

Abstract
It has been recently established that Klotho coreceptors associate with fibroblast growth factor (FGF) receptor tyrosine kinases (FGFRs) to enable signaling by endocrine-acting FGFs. However, the molecular interactions leading to FGF-FGFR-Klotho ternary complex formation remain incompletely understood. Here, we show that in contrast to αKlotho, βKlotho binds its cognate endocrine FGF ligand (FGF19 or FGF21) and FGFR independently through two distinct binding sites. FGF19 and FGF21 use their respective C-terminal tails to bind to a common binding site on βKlotho. Importantly, we also show that Klotho coreceptors engage a conserved hydrophobic groove in the immunoglobulin-like domain III (D3) of the “c” splice isoform of FGFR. Intriguingly, this hydrophobic groove is also used by ligands of the paracrine-acting FGF8 subfamily for receptor binding. Based on this binding site overlap, we conclude that while Klotho coreceptors enhance binding affinity of FGFR for endocrine FGFs, they actively suppress binding of FGF8 subfamily ligands to FGFR.
INTRODUCTION
Fibroblast growth factor (FGF) signaling plays pleiotropic roles in metazoan development and metabolism (5, 24). Based on sequence homology and phylogenetic and structural considerations, the 18 mammalian FGF ligands are grouped into five paracrine-acting subfamilies and one endocrine-acting subfamily comprising FGF19, FGF21, and FGF23 (24, 43). Paracrine FGFs have high affinity for pericellular heparan sulfate (HS) glycosaminoglycans (3, 5) and form distinct morphogenetic gradients in the pericellular matrix (27, 39) to fulfill essential roles during embryonic development (7, 24, 65). In contrast, endocrine FGFs exhibit poor affinity for HS (3, 15) and thus are able to enter the blood circulation to regulate key metabolic processes, including bile acid homeostasis (18, 20, 36) and hepatic glucose and protein metabolism (30, 57) (FGF19), glucose and lipid metabolism (4, 21, 29, 58) (FGF21), and vitamin D and phosphate homeostasis (1, 59, 62) (FGF23). These hormone-like FGFs have taken center stage in drug discovery for a number of inherited and acquired metabolic disorders (5, 6).
Mammalian FGFs signal through four FGF receptor (FGFR) tyrosine kinases (FGFR1 to FGFR4) and their alternatively spliced isoforms (23, 43). The extracellular domain of a prototypical FGFR consists of three immunoglobulin-like domains (D1 to D3), and structural and biochemical studies have established that the region including D2, D3, and the D2-D3 linker comprises the minimal ligand-binding domain (54, 55, 63). The ligand-binding specificity of FGFR1 to FGFR3 is primarily regulated by a tissue-specific splicing in D3 of these receptors that generates “b” and “c” isoforms (9, 26, 42, 52). Structural studies have revealed that this splicing alters the primary amino acid sequence of key ligand-binding loops/pockets in D3 (50, 74).
In addition to mediating paracrine FGF gradient formation in the pericellular matrix, HS is required for enhancing the affinity of paracrine FGF for FGFR and promoting dimerization of ligand-bound receptors (61). In contrast, the crystal structures of FGF19 and FGF23 have shown that the topology of the HS-binding site in these endocrine FGFs deviates from that of paracrine FGFs, accounting for the reduced affinity of these ligands for HS (15). Moreover, compared to paracrine FGFs, endocrine FGFs have intrinsically poor binding affinity for their cognate FGFRs (43). Endocrine FGFs overcome this dual deficiency in binding affinity by relying on α/βKlotho coreceptors that are expressed in their target tissues (32, 33, 46, 68). α/βKlotho coreceptors are single-pass transmembrane proteins with an extracellular domain consisting of tandem type I β-glycosidase domains connected by a short linker (22, 31). α/βKlotho coreceptors constitutively associate with cognate FGFRs of endocrine FGFs to enhance endocrine FGF-FGFR binding (32, 33, 46, 68). αKlotho is the coreceptor for FGF23 (33, 68), and βKlotho is the coreceptor for both FGF19 and FGF21 (32, 46).
We have previously shown that αKlotho binds to FGFR to create a de novo binding site for the C-terminal tail of FGF23 at the composite interface of the FGFR-αKlotho complex (16). Here, we show that in contrast to αKlotho, βKlotho binds FGF19/FGF21 and FGFR independently through two distinct binding sites. In addition, we show that the C-terminal tails of FGF19 and FGF21 mediate binding of these ligands to a common binding site on βKlotho. Importantly, we have also mapped the binding site on FGFR1 for α/βKlotho coreceptors to the D3 domain of the “c” splice isoform of the receptor. Intriguingly, the binding site for α/βKlotho on FGFR1c partially overlaps with the binding site for ligands of the FGF8 subfamily. Hence, we propose that while α/βKlotho coreceptors enhance binding of endocrine FGFs to FGFR, they actively suppress binding of FGF8 subfamily ligands and possibly other paracrine FGFs to FGFR.
MATERIALS AND METHODS
Purification of FGF, FGFR, and Klotho proteins.
The N-terminally hexahistidine-tagged, mature form of human FGF19 (R23 to K216), human FGF21 (H29 to S209), and human FGF23 (Y25 to I251) was refolded in vitro from bacterial inclusion bodies and purified by published protocols (19, 54). In order to minimize proteolysis of FGF23, arginine residues 176 and 179 of the proteolytic cleavage site 176RXXR179 were replaced with glutamine as occurs in the phosphate wasting disorder autosomal dominant hypophosphatemic rickets (1, 70). A chimeric FGF21 protein in which the C-terminal sequence from S191 to S209 of FGF21 had been swapped with the corresponding sequence of FGF19 (FGF2129–190/FGF19197–216) and a chimeric FGF21 protein in which the entire FGF21 C-terminal tail (P168 to S209) had been swapped with that of FGF19 (FGF2129–167/FGF19169–216) were purified by the same protocol as that for wild-type FGF21. The C-terminal tail peptide of FGF19 (M171 to K216, FGF19C-tail; see Fig. 2A) and the C-terminal tail peptide of FGF21 (P168 to S209, FGF21C-tail; see Fig. 2A) were expressed in Escherichia coli as fusion peptides with a 50-residue-long N-terminal tag, including a hexahistidine tag, and purified from the soluble cell lysate fraction by nickel affinity- and ion-exchange chromatographies. Full-length human FGF8b (Q23 to R215), C-terminally truncated FGF8b (Q23 to E186), and full-length human FGF homologous factor 1B (FHF1B; M1 to T181) were purified by published protocols (48, 50). The minimal ligand-binding domain of each of the seven principal human FGFRs, namely, FGFR1b (D142 to E374), FGFR1c (D142 to R365), FGFR2b (A140 to E369), FGFR2c (N149 to E368), FGFR3b (D147 to H356), FGFR3c (D147 to E365), and FGFR4 (Q144 to D355), was refolded in vitro from bacterial inclusion bodies and purified by published protocols (19, 54). Single point mutants of the ligand-binding domain of FGFR1c were purified by the same protocol as that for the wild-type protein. The ectodomain of murine αKlotho (A35 to K982) was purified from culture medium of a HEK293 cell line ectopically expressing the αKlotho ectodomain as a fusion protein with a C-terminal FLAG tag (33, 34). Similarly, the ectodomain of murine βKlotho (F53 to L995) was ectopically expressed in HEK293 cells as a fusion protein with a C-terminal FLAG tag and purified by the same protocol as that for the αKlotho ectodomain. Purified bovine β-glucuronidase was obtained from Sigma-Aldrich.
