ T cells and Tm cells in WT HIV-1-infected mice were comparable to those in vpu-deficient HIV-1-infected mice (Fig. 2F). Taken together, these results suggest that the cytotoxicity induced by WT HIV-1 was similar to that of vpu-deficient HIV-1 in humanized mice.
Reduced production of vpu-deficient HIV-1 particles in the spleen of humanized mice at 7 dpi.
To exclude the possibility of vpu reversion in HIV-1Δvpu-infected mice, we performed PCR targeting the vpu-coding region and confirmed that the 81-bp deletion in vpu was maintained even at 21 dpi (Fig. 3A). This result indicates that vpu reversion did not occur in HIV-1Δvpu-infected mice. We then assessed the proportion of HIV-1-infected cells (i.e., Gag-positive cells) in WT and vpu-deficient HIV-1-infected mice. As previously reported (44), we found that the majority of HIV-1-infected cells in the spleen of WT HIV-1-infected humanized mice were CD3+ CD45RA− Tm cells (Fig. 3B and C). Infected splenic cells of HIV-1 Δvpu-infected mice also consisted of Tm cells, and the level was equal to that of WT HIV-1-infected mice (Fig. 3B and C). These results suggest that vpu deficiency did not affect the preference of target cells.
Fig 3.
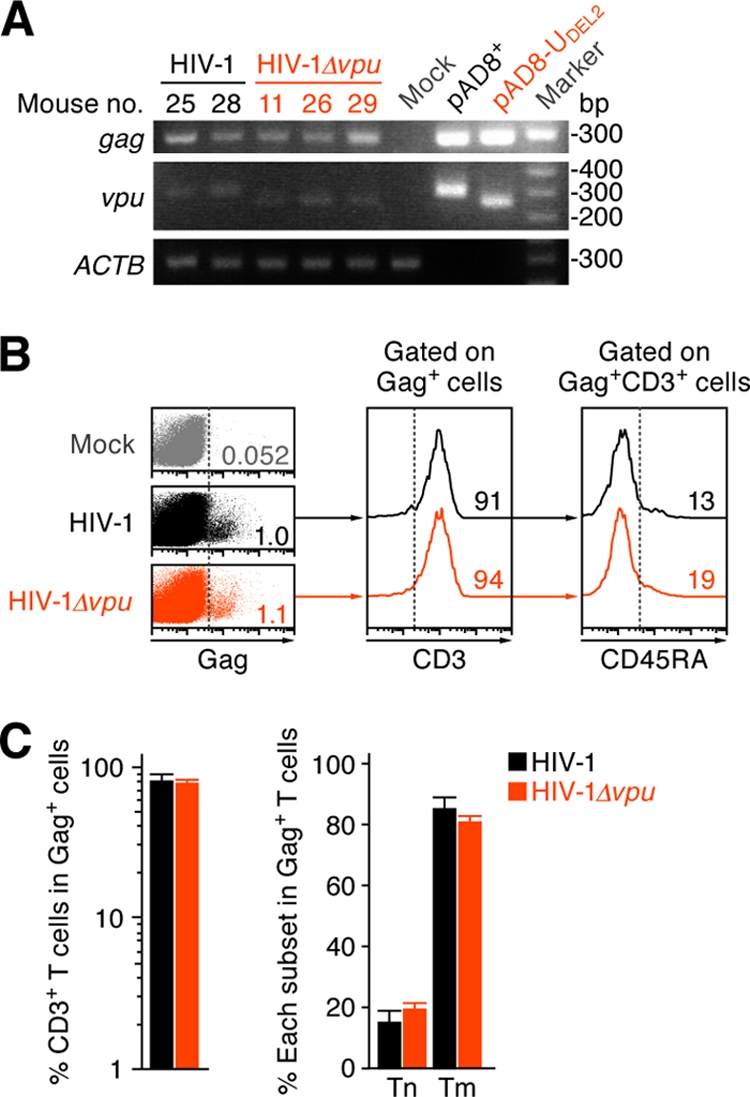
Characteristics of infected cells in humanized mice.(A) PCR analysis of proviral DNA. The regions of gag and vpu in HIV-1 DNA in the spleen of two WT HIV-1-infected mice, three vpu-deficient HIV-1-infected mice, and a mock-infected mouse at 21 dpi were analyzed by PCR. The mouse numbers correspond to those in Table 1. A representative result is shown. As the positive controls for gag and vpu, a WT HIV-1 plasmid (pAD8+) and vpu-deficient HIV-1 plasmid (pAD8-UDEL2) were used. Note that the band size of vpu in vpu-deficient HIV-1-infected mice is smaller than that in WT HIV-1-infected mice because of the 81-bp deletion with frameshift (see Materials and Methods for detail). ACTB (β-actin) was used as the internal control of the assay. (B and C) Characterization of infected cells. Human MNCs in the spleen of WT HIV-1-infected mice, vpu-deficient HIV-1-infected mice, and mock-infected mice were analyzed by flow cytometry. Representative dot plots and histograms are shown (B). The numbers in dot plots and histograms indicate the percentages of positive cells in each parental population, and the vertical broken lines indicate the threshold for the gating of positive cells based on the result from the isotype controls. (C) The percentages of CD3+ T cells in Gag-positive cells (left) and the percentages of Tn cells (CD45RA+) and Tm cells (CD45RA−) in infected T cells (Gag+ CD3+ CD8−cells, right) in the spleen of WT HIV-1-infected mice (n = 7) and vpu-deficient HIV-1-infected mice (n = 8) are shown.
