

Abstract
Hepatitis B virus (HBV) replicates its DNA genome through reverse transcription of a pregenomic RNA (pgRNA) by using a multifunctional polymerase (HP). A critical function of HP is its specific recognition of a viral RNA signal termed ε (Hε) located on pgRNA, which is required for specific packaging of pgRNA into viral nucleocapsids and initiation of viral reverse transcription. HP initiates reverse transcription by using itself as a protein primer (protein priming) and Hε as the obligatory template. We have purified HP from human cells that retained Hε binding activity in vitro. Furthermore, HP purified as a complex with Hε, but not HP alone, displayed in vitro protein priming activity. While the HP-Hε interaction in vitro and in vivo required the Hε internal bulge, but not its apical loop, and was not significantly affected by the cap-Hε distance, protein priming required both the Hε apical loop and internal bulge, as well as a short distance between the cap and Hε, mirroring the requirements for RNA packaging. These studies have thus established new HBV protein priming and RNA binding assays that should greatly facilitate the dissection of the requirements and molecular mechanisms of HP-Hε interactions, RNA packaging, and protein priming.
INTRODUCTION
Hepatitis B virus (HBV), a member of the Hepadnaviridae family, is a worldwide health problem that affects over 350 million chronically infected people and causes a million deaths per year due to hepatic failure, cirrhosis, and hepatocellular carcinoma (40, 57, 58). HBV has a small (ca. 3.2-kb), partially double-stranded DNA genome that replicates through reverse transcription of an RNA intermediate termed pregenomic RNA (pgRNA) (57, 62). The HBV polymerase (HP), a specialized reverse transcriptase (RT), has RNA- and DNA-dependent DNA polymerase activities and contains four domains, including the N-terminal TP (terminal protein) domain followed by a spacer region, the RT domain, and the C-terminal RNase H domain (10, 26, 50, 68). The TP domain is conserved in all hepadnaviruses, but it is not found in any other RT (3, 10, 50). The RT and RNase H domains of HP are conserved with other RTs, including the YMDD RT active site and the RNase H catalytic residues (10, 11, 83).
Initiation of HBV reverse transcription occurs via a novel protein priming mechanism whereby HP itself serves as a protein primer for viral minus-strand DNA synthesis. Efforts to understand the hepadnavirus protein priming mechanism have been facilitated greatly by the development of in vitro priming assays using the polymerase from the avian hepadnavirus duck hepatitis B virus (DHBV). The first in vitro hepadnavirus protein priming system was established by expressing the DHBV polymerase (DP) in a rabbit reticulocyte lysate (RRL) in vitro translation system. This system allowed for the first conclusive identification of DP, specifically Y96 in the TP domain, as the primer for viral minus-strand DNA synthesis (21, 76, 82, 85). DP expressed in yeast (Saccharomyces cerevisiae) cells as a fusion protein using the Ty1 retrotransposon system was also shown to carry out protein-primed reverse transcription (64). Subsequent studies employing the DHBV model also led to the identification of a specific viral RNA signal, called ε (Dε), located on pgRNA and known to be required to direct pgRNA packaging into viral nucleocapsids (20, 49), as the template for protein-primed DNA synthesis as it forms a specific ribonucleoprotein (RNP) complex with DP (49, 65, 67, 75, 77). Specifically, the sequence from the internal bulge of Dε serves as the template for the synthesis of a short (3 to 4 nucleotides long) minus-strand DNA oligomer, which becomes covalently attached to DP via the priming Y96 residue. Distinct requirements for DP sequence and structure indicate that DHBV protein priming can be divided into two different stages (42, 80). Initiation of protein priming involves the addition of the first nucleotide, templated by the last (3′) nucleotide of the Dε internal bulge, to the polymerase protein forming a phosphotyrosyl bond between the initiating nucleotide and the primer Y residue on TP (i.e., DP deoxynucleotidylation), which is followed by DNA polymerization that involves the addition of the subsequent 2 to 3 nucleotides to the initiating nucleotide, templated by the 3′ half of the internal bulge sequence.
Detailed studies of the requirements of DHBV protein priming have also demonstrated that specific host factors, including a cellular chaperone complex consisting of heat shock protein 90 (Hsp90), Hsp70, and other cochaperones, are associated with DP and required for DP to bind Dε and, thus, are required for protein priming and pgRNA packaging (25, 27, 29, 45). We and others have reconstituted DHBV protein priming using purified, bacterially expressed DP, purified Dε, and the eukaryotic chaperone proteins (6, 22, 28, 60). Furthermore, we have been able to construct a severely truncated DP, MiniRT2, that lacks part of TP, the spacer, part of RT, and the entire RNase H domain and is able to carry out authentic Dε-dependent protein priming in vitro independent of the host chaperones (42, 81). It has also become clear that both DP and Dε undergo significant conformational changes upon RNP complex formation that are required for protein priming (5, 59, 63, 66).
Extensive studies using cell culture systems replicating HBV have verified that HBV initiates reverse transcription in vivo by a protein priming mechanism similar to that in DHBV. Thus, HBV minus-strand DNA synthesis is templated by the internal bulge sequence of its cognate ε RNA (Hε) located at the 5′ end of its pgRNA and primed by a specific tyrosine residue, Y63 (Y65), homologous to Y96 in DHBV, within the TP domain of HP (14, 25, 35, 43, 45, 52). It has also been shown that minus-strand DNA synthesis in vivo by HBV, like that by DHBV, arrests following the polymerization of 3 to 4 nucleotides using as the template the internal bulge of the ε located at the 5′ end of pgRNA (43, 52, 67, 75). The covalent polymerase-nascent DNA complex is then translocated, likely following conformational changes in the polymerase and ε RNA that dissociate the RNP complex, to the so-called “direct repeat 1” (DR1) (acceptor site) at the 3′ end of pgRNA, where DNA elongation continues. However, biochemical analysis of HBV protein priming has been hampered by the lack of in vitro systems that can faithfully recapitulate this reaction under cell-free conditions, in contrast to DHBV.
The only reported cell-free system for HBV protein priming utilizes a partially purified HP produced in insect cells with a recombinant baculovirus system (38). Upon purification, a small fraction (less than 1%) of the purified HP showed in vitro priming activity using specifically Y63 as the primer, although it was subsequently found that this insect cell-derived HP was not dependent on the authentic Hε to carry out protein priming in vitro (39). Also consistent with the view that Hε may not be the true template for HBV protein priming in vitro in this system was the observation that the purified HP could utilize all 4 different deoxynucleoside triphosphates (dNTPs) to initiate protein priming (albeit with a preference for TTP and dGTP), in contrast to DP, which displays strict specificity for the initiating nucleotide based on the identity of the last residue of the Dε internal bulge serving as the template (75). Urban et al. subsequently purified HP from a different insect cell line and showed that the coexpression of Hε in those insect cells, either in cis as part of the HP mRNA located downstream of the HP coding sequence or expressed in trans as a separate small RNA, stimulated in vitro DNA synthesis by the purified HP (72). However, the DNA products synthesized in vitro in this later study represented mostly, if not exclusively, DNA chain elongation (far beyond the synthesis of the 3- to 4-nucleotide-long DNA oligomer as would occur during protein priming), and little, if any, in vitro priming actually occurred when the physiologically relevant divalent ion Mg2+ was used. It thus remains to be resolved if HBV protein priming in vitro depends on its cognate Hε RNA.
Attempts to reconstitute protein priming with HP purified from bacteria and the same set of eukaryotic chaperones sufficient to reconstitute DHBV protein priming have been unsuccessful, although chaperone reconstitution did activate HP for specific Hε binding (24). In this reconstituted system, HP-Hε association required the Hε internal bulge but not the apical loop (23, 24). In contrast, HBV (as well as DHBV) pgRNA packaging, which also depends on the polymerase-ε interaction, requires both the ε internal bulge and apical loop (1, 2, 14, 19, 32, 36, 48, 51). Furthermore, Hε is present at both the 5′ and 3′ ends of pgRNA (i.e., within the terminal redundancy of pgRNA), but only the 5′ copy is functional in directing pgRNA packaging due to the apparent requirement for a 5′ cap structure and close proximity of Hε to the cap (31). Similarly, only the 5′, but not the 3′, ε RNA serves as the template for protein priming in vivo (43, 52, 75). In the in vitro reconstituted HP-Hε binding assay, there was no apparent requirement for a 5′ cap structure on Hε for HP binding. It is thus evident that HP-Hε binding, per se, is insufficient for HBV pgRNA packaging or protein priming. In particular, whether HBV protein priming requires the 5′ cap on Hε has not been determined, nor has the specific requirement for the Hε internal bulge or apical loop.
In our efforts to establish an Hε-dependent, cell-free HBV priming system, we decided to isolate HP from human cells that are known to support HBV reverse transcription, as they should contain all of the necessary factors for HBV protein priming. We now report the isolation of HP and the HP-Hε complex from human cells that retained Hε binding and protein priming activity in vitro, respectively. Taking advantage of this newly established HBV priming assay, which was strictly Hε dependent, we have also carried out comparative studies on the requirements for Hε in HP binding, protein priming, and pgRNA packaging.
MATERIALS AND METHODS
Plasmids.
pcDNA-3FHP, expressing a 3×FLAG-tagged HP under the human cytomegalovirus (CMV) promoter in the pcDNA3 (Invitrogen) backbone, was constructed by fusing in-frame three copies of the FLAG epitope tag to the N terminus of the HP coding sequence (strain ayw as in pCMV-HBV) (14). Mutant HP sequences in pcDNA-3FHP-YMHD and pcDNA-3FHP-Y63D were derived from pCMV-HBV-Y63D and pCMV-HBV-YMHD (46). For Hε expression in human cells, pCMV-HE was produced by substituting a 0.5-kb fragment of the CMV promoter plus HBV sequence from positions 1801 to 1993 from pCMV-HBV (NdeI to XbaI) for the CMV and T7 promoter in pcDNA3 (NdeI-to-XbaI fragment). pCMV-HE will produce, upon RNA polymerase II (Pol II) transcription in transfected mammalian cells, a capped and polyadenylated HBV RNA initiating at the authentic pgRNA initiation site (position 1816) and containing the 5′ direct repeat 1 (DR1) (positions 1822 to 1832) and Hε sequences (positions 1845 to 1905). Polyadenylation of this Hε RNA can occur, infrequently, at the native HBV polyadenylation [poly(A)] site (ca. nucleotide 1920), which is inherently weak due to its proximity to the 5′ cap and is normally stimulated by additional HBV poly(A)-enhancing sequences present in pgRNA when Pol II encounters the poly(A) signal the 2nd time (12, 53, 54) and are thus missing in pCMV-HE. More frequently, Hε RNA polyadenylation would occur at the strong bovine growth hormone (BGH) poly(A) site on pcDNA3 ca. 220 nucleotides downstream from the HBV poly(A) site. Overlap extension PCR was used to delete the internal bulge and apical loop sequences from Hε in pCMV-HE. For pCMV-HE-dB, nucleotides 1858 to 1863, which compose the internal bulge of Hε, were removed. For pCMV-HE-dL, nucleotides 1875 to 1880, which compose the apical loop of Hε, were removed. QuikChange site-directed mutagenesis (Statagene) was used to change the last (6th) Hε bulge residue (nucleotide 1863 [B6]) in pCMV-HE, which templates the first nucleotide of reverse transcription, from an rC to a rG (B6G) or rA (B6A). This alteration in the Hε will thus change the first nucleotide covalently attached to HP from dGMP to dCMP or TMP. pCI-HE expresses a 5′-elongated Hε-containing RNA that shares the same 5′ HBV pgRNA sequence as that expressed from pCMV-HE but harbors a 208-nucleotide fragment derived from the pCI (Promega) backbone (from the CMV promoter to the T7 promoter) inserted between the initiation site of the CMV promoter in pCI and the normal initiation site of the HBV pgRNA, as described previously (23). Similar to pCMV-HE, there is a strong simian virus 40 poly(A) site present in the pCI vector ca. 370 nucleotides downstream from the native HBV poly(A) site in pCI-HE, leading to the production of two different RNA species depending on the poly(A) site usage. Synthetic templates for in vitro transcription of Hε and Dε have been described previously (22–24). pCIdA-HBV, which expresses all HBV proteins but lacks the 5′ Hε coding sequence (Hε−), has been described before (45). The DP used in all assays was a SUMO (small ubiquitin-like modifier)- and 3×FLAG-tagged MiniRT2 protein. The MiniRT2 coding sequence (22) was fused in frame downstream of the 6×His and SUMO sequence in pSUMO-T7 (Lifesensors). The 3×FLAG epitope tag was inserted between SUMO and the MiniRT2 sequence.