Fig 2.

The C-terminal tail peptides of FGF19 and FGF21 bind to a common site on βKlotho, albeit with different affinities. (A) Alignment of the C-terminal tail sequences of human FGF19 and FGF21. Residue numbers are in parentheses to the left of the alignment. Gaps (dashes) were introduced to optimize the sequence alignment. Residues that are identical between human FGF19 and FGF21 are shaded gray. Note that the greatest degree of sequence identity (40%) is confined to the 20 most C-terminal residues. (B and C) Overlays of SPR sensorgrams illustrating inhibition by FGF19C-tail (B) or FGF21C-tail (C) of βKlotho binding to FGF19. FGF19 was immobilized on a biosensor chip, and mixtures of a fixed concentration of βKlotho ectodomain with increasing concentrations of either FGF19C-tail or FGF21C-tail were passed over the chip. (D and E) Overlays of SPR sensorgrams illustrating inhibition by FGF21C-tail (D) or FGF19C-tail (E) of βKlotho binding to FGF21. FGF21 was immobilized on a biosensor chip, and mixtures of a fixed concentration of βKlotho ectodomain with increasing concentrations of either FGF19C-tail or FGF21C-tail were passed over the chip. Note that FGF19C-tail is more potent than FGF21C-tail at inhibiting βKlotho binding to FGF19 or FGF21 (compare panels B and E with panels C and D). (F) Overlay of SPR sensorgrams illustrating inhibition by FGF2129–190/FGF19197–216 or FGF21 of βKlotho binding to FGF21 immobilized on a biosensor chip. βKlotho ectodomain alone and 1:1 mixtures of βKlotho ectodomain with either FGF2129–190/FGF19197–216 or FGF21 were passed over a FGF21 chip. Note that the FGF2129–190/FGF19197–216 chimera is a more potent competitor for βKlotho binding than is native FGF21. The data shown in figure panels B to F are representative of two to three independent experiments.
Analysis of α/βKlotho-FGF, α/βKlotho-FGFR, and FGF8b-FGFR interactions by SPR spectroscopy.
Surface plasmon resonance (SPR) experiments were performed on a Biacore 2000 instrument (Biacore AB), and the interaction analyses were carried out at 25°C in HBS-EP buffer (10 mM HEPES-NaOH, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% [vol/vol] polysorbate 20). To study α/βKlotho-FGF and FGF8b-FGFR interactions, FGF chips were prepared by covalent coupling of FGF proteins through their free amino groups on flow channels of research-grade CM5 chips (Biacore AB). The coupling densities (fmol mm−2 of flow channel) were ∼18 to 30 (FGF19, FGF21, and FGF23) and 114 to 126 (FGF8b). To study α/βKlotho-FGFR interactions, the ectodomains of αKlotho and βKlotho were covalently coupled to flow channels of CM5 chips (∼39 to 46 fmol mm−2). Proteins were injected over a CM5 chip at a flow rate of 50 μl min−1, and at the end of each protein injection (180 s), HBS-EP buffer (50 μl min−1) was made to flow over the chip to monitor dissociation for 180 s. The FGF chip surface was regenerated by injecting 50 μl of 2.0 M NaCl in 10 mM sodium acetate, pH 4.5. For α/βKlotho chips, regeneration was achieved by injecting 2.0 M NaCl in 10 mM sodium-potassium phosphate, pH 6.5. To control for nonspecific binding in experiments where an FGF ligand was immobilized on the chip, FHF1B, which shares structural similarity with FGFs but does not exhibit any FGFR binding (48), was coupled to the control flow channel of the chip (∼17 to 101 fmol mm−2). In experiments where the ectodomain of αKlotho or βKlotho was immobilized on the chip, β-glucuronidase was coupled to the control flow channel of the chip (∼43 to 68 fmol mm−2). Like αKlotho and βKlotho, β-glucuronidase is a member of family 1 glycosidases (carbohydrate-active enzymes database at http://www.cazy.org/ [8]) and hence structurally related to each of the two extracellular glycosidase-like domains of αKlotho and βKlotho, respectively. The data were processed with BiaEvaluation software (Biacore AB). For each protein injection over an FGF chip, the nonspecific responses from the FHF1B control flow channel were subtracted from the responses recorded for the FGF flow channel. Similarly, for each protein injection over an α/βKlotho chip, the nonspecific responses from the β-glucuronidase control flow channel were subtracted from the responses recorded for the α/βKlotho flow channel.
To analyze binding of βKlotho to FGF19 and FGF21, increasing concentrations of βKlotho ectodomain were injected over a chip containing immobilized FGF19, FGF21, and, as a control for specificity of the interaction, FGF23. As an additional specificity control, binding of αKlotho to FGF19 and FGF21 was studied.
To examine whether the C-terminal tail peptide of FGF19 or FGF21 can compete with full-length FGF19 or FGF21 for binding to βKlotho, increasing concentrations of either FGF19C-tail or FGF21C-tail were mixed with a fixed concentration of βKlotho ectodomain, and the mixtures were injected over a chip containing immobilized FGF19 and FGF21.
To examine whether the FGF2129–190/FGF19197–216 chimera can compete with FGF21 for binding to βKlotho, increasing concentrations of FGF2129–190/FGF19197–216 were mixed with a fixed concentration of βKlotho ectodomain, and the mixtures were passed over a chip containing immobilized FGF21. As a control, competition of FGF21 in solution with immobilized FGF21 for binding to βKlotho was studied.
To measure binding of αKlotho and βKlotho to each of the seven principal FGFRs, increasing concentrations of the ligand-binding domain of FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c, or FGFR4 were injected over a chip containing immobilized αKlotho and βKlotho ectodomain. Maximal equilibrium binding responses were plotted against the concentrations of FGFR ligand-binding domain, and the equilibrium dissociation constant (KD) was calculated from the fitted saturation binding curve. The fitted binding curve was judged to be accurate based on the distribution of the residuals (even and near zero) and χ2 (<10% of Rmax).
To examine whether point mutations in the ligand-binding domain of FGFR1c affect the binding of the receptor domain to αKlotho or βKlotho, increasing concentrations of FGFR1c wild-type ligand-binding domain and point mutants were injected over a chip containing immobilized αKlotho and βKlotho ectodomain. As a control, binding of FGFR1c wild-type ligand-binding domain and point mutants to FGF8b immobilized on a chip was studied. The maximal binding response obtained for a given concentration of a receptor mutant was expressed as a fraction of the binding of wild-type receptor (25), and the average relative binding response obtained from a range of concentrations was plotted.
Analysis of phosphorylation of FRS2α and 44/42 MAP kinase in hepatoma and epithelial cell lines.