In order to investigate the level of HIV-1 replication in organs, we collected PB/plasma, spleen, and BM of mice infected with 300,000 TCID50 at 7 and 21 dpi and analyzed the specimens by HIV-1 p24 ELISA and flow cytometry. As shown in Fig. 4A, the amounts of cell-free virions in the spleen of both WT and vpu-deficient HIV-1-infected mice were predominantly higher than those in plasma and BM, suggesting that the spleen is a major site for viral replication. At 21 dpi, when the level of viral RNA in the plasma of WT HIV-1-infected mice was similar to that of HIV-1Δvpu-infected mice (Fig. 2B, right panel), the amounts of viral particles (Fig. 4A) and the percentages of HIV-1-infected cells (Fig. 4B) in all tissues we analyzed in both groups of mice were comparable. On the other hand, at 7 dpi when the amount of viral RNA in the plasma of WT HIV-1-infected mice was significantly higher than that of HIV-1Δvpu-infected mice (Fig. 2B, right panel), the percentage of Gag-positive cells in the spleen of WT HIV-1-infected mice was 2.8-fold higher than that of HIV-1Δvpu-infected mice (Fig. 4B). It was of interest that the amount of cell-free viral particles in the spleen of WT HIV-1-infected mice at 7 dpi was profoundly (38.2-fold) higher than that of HIV-1Δvpu-infected mice (Fig. 4A). Furthermore, in the spleen of WT HIV-1-infected mice at 7 dpi, the amount of the cell-free virions significantly correlated with the percentage of infected cells, whereas no correlation in the spleen of HIV-1Δvpu-infected mice was observed (WT HIV-1, r = 0.877 and P = 0.00085; HIV-1Δvpu, r = 0.344 and P = 0.37) (Fig. 4C). These findings indicate that Vpu might enhance the expression of viral proteins, which lead to a robust production of viral particles. To address this possibility, the mean fluorescent intensity (MFI) of HIV-1 Gag in WT and vpu-deficient HIV-1-infected splenic cells was measured. Because the reagents for flow cytometry preferentially permeabilize plasma membranes, the anti-p24 antibody we used had access to the Gag proteins in the cytoplasm (i.e., Pr55Gag), which indicates that the MFI value of Gag can reflect the level of expressed Pr55Gag in proteins in infected cells. However, as shown in Fig. 4D, the levels of Gag MFI in WT and vpu-deficient HIV-1-infected splenic cells were equal. This result suggests that Vpu does not affect the expression of viral proteins leading to the augmentation of virion production.
Downregulation of the surface expression levels of Vpu-associated cellular proteins on infected cells in humanized mice at 7 dpi.
As shown in Fig. 1D to F, we demonstrated that Vpu downregulates CD4, BST2, and SLAMF6 from the surfaces of vpu-proficient HIV-1-infected cells in vitro. To address whether the potential of Vpu to downregulate these three molecules is associated with the decrease of cell-free virions in HIV-1Δvpu-infected mice (Fig. 4A), we focused on the surface expression levels of CD4, BST2, and SLAMF6 on infected cells (i.e., Gag-positive cells). Since the difference in the levels of plasma viral load (Fig. 2B, right panel) and splenic cell-free virions (Fig. 4A) between WT HIV-1-infected mice and HIV-1Δvpu-infected mice is most significant at the higher infection dose (300,000 TCID50) at 7 dpi, we used the splenic specimen of mice infected under these conditions. Because the majority of infected cells were Tm cells (Fig. 3B and C), we analyzed the surface levels of these three proteins on Tm (CD3+ CD8− CD45RA−) cells by flow cytometry. As observed in in vitro experiments (Fig. 1D), we detected a significant downregulation of surface CD4 in both WT and vpu-deficient HIV-1-infected cells (Fig. 5A). Nevertheless, we also found that both the percentage and the MFI of surface CD4 in Gag-positive cells in WT HIV-1-infected mice were significantly lower than those in HIV-1Δvpu-infected mice (Fig. 5A), which suggests that Vpu potently downregulates CD4 even in vivo. Interestingly, BST2 expressed on WT but not on vpu-deficient HIV-1-infected cells was significantly downregulated (Fig. 5B). On the other hand, Vpu-dependent downregulation of SLAMF6 on infected cells was not observed in infected humanized mice (Fig. 5C). Taken together, these results suggest that Vpu downregulates CD4 and BST2 from the surface of infected cells, which leads to the impairment of the release of nascent virions.
Fig 5.