Protein and RNA expression and purification.
HEK293T cells were transfected with pCDNA-3FHP alone, or together with pCMV-HE, pCMV-HE-dB, pCMV-HE-dL, pCMV-HE-B6G, pCMV-HE-B6A, or pCI-HE as described previously (17, 79). Two days after transfection, cells were washed once with phosphate-buffered saline (PBS) and then once more with PBS containing individual protease inhibitors (28 μM E-64, 1 mM phenylmethylsulfonyl fluoride [PMSF], 5 μg/μl leupeptin). Transfected cells were subsequently lysed at 4°C in FLAG lysis buffer (50 mM Tris [pH 7.0], 100 mM NaCl, 50 mM KCl, 10% glycerol, 1% NP-40, 1 mM EDTA) plus 1× complete protease inhibitor cocktail (Roche), 1 mM PMSF, 10 mM β-mercaptoethanol (β-ME), 2 mM dithiothreitol (DTT), and 250 U RNasin Plus RNase inhibitor (Promega) per ml lysis buffer and scraped off the plate. The lysate was centrifuged at 13,000 rpm for 10 min at 4°C. HP was purified from the resulting supernatant using 100 μl protein A/G beads (Pierce) prebound with the M2 anti-FLAG antibody (Sigma) (hereafter referred to as “M2 beads”) per 100-mm dish of transfected cells. Binding of HP to the M2 beads was done at 4°C with rotation overnight. Unbound materials were removed by washing 5 times at 4°C with FLAG lysis buffer containing 10 mM β-ME, 2 mM DTT, individual protease inhibitors, and 10 U/ml RNasin Plus RNase inhibitor. During the final wash, HP bound to the M2 beads was separated into individual single-use aliquots containing ca. 200 ng HP in the wash buffer and stored at −80°C until use. Immunopurified HP and associated factors were eluted from the M2 beads by boiling in 2× sodium dodecyl sulfate (SDS) sample buffer and resolved on an SDS–10% polyacrylamide gel. Bound proteins were visualized by silver staining. Nonspecific binding to the M2 beads was controlled with green fluorescence protein (GFP)-transfected cells in parallel. SUMO-3FMiniRT2 (hereafter referred to as “DP”) was purified using the Escherichia coli expression system by the same procedure as that used for the His-tagged MiniRT2 previously described (8, 22, 41, 42). HP or DP was detected by Western blot analysis using the M2 antibody. Heat shock proteins and other cellular proteins were detected using antibodies against Hsp90 (Sigma AC-16, catalogue no. H1775; 1:2,000), Hsp70 (BB-70; 1:2,000) (9, 73), Hsp60 (Stressgen LK-1, catalogue no. SPA-806; 1:2,000), p60 (Hop; 1:2,000) (9), DDX3 (Biolegend, catalogue no. 639301; 1:500), and p23 (jj3; 1:2,000) (61).
In vitro RNA binding assay.
FLAG lysis buffer from HP purification was removed from aliquots of the HP-bound M2 beads. Binding of the immunopurified HP to the wild-type (WT) or mutant Hε RNA was tested by incubating each aliquot of beads (with ca. 200 ng HP) with 0.5 μg in vitro-transcribed and 32P-labeled ε RNAs in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.05% NP-40) with 1× Complete protease inhibitor cocktail, 2 mM DTT, 1 mM PMSF, and 0.4 U RNasin Plus RNase inhibitor per μl buffer as previously described (24). After 3 h of binding at room temperature with shaking, unbound materials were removed, and the beads were washed in FLAG lysis buffer with 2 mM DTT, individual protease inhibitors, and 10 U RNasin Plus RNase inhibitor per ml buffer. Bound materials were eluted by boiling and resolved on an SDS–15% polyacrylamide gel. The top portion of the gel containing HP was analyzed by Western blotting using the M2 antibody. The bottom portion of the gel containing the 32P-labeled ε RNA was dried and exposed to film to detect the labeled RNA bound to HP.
In vivo RNA binding assay.
To detect HP-Hε interaction in cells, Hε RNA coexpressed and bound to HP in transfected HEK293T cells was coimmunoprecipitated using the M2-bound protein A/G beads as described above for HP purification. RNA bound to HP was extracted from the M2 beads by using 500 μl TRIzol solution (Invitrogen) as instructed by the manufacturer. Total RNA was also extracted from the transfected cells using the same procedure. Formamide gel loading buffer 2 (2×) (Ambion) was added to 1× the purified RNA, and the samples were heated at 65°C for 10 min to denature the RNAs. The RNAs were then resolved on an 8 M urea–6% polyacrylamide gel run in 1× Tris-borate-EDTA (TBE) buffer (89 mM Tris-borate, 2 mM EDTA). The gel was run for 2.5 h at 300 V. The resolved RNAs were then electrotransferred to a Hybond-N nylon membrane (GE Healthcare) for 1.5 h at 300 mA and probed with a 32P-labeled riboprobe specific for the Hε RNA sequence.
In vitro HP priming assay.
FLAG lysis buffer from HP purification was removed from aliquots of the HP-bound beads. TMgNK buffer (20 mM Tris-HCl [pH 7.0], 15 mM NaCl, 10 mM KCl, 4 mM MgCl2) with 1× EDTA-free protease inhibitor cocktail (Roche), 4 mM DTT, 1 mM PMSF, and 1 U RNasin Plus RNase inhibitor per μl buffer was added to the beads (19 μl per aliquot). One microliter of [α-32P]dGTP (10 mCi/ml [3,000 Ci/mmol]; PerkinElmer) was then added, and the reaction mixtures were incubated at 25°C for 4 h with shaking. The beads were then washed in TNK buffer (20 mM Tris-HCl [pH 7], 15 mM NaCl, 10 mM KCl) plus individual protease inhibitors and 10 mM β-ME. The washed beads were then boiled in 2× SDS sample buffer for 10 min. Radiolabeled HP as a result of protein priming was resolved by running the eluate on an SDS–12.5% polyacrylamide gel and detected by autoradiography. DP priming was also performed in TMgNK or TMnNK buffer (the same as TMgNK, except containing 1 mM MnCl2 instead of MgCl2), as described previously (8, 42), and was included as a control. To test the nucleotide specificity of in vitro HBV priming, priming assays were performed using 1 μl [α-32P]dCTP, [α-32P]dATP, [α-32P]TTP, or [α-32P]dGTP (10 mCi/ml [3,000 Ci/mmol]; PerkinElmer). Priming signals were quantified by phosphorimaging.
DNase, pronase, and RNase treatment.
For DNase and pronase treatment, HP-bound beads postpriming were washed once in TNK buffer with individual protease inhibitors and 10 mM β-ME. The beads were then incubated for 30 min at 37°C in 100 μl NEB buffer 3 (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT [pH 7.9]) plus 10 mM CaCl2 with 1 μl (2 U) Turbo DNase (Ambion), and 1 μl (20 μg) pronase or were mock treated (1 μl double-distilled deionized H2O [ddH2O]). Samples were shaken periodically to maintain beads in suspension. Following the digestion, the supernatant was removed, 2× SDS sample buffer was added, and samples were boiled and resolved on an SDS–12.5% polyacrylamide gel. For RNase treatment, HP-bound beads were treated in FLAG lysis buffer with 2 μl (2 μg) DNase-free RNase (Sigma) for 30 min at 37°C. The supernatant was then removed, and priming reactions were conducted with the beads as described above.
5′ Tdp2-mediated release of DNA covalently linked to HP and detection of released DNA.
HBV priming assays were performed as described above, except with 3 μl [α-32P]dGTP or [α-32P]dATP to enhance the priming signal. To allow DNA polymerization, priming assays were performed as described above with the following modifications: 2 μl [α-32P]dGTP was added along with either 10 μM unlabeled dATP, dCTP, and TTP (Invitrogen) or 0.25 μM unlabeled dATP, as indicated. When priming was performed in the presence of two labeled nucleotides, 1.5 μl of each indicated labeled nucleotide was added. Purified DP was bound to the M2-bound protein A/G beads as for HP. Priming reactions with the DP-bound beads were performed as for HP, except with only 1 μl [α-32P]dGTP per reaction and with 0.2% NP-40 (81). For the DP polymerization reaction, 10 μM unlabeled dATP, dCTP, and TTP was also added to the DP priming reactions (42). Primed DP or HP was washed 4 times in TNK buffer with individual protease inhibitors and twice in 1× tyrosyl-DNA phosphodiesterase 2 (Tdp2) buffer (25 mM Tris-HCl [pH 8.0], 130 mM KCl, 1 mM DTT, 10 mM MgCl2) with individual protease inhibitors to remove unincorporated nucleotides. The primed HP or DP was then incubated with 180 ng Tdp2 (Abnova) in 1× Tdp2 buffer with 1× EDTA-free protease inhibitor cocktail for 1.5 h at 37°C with periodic shaking (13, 84). After the Tdp2 treatment, the supernatant containing the released DNA (nucleotide) was collected. Formamide gel loading buffer 2 (2×) was added to 1× to the supernatant, and the mixture was boiled for 5 min. The samples were then loaded onto an 8 M urea–20% polyacrylamide gel (42 cm by 20 cm, high resolution) and run in 1× TBE buffer at 2,500 V for approximately 2 h. The labeled HP or DP bound to the beads after Tdp2 cleavage was also analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) as described above.
S1 nuclease treatment of primed DP and HP.
Priming reactions were conducted with DP and HP bound to M2 beads as described above, with the following modifications. For DP priming with the dGTP- plus-[α-32P]TTP reaction mixture, DP was first incubated in TMnNK buffer for 2 h with 1.5 μM unlabeled dGTP. One microliter of [α-32P]TTP was then added, and the incubation was continued for another 2 h. For HP priming with unlabeled dGTP and [α-32P]dATP, 0.75 μM dGTP and 1 μl [α-32P]dATP were used, and the reaction mixture was incubated for 4 h. After priming, samples were washed once in TNK buffer with the individual protease inhibitors and 10 mM β-ME. Twenty microliters of 1× S1 nuclease buffer (50 mM sodium acetate [pH 4.5], 280 mM NaCl, 4.5 mM ZnSO4) (Promega) was added to beads along with 1× EDTA-free protease inhibitor cocktail, 1 μl (98 U) S1 nuclease (Promega), or 1 μl ddH2O as a mock treatment. Samples were incubated for 45 min at 37°C with shaking. After the S1 treatment, the supernatant was removed, and 2× SDS sample buffer was added to the beads. Samples were boiled for 10 min and loaded onto an SDS–12.5% polyacrylamide gel. For sequential S1 and Tdp2 digestion, DP priming products were first digested with S1 as described above. The S1 nuclease-treated samples were then washed 5 times in 1× Tdp2 buffer with the individual protease inhibitors and treated with Tdp2 as described above. In order to verify protease contamination in the commercial S1 nuclease preparation, HP or DP bound to M2 beads was washed in TNK buffer with individual protease inhibitors and 10 mM β-ME and then incubated with S1 as described above or mock treated (buffer alone). The treated HP or DP was then detected by Western blotting using the M2 antibody.
TdT treatment of DP and HP priming products.
DP bound to the M2 beads was mock primed (with no dNTP) or “cold” primed (with no labeled nucleotides) with 0.25 μM dGTP and 10 μM dATP, dCTP, and TTP in TMnNK buffer. DP was then washed 4 times in 1× TNK buffer with individual protease inhibitors and 10 mM β-ME. HP isolated from HEK293T cells, without any in vitro reaction, and the in vitro “cold”-primed or mock-primed DP were RNase treated as described above to prevent any further priming. Subsequently, HP and DP were washed once in 1× TNK buffer with individual protease inhibitors and 10 mM β-ME and then once in NEB buffer 4 (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM DTT [pH 7.9]) with individual protease inhibitors. HP or DP was then resuspended in 50 μl 1× NEB buffer 4 with 0.5 mM CoCl2, individual protease inhibitors, 1 μl [α-32P]cordycepin triphosphate (10 mCi/ml [5,000 Ci/mmol]; PerkinElmer) and either 50 U terminal deoxynucleotidyl transferase (TdT; New England BioLabs) or an equal volume of ddH2O as a mock treatment. Samples were incubated for 1 h with periodic shaking at 37°C. Unincorporated nucleotides were removed by washing 4 times in 1× TNK buffer with individual protease inhibitors and 10 mM β-ME and then twice in 1× Tdp2 buffer with EDTA-free protease inhibitor cocktail. Tdp2 treatment was performed, and the reaction products were resolved using urea-PAGE and SDS-PAGE as described above.