To test whether the C-terminal tail peptides of FGF19 and FGF21 are interchangeable in inhibiting the signaling of FGF19, H4IIE rat hepatoma cells, which endogenously express βKlotho and FGFR4 (32), were serum starved overnight and then pretreated for 60 min with either FGF19C-tail (10 to 1,000 ng ml−1) or FGF21C-tail (10 to 1,000 ng ml−1) prior to stimulation with FGF19 (30 ng ml−1) for 10 min. Cell stimulation with FGF19 (3 to 300 ng ml−1), FGF19C-tail (10 to 1,000 ng ml−1), or FGF21C-tail (10 to 1,000 ng ml−1) alone served as controls. After stimulation, the cells were lysed (34), and cellular proteins were resolved on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. The protein blots were probed with antibodies to phosphorylated FGFR substrate 2α (FRS2α), phosphorylated 44/42 mitogen-activated protein (MAP) kinase, and total (phosphorylated and nonphosphorylated) 44/42 MAP kinase. All antibodies were from Cell Signaling Technology.
Analysis of Egr1 protein expression in an epithelial cell line.
The ability of the FGF2129–167/FGF19169–216 chimera to activate FGFR in a βKlotho-dependent fashion was studied using induction of early growth response 1 (Egr1) expression as readout for FGFR activation. HEK293 cells transfected with murine βKlotho were serum starved overnight and then stimulated for 90 min with FGF2129–167/FGF19169–216 or FGF21 (3 to 300 ng ml−1). After stimulation, the cells were lysed (34), and cellular proteins were resolved on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. The protein blots were probed with antibodies to Egr1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The anti-Egr1 antibody was from Cell Signaling Technology, and the anti-GAPDH antibody was from Abcam.
Analysis of Egr1 promoter activation in an epithelial cell line.
The effect of βKlotho expression on the ability of FGF8b to activate FGFR was studied using induction of Egr1 gene promoter activity as readout for FGFR activation. Subconfluent HEK293 cells were cotransfected with Egr1 promoter-luciferase reporter (10) and either βKlotho or empty vector. At 16 h after transfection, the cells were serum starved for 10 h and then stimulated for 16 h with FGF8b (0 to 20 nM). Thereafter, the cells were lysed (10), and luciferase activity was measured using a luciferase assay system (Promega). Luciferase activity of cells expressing βKlotho was normalized to that of cells transfected with empty vector. Using GraphPad Prism 5 software, a dose-response curve was fitted from the mean values of three independent experiments each performed in triplicate.
Insulin tolerance test in mice.
The ability of the FGF2129–167/FGF19169–216 chimera to potentiate the hypoglycemic effect of insulin was used as a readout for FGF21-like metabolic activity (47). Eight- to 12-week-old mice of the C57BL/6 strain were fasted for 4 h and then bled from the cheek pouch for measuring fasting blood glucose levels. Thereafter, mice were intraperitoneally administered insulin (0.5 units kg body weight−1) alone or insulin (0.5 units kg body weight−1) plus FGF2129–167/FGF19169–216 (0.3 mg kg body weight−1). As controls, mice were injected with vehicle alone or coinjected with insulin plus FGF21. At the indicated time points after the injection (see Fig. 4B), blood was drawn from the tail vein. Glucose concentrations in the blood samples were determined using Bayer Contour blood glucose test strips (Bayer Corp.). Insulin was obtained from Eli Lilly & Co. The experiments were approved by the Harvard University Animal Care and Research committee board and complied with the Guide for the Care and Use of Laboratory Animals (45).
Fig 4.

A chimeric FGF21 protein in which the FGF21 C-terminal tail had been swapped with that of FGF19 exhibits FGF21-like activity. (A) Immunoblot analysis of Egr1 expression in HEK293-βKlotho cells stimulated with FGF21 or FGF2129–167/FGF19169–216. Numbers above the lanes give the amounts of protein added in ng ml−1. To control for equal sample loading, the protein blots were probed with an antibody to GAPDH. Note that the FGF2129–167/FGF19169–216 chimera is more potent than native FGF21 at inducing Egr1 expression, which is consistent with the SPR data shown in Fig. 2F. The data are representative of two independent experiments. (B) Analysis of blood glucose concentrations in mice before and at the indicated time points after intraperitoneal injection of insulin alone, insulin plus FGF2129–167/FGF19169–216, insulin plus FGF21, or vehicle alone. Insulin and FGF21 ligand were injected at 0.5 units per kg of body weight and 0.3 mg per kg of body weight, respectively. Blood glucose concentrations are expressed as percentages of preinjection values. Data are presented as means ± SEM. (C) Changes in plasma insulin concentrations in mice in response to a single intraperitoneal injection of FGF2129–167/FGF19169–216, FGF21, or vehicle. Three different doses of protein were tested, and the numbers below the x axis give each dose of protein injected in mg per kg of body weight. Plasma insulin concentrations are expressed as percentages of preinjection values. Data are presented as means ± SEM. *, P < 0.05; **, P < 0.01 (both by Student's t test).
Measurement of plasma insulin concentrations in mice.
The ability of the FGF2129–167/FGF19169–216 chimera to reduce circulating insulin levels was used as another readout for FGF21-like metabolic activity (11). Twelve-week-old mice of the C57BL/6 strain were fasted for 3 h and then given intraperitoneally a single dose (0.1, 0.3, or 1.0 mg kg body weight−1) of FGF2129–167/FGF19169–216 or FGF21. Blood was drawn from the tail vein before the injection. One hour after the injection, the mice were sacrificed and another blood sample was taken. Insulin concentrations in blood plasma were measured by enzyme-linked immunosorbent assay (ELISA; ultrasensitive mouse insulin ELISA kit; Crystal Chem Inc.). The experiments were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center at Dallas and complied with the Guide for the Care and Use of Laboratory Animals (45).
Statistical analysis.
Data from the experiments in mice are expressed as means ± standard errors of the means (SEM). Student's t test or analysis of variance (ANOVA) was used as appropriate to make statistical comparisons. A P value of <0.05 was considered significant.
RESULTS
Klotho coreceptors use different mechanisms to promote binding of endocrine FGF ligands to cognate FGFRs.
We have previously characterized the protein-protein interactions leading to the formation of the ternary complex between FGF23, FGFR1c, and αKlotho (16). We showed that the ectodomain of αKlotho possesses a high-affinity binding site for the ligand-binding domain of FGFR1c but not for the FGF23 ligand (16) and that the preformed binary FGFR1c-αKlotho complex binds avidly to FGF23 (16). We concluded that FGF23 binds to a de novo binding site generated at the composite FGFR1c-αKlotho interface. The region on FGF23 that binds to this site was mapped to the C-terminal tail that follows the β-trefoil core domain (16). Here, we explored whether βKlotho uses the same mechanism to promote binding of FGF19 and FGF21 to FGFR4 and FGFR1c, the principal cognate FGFRs of these ligands.