Surface expression profile of CD4, BST2, and SLAMF6 in infected humanized mice at 7 dpi. Tm cells (CD3+ CD8− CD45RA−) in the spleen of WT HIV-1-infected mice (300,000 TCID50; n = 7), vpu-deficient HIV-1-infected mice (300,000 TCID50; n = 6), and mock-infected mice (n = 7) at 7 dpi were classified into infected (Gag-positive) or uninfected (Gag-negative) populations by using an anti-p24 antibody. Representative histograms (left), the positive percentages (middle), and the MFIs (right) of CD4 (A), BST2 (B), and SLAMF6 (C) in each population are shown. In the left panels, the numbers on each histogram indicate the MFI values, and the vertical broken lines indicate the thresholds for the gating of positive cells based on the result from the isotype controls. The statistical difference is determined by Welch's t test. Statistically significant differences are shown as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SEMs. NS, no statistical significance.
In order to investigate the dynamics of CD4 and BST2 in depth, we classified infected and uninfected cells into four types: CD4+ BST2+, CD4+ BST2−, CD4− BST2+, and CD4− BST2−. In Gag-negative Tm cells (CD3+ CD8− CD45RA− Gag−) of mock-infected, WT HIV-1-infected, and HIV-1Δvpu-infected mice, most of the cells expressed CD4, while approximately 60% of the cells were positive for BST2 (Fig. 6A to D). On the other hand, in Gag-positive Tm cells (CD3+ CD8− CD45RA− Gag+) of WT HIV-1-infected mice, we detected a significant level of the cells negative for both CD4 and BST2, and the percentage of CD4− BST2− cells was significantly higher than that of CD4− BST2+ cells (CD4− BST2− cells, 36.6% ± 3.8%; CD4− BST2+ cells, 3.5% ± 1.4%) (Fig. 6B and E). In contrast, in Gag-positive Tm cells of HIV-1Δvpu-infected mice, the percentage of CD4− BST2− cells was significantly lower than that of CD4− BST2+ cells (CD4− BST2− cells, 5.3% ± 0.8%; CD4− BST2+ cells, 14.2% ± 0.9%) (Fig. 6B and E). Furthermore, the percentage of CD4− BST2− Tm cells positive for Gag in WT HIV-1-infected mice was significantly higher than that in HIV-1Δvpu-infected mice (Fig. 6E). Taken together, these results suggest that Vpu concomitantly downregulates the cell surface expression of CD4 and BST2 in infected cells.
Fig 6.

Codownregulation of CD4 and BST2 from the surface of WT HIV-1-infected cells. Surface expression levels of CD4 and BST2 on the surface of Gag-positive Tm cells (CD3+ CD8− CD45RA− Gag+) or Gag-negative Tm cells (CD3+ CD8− CD45RA− Gag−) in the spleen of WT HIV-1-infected mice (300,000 TCID50; n = 6), vpu-deficient HIV-1-infected mice (300,000 TCID50; n = 5), and mock-infected mice (n = 7) at 7 dpi were analyzed by flow cytometry. Representative dot plots (A to C) and the summarized results (D and E) are shown. In panels A to C, the numbers indicate the percentages of the cells in the respective quadrant. The statistical difference is determined by Welch's t test (*, P < 0.05). Error bars represent SEMs.
Modulation of the surface expression levels of Vpu-associated cellular proteins in infected humanized mice at 21 dpi.
Although a marked difference in the levels of plasma viral load and cell-free virions in the spleen of WT HIV-1-infected and HIV-1Δvpu-infected mice was observed at 7 dpi, the viral loads became comparable at 21 dpi (Fig. 2B, right panel). To explore the possibility that Vpu loses its ability to downregulate CD4 and BST2 at 21 dpi, we used the splenic specimens at this time point and analyzed the surface levels of CD4, BST2, and SLAMF6 by flow cytometry. In contrast to this hypothesis, we detected significant downregulation of CD4 and BST2 on WT HIV-1-infected Tm cells (Fig. 7A and B). Although a significant downregulation of SLAMF6 was also observed in infected Tm cells, it was independent on Vpu (Fig. 7C). Unexpectedly, we found that BST2 was upregulated on Gag-negative Tm cells of HIV-1Δvpu-infected mice (Fig. 7B). Since the upregulation of BST2 was observed in Gag-negative Tm cells of not only HIV-1Δvpu-infected mice but also WT HIV-1-infected mice (Fig. 7B), we speculated that this increase was independent of Vpu and is indirectly induced by HIV-1 infection. To address this issue, we analyzed the expression level of BST2 on the other human leukocytes, which were less susceptible or resistant to HIV-1 infection. As shown in Fig. 7D and E, the levels of surface BST2 on Tn cells and CD8+ T cells of WT HIV-1- and HIV-1Δvpu-infected mice were significantly higher than those of mock-infected mice. These findings suggest that CD4 and BST2 were still downregulated by Vpu at 21 dpi, while BST2 is upregulated on uninfected cells in infected mice.
Fig 7.