RNA packaging assay.
HEK293T cells were transfected with pCMV-HE, pCMV-HE-dB, pCMV-HE-dL, pCI-HE, or pcDNA3, together with pCIdA-HBV (Hε−) (45) to complement RNA packaging. Five days after transfection, cells were washed with PBS and lysed using 50 mM Tris [pH 8.0], 1% NP-40, and 1× Complete protease inhibitor cocktail. Micrococcal nuclease-treated lysate was prepared, and RNA packaging was measured by resolving the capsids on a native agarose gel followed by Southern transfer and probing with the Hε-specific riboprobe, as described previously (45, 46). For a positive control, micrococcal nuclease-treated HBV capsids from induced HepAD38 cells were included. The HepAD38 cell line was derived from HepG-2, and upon removal of tetracycline replicates HBV (37).
Apyrase treatment.
[α-32P]dGTP was mock treated (with 1 μl ddH2O) or treated with 1 μg apyrase (Sigma) for 1 min at 25°C in 150 mM Tris (pH 6.0) and 5 mM CaCl2. The treated samples were then resolved on a urea–20% polyacrylamide gel.
HPLC verification of Tdp2-released dGMP from primed DP.
DP was primed in TMgNK buffer and Tdp2 treated as described above. The supernatant was collected after Tdp2 treatment and filtered through an Amicon Ultra 10K filter (Millipore) to remove Tdp2. As standards, unlabeled dGMP (Sigma) and dGTP (Invitrogen) were spiked into each sample and run concurrently as markers. The filtrate and the standards were analyzed by high-performance liquid chromatography (HPLC) using a Waters Breeze HPLC system consisting of a Waters 1525 binary HPLC pump, Waters 2487 dual λ absorbance detector, Waters 717 Plus autosampler, and an INUS β-RAM model 4 radioactive detector equipped with a 250-μl Cerenkov cell. The UV detector was operated at 260 nm, and the window for the radioactivity was 0 to 1,000. The samples were separated using a 4.6-mm by 150-mm 5 μ Gemini-NX C18 column (Phenomenex, Torrance, CA) with a linear gradient from 0 to 20% CH3OH in 50 mM Et3N-HOAc (pH 7.0). The data were analyzed with LauraLite version 3 software.
RESULTS
Expression and purification of HP in association with host factors using human cells.
To express HP in a mammalian system, we chose HEK293T cells as they support high levels of HBV reverse transcription (17, 55, 79) and can be transfected at high efficiency. To facilitate purification, HP was tagged with a triple-FLAG tag, which is fully functional in complementing an HP-defective HBV replicon (78) (data not shown). The tagged HP was purified via immunoaffinity using anti-FLAG (M2)-coupled protein A/G beads. The full-length HP ran at approximately 90 kDa (just above Hsp90) on the SDS-PAGE gel and was associated with several factors previously shown to bind HP or facilitate HP-Hε interaction, including the Hsp90 chaperone components (Hsp90, Hsp70, p60, and p23), Hsp60, and DDX3, as detected by silver staining and Western blot analysis (Fig. 1A and B) (22, 24, 47, 78). Each purified aliquot (bound to the antibody-coupled beads) contained ca. 200 ng of HP (corresponding to 1/5 the purified products from one 100-mm dish of transfected HEK293T cells), as measured by quantitative Western blotting using the purified 3×FLAG-tagged DP from E. coli as standards (data not shown). We hypothesized that coexpression of the Hε RNA along with HP might stimulate its activity and allow it to bind to additional host factors that could facilitate HP functions. Cells were thus cotransfected with constructs expressing HP and Hε. In order to determine if HP was associated with additional cellular factors when HP was coexpressed with Hε, the purified HP, with or without coexpression of Hε, was resolved by SDS-PAGE and analyzed by silver staining in parallel. No additional factors bound to HP in the presence of Hε were apparent compared to in its absence (Fig. 1A, lane 4 versus lane 3). However, it was later found by Northern blotting that only pg amounts of Hε were bound to 200 ng of purified HP (i.e., only pg amounts of the RNP complex were loaded per lane) (see Fig. 3) (data not shown). Thus, if a novel host factor was bound to the HP-Hε complex but not HP alone, it would have been below the limit of detection of the silver stain.
Fig 1.

Purification of HBV polymerase (HP) in association with host factors. The 3×FLAG-tagged HP was purified with the M2 (anti-FLAG [α-FLAG])-bound protein A/G beads from transiently transfected HEK293T cells (A, lane 3, and B, lane 2). HP + Hε represents purification of HP from cells that were coexpressing the Hε RNA (A, lane 4). Control purification in parallel was performed with GFP-transfected cells (A, lane 2; B, lane 1). (A) Purified HP and bound cellular proteins were resolved on an SDS–10% polyacrylamide gel and visualized by silver staining. (B) The identity of HP and selected host proteins was verified by Western blotting using the indicated antibodies (α-). The positions of the protein molecular mass markers (in kDa) are indicated (lane M), as are the various host proteins and the antibody heavy and light chains in panel A. The symbol * in panel B indicates the antibody heavy chain that cross-reacted weakly with anti-Hsp60.
Fig 3.
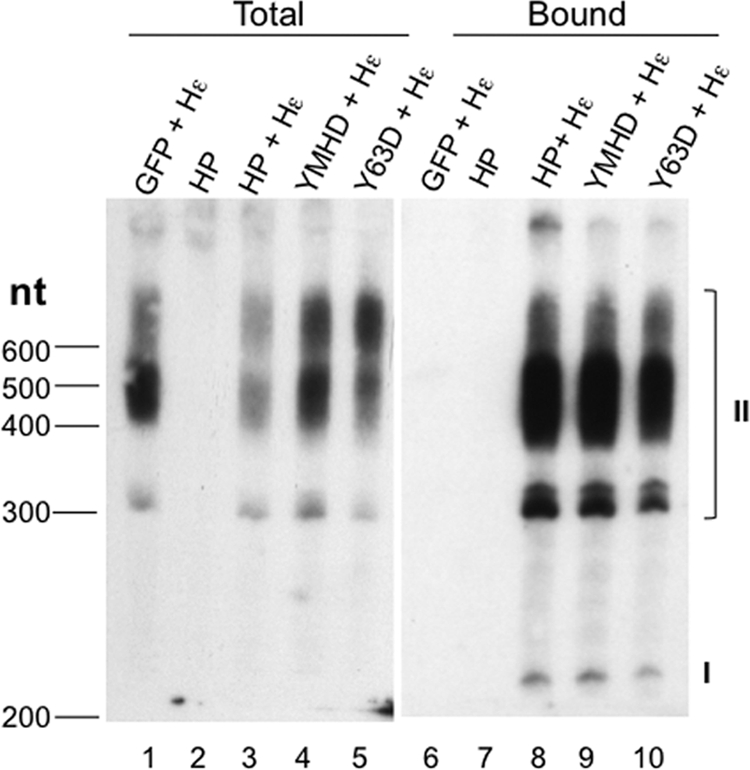
Detection of HP-Hε interaction in vivo. Total RNA was extracted from HEK293T cells transfected with the indicated construct(s) (lanes 1 to 5). HP-bound RNA was extracted from immunoaffinity-purified HP (lanes 6 to 10). The purified RNAs were resolved on an 8 M urea–6% polyacrylamide gel, electrotransferred to membrane, and probed with a 32P-labeled riboprobe specific for the Hε RNA sequence. The capped, polyadenylated Hε species are indicated (I and II): I, ca. 200 nucleotides long, representing polyadenylation at the weak native HBV poly(A) site that is ca. 100 nucleotides downstream of the 5′ cap; II, from 300 to 600 nucleotides long, representing polyadenylation at the strong bovine growth hormone (BGH) poly(A) site ca. 220 nucleotides downstream from the HBV poly(A) site on the pcDNA3 vector (see also Materials and Methods). The sizes (in nucleotides [nt]) of the marker (denatured DNA) are indicated on the left.
HP was able to bind Hε in vitro and in vivo.
Because HP priming and RNA packaging are dependent on HP association with Hε, an in vitro Hε binding assay was developed to test the RNA binding activity of the purified HP. Labeled Hε and Dε RNAs were prepared by in vitro transcription and incubated with purified HP bound to the beads. HP and any bound ε RNA were resolved by SDS-PAGE, as previously described (22). The labeled RNAs were detected by autoradiography (Fig. 2A) and HP by Western blotting using anti-FLAG (Fig. 2B). This in vitro RNA binding assay showed that the purified HP was able to specifically associate with Hε, and not the heterologous Dε. Binding assays performed using mutant Hε RNA constructs further showed that the requirements for Hε binding to the HP purified from mammalian cells were similar to those defined previously using truncated HP proteins purified from bacteria and reconstituted with the Hsp90 chaperone complex in vitro (23, 24), including the necessity of the Hε internal bulge and the dispensability of its apical loop (Fig. 2A).
Fig 2.
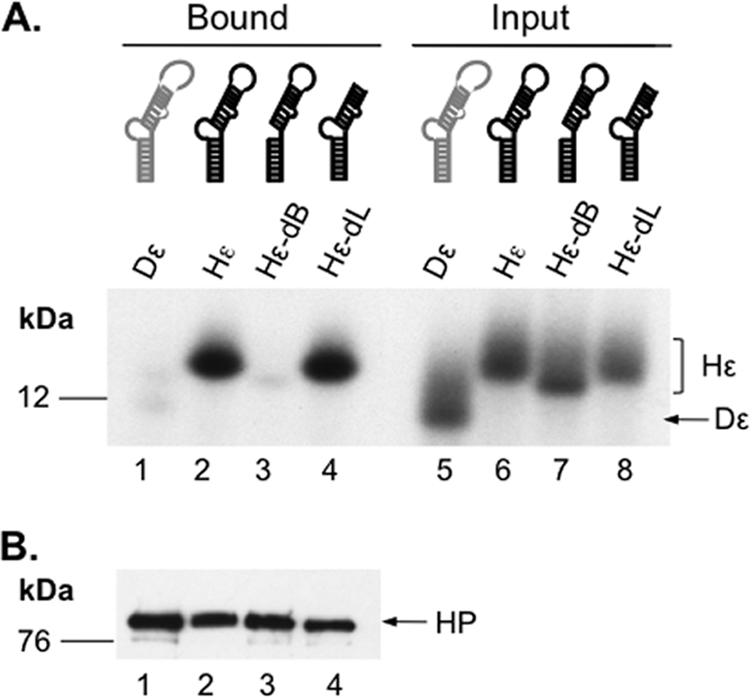
Detection of HP-Hε interaction in vitro. Immunoaffinity-purified HP was incubated with 32P-labeled, in vitro-transcribed WT or mutant Hε or Dε RNA. After removing unbound RNA, the bound RNA and HP were resolved on an SDS–15% polyacrylamide gel. (A) The bottom portion of the gel, which contained the 32P-labeled ε RNA, was dried and analyzed by autoradiography (lanes 1 to 4). Input representing 1% of the amount of the indicated ε RNA added to each binding reaction mixture was also analyzed in parallel (lanes 5 to 8). (B) The top portion of the gel containing HP was analyzed by Western blotting for HP in the binding reactions using the M2 anti-FLAG antibody. The Hε and Dε RNAs and HP are indicated, as are the positions of the protein molecular mass markers (in kDa).