To determine whether βKlotho contains a high-affinity binding site for FGFR1c and FGFR4, βKlotho ectodomain was immobilized on an SPR biosensor chip, and increasing concentrations of the ligand-binding domain of either FGFR1c or FGFR4 were passed over the chip. βKlotho bound the two receptors with comparably high affinities (Fig. 1A and B), demonstrating that, similarly to αKlotho, βKlotho contains a high-affinity binding site for its cognate FGFRs.
Fig 1.

βKlotho possesses distinct high-affinity binding sites for FGF19/21 and FGFR. (A and B) Overlays of SPR sensorgrams illustrating binding of FGFR1c (A) and FGFR4 (B) to βKlotho and fitted saturation binding curves. βKlotho ectodomain was immobilized on a biosensor chip, and increasing concentrations of the ligand-binding domain of FGFR1c or FGFR4 were passed over the chip. The dissociation constants (KDs) were calculated from the saturation binding curve. (C and D) Overlays of SPR sensorgrams illustrating binding of βKlotho to FGF19 (C) and FGF21 (D). FGF19 and FGF21 were immobilized on a biosensor chip, and increasing concentrations of βKlotho ectodomain were passed over the chip. Note that for any given concentration of βKlotho, the binding response is greater on the FGF19 chip surface than on the FGF21 chip surface. Also note that the FGF19-βKlotho complex dissociates more slowly than the FGF21-βKlotho complex (compare the dissociation phases of the sensorgrams shown in panels C and D). (E) Overlay of SPR sensorgrams showing no interaction between βKlotho and FGF23. FGF23 was immobilized on a biosensor chip, and increasing concentrations of βKlotho ectodomain were passed over the chip. (F and G) Overlays of SPR sensorgrams showing no interaction between αKlotho and FGF19 (F) or FGF21 (G). FGF19 and FGF21 were immobilized on a biosensor chip, and increasing concentrations of αKlotho ectodomain were passed over the chip. The data shown in each figure panel are representative of two to three independent experiments.
To examine whether βKlotho also contains a high-affinity binding site for FGF19 and FGF21, these two ligands and, as a specificity control, FGF23 were immobilized on an SPR biosensor chip, and increasing concentrations of βKlotho ectodomain were passed over the chip. Both FGF19 and FGF21 bound strongly to βKlotho (Fig. 1C and D), whereas no interaction was observed between FGF23 and βKlotho (Fig. 1E). To further confirm the specificity of the interaction, increasing concentrations of αKlotho ectodomain were passed over the chip. Neither FGF19 nor FGF21 bound to αKlotho (Fig. 1F and G). Our data show that in contrast to αKlotho, βKlotho possesses distinct high-affinity binding sites for cognate endocrine FGF ligand and FGFR, indicating that βKlotho promotes ternary complex formation by engaging FGF ligand and FGFR independently.
FGF19 and FGF21 bind to a common binding site on βKlotho, albeit with different affinities.
Since both FGF19 and FGF21 bind to βKlotho, it raised the question whether these ligands bind to a shared site on βKlotho or whether each ligand has its own distinct binding site. Previous studies have suggested that, reminiscent of FGF23, FGF19 and FGF21 use their C-terminal tail to bind βKlotho (41, 73, 75). Based on this knowledge, we set up an SPR-based competition binding assay to examine whether the isolated C-terminal tail peptide of FGF19 (FGF19C-tail [Fig. 2A]) can compete with full-length FGF21 for binding to βKlotho and, conversely, whether the C-terminal tail peptide of FGF21 (FGF21C-tail [Fig. 2A]) can compete with full-length FGF19 for binding to βKlotho. As shown in Fig. 2E, FGF19C-tail effectively competed with FGF21 for binding to βKlotho. Similarly, FGF21C-tail was capable of inhibiting βKlotho binding to FGF19 (Fig. 2C). These data show that FGF19 and FGF21 have overlapping binding sites on βKlotho. As expected, each of the two C-terminal tail peptides competed with its respective full-length ligand for binding to βKlotho (Fig. 2B and D).
Interestingly, a quantitative analysis of the SPR data shows that the FGF19C-tail peptide is more potent than the FGF21C-tail peptide at inhibiting binding of βKlotho to full-length FGF19 or FGF21. Specifically, an equimolar amount of FGF19C-tail relative to βKlotho already yielded nearly complete inhibition of βKlotho binding to FGF19 or FGF21 (Fig. 2B and E), whereas a 10- to 20-fold molar excess of FGF21C-tail over βKlotho was needed to achieve a similar effect (Fig. 2C and D). These data indicate that the C-terminal tail of FGF19 binds βKlotho with greater affinity than does the C-terminal tail of FGF21, suggesting that primary sequence differences at this region account for the observed difference in binding affinity of the two ligands for βKlotho. Comparison of the C-terminal tail sequences of FGF19 and FGF21 shows a significant degree of sequence similarity (40% amino acid identity) only in the last 20 residues (Fig. 2A), pointing to these residues as the major binding epitope for βKlotho. To test this possibility, we swapped the 19 most C-terminal residues in FGF21 with the corresponding residues of FGF19, including a one-residue insertion, and examined, by SPR spectroscopy, whether the chimeric FGF21 protein (FGF2129–190/FGF19197–216) is more potent than wild-type FGF21 at inhibiting binding of βKlotho to FGF21 immobilized on a biosensor chip. As shown in Fig. 2F, an equimolar amount of the FGF2129–190/FGF19197–216 chimera relative to βKlotho already yielded nearly complete inhibition of βKlotho binding to immobilized FGF21, whereas the same molar ratio of wild-type FGF21 to βKlotho produced at best half-maximum inhibition. Taken together, our SPR data show that FGF19 and FGF21 bind with their C-terminal tail to a common binding site on βKlotho, albeit with different affinities, which is primarily due to differences in amino acid sequence between FGF19 and FGF21 at the distal portion of the C-terminal tail.
To provide biological evidence for our in vitro finding that FGF19 and FGF21 bind to a shared binding site on βKlotho, we next examined whether the FGF19C-tail peptide and the FGF21C-tail peptide are both able to block FGF19 signaling in cells. H4IIE hepatoma cells, which endogenously express βKlotho and FGFR4 (32), were pretreated with FGF19C-tail or FGF21C-tail and then stimulated with FGF19. As shown in Fig. 3A and B, both FGF19C-tail and FGF21C-tail inhibited, in a dose-dependent fashion, FGF19-induced tyrosine phosphorylation of FRS2α and downstream activation of MAP kinase cascade. As expected, neither of the two peptides elicited any signaling response when applied alone (Fig. 3A and B). These data show that the C-terminal tail peptides of FGF19 and FGF21 are interchangeable in inhibiting the signaling of FGF19. Consistent with these data, a chimeric FGF21 molecule in which the FGF21 C-terminal tail had been swapped with that of FGF19 (FGF2129–167/FGF19169–216) was able to induce protein expression of the transcription factor Egr1, just like native FGF21 (Fig. 4A). Together, the data provide cell-based evidence that FGF19 and FGF21 share a binding site on βKlotho. To enhance this finding, we examined whether the FGF2129–167/FGF19169–216 chimera acts as an FGF21-like hormone in vivo. Potentiation of the hypoglycemic effect of insulin (47) and downregulation of circulating insulin levels (11) were used as readouts for FGF21-like metabolic activity. Mice were injected with insulin plus FGF2129–167/FGF19169–216, insulin plus FGF21, or insulin alone, and blood glucose concentrations were monitored for up to 1 h after the injection. Similarly to FGF21, the FGF2129–167/FGF19169–216 chimera enhanced the hypoglycemic effect of insulin (Fig. 4B). Likewise, when injected alone into mice, the chimera was as potent as native FGF21 at reducing plasma levels of endogenous insulin (Fig. 4C). These data confirm in vivo that FGF19 and FGF21 have a common binding site on βKlotho.