Surface expression profile of CD4, BST2, and SLAMF6 in infected humanized mice at 21 dpi. Tm cells (CD3+ CD8−CD45RA−; A to C), Tn cells (CD3+ CD8− CD45RA+; D), and CD8+ T cells (CD3+ CD8+; E) in the spleen of WT HIV-1-infected mice (300,000 TCID50; n = 5), vpu-deficient HIV-1-infected mice (300,000 TCID50; n = 7), and mock-infected mice (n = 7) at 21 dpi were analyzed by flow cytometry. Tm cells were further classified into infected (Gag-positive) or uninfected (Gag-negative) populations by using an anti-p24 antibody. Representative histograms (left), the positive percentages (middle), and the MFIs (right) of CD4 (A), BST2 (B, D, and E), and SLAMF6 (C) in each population are shown. In the left panels, the numbers on the histograms indicate the MFI values, and the vertical broken lines indicate the thresholds for the gating of positive cells based on the result from the isotype controls. The statistical difference is determined by Welch's t test. Statistically significant differences are shown as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SEMs. NS, no statistical significance.
Cell-to-cell contact of infected cells in the spleen of infected mice.
To investigate why the viral loads were equal at 21 dpi (Fig. 2B, right panel), we performed immunostaining analysis in the spleen of WT HIV-1- and HIV-1Δvpu-infected mice (Fig. 8A). Interestingly, results (Fig. 8B) revealed that most of the Gag-positive cells were in contact with the other adjunct Gag-positive cell(s) in the spleen of WT HIV-1- and HIV-1Δvpu-infected mice (WT HIV-1, 84.2%; HIV-1Δvpu, 82.4%) (Fig. 8C). These findings suggest that cell-to-cell viral transmission frequently occurs in the spleens of WT HIV-1- and HIV-1Δvpu-infected mice.
Fig 8.

Immunostaining for HIV-1 Gag protein.(A) Representatives of the spleen of WT HIV-1-infected mice (left) and vpu-deficient HIV-1-infected mice (right). HIV-1 Gag is shown in red, and nuclei are shown in blue by staining with Hoechst 33342. Boxed areas are enlarged in the bottom right of each panel. Scale bar, 50 μm. (B) A representative of the Gag-positive cell contacted with (solid-line box) or without (broken-line box) the other adjunct Gag-positive cell in spleen is shown (top). The middle panel shows enlargements of the boxed areas. (bottom) The fluorescent intensities of Hoechst (blue) and Gag (red) indicated by arrowheads in the middle panels are shown. AU, arbitrary unit. (C) The percentages of the Gag-positive cells that are contacted with the other adjunct Gag-positive cells in the spleen of WT HIV-1-infected mice (n = 272) and vpu-deficient HIV-1-infected mice (n = 448) are shown.
The different effects of CD4 and BST2 on cell-free and cell-to-cell HIV-1 infection in vitro.
To directly demonstrate the potential of CD4 and BST2 to inhibit cell-free and cell-to-cell infections of HIV-1, pAD8+ or pAD8-UDEL2 was cotransfected with or without CD4 or BST2 expression plasmids into HEK293 cells in which neither CD4 nor BST2 is endogenously expressed (56). As observed in infected humanized mice (Fig. 5 and 6), ectopically expressed CD4 and BST2 were downregulated in a Vpu-dependent manner (Fig. 9A and B). In addition, in the cells cotransfected with CD4 or BST2 expression plasmids, the percentage of the cells negative for both CD4 and BST2 in pAD8+-transfected cells was significantly higher than percentages in pAD8-UDEL2-transfected and mock-transfected cells (Fig. 9C). These results suggest that CD4 and BST2 were concurrently downregulated by Vpu, reflecting observations in our humanized mouse model (Fig. 6).
We then measured the yield of the released virions in the supernatant and found that the release of vpu-deficient HIV-1 was suppressed by CD4 and BST2 (Fig. 9D). Interestingly, the yield of vpu-deficient HIV-1 in the supernatant of the cells cotransfected with both CD4 and BST2 expression plasmids was significantly lower than that of the cells transfected solely with either CD4 or BST2 expression plasmid alone (Fig. 9D), suggesting that CD4 and BST2 impair the production of nascent virions through distinct manners.
Finally, we quantified the infectious potential of transfected cells by coculturing them with TZM-bl cells. Interestingly, the infectious potential of pAD8-UDEL2-transfected cells was not suppressed by ectopic expression of either CD4 or BST2 either alone or together and was comparable to that of pAD8+-transfected cells (Fig. 9E). Taken together, these findings suggest that CD4 and BST2 do not have the potential to attenuate cell-to-cell HIV-1 infection and that Vpu slightly associates with the efficiency of cell-to-cell viral transmission.
DISCUSSION
The significance of Vpu for HIV-1 infection, especially in terms of its interplay with BST2, has been extensively investigated in cell culture experiments. However, its importance for HIV-1 propagation in vivo remains unclear and needs to be addressed (7, 26). In this study, we used vpu-deficient CCR5-tropic HIV-1 to infect a humanized mouse model, which allowed us to track the pathogenesis of HIV-1 from the onset of infection, and elucidated the role of Vpu in HIV-1 propagation in vivo. Our results demonstrate that Vpu downregulates CD4 and BST2 on the surface of infected cells and enhances viral dissemination during the initial phase of infection in vivo. This is the first report directly implicating the importance of Vpu during HIV-1 infection in vivo.