When HP and Hε were coexpressed in cells, it was possible that HP would be able to associate with Hε inside the cells. To detect Hε bound to HP in cells, RNA was extracted from the purified HP coexpressed with Hε and was resolved by urea-PAGE and detected by Northern blotting. Indeed, HP was found to bind the coexpressed Hε (Fig. 3, lane 8). The identification of the two different species of Hε RNA (labeled as I and II in Fig. 3), in particular, the presence of the pcDNA3-derived vector sequences in species II but not I, was confirmed by probing with the vector sequences between the HBV poly(A) site and downstream, vector-derived BGH poly(A) site (see Materials and Methods) (data not shown). It was estimated, based on quantification using RNA standards, that each purified HP pellet aliquot (ca. 200 ng HP) contained ca. 200 pg of bound Hε (data not shown). The HP YMHD or Y63D mutation, which abolishes HP DNA polymerase or primer function, respectively, did not affect HP binding to Hε (Fig. 3, lanes 9 and 10), suggesting that Hε binding was not dependent on HP enzymatic activity or primer function, similar to what has been found for the DHBV polymerase (49, 66, 77).
In vitro Hε-dependent protein priming activity of purified HP.
Because the purified HP was associated with the cellular chaperone proteins known to facilitate polymerase function in ε RNA binding (in DHBV and HBV) (22–24, 27, 29) and protein priming (in DHBV) (22, 27, 28) and was active in binding Hε in vitro and in vivo, we were interested in determining if the purified HP was indeed active in protein priming in vitro under conditions similar to those that support DP priming. When HP purified from cells that were also coexpressing Hε was tested for in vitro priming activity in the presence of [α-32P]dGTP, 32P-labeled HP, presumably as a result of the covalent linkage of the 32P-labeled dGMP residue to HP (verified below) and thus indicative of in vitro protein priming, was detected (Fig. 4A, lane 4). However, without coexpression of Hε, the purified HP was inactive in priming in vitro (Fig. 4A, lane 3), indicating that Hε was required for priming. Since the in vivo RNA binding results above indicated that most of the purified HP had no Hε bound even in the cotransfection setting (Fig. 3), the small amounts (ca. 100 pg loaded in Fig. 4A, lane 4) of purified HP bound to Hε were highly competent in the priming assay, being as active (95% or more) as the purified DHBV MiniRT2 (DP; 3 ng) used as a control (Fig. 4A, lane 1), whose specific activity (in the presence of Mn2+) was estimated to be ca. 50% (28, 42, 81). As expected, the YMHD HP mutant, lacking the polymerase catalytic active site, was completely inactive in priming (Fig. 4A, lane 5), as was the Y63D mutant lacking the primer Y63 residue (Fig. 4A, lane 6). Sensitivity to pronase, but not DNase, verified that the priming signal detected was indeed a labeled protein (Fig. 4B). Consistent with the template for protein priming in vitro being RNA, the HP priming reaction was eliminated by RNase pretreatment (Fig. 4C).
Fig 4.

Detection of in vitro protein priming by purified HP. Priming reactions were performed by incubating immunoaffinity-purified HP with TMgNK buffer and [α-32P]dGTP (A to C) or another labeled nucleotide as indicated (D and E). After priming, the beads were washed, and the labeled HP was resolved on an SDS–12.5% polyacrylamide gel. A priming reaction was also performed with the DHBV MiniRT2 (DP) in TMnNK buffer and resolved on the same gel for comparison (A, lane 1). Labeled HP and DP priming products were detected by autoradiography after SDS-PAGE. (A) In vitro priming reactions with WT (lanes 3 and 4) or mutant (lanes 5 and 6) HP with (lanes 4 to 6) or without Hε (lane 3) coexpression in cells. GFP + Hε (lane 2) represents priming using the control purification product from cells cotransfected with GFP and the Hε-expressing plasmid. (B) After protein priming, primed HP was untreated (−; lane 1) or treated with DNase I (D; lane 2) or pronase (P; lane 3) before analysis by SDS-PAGE. (C) The purified HP was mock treated (lane 1) or RNase treated (lane 2) before being used in protein priming. Labeled HP was detected by autoradiography after SDS-PAGE (top), and HP protein levels were measured by Western blotting using the anti-FLAG (α-Flag) antibody (bottom). (D) HP purified either with (lanes 5 to 8) or without (lanes 1 to 4) the coexpressed Hε was assayed for priming activity in the presence of [α-32P]dGTP (G; lanes 2 and 6), [α-32P]TTP (T; lanes 1 and 5), [α-32P]dCTP (C; lanes 3 and 7), or [α-32P]dATP (A; lanes 4 and 8). Priming signals were quantified via phosphorimaging, normalized to the highest signal (dGTP priming, set as 100%), and denoted below the lane numbers (as a percentage of dGTP signal). The labeled HP and DP priming products are indicated. (E) Shown on the top is a schematic diagram of the mutant Hε RNAs, with the last 4 nucleotides of the internal bulge and part of the upper stem, including its bottom A-U base pair. In Hε-B6G (left), the last (6th) bulge residue (i.e., B6) was changed (from rC in the WT) to rG and in Hε-B6A (right), the same residue was changed to rA. The mutated residues are highlighted in bold. Shown at the bottom are priming products obtained with the mutant Hε RNAs. The Hε-B6G (lanes 1 and 2) or -B6A (lanes 3 and 4) mutant was coexpressed with HP, and the purified HP-Hε complex was assayed for protein priming in vitro in the presence of the indicated 32P-labeled nucleotide. The labeled HP priming products are indicated, as is the position of the protein molecular mass marker (in kDa).
As an initial test for the template specificity during the in vitro HP priming reaction, different labeled dNTPs were used individually in the priming assay. dGTP was by far the preferred nucleotide utilized for priming (Fig. 4D, lane 6). Much less priming was detected with TTP and dATP, and no signal was detected with dCTP (Fig. 4D). As will be described below, the dGTP priming signal most likely represented initiation of priming (i.e., covalent linkage of the dGMP residue to HP) templated by the last nucleotide (an rC residue) of the internal bulge of Hε. The weak TTP and dATP priming signals could represent polymerization of initiated species (i.e., HP already covalently attached to dGMP) that accumulated in the cell (see below) or possibly low levels of initiation at alternative sites or misincorporation in vitro. Again, without coexpression of Hε in vivo, no priming signal was detected with any dNTP in vitro (Fig. 4D, lanes 1 to 4).
To verify that initiation of protein priming was occurring with the purified HP, and specifically from the last internal bulge (the 6th residue of the bulge or B6) residue of Hε, we changed that single residue of Hε from rC to rG (B6G) or rA (B6A) (Fig. 4E) such that the first nucleotide that would be covalently linked to Y63 of HP during protein priming would be dCMP or TMP, respectively, instead of dGMP. Indeed, when HP coexpressed with the bulge-mutated Hε was purified, it lost any labeling when priming was conducted with [α-32P]dGTP, and instead was labeled strongly with [α-32P]dCTP (when Hε-B6G was cotransfected) or [α-32P]TTP (when Hε-B6A was cotransfected), just as predicted (Fig. 4E).
The HP-DNA linkage formed during in vitro priming was sensitive to Tdp2 cleavage: characterization of in vitro HP priming products.
In order to analyze further the HP-DNA linkage and DNA product(s) linked to HP as a result of in vitro protein priming, the labeled HP priming products were treated with Tdp2, a recently identified enzyme that is able to specifically break the phosphotyrosyl linkage between a tyrosine residue in a protein and the 5′ phosphate of a covalently attached DNA (13, 84). Since the same linkage would be found in the primed HP between Y63 of HP and the covalently attached nucleotide or DNA, treatment of the priming reaction products should decrease the labeled HP signal due to the release of the labeled nucleotide or DNA from HP (as detected by SDS-PAGE), and furthermore, the released and labeled nucleotide or DNA fragments can be detected by urea-PAGE. This method would also allow the different DNA (nucleotide) species attached to HP to be visualized simultaneously. As a control for the Tdp2 cleavage reaction, the DP priming initiation product (a single dGMP residue attached to Y96 of DP, when priming was carried out in the presence of Mg2+) was treated with Tdp2. We indeed observed the anticipated decrease in the labeled DP signal by SDS-PAGE (Fig. 5A, lane 2) as well as the Tdp2-released, labeled dGMP by urea-PAGE (Fig. 5B, lane 6). The identification of dGMP was confirmed by treatment of [α-32P]dGTP with apyrase (Fig. 5C), which converts nucleotide triphosphates to nucleotide monophosphates (34, 74), and by HPLC analysis of the Tdp2-released nucleotides along with nucleotide standards (Fig. 5D). The decrease in DP (and also HP, as described below) priming signal by Tdp2 was only partial, likely a result of incomplete Tdp2 cleavage of the polymerase-dGMP (or DNA oligomer) linkage and possibly suggesting that a single nucleotide or very short DNA oligomers attached to a protein may not be an optimal substrate for Tdp2. As we reported recently (8, 42), the DHBV MiniRT2 protein (i.e., “DP” as referred to here) is able to carry out polymerization in the presence of Mn2+ (but not Mg2+) to generate short DNA oligomers attached to DP. When the DP Mn2+ priming products were treated with Tdp2, we again observed the expected release of the DNA oligomers from DP that could be readily visualized after urea-PAGE (Fig. 5B, lane 8). Although the DP priming reaction is expected to generate DNA oligomers 3 to 4 nucleotides long under in vivo conditions (75, 76), we detected Tdp2-released DNA fragments up to 10 nucleotides in length. It is possible that polymerase stuttering, as observed earlier during HP priming in vivo (43), may have led to the synthesis of DNA oligomers longer than 4 nucleotides. It is also possible that the reduced template specificity in the Mn2+ condition used here allowed DP to extend the DNA further along the Dε template in the in vitro priming reaction (8, 42).
Fig 5.

Analysis of DP protein priming products by Tdp2 cleavage of the phosphotyrosyl bond between DNA and protein. Purified DP bound to M2 antibody affinity beads was assayed for protein priming. Free nucleotides were then removed with extensive washing, and priming products were mock treated (−) or treated with Tdp2 (+) to cleave the phosphotyrosyl-DNA linkages between DP and the linked nucleotides or DNA oligomers. The supernatant, which contained the released nucleotides/DNA, was collected and resolved on a urea–20% polyacrylamide gel (B). The beads, which contained the primed DP, were processed for SDS-PAGE to visualize the labeled DP (A). Radiolabeled proteins and nucleotides/DNA were detected by autoradiography. Priming was done in the presence of TMgNK buffer and [α-32P]dGTP (A, lanes 1 and 2; B, lanes 5 and 6) or TMnNK buffer and [α-32P]dGTP plus the unlabeled dCTP, TTP, and dATP (A, lanes 3 and 4; B, lanes 7 and 8). (C) [α-32P]dGTP stock was mock (lane 4) or apyrase treated (lane 5). The DP priming product obtained in TMgNK buffer and [α-32P]dGTP was either mock treated (lane 2) or Tdp2 treated (lane 3), which released dGMP from the DP-dGMP phosphotyrosyl linkage. Samples were resolved on a urea–20% polyacrylamide gel. The positions of 32P-labeled 10-nucleotide marker (Invitrogen) (B) and DNA oligomers (dTG, dTGA, and dTGAA in panels B and C) are indicated, as are the positions of dGTP and dGMP. (D) HPLC analysis of dGTP and dGMP. (Panel 1) UV (A260) detection showing retention times of unlabeled dGMP and dGTP. (Panel 2) Detection of 32P radioactivity from mock-treated DP priming products (−Tdp2), showing the absence of dGMP and the presence of residual dGTP substrate input. (Panel 3) Detection of 32P radioactivity from Tdp2-treated DP priming products (+Tdp2), showing the presence of dGMP released by Tdp2 from DP and again some residual dGTP substrate input. The positions of dGMP and dGTP are indicated.