Fig 3.

The C-terminal tail peptides of FGF19 and FGF21 are interchangeable in inhibiting the signaling of FGF19. (A) Immunoblot analysis of phosphorylation of FRS2α (pFRS2α) and 44/42 MAP kinase (p44/42 MAPK) in the rat hepatoma cell line H4IIE, which had been pretreated with FGF19C-tail and then stimulated with FGF19. As controls, cells were stimulated with FGF19 or FGF19C-tail alone. (B) Immunoblot analysis of phosphorylation of FRS2α (pFRS2α) and 44/42 MAP kinase (p44/42 MAPK) in the rat hepatoma cell line H4IIE, which had been pretreated with FGF21C-tail and then stimulated with FGF19. As controls, cells were stimulated with FGF19 or FGF21C-tail alone. Numbers above the lanes give the amounts of protein/peptide added in ng ml−1. To control for equal sample loading, the protein blots were probed with an antibody recognizing both phosphorylated and nonphosphorylated 44/42 MAP kinase (44/42 MAPK). The data shown in each figure panel are representative of two independent experiments.
The binding site for Klotho coreceptors on FGFRc isoforms and FGFR4 overlaps with the binding site for FGF8 subfamily ligands.
The binding site(s) for Klotho proteins on FGFR has not been identified to date. To explore this, we first measured, by SPR spectroscopy, binding affinities between the ectodomain of Klotho proteins and the ligand-binding domain of each of the seven principal FGFRs (FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c, and FGFR4). αKlotho exhibited greatest affinity for FGFR1c (KD, 72 nM [16]) followed by FGFR3c (KD, 82 nM) and FGFR4 (KD, 123 nM) (Fig. 5). βKlotho exhibited greatest affinity for FGFR4 (KD, 84 nM) followed by FGFR1c (KD, 124 nM) and FGFR2c (KD, 170 nM) (Fig. 1A and B and Fig. 5). Interestingly, αKlotho bound poorly to FGFR2c whereas βKlotho bound poorly to FGFR3c (Fig. 5). Neither of the two Klotho proteins interacted with FGFR1b, FGFR2b, or FGFR3b (Fig. 5). This is the first quantitative analysis of α/βKlotho-FGFR binding that conclusively shows that αKlotho and βKlotho preferentially bind to the “c” splice isoforms of FGFR1 to FGFR3 and to FGFR4. However, they still exhibit differences in selectivity for FGFR2c and FGFR3c. Our SPR data are consistent with previous coimmunoprecipitation studies (28, 33, 46, 68).
Fig 5.

Comprehensive analysis of FGFR binding specificity of Klotho coreceptors. (A and B) Overlays of SPR sensorgrams illustrating the FGFR binding specificity profile of αKlotho (A) and βKlotho (B). The ectodomains of αKlotho and βKlotho were immobilized on a biosensor chip, and increasing concentrations of the ligand-binding domain of each of the seven principal FGFRs were passed over the chip. Where possible, equilibrium dissociation constants (KDs) were derived from fitted saturation binding curves. The data are representative of two to five independent experiments. To give a complete view of the FGFR binding specificity of Klotho coreceptors, the sensorgrams illustrating βKlotho binding to FGFR1c and FGFR4 from Fig. 1 are shown again in panel B of this figure.
Interestingly, the receptor-binding specificity of αKlotho and βKlotho resembles that of ligands of the FGF4, FGF8, and FGF9 subfamilies, which preferentially bind the “c” splice isoforms of FGFR1 to FGFR3 (19, 50, 51, 77). This led us to speculate that the binding sites for these paracrine FGFs and Klotho proteins on the D3 domain of receptor might overlap. We have previously solved the crystal structure of FGF8b, a member of the FGF8 subfamily, in complex with FGFR2c (Protein Data Bank identifier [PDB ID], 2FDB [50]). In the structure, hydrophobic residues from the N-terminal helix and the β4-β5 loop of FGF8b engage a hydrophobic groove on receptor D3, which is present only in the “c” isoforms of FGFR1 to FGFR3 and in FGFR4 (PDB IDs, 1CVS, 1DJS, 1EVT, 1EV2, and 1RY7 [49, 54, 55, 63]) (Fig. 6). The hydrophobic residues of FGF8b that engage the hydrophobic groove are conserved in FGF17 and FGF18, the other two members of the FGF8 subfamily, and therefore, the FGF8b-FGFR2c structure is representative of the mode of receptor binding of the entire FGF8 subfamily. Since FGFR1c is the only receptor among the “c” isoforms that binds avidly to both Klotho proteins, we built a model of the FGF8b-FGFR1c complex using the FGF8b-FGFR2c structure (PDB ID, 2FDB [50]) (Fig. 7A). The FGF8b-FGFR1c model shows that, reminiscent of the interaction of FGF8b with FGFR2c (PDB ID, 2FDB [50]), hydrophobic residues from the N-terminal helix and the β4-β5 loop of FGF8b would engage the hydrophobic groove on receptor D3 formed by L290, L305, P306, and V308 of the constant region of D3 and T340, L342, L349, and H351 of the alternatively spliced region (Fig. 7A). The validity of the FGF8b-FGFR1c model is supported by our findings that mutation of L342 in FGFR1c to serine as occurs in Kallmann syndrome diminishes the binding affinity of FGFR1c for FGF8b by nearly 20-fold (53) and also abrogates FGF17b signaling through FGFR1c (unpublished data).
Fig 6.

Alignment of the D3 domain sequences of the seven principal human FGFRs. Residue numbers are in parentheses to the left of the alignment. Gaps (dashes) were introduced to optimize the alignment. Bars on top of the alignment indicate the location of the β strands in FGFR1c. A dashed line across the alignment marks the junction between the constant N-terminal half and the alternatively spliced C-terminal half of the D3 domain. Residues that form the hydrophobic groove in D3 of FGFR1c are shaded gray. Note that four of these residues map to the alternatively spliced region of D3. The positions of pathogenic mutations in the D3 domain of FGFR1 are indicated by boxes.
Fig 7.