Although our findings suggest that Vpu is weakly associated with the viral cytotoxicity of HIV-1 infection in vivo, we hypothesize that its involvement in early viral replication kinetics may be critical. In our humanized mouse model, the preference of target cells (Fig. 3B and C), the level of viral protein expression (Fig. 4D), and the cytopathic effect (Fig. 2C to F) were nearly identical between WT and vpu-deficient HIV-1-infected mice. However, the kinetics of early viral expansion in WT HIV-1-infected mice was significantly higher than in HIV-1Δvpu-infected mice (Fig. 2B). In addition, although the percentage of Gag-positive cells in the spleen of WT HIV-1-infected mice at 7 dpi was only 2.8-fold higher than that of HIV-1Δvpu-infected mice (Fig. 4B), the amount of cell-free virions in WT HIV-1-infected spleens was 38.2-fold higher (Fig. 4A). Moreover, the levels of cell-free virions positively and significantly correlated with infected cells in the spleen of WT HIV-1-infected mice but not in HIV-1Δvpu-infected mice (Fig. 4C). Interestingly, significant downregulation of BST2 by WT HIV-1 appeared to be important for this virus dissemination to occur (Fig. 5B). Taken together, our findings suggest that the Vpu antagonism of BST2 closely associates with the efficiency of virus expansion in vivo, especially during the early phase of infection.
The amount of cell-free virions in the spleen of WT HIV-1-infected mice was prominently (38.2-fold) higher than that in HIV-1Δvpu-infected mice (Fig. 4A); however, statistical significance was not determined. To obtain the specimens of cell suspensions and cell-free virions from the spleen of infected mice, the spleen was mechanically dissociated according to a conventional method. In carrying out this procedure, it is possible that the virions tethered by BST2 on the surface of HIV-1Δvpu-infected cells were partially dissociated. In fact, one study has suggested that the virions tethered by BST2 can be partially dissociated by mechanical agitation using a vortex (38). Therefore, it might be possible to assume that the virions tethered by BST2 on the surface of vpu-deficient HIV-1-infected cells partially dissociated by our procedure, which led to no statistical difference.
In comparing the level of Gag expression in HIV-1-infected cells, we noticed differences depending on the method used for detection. Flow cytometry analyses of Pr55Gag expression in vpu-deficient HIV-1-producing cells produced results comparable to those in WT HIV-1-producing cells in the transfected HeLa cells (data not shown), infected primary human CD4+ T cells (data not shown), and infected humanized mice (Fig. 4D), suggesting equal levels of virus production. On the other hand, Western blotting analyses revealed that the amount of p24 in the pAD8-UDEL2-transfected HeLa cells was clearly higher than that in the pAD8+-transfected HeLa cells (Fig. 1B) although the expression levels of Pr55Gag were equal (Fig. 1B). These data indicate that the release of virions in the absence of Vpu was impaired. HIV-1 particles tethered on the plasma membrane are rapidly endocytosed and accumulate in endosomal compartments (40). Thus, the anti-p24 antibody used for flow cytometry may have underestimated the total amount of Gag proteins because only intracellular Pr55Gag was detected as p24 of virions in the endosomal compartment was not accessible.
As shown in Fig. 2B, HIV-1 propagated more rapidly in humanized mice at the highest dose than at the other doses. These results suggest that the kinetics of viral growth is dependent on the dose of inoculated viruses. On the other hand, the cytopathic effect was observed in the PB of infected mice at 14 and 21 dpi at the highest dose but not at the other doses (Fig. 2C to E). In this regard, we have previously observed that the viral load of HIV-1-infected humanized mice reached a plateau at 3 to 4 weeks postinfection and that peripheral CD4+ T cells, especially memory CD4+ T cells, in infected humanized mice gradually decreased at later time points (after 9 weeks postinfection) (44, 52). Therefore, it is conceivable that the cytopathic effect (i.e., the decrease of peripheral CD4+ T cells) in infected mice at the highest dose at 14 and 21 dpi (Fig. 2C and D) is caused by faster viral dissemination than at the other doses and that the cytopathic effect would be observed at later time points in infected mice at lower doses.
It was reported that CD4, BST2, and SLAMF6 are downregulated from the surface of HIV-1-infected cells by Vpu in cell culture systems (7, 9, 31, 64). Here, we demonstrated that the Vpu encoded by strain AD8 is able to downregulate these three molecules from the surface of infected cells in vitro (Fig. 1D to F). However, the magnitude of Vpu-mediated downregulation of each cellular protein in vivo remains unknown. We observed that CD4 and BST2 were significantly downregulated by Vpu expressed in infected cells at both 7 dpi (Fig. 5) and 21 dpi (Fig. 7). We also demonstrated that both molecules were downregulated from the surface of the same infected cells in a Vpu-dependent manner (Fig. 6). In contrast to CD4 and BST2, Vpu-dependent SLAMF6 downregulation was not observed in infected humanized mice (Fig. 5C and 6C). Therefore, our results suggest that Vpu downregulates cell surface CD4 and BST2 both in vitro and in vivo, while that the sequestration of SLAMF6 from the surface of infected cells is not sufficiently elicited by Vpu in vivo.