Similarly, the HP in vitro priming products were found to be susceptible to Tdp2 cleavage, as represented by a decrease in labeled HP after treatment upon SDS-PAGE analysis (Fig. 6A). Again, as for DP, Tdp2 only partially removed the HP priming signal, presumably due to the limited length of the DNA (or single nucleotide) covalently linked to the protein. However, urea-PAGE analysis revealed a prominent background band appearing in both mock- and Tdp2-treated samples where dGMP would migrate (Fig. 6B, lanes 3 and 4, C, and D). This background signal could not be removed by extensive washing (data not shown). The background band was absent in the DHBV priming reactions conducted in parallel with DP-bound M2 beads (Fig. 5B, lanes 5 to 8). The origin of this dGMP background signal (i.e., Tdp2 independent) in the HP reaction but not DP reaction remained unclear but might be related to the fact that DP was purified from bacteria while HP was purified from human cells and associated with a number of host proteins (the Hsp90 complex, Hsp60, DDX3, and possibly other factors) (Fig. 1). It was possible that one or more these factors might bind tightly, but noncovalently, to dGMP, which was present in the [α-32P]dGTP stock (Fig. 5C, lane 4) and was released during the incubation period in the Tdp2 buffer (but requiring no actual Tdp2 cleavage). We also considered the possibility that the apparent Tdp2-independent dGMP release from HP might be due to HP self-cleavage of the priming products (i.e., a reversal of protein priming). However, this possibility was eliminated by the detection of similar dGMP background signals with the priming-incompetent YMHD mutant (data not shown).
Fig 6.

Analysis of HP protein priming products by Tdp2 cleavage of the phosphotyrosyl bond between DNA and protein. Purified HP bound to M2 antibody affinity beads was assayed for protein priming. Free nucleotides were then removed with extensive washing, and priming products were mock treated (−) or treated with Tdp2 (+) to cleave the phosphotyrosyl-DNA linkages between HP and the linked nucleotides or DNA oligomers. The supernatant, which contained the released nucleotides/DNA, was collected and resolved on a urea–20% polyacrylamide gel (B to D). The beads, which contained the primed HP, were processed for SDS-PAGE to visualize the labeled HP (A). Radiolabeled proteins and nucleotides/DNA were detected by autoradiography. Priming was done in the presence of [α-32P]dGTP (A, lanes 1 and 2; B, lanes 3 and 4), [α-32P]dATP (A, lanes 3 and 4; B, lanes 5 and 6), [α-32P]dGTP plus [α-32P]dATP (A, lanes 5 and 6; B, lanes 1 and 2; D, lanes 1 and 2), [α-32P]dGTP plus [α-32P]dTTP (D, lanes 3 and 4), [α-32P]dGTP plus unlabeled dATP (C, lanes 3 and 4), or the other three unlabeled dNTPs (C, lanes 5 and 6; denoted as N). Unlabeled dNTPs are denoted with parentheses in panel C. The positions of the 32P-labeled 10-nucleotide marker (Invitrogen) (C) and DNA oligomers (dGA, dGAA, and dGAAA in panels B to D and dTG, dTGA, and dTGAA in panel C) are indicated, as are the positions of dGTP and dGMP. (E) The top diagram depicts the HP priming product, i.e., the dGAA DNA oligomer that is covalently attached to HP via Y63 and templated by the last three nucleotides (rUUC) of the internal bulge of Hε. Part of the upper stem of Hε, with its bottom A-U base pair, is also shown. The phosphotyrosyl protein-DNA linkage is specifically cleaved by Tdp2 as shown. The bottom diagram depicts DNA strand elongation following primer transfer, whereby the HP-dGAA complex is translocated from Hε to DR1, and the dGAA oligomer is further extended, potentially up to dGAAAAA in the presence of only dGTP and dATP. The putative dGAAAA or dGAAAAA product released by Tdp2 from HP is also denoted by “GAAAA(?)” in panel D.
The Tdp2-independent dGMP background on urea-PAGE rendered it impossible to detect, reliably, the anticipated Tdp2-dependent release of dGMP from the initiated HP (i.e., the HP-dGMP covalent complex) primed in the presence of labeled dGTP alone (Fig. 6B, lanes 3 and 4). However, the absence of any detectable polymerization products (DNA oligomers 2 nucleotides long or longer, which would have migrated above dGMP) provided indirect evidence that the strong HP signal detected on SDS-PAGE was due to initiation only, and no polymerization occurred when only [α-32P]dGTP was provided. The altered, and predictable, nucleotide specificity in priming intiation as a result of Hε bulge mutations (B6G and B6A) shown above (Fig. 4E) also supported the notion that in vitro HP priming initiated from the last position of the Hε internal bulge. In order to demonstrate further that initiation of priming was indeed occurring with the wild-type (WT) Hε, we decided to assay if we could elongate the nascent DNA attached to HP, thus allowing for its detection by urea-PAGE following Tdp2 cleavage, by providing additional dNTP substrates. Based on the predicted template specificity (i.e., the Hε internal bulge, 5′ rUUC 3′) for HP priming, the next nucleotide(s) that should be added after dGMP would be one or two dAMP residues. After priming with dGTP and dATP (both 32P labeled), we could indeed detect Tdp2-dependent release from the primed HP of DNA oligomers (polymerization products) comigrating with the 3 (dGAA sequence) and 4 (dGAAA sequence) nucleotide markers (Fig. 6B, lane 2). The release of the prominent dGAA oligomer by Tdp2 cleavage was fully consistent with the authentic HP priming mechanism, whereby priming initiation and DNA polymerization are templated by the 3′ half of the internal bulge of Hε, and a Y residue (i.e., Y63) serves as the primer for protein priming. The detection of longer DNA oligomers migrating above dGAA (including the 4-mer dGAAA and even longer, less abundant species) (Fig. 6B to D) suggested that some primer translocation might have occurred in the in vitro reaction, whereby the HP-dGAA complex was transferred to the DR1 sequence (equivalent to the 5′ DR1 in the full-length pgRNA), which is present upstream of Hε in the coexpressed RNA (see Materials and Methods for details). This would allow further elongation of the DNA oligomers to produce dGAAA, dGAAAA, and dGAAAAA in the presence of only dGTP and dATP substrates (Fig. 6E). Without primer translocation, further DNA polymerization along the internal bulge of Hε would be impossible in the presence of only dGTP and dATP (without a mismatch) since the next nucleotide to be added after dGAA would be a dCMP.
The fact that no 2-mer species (i.e., dGA) was detected (Fig. 6B) was likely due to rapid polymerization after priming initiation, producing the observed predominant 3-mer species. Priming with 32P-labeled dGTP and unlabeled dATP verified that the Tdp2-released DNAs included (the only labeled) dGMP (Fig. 6C, lane 4). A priming reaction with 32P-labeled dGTP and all three other unlabeled nucleotides did not produce additional detectable extension of the nascent DNA (Fig. 6C, lane 6), indicating the absence of dCMP or TMP incorporation during priming in vitro and the presence of no DNA elongation beyond dGAA at the Hε bulge (which would have added a dCMP residue templated by rG, the 3rd position of the Hε bulge). Since dGMP was likely the 1st nucleotide attached to HP and there was no 2-mer (dGG) produced with dGTP alone, the 3-mer produced with dGTP plus dATP thus most likely had the sequence of dGAA, further supporting the notion that HP protein priming in vitro was templated by the Hε bulge template sequence (UUC) (Fig. 6E). Moreover, the Tdp2-released 3-mer comigrated with the dGAA marker, but not the dTGA marker (Fig. 6C). No labeled DNA species was released by Tdp2 when 32P-labeled dGTP and TTP were added together (i.e., other than the presumed dGMP that would be released but masked by the background dGMP signal, as discussed above) (Fig. 6D), and none of the released species that were detected with all four dNTPs added comigrated with the dTG, dTGA, or dTGAA markers (Fig. 6C), further indicating that TMP was not part of the priming products. These results thus indicated that HBV priming was unlikely to be initiated with TMP (i.e., templated by the rA residue at the start of the upper stem of Hε) (Fig. 6E) (see Discussion later).
Initiation of HP priming in vitro as demonstrated by S1 nuclease digestion.
To further verify that initiation of protein priming (i.e., the covalent attachment of the first nucleotide, dGMP, to HP) was occurring in the in vitro priming reaction, priming products were treated with S1 nuclease, which degrades single-stranded DNA but is unable to break the phosphotyrosyl linkage between protein and DNA. S1 nuclease resistance in this context thus signifies a direct linkage of a nucleotide to a protein. To verify the S1 cleavage specificity, DP priming reactions were conducted with [α-32P]dGTP and the other 3 unlabeled dNTPs (with Mn2+) to allow for DNA polymerization. The polymerization products attached to DP were treated sequentially with S1 followed by Tdp2, which would release the S1 digestion products from DP to be visualized by urea-PAGE. As expected, the DP-linked DNA ladder components (heterogeneous polymerization products) were efficiently degraded by S1 down to a 2-mer and mostly to a single nucleotide, thus directly confirming that S1 could degrade the single-stranded DNA to a single nucleotide but did not break the dGMP-DP linkage (Fig. 7A and F). Also, when the DHBV priming initiation product (DP-dGMP) generated in the presence of dGTP alone (Fig. 5B) was treated with S1, no significant decrease in priming signal was observed (Fig. 7B, lane 2, C, lane 2, and D), again indicating that S1 did not break the phosphotyrosyl linkage between the labeled dGMP and DP. The apparent slight decrease (ca. 20 to 30%) in DP labeling was attributed to protease contamination in the S1 nuclease, which generated heterogeneous signals below the intact DP on SDS-PAGE detectible by autoradiography (following protein priming) or by Western blotting (Fig. 7B, lane 2, C, lane 2, and E, lane 2). When the DP priming reaction was initiated with unlabeled dGTP first and then [α-32P]TTP was added to extend the dGMP primer (under Mn2+ priming conditions), S1 nuclease treatment removed most (80 to 90%) of the label (i.e., labeled TMP) from DP (Fig. 7B, lanes 3 and 4, and D), as expected from the cleavage of the internucleotide (dGMP-TMP) bond by S1. Residual signal was likely due to incomplete S1 digestion (Fig. 7B).
Fig 7.

Differentiation of priming initiation from DNA polymerization by S1 nuclease digestion. (A) Protein priming was conducted with DP bound to M2 affinity beads in TMnNK buffer, in the presence of [α-32P]dGTP and unlabeled dCTP, dATP, and TTP. Priming products were either mock treated (−; lanes 5 and 6) or S1 treated (+; lanes 7 and 8), followed by mock treatment (−; lanes 5 and 7) or Tdp2 treatment (+; lanes 6 and 8), as described in Materials and Methods. Released nucleotides or DNAs were resolved by urea-PAGE and detected by autoradiography. The 10-nucleotide marker, the dTG, dTGA, and dTGAA DNA oligomers, and dGMP positions are indicated, as is the priming initiation product (I; i.e., the single dGMP residue released by Tdp2 from DP) or polymerization products (P; DNA polymerization from the first dGMP residue). (B) Protein priming was performed with DP in TMnNK buffer with [α-32P]dGTP (lanes 1 and 2) or with unlabeled dGTP (unlabled dNTP denoted by parentheses) followed by the addition of [α-32P]TTP to extend the unlabeled DP-dGMP initiation product (lanes 3 and 4). The priming products were then mock treated (−; lanes 1 and 3) or treated with S1 nuclease (+; lanes 2 and 4), resolved by SDS-PAGE, and detected by autoradiography. (C) Priming was performed with DP (lanes 1 and 2) or HP (lanes 3 to 6) in TMgNK buffer with [α-32P]dGTP (lanes 1 to 4) or with unlabeled dGTP first followed by addition of [α-32P]dATP to extend the unlabeled HP-dGMP initiation product (lanes 5 and 6). The priming products were either mock treated (−; lanes 1, 3, and 5) or S1 treated (+; lanes 2, 4, and 6), resolved by SDS-PAGE, and detected by autoradiography. (D) The percent decreases in DP and HP priming signals as a result of S1 nuclease treatment are represented. Mock-treated DP initiation reaction in the presence of [α-32P]dGTP alone, with either TMnNK or TMgNK buffer, was set as 100%, and the other reaction conditions, as explained in panels B and C, were normalized to this. The decrease in priming signal due to proteolytic degradation (unrelated to S1 nuclease cleavage of internucleotide linkages) was subtracted from the calculations. (E) DP or HP was incubated with or without S1 nuclease as described above. Protease degradation was monitored by Western blotting using the M2 anti-Flag antibody. HC, antibody heavy chain. The symbol * in panels B, C, and E represents DP and HP degradation products caused by contaminating protease activity in S1. Note that only some proteolytic degradation products detected by the Western blot (E) appeared to match the 32P-labeled degradation products (B and C) since the labeled products must have contained the priming site(s), whereas the Western blot detected only fragments containing the N-terminal FLAG tag. Also, some labeled degradation products might be present at such low levels that they were undetectable by Western blotting. Note also that the appearance of the proteolytic degradation products was accompanied by the decrease of the full-length HP or DP in panels B, C, and E. (F) The diagram depicts the cleavage of the internucleotide linkages, but not the HP-dGMP linkage, by S1.