Klotho proteins and FGF8b engage the same hydrophobic groove in D3 of FGFR1c. (A) Structural model of the FGF8b-FGFR1c complex. The model was created by superimposing the ligand-binding domain of FGFR1c from the FGF2-FGFR1c crystal structure (PDB ID, 1CVS [55]) onto the ligand-binding domain of FGFR2c in the FGF8b-FGFR2c crystal structure (PDB ID, 2FDB [50]). On the left is a view of the whole model, and on the right is a close-up view of the ligand-receptor D3 interface. FGF8b is shown as a ribbon diagram, and FGFR1c is shown as a space-filling molecular surface. Note that F32 and V36 of the N-terminal g helix and F93 of the β4-β5 loop of FGF8b bind to a hydrophobic groove in the D3 domain of FGFR1c formed by L290, L305, P306, V308, T340, L342, L349, and H351. NT and CT denote N and C termini of FGF8b and FGFR1c, respectively. (B, C, and D) Overlays of SPR sensorgrams illustrating binding of wild-type and mutant FGFR1c proteins to αKlotho (B), βKlotho (C), and FGF8b (D). αKlotho ectodomain, βKlotho ectodomain, or FGF8b was immobilized on biosensor chips, and increasing concentrations of either wild-type or mutant FGFR1c ligand-binding domain were passed over the chips. Maximal binding responses of FGFR1c mutants relative to wild-type protein were plotted (25). As we have previously reported (53), the L342S mutation causes a major loss in binding affinity of FGFR1c for FGF8b.
We first examined, by SPR spectroscopy, whether the L342S Kallmann syndrome mutation negatively affects the interaction of FGFR1c with Klotho proteins as in the case of FGF8b. As shown in Fig. 7B and C, the L342S mutation greatly reduced the ability of FGFR1c to bind either of the two Klotho proteins. The receptor's interaction with βKlotho was more affected than its interaction with αKlotho. These data reveal that L342 from the hydrophobic groove on FGFR1c D3 is essential for FGFR1c binding to both αKlotho and βKlotho, and hence, the data provide the first biochemical evidence for an overlap of the binding sites for FGF8b and Klotho proteins on receptor D3.
Using the FGF8b-FGFR1c model as a guide, we then mutated five additional constituents of the hydrophobic D3 groove and analyzed the impact of the mutations on binding of FGFR1c to αKlotho and βKlotho. As a control, binding of the receptor mutants to FGF8b was studied. L290 and V308 of the constant region of D3 were mutated to threonine, and T340, L349, and H351 from the alternatively spliced region were replaced with the corresponding residues of the “b” isoform of FGFR1, namely, valine, glutamate, and asparagine (Fig. 6). Each of the five mutations reduced the binding affinity of FGFR1c for both αKlotho and βKlotho, albeit to different degrees (Fig. 7B and C). The threonine substitutions at L290 and V308 caused a greater loss in binding affinity than did the T340V, L349E, and H351N mutations. The L290T, V308T, L349E, and H351N mutations also reduced the binding affinity of FGFR1c toward FGF8b (Fig. 7D), and as observed for the receptor's interaction with Klotho proteins, the replacement of L290 or V308 with threonine had a more pronounced effect than did the L349E and H351N mutations. Taken together, our findings show that the hydrophobic D3 groove of FGFR1c represents a shared binding pocket for Klotho proteins and ligands of the FGF8 subfamily. Importantly, as already mentioned above, this hydrophobic groove is conserved in the “c” isoforms of FGFR1 to FGFR3 and in FGFR4 (PDB IDs, 1CVS, 1DJS, 1EVT, 1EV2, and 1RY7 [49, 54, 55, 63]) (Fig. 6). This suggests that association of Klotho proteins with FGFRc isoforms and FGFR4 should obscure the binding site for FGF8 subfamily ligands on the receptor, resulting in inhibition of signaling by FGF8 subfamily ligands through FGFRc isoforms and FGFR4. To test this possibility, we studied the impact of βKlotho expression on the ability of FGF8b to activate FGFR in cells. HEK293 cells, which naturally express the “c” isoforms of FGFR1 to FGFR3 (33), were transfected with βKlotho or empty-vector control and then stimulated with FGF8b. As shown in Fig. 8, induction of Egr1 promoter activity by FGF8b was markedly reduced in cells expressing βKlotho compared to cells transfected with empty vector. These data show that βKlotho inhibits FGF8b signaling, thus providing biological evidence that the binding site for Klotho coreceptors on FGFR1c overlaps with the binding site for FGF8 subfamily ligands.
Fig 8.
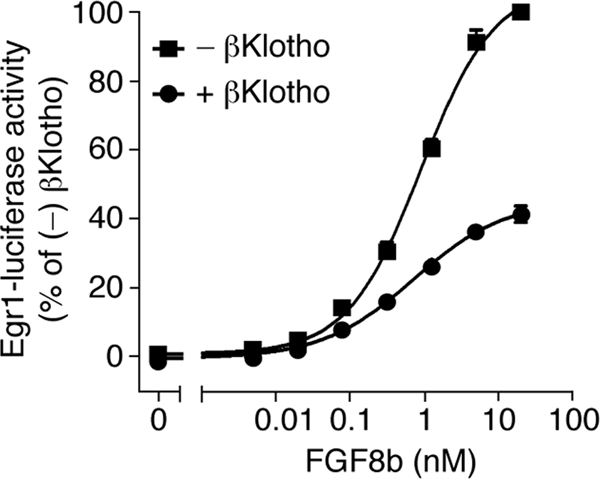
βKlotho inhibits FGF8b signaling. Shown are dose-response curves for Egr1 promoter-driven induction of luciferase activity by FGF8b in HEK293 cells transfected with βKlotho (+) or empty vector (−). Luciferase activity of cells expressing βKlotho was normalized to that of cells transfected with empty vector. Data points represent mean values of three independent experiments each performed in triplicate, and error bars denote standard deviations.
DISCUSSION
Our study shows that Klotho coreceptors utilize different mechanisms to promote binding of endocrine FGF ligands to their cognate FGFRs. αKlotho binds to FGFR to create a de novo binding site for FGF23 (16), whereas βKlotho binds FGFR and FGF19/FGF21 independently through two distinct binding sites. We also show that FGF19 and FGF21 use their respective C-terminal tails to bind to a common binding site on βKlotho. This binding site overlap may provide a molecular mechanism for why transgenic expression or therapeutic administration of FGF19 produces beneficial effects on glucose and lipid metabolism resembling those elicited by FGF21 (12, 66). Interestingly, FGF19 binds βKlotho with greater affinity than FGF21, implying that FGF19 would outcompete FGF21 for βKlotho, if both ligands were present in target tissue at the same time. Under physiological conditions, FGF19 and FGF21 do not appear to equally coexist in the blood circulation (4, 14, 18, 20, 21, 67). We speculate that the high-affinity interaction between βKlotho and FGF19, together with the binding preference of βKlotho for FGFR4, ensures that most of the postprandially secreted FGF19 acts on the liver (and the gallbladder) and hence becomes trapped in the enterohepatic circulation.