It has been reported that CD4 expressed on the surface of infected cells has the potential to impair HIV-1 replication by (i) inhibiting the release of progeny virions (48), (ii) impairing the incorporation of Env into nascent virions (6, 29), and (iii) sequestering Env from the virion budding site (3, 6, 29, 31, 32). We confirmed that CD4 suppresses the production of infectious virions (Fig. 9D). Moreover, we provide the first evidence demonstrating that CD4 and BST2 synergistically suppress the production of infectious virions (Fig. 9D). Therefore, in accordance with previous reports (29, 31), our results suggest that not only BST2 but also CD4 possesses the potential to impair the production of infectious nascent virions, which can be counteracted by Vpu.
Even though Vpu downregulated CD4 and BST2 at 21 dpi (Fig. 7), the levels of plasma viral load and splenic cell-free virion in vpu-deficient HIV-1-infected mice became equal to those in WT HIV-1-infected mice (Fig. 2B and 3A). These findings raise the possibility that vpu-deficient HIV-1 is able to overcome the restriction mediated by CD4 and BST2 at this point. Interestingly, it has been reported that HIV-1 can be transmitted through cell-to-cell contact (17–22, 36, 57). In addition, although it is still controversial (5, 28), one study has recently demonstrated that the cell-to-cell HIV-1 spread is not restricted by BST2 in a T cell culture system (16). By using an in vitro transfection system, we demonstrated that CD4 and BST2 did not associate with cell-to-cell infection and that the efficiency of cell-to-cell transmission was not enhanced by Vpu (Fig. 9E). Moreover, we found that the majority of Gag-positive cells in the spleens of WT and vpu-deficient HIV-1-infected mice were in contact with each other (Fig. 8). Taken together, our findings, in agreement with previous reports, strongly suggest that cell-to-cell virus transmission efficiently occurs in humanized mice at 21 dpi and is not restricted by BST2 and CD4.
As mentioned above, viruses are propagated by two modes: cell-free virus-mediated transmission and cell-to-cell transmission. In comparisons of these two modes of transmission, it has been reported that cell-to-cell HIV-1 spread is more efficient than cell-free infection in in vitro cell culture systems (4, 50). Here, we found that the infectivity of transfected cells per culture was markedly higher than that of the supernatant per culture (Fig. 9D and E). Given that leukocytes, including CD4+ T cells, exist in dense clumps and are in contact with each other in lymphoid tissues (e.g., spleen), it is feasible to assume that HIV-1 dissemination is more likely to occur through cell-to-cell contact in vivo. Moreover, previous reports have demonstrated that BST2 potently impairs the release of Orthomyxoviridae (influenza A virus) and Filoviridae (Ebola virus and Marburg virus); however, BST2 does not suppress the spread of these viruses in in vitro cell cultures (47, 69). Therefore, it is conceivable that BST2 does not impair the cell-to-cell spread of viruses even if the cell-free virus-mediated infection of these viruses is restricted by BST2.
Andrew and Strebel have recently suggested that endogenous BST2 is not a “restrictive factor” that limits HIV-1 spread but, rather, a “modulator” that shifts the mode of HIV-1 infection from cell-free spread to cell-to-cell transmission (2). Building upon this hypothesis, we used our current findings, together with previous reports, to propose a model regarding the role of Vpu in HIV-1 infection in vivo. During the acute phase of infection (until 7 dpi in humanized mice), when the level of infected cells in the body is low (Fig. 4B), cell-free virus-mediated infection serves as the major route of virus spread in vivo. Therefore, Vpu-mediated counteraction of BST2 (and CD4) has a crucial role for the efficient virus amplification in vivo during this phase. From our data, the reduced infectivity of vpu-deficient HIV-1 compared to WT HIV-1 in humanized mice (Fig. 2A) further suggests the importance of a boost in the amount of cell-free viruses necessary for the establishment of virus production in vivo. However, at a later stage of infection when the level of infected cells achieves a certain threshold (21 dpi in humanized mice), HIV-1 would be mainly propagated through cell-to-cell transmission, which is resistant to BST2-mediated restriction. Therefore, the antagonistic behavior of Vpu would become dispensable for efficient virus spread at this point. In summary, consistent with the concept proposed by Andrew and Strebel (2), our findings suggest that endogenous BST2 is not a restrictive but a modulatory factor for HIV-1 spread in vivo.
ACKNOWLEDGMENTS
We thank Peter Gee (Laboratory of Viral Pathogenesis, Institute for Virus Research, Kyoto University) for proofreading the manuscript, and Takao Ueno (Graduate School of Mathematical Sciences, the University of Tokyo), Mariko Horiike (Institute for Virus Research, Kyoto University), Takashi Nakano, and Kouichi Sano (Osaka Medical College) for their generous help in our study. We also thank Klaus Strebel (National Institute of Allergy and Infectious Diseases, National Institutes of Health) for providing HIV-1-producing plasmids (pAD8+ and pAD8-UDEL2) and a rabbit anti-Vpu polyclonal antibody (U2-3), Yuetsu Tanaka (University of the Ryukyus) for providing a rat anti-p24 monoclonal antibody (clone 2C2), and Masayuki Miyasaka (Osaka University Graduate School of Medicine) for providing a human CD4 expression plasmid (pBCMGSNeo-human CD4). We appreciate Kotubu Misawa's dedicated support.