Similarly, the HP priming initiation product (the single dGMP attached to HP) was found to be S1 resistant after the contaminating proteolytic degradation was taken into account (Fig. 7C, lane 4, E, lane 4, and D). In contrast, when the HP priming polymerization product was created with the unlabeled dGTP and [α-32P]dATP (i.e., HP-dGMP-[32P]dAMP2–3) and was treated with S1, most (91%) of the label was eliminated due to the removal of the 2nd (and subsequent) labeled dAMP from the polymerized DNA oligomers (Fig. 7C, lane 6, and D). The strong signal obtained with [α-32P]dATP and unlabeled dGTP—even stronger than that with [α-32P]dGTP alone (Fig. 7C, lane 5 versus lane 3)—in contrast with the very weak signal obtained with [α-32P]dATP alone (Fig. 6A, lane 3 versus lane 1) also indicated that dAMP was mostly added after the dGMP linkage to HP, again supporting the identification of the DNA polymerization product as dGAn.
Detection of in vivo HP priming products.
As HP was shown to bind Hε in the transfected cells (Fig. 3), we decided to test if protein priming might also be occurring in HEK293T cells when HP and Hε were coexpressed. To detect these putative HP priming products (i.e., HP covalently attached to short DNA oligomers) formed in the cells, we took advantage of the ability of TdT to extend from short DNA oligomers (but not from protein). The same approach was previously utilized to visualize short DNA oligomers covalently attached to a DNA repair protein (44). To verify that TdT extended from DNA oligomers but not from protein, control DP polymerization products were produced by incubating DP with all 4 unlabeled dNTPs. In parallel, a mock priming reaction was performed without any nucleotide substrate. After priming, RNase treatment was performed to degrade Dε so as to prevent any further protein priming (Fig. 4C) after the labeled nucleotide was added, in order to ensure that any labeled DNA products formed during the subsequent TdT reaction were indeed generated only by TdT but not DP. Following DP priming and RNase treatment, the priming products were incubated with TdT and [α-32P]cordycepin triphosphate, a dATP analog chain terminator (71). The advantage of using this nucleotide analog is that the exact size of in-cell primed HP DNA oligomer length can be determined (i.e., one nucleotide shorter than the TdT product). Indeed, the DP priming (polymerization) products that were labeled (and extended by one nucleotide) by TdT could be detected (Fig. 8A, lane 5), while no labeling was detected on DP itself in the mock priming reaction with no nucleotides (Fig. 8A, lane 3). Also, no labeled product was detected if TdT was omitted, even if DP priming products were formed (Fig. 8A, lane 1), confirming that DP itself did not incorporate the labeled cordycepin monophosphate.
Fig 8.

Detection of in vivo protein priming by TdT-mediated labeling of HP-linked DNA oliogomers. DP bound to anti-FLAG beads was either mock primed (without dNTPs [−dNTPs]; lanes 3 and 4) or primed in the presence of all 4 unlabeled dNTPs (+dNTPs; lanes 1, 2, 5, and 6). DP was then washed to remove the free nucleotides. WT (A, lanes 7 to 12; B, lanes 7, 8, and 11 to 14) or mutant (YMHD) (A, lanes 13 and 14; B, lanes 9 and 10) HP, with (A, lanes 7, 8, and 11 to 14; B, lanes 7 to 10, 13, and 14) or without (A, lanes 9 and 10; B, lanes 11 and 12) Hε coexpression, was immunoaffinity purified from transfected cells. To prevent further priming in vitro in the presence of the labeled nucleotide, primed DP and HP were treated with RNase to degrade the Hε and Dε RNA template and inactivate priming activity (Fig. 4C). Subsequently, DP and HP were either mock treated (lanes 1, 2, 7, and 8) or TdT treated (lanes 3 to 6 and 9 to 14) in the presence of [α-32P]cordycepin triphosphate to extend (by one nucleotide only) and label the DP/HP-linked DNAs. A portion of the TdT reaction mixtures were further treated with Tdp2 to release the TdT-labeled DNAs (+; even-numbered lanes) or mock treated (−; odd-numbered lanes). TdT and Tdp2 reaction products were then analyzed by SDS-PAGE (A) or urea-PAGE (B) and detected by autoradiography. The symbol * in panel A represents TdT labeling of an unidentified substrate unrelated to HP or DP but which appeared to comigrate with DP on SDS-PAGE. Also indicated are the positions of the protein molecular mass markers (kDa) (A) and the dGA, dGAA, dGAAA, and 10-nucleotide DNA marker (B).
Tdp2 treatment of the TdT-labeled DP eliminated most of the DP signal indicating that the TdT-extended DNA oligomer(s) was covalently linked to DP via a tyrosine residue (i.e., Y96) (Fig. 8A, lane 6). Furthermore, urea-PAGE analysis of the Tdp2-released DNA from DP detected labeled DNA oligomers ca. 5 to 10 nucleotides long (Fig. 8B, lane 6), consistent with the DP priming (polymerization) products being mostly 4 to 9 nucleotides long (Fig. 5B and 7A). When HP expressed and purified with or without Hε was treated with TdT, only the HP expressed in the presence of Hε, but not in its absence, showed detectable labeling, both as a labeled HP species on SDS-PAGE that was sensitive to Tdp2 treatment (Fig. 8A, lanes 9 to 12) and as the Tdp2-released DNA oligomers ca. 5 to 7 nucleotides in length on urea-PAGE (Fig. 8B, lanes 11 to 14). As expected, the polymerase-inactive HP mutant YMHD showed no in vivo priming even in the presence of the coexpressed Hε (Fig. 8A, lanes 13 and 14, and B, lanes 9 and 10). The control reaction where TdT was omitted verified the labeling of HP was not due to HP priming activity in vitro (which was inactivated by the RNase treatment before addition of the labeled nucleotide analog) (Fig. 8A and B, lanes 7 and 8). These results thus indicated that some HP was indeed able to carry out protein priming in cells when coexpressed with Hε to synthesize DNA oligomers ca. 4 to 6 nucleotides long that were covalently attached to HP via a tyrosine residue (Y63). The presence of even shorter DNA oligomers attached to HP could not be excluded as TdT may not extend DNAs less than 3 nucleotides long (33).
Hε requirements for protein priming in vitro mirrored those for RNA packaging in vivo.
To begin to analyze viral and host determinants required for HBV priming using our newly developed in vitro HP priming system, different Hε mutants were coexpressed with HP. HP or HP-Hε RNP complexes were purified, depending on the capacity of the Hε mutants to associate with HP in cells. The in vivo RNA binding assay showed that the dL Hε mutant, missing the apical loop, bound HP almost (65%) as efficiently as the WT Hε (Fig. 9A, lane 9 versus lane 7). In contrast, the Hε mutant missing the internal bulge (dB) did not associate with HP at all (Fig. 9A, lane 8). These results were thus in excellent agreement with those obtained with the in vitro RNA binding assay using purified HP and in vitro transcribed Hε (Fig. 2). Hε/Ins, a variant Hε that extended the distance between the 5′ cap and the Hε sequence by 208 nucleotides (see Materials and Methods for details), exhibited ca. 40% HP binding compared to the WT Hε (Fig. 9A, lane 10), suggesting that the distance between the cap and Hε, or the cap itself, was not critical for HP binding, although the cap may enhance HP-Hε interaction in vivo. This result was also consistent with that obtained with the in vitro RNA binding assay since the in vitro-transcribed Hε RNAs were not capped but capable of binding HP (Fig. 2).
Fig 9.

Correlation between HP-Hε association in vivo, protein priming in vitro, and RNA packaging in vivo directed by Hε mutants. (A and B) HP was coexpressed with the indicated WT (Hε) or mutant (dB, dL, or Hε/Ins) Hε-expressing constructs in HEK293T cells. HP, with any associated Hε RNA, was purified by immunoaffinity as described in the legend to Fig. 1. The WT or mutant Hε RNA associated with the purified HP (lanes 6 to 10), as well as total RNA extracted from the transfected cells (lanes 1 to 5), was detected by Northern blotting as described in the legend to Fig. 3 (A). The positions of the RNAs are indicated. The different size species of the Hε RNAs are due to polyadenylation at the two different poly(A) sites, while the Hε/Ins species is ca. 200 nucleotides longer than the Hε species (see Fig. 3 and Materials and Methods for details). In vitro protein priming activity of the purified HP, in association with the different RNAs, was assayed as described in the legend to Fig. 4B. (C) The ability of the different Hε RNAs to direct RNA packaging into HBV nucleocapsids was determined by the RNA packaging assay. HEK293T cells were transfected with pCIdA-HBV (Hε− HBV) (lanes 3 to 7), expressing all HBV proteins but lacking the Hε coding sequence, together with pCMV-HE (Hε; lane 4), pCMV-HE-dB (dB; lane 5), pCMV-HE-dL (dL; lane 6), or pCI-HE (Hε/Ins; lane 7) to express the WT or mutant Hε or with pcDNA3 (lane 3) as a negative control. A GFP control transfection was also included in which neither HBV protein nor Hε was expressed (lane 2). Viral RNA associated with the nucleocapsids was detected by resolution of the capsids on an agarose gel followed by transfer to nitrocellulose membrane, probing with a riboprobe specific for Hε (RNA, autoradiography; top), and subsequent reprobing of the same membrane with the anti-HBV core antibody (capsid, chemiluminescence; bottom). For a positive control, HBV nucleocapsids harvested from the induced HepAD38 cells were included (lane 1).
We then tested the in vitro priming activity of the purified HP in association with the different Hε mutants. Although it was expected that the dB mutant would not be able to support priming because of its lack of HP association (Fig. 9B, lane 3), the dL mutant that was competent in HP binding did not support any protein priming either (Fig. 9B, lane 4). Furthermore, increasing the distance between the cap and Hε, while still allowing nearly WT levels of HP association, almost completely abolished priming (a 33-fold reduction) (Fig. 9B, lane 5). In agreement with previous reports (2, 14, 31, 36, 48, 69, 70), Hε mutants deleting either the internal bulge or apical loop, or with increased distance between the 5′ cap and Hε (Hε/Ins), were all defective in directing RNA packaging (Fig. 9C), indicating that HP protein priming, as detected in the newly developed in vitro assay, shared the same Hε RNA requirements as those for in vivo RNA packaging.
DISCUSSION
Protein-primed initiation of reverse transcription is a novel and essential step in HBV replication. Efforts to unravel the mechanisms and requirements of HBV protein priming have been hampered by the lack of appropriate cell-free systems that can faithfully recapitulate this reaction in vitro. By using a human cell line that can support HBV reverse transcription, we have now expressed and purified HP that was active in vivo and in vitro in binding its specific viral RNA ligand Hε (Fig. 2 and 3), which is the obligatory template for protein priming. Furthermore, by purifying the HP-Hε RNP complex from the human cells, we have established the first cell-free HBV protein priming system that recapitulated all of the essential features of HBV protein priming, including the absolute requirement for the Hε RNA template, the polymerase active site in the HP RT domain, and the specific Y63 primer in the HP TP domain (Fig. 4). The purified HP-Hε complex was able to carry out both the initiation and polymerization stages of protein priming in vitro with the appropriate nucleotide specificity, as determined by the authentic Hε internal bulge template sequence (Fig. 4, 6, and 7). Furthermore, we found that the requirements of Hε for HBV protein priming in vitro were remarkably similar to those defined for HBV pgRNA packaging in vivo, including the need for both its internal bulge and apical loop as well as close proximity between the 5′ cap structure and Hε (Fig. 9 and 10).
Fig 10.