Importantly, we have mapped for the first time the binding sites for αKlotho and βKlotho on the D3 domain of FGFR1c and shown that the binding site for α/βKlotho on FGFR1c partially overlaps with the binding site for FGF8 subfamily ligands. The binding site overlap indicates that α/βKlotho coreceptors would inhibit signaling by FGF8 subfamily ligands, if α/βKlotho coreceptors and FGF8 subfamily ligands were coexpressed in the same tissue at the same time. Indeed, in the liver, the major target tissue of FGF19, FGF18 protein is coexpressed with βKlotho (2, 32), and similarly, in the kidney, the major target tissue of FGF23, FGF17 protein is coexpressed with αKlotho (35, 60, 68). Protein coexpression of FGF8 subfamily ligands and Klotho coreceptors also occurs in mature ovarian follicles (35, 38, 56) and in breast tissue (64, 72, 76). It should also be noted that other paracrine FGFs, including ligands of the FGF4 and FGF9 subfamilies, also coexist with α/βKlotho coreceptors in target tissues of endocrine FGFs such as pancreas (17) and adipose tissue (13). Based on these data, we propose that the association of Klotho proteins with FGFR not only enhances the binding affinity of the receptor for endocrine FGFs but at the same time also suppresses the binding and activation of FGFR by FGF8 subfamily ligands and possibly other paracrine FGFs. In essence, Klotho proteins modify the ligand-binding specificity of FGFR in favor of endocrine FGFs. While it is well established that coreceptors of a growth factor/cytokine receptor kinase serve to enhance the binding affinity of a given receptor for its cognate ligand (40), our study is the first to imply that a coreceptor can modify ligand-binding specificity of its receptor per se.
This function of Klotho coreceptors probably evolved to dedicate the FGFR in target cells of endocrine FGFs solely to endocrine FGFs without interference by paracrine FGFs. The inhibition of paracrine FGF signaling in target cells of endocrine FGFs also is consistent with the notion that paracrine FGFs possess much greater affinity for FGFR than do endocrine FGFs and therefore would outcompete endocrine FGFs for the receptor in the absence of a Klotho coreceptor. By inhibiting paracrine FGF signaling, Klotho proteins might also downregulate the paracrine FGF-mediated growth of target tissues of endocrine FGFs. Hence, we speculate that Klotho protein expression might be reduced in situations demanding tissue repair and regeneration to allow for more paracrine FGF signaling and restored to normal levels upon completion of tissue remodeling. Importantly, the inhibition of paracrine FGF signaling might represent a mechanism underlying the reported tumor-suppressive activity of Klotho proteins (37, 72). It should also be noted that αKlotho and βKlotho bind to a shared binding pocket on D3, suggesting that the two Klotho proteins would compete for FGFR, if coexpressed in the same tissue at the same time. In such a case, the expression level of αKlotho would affect the signaling through FGFR-βKlotho complex and vice versa. This might provide an explanation for the previously reported enhancement of putatively βKlotho-dependent metabolic effects when αKlotho is knocked out (44, 47, 69).
Our findings also have important pathophysiological implications. As introduced earlier, L342 of FGFR1c, a residue of the binding pocket shared by Klotho proteins and FGF8b, is the location of a pathogenic mutation. Specifically, substitution of L342 for serine causes Kallmann syndrome (53), a congenital disorder characterized by hypogonadotropic hypogonadism and anosmia. Our data show that the mutation not only disrupts FGFR1c interaction with FGF8b but also dramatically reduces the affinity of FGFR1c for both αKlotho and βKlotho. While previous investigations into Kallmann syndrome have identified impaired FGF8 signaling through the mutant FGFR1c as a pathogenetic mechanism (53), our data point to the possibility of deficiencies in endocrine FGF signaling and hence call for a clinical reevaluation of Kallmann syndrome patients carrying the L342S mutation. Our data also call for the reevaluation of patients carrying other pathogenic mutations in the binding pocket shared by Klotho proteins and paracrine FGFs, such as craniosynostosis mutations (71).
ACKNOWLEDGMENTS
This work was funded by U.S. National Institutes of Health grants DE13686 (M.M.), DK077276 (M.S.R.), and AG019712 and DK091392 (M.K.) and the Texas Higher Education Coordinating Board grant NHRP 010019-0043-2009 (M.K.). X.D. was supported by U.S. National Institutes of Health grant DK067158. Support for the Biacore 2000 SPR instrument was provided by grant P30 NS050276.
Footnotes
Published ahead of print 26 March 2012
REFERENCES
- 1. ADHR Consortium 2000. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 26:345–348 [DOI] [PubMed] [Google Scholar]
- 2. Antoine M, et al. 2006. Fibroblast growth factor 16 and 18 are expressed in human cardiovascular tissues and induce on endothelial cells migration but not proliferation. Biochem. Biophys. Res. Commun. 246:224–233 [DOI] [PubMed] [Google Scholar]
- 3. Asada M, et al. 2009. Glycosaminoglycan affinity of the complete fibroblast growth factor family. Biochim. Biophys. Acta 1790:40–48 [DOI] [PubMed] [Google Scholar]
- 4. Badman MK, et al. 2007. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5:426–437 [DOI] [PubMed] [Google Scholar]
- 5. Beenken A, Mohammadi M. 2009. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8:235–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beenken A, Mohammadi M. 2011. The structural biology of the FGF19 subfamily, p 1–24 In Kuro-o M. (ed), Endocrine FGFs and Klothos, 1st ed Landes Bioscience, Austin, TX [Google Scholar]
- 7. Bottcher RT, Niehrs C. 2005. Fibroblast growth factor signaling during early vertebrate development. Endocr. Rev. 26:63–77 [DOI] [PubMed] [Google Scholar]
- 8. Cantarel BL, et al. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chellaiah AT, McEwen DG, Werner S, Xu J, Ornitz DM. 1994. Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. J. Biol. Chem. 269:11620–11627 [PubMed] [Google Scholar]
- 10. Cole LW, et al. 2008. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J. Clin. Endocrinol. Metab. 93:3551–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fisher FM, et al. 2011. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology 152:2996–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fu L, et al. 2004. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145:2594–2603 [DOI] [PubMed] [Google Scholar]
- 13. Gabrielsson BG, et al. 2002. Depot-specific expression of fibroblast growth factors in human adipose tissue. Obes. Res. 10:608–610 [DOI] [PubMed] [Google Scholar]
- 14. Galman C, et al. 2008. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 8:169–174 [DOI] [PubMed] [Google Scholar]
- 15. Goetz R, et al. 2007. Molecular insights into the Klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 27:3417–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goetz R, et al. 2010. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. U. S. A. 107:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hart AW, Baeza N, Apelqvist A, Edlund H. 2000. Attenuation of FGF signalling in mouse β-cells leads to diabetes. Nature 408:864–868 [DOI] [PubMed] [Google Scholar]
- 18. Holt JA, et al. 2003. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 17:1581–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ibrahimi OA, et al. 2004. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum. Mol. Genet. 13:2313–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Inagaki T, et al. 2005. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2:217–225 [DOI] [PubMed] [Google Scholar]
- 21. Inagaki T, et al. 2007. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5:415–425 [DOI] [PubMed] [Google Scholar]
- 22. Ito S, et al. 2000. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech. Dev. 98:115–119 [DOI] [PubMed] [Google Scholar]
- 23. Itoh N, Ornitz DM. 2004. Evolution of the Fgf and Fgfr gene families. Trends Genet. 20:563–569 [DOI] [PubMed] [Google Scholar]