This work was supported in part by grants from the following: Grants-in-Aid for Scientific Research B21390137 (to Y.K.) and S22220007 (to M.I. and Y.K.) and a Grant-in-Aid for Young Scientists B23790500 (to K.S.) from the Japan Society for the Promotion of Science; grants from Research on Emerging and Reemerging Infectious Diseases (to Y.K.) and Research on HIV/AIDS (to Y.K.) from the Ministry of Health, Labor and Welfare of Japan; a grant from the Uehara Memorial Foundation (to K.S.); JST PRESTO program (to S.I.); and a UCLA CFAR grant 5P30AI028697 (to D.S.A).
Footnotes
Published ahead of print 22 February 2012
REFERENCES
- 1. An DS, et al. 2007. Use of a novel chimeric mouse model with a functionally active human immune system to study human immunodeficiency virus type 1 infection. Clin. Vaccine Immunol. 14:391–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrew A, Strebel K. 2011. The interferon-inducible host factor bone marrow stromal antigen 2/tetherin restricts virion release, but is it actually a viral restriction factor? J. Interferon Cytokine Res. 31:137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arganaraz ER, Schindler M, Kirchhoff F, Cortes MJ, Lama J. 2003. Enhanced CD4 down-modulation by late stage HIV-1 nef alleles is associated with increased Env incorporation and viral replication. J. Biol. Chem. 278:33912–33919 [DOI] [PubMed] [Google Scholar]
- 4. Carr JM, Hocking H, Li P, Burrell CJ. 1999. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 265:319–329 [DOI] [PubMed] [Google Scholar]
- 5. Casartelli N, et al. 2010. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 6:e1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cortes MJ, Wong-Staal F, Lama J. 2002. Cell surface CD4 interferes with the infectivity of HIV-1 particles released from T cells. J. Biol. Chem. 277:1770–1779 [DOI] [PubMed] [Google Scholar]
- 7. Douglas JL, et al. 2010. The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 6:e1000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Douglas JL, et al. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J. Virol. 83:7931–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evans DT, Serra-Moreno R, Singh RK, Guatelli JC. 2010. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 18:388–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Freed EO, Martin MA. 2007. HIVs and their replication, p 2107–2185 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 11. Goffinet C, et al. 2009. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5:285–297 [DOI] [PubMed] [Google Scholar]
- 12. Gottlinger HG, Dorfman T, Cohen EA, Haseltine WA. 1993. Vpu protein of human immunodeficiency virus type 1 enhances the release of capsids produced by gag gene constructs of widely divergent retroviruses. Proc. Natl. Acad. Sci. U. S. A. 90:7381–7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hauser H, et al. 2010. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito M, et al. 2002. NOD/SCID/γcnull mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100:3175–3182 [DOI] [PubMed] [Google Scholar]
- 15. Jia B, et al. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5:e1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jolly C, Booth NJ, Neil SJ. 2010. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 84:12185–12199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jolly C, Mitar I, Sattentau QJ. 2007. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 81:13916–13921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jolly C, Mitar I, Sattentau QJ. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 81:5547–5560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jolly C, Sattentau QJ. 2007. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J. Virol. 81:7873–7884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jolly C, Sattentau QJ. 2005. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J. Virol. 79:12088–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jolly C, Sattentau QJ. 2007. Regulated secretion from CD4+ T cells. Trends Immunol. 28:474–481 [DOI] [PubMed] [Google Scholar]
- 22. Jolly C, Sattentau QJ. 2004. Retroviral spread by induction of virological synapses. Traffic 5:643–650 [DOI] [PubMed] [Google Scholar]
- 23. Jouvenet N, et al. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83:1837–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 106:2886–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kawano Y, et al. 1997. Mutational analysis of human immunodeficiency virus type 1 (HIV-1) accessory genes: requirement of a site in the nef gene for HIV-1 replication in activated CD4+ T cells in vitro and in vivo. J. Virol. 71:8456–8466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirchhoff F. 2010. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8:55–67 [DOI] [PubMed] [Google Scholar]
- 27. Kobayashi T, et al. 2011. Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85:932–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuhl BD, et al. 2010. Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology 7:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lama J, Mangasarian A, Trono D. 1999. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 9:622–631 [DOI] [PubMed] [Google Scholar]
- 30. Le Tortorec A, Neil SJ. 2009. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83:11966–11978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levesque K, Finzi A, Binette J, Cohen EA. 2004. Role of CD4 receptor down-regulation during HIV-1 infection. Curr. HIV Res. 2:51–59 [DOI] [PubMed] [Google Scholar]
- 32. Levesque K, Zhao YS, Cohen EA. 2003. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J. Biol. Chem. 278:28346–28353 [DOI] [PubMed] [Google Scholar]
- 33. Maldarelli F, Chen MY, Willey RL, Strebel K. 1993. Human immunodeficiency virus type 1 Vpu protein is an oligomeric type I integral membrane protein. J. Virol. 67:5056–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mansouri M, et al. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 83:9672–9681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Margottin F, et al. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1:565–574 [DOI] [PubMed] [Google Scholar]