Summary of the requirements for HP-Hε interaction, protein priming, and RNA packaging. For RNP formation (i.e., HP-Hε binding) (left), either in vitro or in vivo, the Hε bulge, but not the apical loop, is required. The 5′ cap of the Hε-containing RNA is unnecessary (denoted by the unfilled circle), and the distance between the cap and Hε is not critical (as denoted by the hatched line). For protein priming (middle) and RNA packaging (right), both the Hε bulge and apical loop are required, as is the short distance (solid line) between the cap (filled circle) and Hε. One potential role for the Hε apical loop and the nearby 5′ cap of the RNA in protein priming and RNA packaging is to bind putative host factors (HF) required for these reactions. HSP, heat shock protein. See text for details.
Both stages of protein priming (i.e., initiation involving the covalent attachment of the first nucleotide of the viral minus-strand DNA to HP and polymerization involving the subsequent addition of the few nucleotides to the initiating nucleotide) were demonstrated in vitro using a number of different approaches. First, when each dNTP was used as the only nucleotide substrate in the priming reaction, dGTP was shown to be, by far, the preferred nucleotide, as expected if initiation of protein priming occurred opposite from (i.e., was templated by) the last (3′) nucleotide (rC) in the internal bulge of Hε. As will be discussed in more detail later, this is in excellent agreement with results obtained in cell cultures replicating HBV as well as in vitro and in vivo results obtained with the related DHBV for which the protein priming mechanism has been more thoroughly characterized (see the introduction). The much weaker labeling of HP with dATP or TTP alone was possibly due to in vitro extension of in vivo priming products (see below for further discussion on in vivo priming) and/or low-efficiency priming initiation at an alternative site(s) (see later discussion also). Second, mutating the template residue of Hε for priming initiation (i.e., the last nucleotide of its internal bulge) from rC to rG or rA changed the first nucleotide attached to HP from dGMP to dCMP or TTP, respectively, as predicted, strongly indicating that priming initiation occurred in vitro and specifically from the last nucleotide of the Hε internal bulge.
Additional evidence for dGTP as the initiating nucleotide was also obtained by a third approach, which was to extend the single, HP-attached dGMP residue by including additional dNTP substrates so that the extended DNA oligomers that were synthesized by the second stage of protein priming (i.e., DNA polymerization) and covalently attached to HP could be directly visualized. Thus, inclusion of a second nucleotide substrate, dATP, led to the detection of a prominent, 3-nucleotide-long DNA oligomer that was released by Tdp2 from the primed HP and comigrated with a dGAA marker (and not a dTGA marker), representing DNA polymerization using, as expected, the 3′ half of the Hε internal bulge (rUUC) as the template. The lack of detectable DNA polymerization products (2-mer or longer) when priming was carried out with dGTP alone (despite the strong labeling of HP by dGTP, described above) or in combination with TTP is also consistent with the identification of the predominant polymerization product as dGAA, as is the fact that further inclusion of dCTP and TTP did not change the polymerization products, suggesting that neither dCMP nor TMP was part of the polymerization products.
Fourth, HP primed with dGTP alone was S1 nuclease resistant, just like the control DHBV priming initiation product (i.e., DP-dGMP complex), as expected from the inability of S1 to cleave the HP (DP)-dGMP bond. On the other hand, HP priming products with unlabeled dGTP and labeled dATP were S1 nuclease sensitive, as expected from S1 cleavage of internucleotide bonds (dGMP-dAMP and dAMP-dAMP) and thus the removal of labeled dAMP residue(s) from the initiating (unlabeled) dGMP. These results thus reinforce the identification of the polymerization products during in vitro HP priming as dGMP followed by one or more dAMP residues. That dAMP was not the initiating nucleotide but was added only following dGMP was also supported by the very weak labeling of HP by dATP alone (which may or may not represent inefficient priming initiation at an alternative site, see later discussion) but much stronger labeling when unlabeled dGTP was also included. The sensitivity of the HP-dGMP (or dGAA) linkage to Tdp2, together with the loss of protein priming in the Y63D HP mutant, established that during the in vitro HP priming reaction, dGMP and the DNA oligomers were in fact attached to Y63 in the TP domain, the same site used during authentic HBV replication in vivo and during in vitro priming using HP purified from the insect cells (35, 39, 45).
Previous efforts to map the initiation site of the HBV minus-strand DNA in vivo indicated that most minus-strand DNA initiates from the last position (an rC) of the Hε internal bulge (43, 56), as we found here during in vitro priming. However, a minor initiation site at the first position of Hε upper stem (rA) could not be excluded, although it could be attributed to nontemplated addition of an extra nucleotide, an artifact of the primer extension assay used to map the initiation site. An in vitro priming assay using HP purified from insect cells did not help to resolve this uncertainty. The insect cell-derived HP appeared to show some preference for TTP over dGTP during in vitro priming (38). However, the Hε independence of protein priming in the insect cell system (39) and the lack of detailed characterization of the very short DNA polymerization products (ca. 3 nucleotides long) (38, 72) have made it rather difficult to determine the authentic minus-strand DNA initiation site on Hε based on this system. In contrast, the in vitro HBV priming assay established here using HP purified from human cells demonstrated a clear and strong preference for dGTP as the initiating nucleotide. Furthermore, two different Hε mutants with substitutions of the last bulge residue changed the nucleotide used for priming initiation in a predictable fashion. Third, the predominant DNA polymerization product synthesized during priming in vitro here had the sequence dGAA, as expected from the template sequence on the Hε internal bulge. These results thus strongly support the notion that HBV minus-strand DNA synthesis is initiated from the last position of the Hε internal bulge and extended 2 more nucleotides along the bulge, as anticipated from the analysis of HBV protein priming in vivo and similar results obtained in vitro and in vivo with DHBV protein priming. A weak signal was obtained with 32P-labeled TTP or dATP alone in our in vitro HP priming assay. The weak dATP labeling could represent in vitro extension of DNA oligomers that were already attached to the purified HP as a result of priming in vivo (see below) rather than true initiation, although we could not exclude completely the possibility of a low level of alternative priming initiation from an rA residue on Hε, such as the first position of the upper stem. The weak labeling with TTP alone could also represent either alternative initiation or extension or even a low level of misincorporation. Due to the low levels of priming signal obtained with TTP or dATP alone, it was difficult to characterize these priming products in further detail.
We also developed a method that allows the monitoring of in vivo HBV protein priming and the very early stage of minus-strand DNA synthesis by TdT-mediated visualization of very short (ca. 4 nucleotides long) DNA strands covalently attached to the purified HP and released by Tdp2. With this assay, we detected short DNA oligomers (4 to 6 nucleotides long) covalently attached to HP via a phosphotyrosyl linkage that were synthesized in the cells only in the presence, but not the absence, of the coexpressed Hε, demonstrating that protein priming in vivo, as in vitro, was Hε dependent. In contrast to the Hε-dependent synthesis of very short (4 to 6 nucleotides) DNA strands by HP priming, either in vivo or in vitro, in our human cell system, the insect cell-expressed HP was shown to synthesize much longer DNA in the insect cells (up to 500 nucleotides long) and also in vitro (more than 60 nucleotides long) (38). A portion of the HP-attached DNA synthesized in the insect cells could be mapped to DR1, but it was uncertain if Hε was indeed serving as the template for protein priming in the insect cells with further DNA elongation occurring following primer translocation to DR1. It is possible that the difference in the length of the DNA synthesized by HP expressed in human cells versus that in insect cells can be in part explained by the much higher levels of HP expression, and hence the increased potential of detecting very minor products, using the insect system compared to the relatively low levels of HP expression in the human cells. However, we favor the explanation that HP expressed in human cells and insect cells likely adopts different conformations, which is indicated in part by the differences observed in Hε dependence and processivity of DNA synthesis. Extensive studies conducted using the related DP have clearly demonstrated that the hepadnavirus polymerase can show remarkable structural and functional flexibility that is influenced by factors such as cellular chaperones (23, 24, 27, 29, 59), metal ions (4, 8, 42), and likely other yet-to-be-identified factors.
Our ability to assess specific Hε RNA binding, protein priming, and pgRNA packaging using the same HP protein expressed in the human cells affords the opportunity, for the first time, to directly compare the requirements for these three critical and related steps in HBV replication. We have begun these comparative studies by taking a first look at the requirements from Hε for these reactions. It is clear that physical association between the polymerase and the ε RNA is insufficient to allow protein priming or pgRNA packaging, as demonstrated previously for DHBV (5, 30, 49, 63, 66) but only inferred for HBV (23, 24). For association with HP, the Hε internal bulge, but not apical loop, was required, whether HP was expressed in human cells and tested for Hε binding either in vitro or in vivo (this study) or purified from bacteria (with functional reconstitution in vitro using the Hsp90 chaperone complex) (23, 24). Also, there was no apparent requirement for the 5′ cap on the Hε RNA as in vitro-transcribed, uncapped Hε was able to bind HP purified from human cells or bacteria and extending the distance between the cap and Hε sequence did not significantly affect HP binding in vivo. In contrast, we could confirm here that extending this distance abolished pgRNA packaging in vivo, as reported before (31), and also, importantly, almost totally eliminated protein priming. Interestingly, DHBV priming in vitro does not require the presence of the cap structure on Dε (76, 77), suggesting that important differences exist between these two viruses, even though the general mechanisms of protein priming are very similar in both cases. Furthermore, we have found here that both the internal bulge and apical loop are required for protein priming, similar to their role in pgRNA packaging, as reported earlier (14, 36, 48, 51, 70).
It remains to be determined what specific functions of the Hε apical loop or the 5′ cap play in protein priming or RNA packaging as they are not required for HP binding. Limited structural information obtained by nuclear magnetic resonance on Hε and Dε suggests that these RNAs have complex and dynamic structures that may affect not only their association with the polymerase protein but also its functions in pgRNA packaging and protein priming (15, 16, 18). As we proposed earlier (23), the apical loop may bind to an important cellular protein that is not required for HP-Hε association but is necessary for priming and RNA packaging. Another possibility would be that the apical loop provides Hε some structural flexibility that is necessary for Hε to undergo a conformational change required to support RNA packaging or protein priming, as suggested for DHBV (5, 7). The apical loop of Hε could also be involved in inducing, either directly or indirectly, a putative conformational change in HP that is in turn needed for RNA packaging or protein priming as reported for DHBV (63, 66). Similar roles can be envisaged for the 5′ cap of pgRNA or a cap binding protein(s) in supporting viral RNA packaging or protein priming. The requirement for a short distance between the cap and Hε in pgRNA packaging and protein priming further suggests that the putative cap binding factor and the Hε host binding factor or the polymerase somehow interact with each other in a distance-dependent manner to coordinate both of these reactions. On the other hand, Hε can apparently function to stimulate DNA synthesis by HP expressed in insect cells when placed thousands of nucleotides away from the 5′ cap (and presumably functioned independently of the cap) (39, 72). However, it was uncertain if Hε indeed stimulated protein priming or DNA strand elongation in those experiments. Interestingly, it was also reported that Dε, uncapped, could stimulate DNA synthesis by DP that was not templated by Dε and not protein primed (in the so-called “trans reaction”) (66). These results thus suggest that Hε can under certain circumstances stimulate polymerase activity independently of the 5′ cap, but for it to stimulate authentic protein priming or pgRNA packaging, the cap is apparently needed.
Other than the insect cell (38) and the current human cell expression systems, attempts to obtain a priming active HP protein using a number of expression systems that can support DP priming, such as the RRL in vitro translation system (76) and purification from bacteria followed by chaperone reconstitution (23, 24, 28), have been unsuccessful. It is possible that there is a dominant inhibitor present or a positive facilitator missing specifically for HBV, but not DHBV, protein priming in these other systems. The in vitro and in vivo HBV RNA binding and protein priming assays established here should facilitate future efforts to identify these putative factors as well as to probe the molecular mechanisms and structural dynamics of both HP and Hε during RNP complex formation and protein priming, endeavors that have been highly successful for the DHBV model system but have remained unfruitful for HBV until now. As illustrated here, the ability to carry out comparative studies on the requirements for HP-Hε binding, protein priming, and RNA packaging with both in vitro and in vivo assays using the same expression system should greatly facilitate efforts to dissect the requirements for these complex and related reactions. Furthermore, the availability of these assays should also facilitate efforts to develop specific inhibitors targeted at these reactions that are essential for HBV replication but remain largely unexplored by current antiviral regimens.
ACKNOWLEDGMENTS
We thank Christina Adams and Laurie Mentzer for excellent technical assistance. We thank David Smith for the antibody against p60 and David Toft for the antibodies against Hsp70 and p23.