- 24. Itoh N, Ornitz DM. 2011. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J. Biochem. 149:121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang J, Prasad K, Lafer EM, Sousa R. 2005. Structural basis of interdomain communication in the Hsc70 chaperone. Mol. Cell 20:513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson DE, Lu J, Chen H, Werner S, Williams LT. 1991. The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol. 11:4627–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalinina J, et al. 2009. Homodimerization controls the fibroblast growth factor 9 subfamily's receptor binding and heparan sulfate-dependent diffusion in the extracellular matrix. Mol. Cell. Biol. 29:4663–4678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kharitonenkov A, et al. 2008. FGF-21/FGF-21 receptor interaction and activation is determined by betaKlotho. J. Cell. Physiol. 215:1–7 [DOI] [PubMed] [Google Scholar]
- 29. Kharitonenkov A, et al. 2005. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115:1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kir S, et al. 2011. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 331:1621–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuro-o M, et al. 1997. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51 [DOI] [PubMed] [Google Scholar]
- 32. Kurosu H, et al. 2007. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 282:26687–26695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kurosu H, et al. 2006. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 281:6120–6123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kurosu H, et al. 2005. Suppression of aging in mice by the hormone Klotho. Science 309:1829–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li SA, et al. 2004. Immunohistochemical localization of klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct. Funct 29:91–99 [DOI] [PubMed] [Google Scholar]
- 36. Lundasen T, Galman C, Angelin B, Rudling M. 2006. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 260:530–536 [DOI] [PubMed] [Google Scholar]
- 37. Luo Y, et al. 2010. Metabolic regulator betaKlotho interacts with fibroblast growth factor receptor 4 (FGFR4) to induce apoptosis and inhibit tumor cell proliferation. J. Biol. Chem. 285:30069–30078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Machado MF, et al. 2009. Regulation and action of fibroblast growth factor 17 in bovine follicles. J. Endocrinol. 202:347–353 [DOI] [PubMed] [Google Scholar]
- 39. Makarenkova HP, et al. 2009. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci. Signal. 2:ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Massague J. 1998. TGF-beta signal transduction. Annu. Rev. Biochem. 67:753–791 [DOI] [PubMed] [Google Scholar]
- 41. Micanovic R, et al. 2009. Different roles of N- and C-termini in the functional activity of FGF21. J. Cell. Physiol. 219:227–234 [DOI] [PubMed] [Google Scholar]
- 42. Miki T, et al. 1992. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc. Natl. Acad. Sci. U. S. A. 89:246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mohammadi M, Olsen SK, Ibrahimi OA. 2005. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16:107–137 [DOI] [PubMed] [Google Scholar]
- 44. Mori K, et al. 2000. Disruption of klotho gene causes an abnormal energy homeostasis in mice. Biochem. Biophys. Res. Commun. 278:665–670 [DOI] [PubMed] [Google Scholar]
- 45. National Research Council 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC [Google Scholar]
- 46. Ogawa Y, et al. 2007. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. U. S. A. 104:7432–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ohnishi M, Kato S, Akiyoshi J, Atfi A, Razzaque MS. 2011. Dietary and genetic evidence for enhancing glucose metabolism and reducing obesity by inhibiting klotho functions. FASEB J. 25:2031–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olsen SK, et al. 2003. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 278:34226–34236 [DOI] [PubMed] [Google Scholar]
- 49. Olsen SK, et al. 2004. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. U. S. A. 101:935–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Olsen SK, et al. 2006. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 20:185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ornitz DM, et al. 1996. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271:15292–15297 [DOI] [PubMed] [Google Scholar]
- 52. Orr-Urtreger A, et al. 1993. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev. Biol. 158:475–486 [DOI] [PubMed] [Google Scholar]
- 53. Pitteloud N, et al. 2007. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Invest. 117:457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. 2000. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 101:413–424 [DOI] [PubMed] [Google Scholar]
- 55. Plotnikov AN, Schlessinger J, Hubbard SR, Mohammadi M. 1999. Structural basis for FGF receptor dimerization and activation. Cell 98:641–650 [DOI] [PubMed] [Google Scholar]
- 56. Portela VM, et al. 2010. Expression and function of fibroblast growth factor 18 in the ovarian follicle in cattle. Biol. Reprod. 83:339–346 [DOI] [PubMed] [Google Scholar]
- 57. Potthoff MJ, et al. 2011. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab. 13:729–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Potthoff MJ, et al. 2009. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. U. S. A. 106:10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Razzaque MS. 2009. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat. Rev. Endocrinol. 5:611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. R&D Systems Immunohistochemistry Laboratory 2011. FGF17 in human kidney. R&D Systems, Minneapolis, MN: www.rndsystems.com/pdf/mab319/pdf [Google Scholar]
- 61. Schlessinger J, et al. 2000. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6:743–750 [DOI] [PubMed] [Google Scholar]
- 62. Shimada T, et al. 2004. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 113:561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stauber DJ, DiGabriele AD, Hendrickson WA. 2000. Structural interactions of fibroblast growth factor receptor with its ligands. Proc. Natl. Acad. Sci. U. S. A. 97:49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tanaka A, et al. 1998. High frequency of fibroblast growth factor (FGF) 8 expression in clinical prostate cancers and breast tissues, immunohistochemically demonstrated by a newly established neutralizing monoclonal antibody against FGF8. Cancer Res. 58:2053–2056 [PubMed] [Google Scholar]
- 65. Thisse B, Thisse C. 2005. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev. Biol. 287:390–402 [DOI] [PubMed] [Google Scholar]
- 66. Tomlinson E, et al. 2002. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 143:1741–1747 [DOI] [PubMed] [Google Scholar]
- 67. Tong X, et al. 2010. Transcriptional repressor E4-binding protein 4 (E4BP4) regulates metabolic hormone fibroblast growth factor 21 (FGF21) during circadian cycles and feeding. J. Biol. Chem. 285:36401–36409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Urakawa I, et al. 2006. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774 [DOI] [PubMed] [Google Scholar]
- 69. Utsugi T, et al. 2000. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism 49:1118–1123 [DOI] [PubMed] [Google Scholar]
- 70. White KE, et al. 2001. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 60:2079–2086 [DOI] [PubMed] [Google Scholar]
- 71. Wilkie AO. 2005. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 16:187–203 [DOI] [PubMed] [Google Scholar]
- 72. Wolf I, et al. 2008. Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 27:7094–7105 [DOI] [PubMed] [Google Scholar]
- 73. Wu X, et al. 2008. C-terminal tail of FGF19 determines its specificity toward Klotho co-receptors. J. Biol. Chem. 283:33304–33309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yeh BK, et al. 2003. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc. Natl. Acad. Sci. U. S. A. 100:2266–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yie J, et al. 2009. FGF21 N- and C-termini play different roles in receptor interaction and activation. FEBS Lett. 583:19–24 [DOI] [PubMed] [Google Scholar]
- 76. Zammit C, et al. 2002. Fibroblast growth factor 8 is expressed at higher levels in lactating human breast and in breast cancer. Br. J. Cancer. 86:1097–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang X, et al. 2006. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281:15694–15700 [DOI] [PMC free article] [PubMed] [Google Scholar]