- 36. Martin N, Sattentau Q. 2009. Cell-to-cell HIV-1 spread and its implications for immune evasion. Curr. Opin. HIV AIDS 4:143–149 [DOI] [PubMed] [Google Scholar]
- 37. Mitchell RS, et al. 2009. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via β-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5:e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miyagi E, Andrew AJ, Kao S, Strebel K. 2009. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U. S. A. 106:2868–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moyer CL, Wiethoff CM, Maier O, Smith JG, Nemerow GR. 2011. Functional genetic and biophysical analyses of membrane disruption by human adenovirus. J. Virol. 85:2631–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Neil SJ, Eastman SW, Jouvenet N, Bieniasz PD. 2006. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD. 2007. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430 [DOI] [PubMed] [Google Scholar]
- 43. Ni H, Barrett AD. 1998. Attenuation of Japanese encephalitis virus by selection of its mouse brain membrane receptor preparation escape variants. Virology 241:30–36 [DOI] [PubMed] [Google Scholar]
- 44. Nie C, et al. 2009. Selective infection of CD4+ effector memory T lymphocytes leads to preferential depletion of memory T lymphocytes in R5 HIV-1-infected humanized NOD/SCID/IL-2Rγnull mice. Virology 394:64–72 [DOI] [PubMed] [Google Scholar]
- 45. Okuma K, et al. 2008. Interleukin-4-transgenic hu-PBL-SCID mice: a model for the screening of antiviral drugs and immunotherapeutic agents against X4 HIV-1 viruses. J. Infect. Dis. 197:134–141 [DOI] [PubMed] [Google Scholar]
- 46. Pardieu C, et al. 2010. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 6:e1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Radoshitzky SR, et al. 2010. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 84:10569–10580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ross TM, Oran AE, Cullen BR. 1999. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol. 9:613–621 [DOI] [PubMed] [Google Scholar]
- 49. Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. 2009. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83:2382–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sato H, Orenstein J, Dimitrov D, Martin M. 1992. Cell-to-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 186:712–724 [DOI] [PubMed] [Google Scholar]
- 51. Sato K, et al. 2008. Modulation of human immunodeficiency virus type 1 infectivity through incorporation of tetraspanin proteins. J. Virol. 82:1021–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sato K, et al. 2010. Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice. J. Virol. 84:9546–9556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sato K, Koyanagi Y. 2011. The mouse is out of the bag: insights and perspectives on HIV-1-infected humanized mouse models. Exp. Biol. Med. (Maywood) 236:977–985 [DOI] [PubMed] [Google Scholar]
- 54. Sato K, et al. 2011. A novel animal model of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in humanized mice. Blood 117:5663–5673 [DOI] [PubMed] [Google Scholar]
- 55. Sato K, et al. 2010. Dynamics of memory and naive CD8+ T lymphocytes in humanized NOD/SCID/IL-2Rγnull mice infected with CCR5-tropic HIV-1. Vaccine 28(Suppl 2):B32–B37 [DOI] [PubMed] [Google Scholar]
- 56. Sato K, et al. 2009. Comparative study on the effect of human BST-2/Tetherin on HIV-1 release in cells of various species. Retrovirology 6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815–826 [DOI] [PubMed] [Google Scholar]
- 58. Sauter D, et al. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schindler M, et al. 2010. Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo human lymphoid tissue. Retrovirology 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schubert U, et al. 1998. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 72:2280–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schubert U, et al. 1996. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 70:809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schubert U, Bour S, Willey RL, Strebel K. 1999. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either Vpu or Env. J. Virol. 73:887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schubert U, Clouse KA, Strebel K. 1995. Augmentation of virus secretion by the human immunodeficiency virus type 1 Vpu protein is cell type independent and occurs in cultured human primary macrophages and lymphocytes. J. Virol. 69:7699–7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shah AH, et al. 2010. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 8:397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Strebel K, Klimkait T, Martin MA. 1988. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science 241:1221–1223 [DOI] [PubMed] [Google Scholar]
- 66. Theodore TS, et al. 1996. Construction and characterization of a stable full-length macrophage-tropic HIV type 1 molecular clone that directs the production of high titers of progeny virions. AIDS Res. Hum. Retroviruses 12:191–194 [DOI] [PubMed] [Google Scholar]
- 67. Van Damme N, et al. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Varthakavi V, Smith RM, Bour SP, Strebel K, Spearman P. 2003. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc. Natl. Acad. Sci. U. S. A. 100:15154–15159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Watanabe R, Leser GP, Lamb RA. 2011. Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology 417:50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wei X, et al. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weidner JM, et al. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 84:12646–12657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 66:7193–7200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang F, et al. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]