This work was supported by a Public Health Service grant (R01 AI043453 to J.H.) from the National Institutes of Health. S.A.J. was supported by Viruses and Cancer Training Grant 2 T32 CA60395 from the National Cancer Institute.
Footnotes
Published ahead of print 29 February 2012
REFERENCES
- 1. Bartenschlager R, Junker-Niepmann M, Schaller H. 1990. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 64:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 11:3413–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartenschlager R, Schaller H. 1988. The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein (genome-linked protein) believed to prime reverse transcription. EMBO J. 7:4185–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beck J, Nassal M. 2011. A Tyr residue in the reverse transcriptase domain can mimic the protein-priming Tyr residue in the terminal protein domain of a hepadnavirus P protein. J. Virol. 85:7742–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beck J, Nassal M. 1998. Formation of a functional hepatitis B virus replication initiation complex involves a major structural alteration in the RNA template. Mol. Cell. Biol. 18:6265–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beck J, Nassal M. 2001. Reconstitution of a functional duck hepatitis B virus replication initiation complex from separate reverse transcriptase domains expressed in Escherichia coli. J. Virol. 75:7410–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beck J, Nassal M. 1997. Sequence- and structure-specific determinants in the interaction between the RNA encapsidation signal and reverse transcriptase of avian hepatitis B viruses. J. Virol. 71:4971–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boregowda RK, Lin L, Zhu Q, Tian F, Hu J. 2011. Cryptic protein priming sites in two different domains of duck hepatitis B virus reverse transcriptase for initiating DNA synthesis in vitro. J. Virol. 85:7754–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carrigan PE, et al. 2004. Multiple domains of the co-chaperone Hop are important for Hsp70 binding. J. Biol. Chem. 279:16185–16193 [DOI] [PubMed] [Google Scholar]
- 10. Chang LJ, Hirsch RC, Ganem D, Varmus HE. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 64:5553–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y, Robinson WS, Marion PL. 1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J. Virol. 68:5232–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cherrington J, Russnak R, Ganem D. 1992. Upstream sequences and cap proximity in the regulation of polyadenylation in ground squirrel hepatitis virus. J. Virol. 66:7589–7596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. 2009. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461:674–678 [DOI] [PubMed] [Google Scholar]
- 14. Fallows DA, Goff SP. 1995. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69:3067–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Flodell S, et al. 2006. Solution structure of the apical stem-loop of the human hepatitis B virus encapsidation signal. Nucleic Acids Res. 34:4449–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Flodell S, et al. 2002. The apical stem-loop of the hepatitis B virus encapsidation signal folds into a stable tri-loop with two underlying pyrimidine bulges. Nucleic Acids Res. 30:4803–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J. Virol. 81:6164–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Girard FC, Ottink OM, Ampt KA, Tessari M, Wijmenga SS. 2007. Thermodynamics and NMR studies on duck, heron and human HBV encapsidation signals. Nucleic Acids Res. 35:2800–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirsch RC, Lavine JE, Chang LJ, Varmus HE, Ganem D. 1990. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature 344:552–555 [DOI] [PubMed] [Google Scholar]
- 20. Hirsch RC, Loeb DD, Pollack JR, Ganem D. 1991. cis-acting sequences required for encapsidation of duck hepatitis B virus pregenomic RNA. J. Virol. 65:3309–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu J. 2004. Studying DHBV polymerase by in vitro transcription and translation. Methods Mol. Med. 95:259–269 [DOI] [PubMed] [Google Scholar]
- 22. Hu J, Anselmo D. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol. 74:11447–11455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu J, Boyer M. 2006. Hepatitis B virus reverse transcriptase and epsilon RNA sequences required for specific interaction in vitro. J. Virol. 80:2141–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 78:13122–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu J, Lin L. 2009. RNA-protein interactions in hepadnavirus reverse transcription. Front. Biosci. 14:1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu J, Seeger C. 1996. Expression and characterization of hepadnavirus reverse transcriptases. Methods Enzymol. 275:195–208 [DOI] [PubMed] [Google Scholar]
- 27. Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 93:1060–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu J, Toft D, Anselmo D, Wang X. 2002. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol. 76:269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu K, Beck J, Nassal M. 2004. SELEX-derived aptamers of the duck hepatitis B virus RNA encapsidation signal distinguish critical and non-critical residues for productive initiation of reverse transcription. Nucleic Acids Res. 32:4377–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jeong JK, Yoon GS, Ryu WS. 2000. Evidence that the 5′-end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J. Virol. 74:5502–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Junker-Niepmann M, Bartenschlager R, Schaller H. 1990. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 9:3389–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kato KI, Goncalves JM, Houts GE, Bollum FJ. 1967. Deoxynucleotide-polymerizing enzymes of calf thymus gland. II. Properties of the terminal deoxynucleotidyltransferase. J. Biol. Chem. 242:2780–2789 [PubMed] [Google Scholar]
- 34. Kettlun AM, Espinosa V, Garcia L, Valenzuela MA. 2005. Potato tuber isoapyrases: substrate specificity, affinity labeling, and proteolytic susceptibility. Phytochemistry 66:975–982 [DOI] [PubMed] [Google Scholar]
- 35. Kim HY, et al. 2004. Oligomer synthesis by priming deficient polymerase in hepatitis B virus core particle. Virology 322:22–30 [DOI] [PubMed] [Google Scholar]
- 36. Knaus T, Nassal M. 1993. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res. 21:3967–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ladner SK, et al. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lanford RE, Notvall L, Beames B. 1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 69:4431–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lanford RE, Notvall L, Lee H, Beames B. 1997. Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J. Virol. 71:2996–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee W. 1997. Hepatitis B virus infection. N. Engl. J. Med. 337:1733–1745 [DOI] [PubMed] [Google Scholar]
- 41. Lin L, Hu J. 2008. Inhibition of hepadnavirus reverse transcriptase-epsilon RNA interaction by porphyrin compounds. J. Virol. 82:2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin L, Wan F, Hu J. 2008. Functional and structural dynamics of hepadnavirus reverse transcriptase during protein-primed initiation of reverse transcription: effects of metal ions. J. Virol. 82:5703–5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nassal M, Rieger A. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 70:2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neale MJ, Pan J, Keeney S. 2005. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436:1053–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 81:4465–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nguyen DH, Hu J. 2008. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J. Virol. 82:6852–6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Park SG, Jung G. 2001. Human hepatitis B virus polymerase interacts with the molecular chaperonin Hsp60. J. Virol. 75:6962–6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pollack JR, Ganem D. 1993. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol. 67:3254–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pollack JR, Ganem D. 1994. Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol. 68:5579–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Radziwill G, Tucker W, Schaller H. 1990. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol. 64:613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rieger A, Nassal M. 1995. Distinct requirements for primary sequence in the 5′- and 3′-part of a bulge in the hepatitis B virus RNA encapsidation signal revealed by a combined in vivo selection/in vitro amplification system. Nucleic Acids Res. 23:3909–3915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rieger A, Nassal M. 1996. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5′ encapsidation signal and the 3′-proximal direct repeat DR1. J. Virol. 70:585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Russnak R, Ganem D. 1990. Sequences 5′ to the polyadenylation signal mediate differential poly(A) site use in hepatitis B viruses. Genes Dev. 4:764–776 [DOI] [PubMed] [Google Scholar]
- 54. Russnak RH. 1991. Regulation of polyadenylation in hepatitis B viruses: stimulation by the upstream activating signal PS1 is orientation-dependent, distance-independent, and additive. Nucleic Acids Res. 19:6449–6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scaglioni PP, Melegari M, Wands JR. 1997. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J. Virol. 71:345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Seeger C, Maragos J. 1991. Identification of a signal necessary for initiation of reverse transcription of the hepadnavirus genome. J. Virol. 65:5190–5195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seeger C, Mason WS, Zoulim F. 2007. Hepadnaviruses, p 2977–3029 In Knipe DM, Howley PM. (ed), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 59. Stahl M, Beck J, Nassal M. 2007. Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C-proximal region in the terminal protein domain that contributes to epsilon RNA binding. J. Virol. 81:13354–13364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stahl M, Retzlaff M, Nassal M, Beck J. 2007. Chaperone activation of the hepadnaviral reverse transcriptase for template RNA binding is established by the Hsp70 and stimulated by the Hsp90 system. Nucleic Acids Res. 35:6124–6136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sullivan WP, Owen BA, Toft DO. 2002. The influence of ATP and p23 on the conformation of hsp90. J. Biol. Chem. 277:45942–45948 [DOI] [PubMed] [Google Scholar]
- 62. Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415 [DOI] [PubMed] [Google Scholar]
- 63. Tavis JE, Ganem D. 1996. Evidence for activation of the hepatitis B virus polymerase by binding of its RNA template. J. Virol. 70:5741–5750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tavis JE, Ganem D. 1993. Expression of functional hepatitis B virus polymerase in yeast reveals it to be the sole viral protein required for correct initiation of reverse transcription. Proc. Natl. Acad. Sci. U. S. A. 90:4107–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tavis JE, Ganem D. 1995. RNA sequences controlling the initiation and transfer of duck hepatitis B virus minus-strand DNA. J. Virol. 69:4283–4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tavis JE, Massey B, Gong Y. 1998. The duck hepatitis B virus polymerase is activated by its RNA packaging signal, epsilon. J. Virol. 72:5789–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tavis JE, Perri S, Ganem D. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Toh H, Hayashida H, Miyata T. 1983. Sequence homology between retroviral reverse transcriptase and putative polymerases of hepatitis B virus and cauliflower mosaic virus. Nature 305:827–829 [DOI] [PubMed] [Google Scholar]
- 69. Tong SP, Li JS, Vitvitski L, Kay A, Treepo C. 1993. Evidence for a base-paired region of hepatitis B virus pregenome encapsidation signal which influences the patterns of precore mutations abolishing HBe protein expression. J. Virol. 67:5651–5655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tong SP, Li JS, Vitvitski L, Trepo C. 1992. Replication capacities of natural and artificial precore stop codon mutants of hepatitis B virus: relevance of pregenome encapsidation signal. Virology 191:237–245 [DOI] [PubMed] [Google Scholar]
- 71. Tu CP, Cohen SN. 1980. 3′-end labeling of DNA with [alpha-32P]cordycepin-5′-triphosphate. Gene 10:177–183 [DOI] [PubMed] [Google Scholar]
- 72. Urban M, et al. 1998. In vitro activity of hepatitis B virus polymerase: requirement for distinct metal ions and the viral epsilon stem-loop. J. Gen. Virol. 79:1121–1131 [DOI] [PubMed] [Google Scholar]
- 73. Uzawa M, Grams J, Madden B, Toft D, Salisbury JL. 1995. Identification of a complex between centrin and heat shock proteins in CSF-arrested Xenopus oocytes and dissociation of the complex following oocyte activation. Dev. Biol. 171:51–59 [DOI] [PubMed] [Google Scholar]
- 74. Valenzuela M, et al. 1988. The effect of bivalent metal ions on ATPase-ADPase activities of apyrase from Solanum tuberosum. Phytochemistry 27:1981–1985 [Google Scholar]
- 75. Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670 [DOI] [PubMed] [Google Scholar]
- 77. Wang GH, Zoulim F, Leber EH, Kitson J, Seeger C. 1994. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J. Virol. 68:8437–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang H, Kim S, Ryu WS. 2009. DDX3 DEAD-box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J. Virol. 83:5815–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang X, Grammatikakis N, Hu J. 2002. Role of p50/CDC37 in hepadnavirus assembly and replication. J. Biol. Chem. 277:24361–24367 [DOI] [PubMed] [Google Scholar]
- 80. Wang X, Hu J. 2002. Distinct requirement for two stages of protein-primed initiation of reverse transcription in hepadnaviruses. J. Virol. 76:5857–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang X, Qian X, Guo HC, Hu J. 2003. Heat shock protein 90-independent activation of truncated hepadnavirus reverse transcriptase. J. Virol. 77:4471–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Weber M, et al. 1994. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol. 68:2994–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Xiong Y, Eickbush TH. 1990. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9:3353–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zeng Z, Cortes-Ledesma F, El Khamisy SF, Caldecott KW. 2011. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 286:403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zoulim F, Seeger C. 1994. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]