

Abstract
Melioidosis, caused by the Gram-negative bacterium Burkholderia pseudomallei, is an important cause of community-acquired sepsis in Southeast Asia and northern Australia. An important controller of the immune system is the pleiotropic cytokine transforming growth factor β (TGF-β), of which Smad2 and Smad3 are the major signal transducers. In this study, we aimed to characterize TGF-β expression and function in experimental melioidosis. TGF-β expression was determined in 33 patients with culture-proven infection with B. pseudomallei and 30 healthy controls. We found that plasma TGF-β concentrations were strongly elevated during melioidosis. In line with this finding, TGF-β expression in C57BL/6 mice intranasally inoculated with B. pseudomallei was enhanced as well. To assess the role of TGF-β, we inhibited TGF-β using a selective murine TGF-β antibody. Treatment of mice with anti-TGF-β antibody resulted in decreased lung Smad2 phosphorylation. TGF-β blockade appeared to be protective: mice treated with anti-TGF-β antibody and subsequently infected with B. pseudomallei showed diminished bacterial loads. Moreover, less distant organ injury was observed in anti-TGF-β treated mice as shown by reduced blood urea nitrogen (BUN) and aspartate transaminase (AST) values. However, anti-TGF-β treatment did not have an effect on survival. In conclusion, TGF-β is upregulated during B. pseudomallei infection and plays a limited but proinflammatory role during experimental melioidosis.
INTRODUCTION
The pleiotropic cytokine transforming growth factor β (TGF-β) is regarded as a key regulator of the immune response (30). TGF-β is produced by many different body cells, including every leukocyte lineage, of both the innate and adaptive immune systems. Its expression serves in both autocrine and paracrine modes to control the differentiation, proliferation, and state of activation of these immune cells (7). TGF-β1, -β2, and -β3 are the three isoforms that have been identified in mammals, of which TGF-β1 is predominantly expressed (1, 2). TGF-β is recognized by the transmembranous TGF-β receptor complex, after which phosphorylation of Smad2 and Smad3 will result in the formation of hetero-oligomeric complexes with Smad4. This complex then translocates to the nucleus and regulates transcriptional responses together with DNA binding cofactors (1, 2). TGF-β has multiple immunosuppressive properties, as evidenced by the observation that TGF-β knockout (KO) mice develop multiorgan autoimmune inflammatory disease and die shortly after birth (6, 14). In addition, it is known that gamma interferon (IFN-γ) signaling pathways are dysregulated in the absence of TGF-β (13). Of interest, macrophages become important producers of TGF-β upon activation by phagocytosis of apoptotic cells which could constitute a mechanism involving the anti-inflammatory effect of apoptotic cells (9). Recent studies have intriguingly revealed additional potential proinflammatory roles of TGF-β in the inflammatory response. Interestingly, TGF-β can induce interleukin-17 (IL-17) producing proinflammatory Th17 cells in the presence of IL-6 (17, 30) and functions as a chemoattractant for monocytes and granulocytes (9, 10).
It has been known for many years that TGF-β levels are elevated in patients with sepsis (11). However, the function of increased TGF-β expression in the host response against invading bacteria during sepsis remains unknown. Recent evidence suggests that TGF-β is vital in the regulation of the balance between the initial Toll-like receptor (TLR)-induced protective response and the dysregulated immune response that sometimes follows in sepsis (12). Melioidosis, caused by the Gram-negative bacillus Burkholderia pseudomallei, is an important cause of community-acquired sepsis in Southeast Asia and northern Australia and regarded as a good model to study Gram-negative sepsis given both the homogeneity and severity of its clinical picture (1, 15, 22). More than half of the cases of melioidosis present with pneumonia, frequently associated with bacterial dissemination to distant sites (1, 2, 22). A recent gene expression study found that the TGF-β/Smad pathway is highly differentially regulated in melioidosis, ranking above even the IL-1, TLR, and Nod-like receptor (NLR) pathways (5) (G. C. K. W. Koh, personal communication). It has been recognized that severe melioidosis probably can be seen as the clinical manifestation of a TLR-mediated dysregulation of the immune response to invading B. pseudomallei (20, 24, 25, 27).
In the present study, we aimed to characterize the expression and function of TGF-β in melioidosis. To do so, we analyzed TGF-β expression patterns in patients with melioidosis and in a mouse model of B. pseudomallei infection. TGF-β function was investigated in experimental murine melioidosis using anti-TGF-β antibodies.
MATERIALS AND METHODS
Ethics statement.
The patient study was approved by both the Ministry of Public Health, Royal Government of Thailand, and the Oxford Tropical Research Ethics Committee, University of Oxford, England. We obtained written informed consent from all subjects before the study. The Animal Care and Use Committee of the University of Amsterdam approved all mouse experiments.
Patients.
We included 33 individuals with sepsis caused by B. pseudomallei and 30 healthy controls in this study. Individuals were recruited prospectively at Sapprasithiprasong Hospital, Ubon Ratchathani, Thailand, in 2004. Sepsis due to melioidosis was defined as culture positivity for B. pseudomallei from any clinical sample plus a systemic inflammatory response syndrome (SIRS) (8). Study design and subjects have been described in detail (24).
Murine melioidosis.
Male C57BL/6 mice (age 8 to 10 weeks) were purchased from Harlan Sprague Dawley Inc. (Horst, The Netherlands). Age-matched animals were used in each experiment. For the inoculum, B. pseudomallei strain 1026b was used and prepared as described previously (23, 26). Pneumonia was induced by intranasal inoculation of 3.0 × 102 or 7.5 × 102 CFU (CFU/50 μl). Seventy-two hours after infection, mice were anesthetized and sacrificed by bleeding from the vena cava inferior (24). Organs were harvested and homogenized, after which CFU were determined from serial dilutions of homogenates as described previously (24). In depletion experiments, mice were injected intraperitoneally with 200 μg of a monoclonal anti-TGF-β (1D11, anti-TGF-β1, -β2, and -β3; Bioceros) or an isotype-matched control IgG antibody (anti-β-galactosidase, G113; Bioceros) 2 h before and 48 h after bacterial inoculation (16, 19).
Assays.
For cytokine measurements, lung and spleen homogenates were diluted 1:2 in lysis buffer containing 300 mM NaCl, 30 mM Tris, 2 mM MgCl2, 2 mM CaCl2, 1% Triton X-100, and AEBSF (4-(2-aminoethyl-benzenesulfonyl fluoride), EDTA-Na2, pepstatin, and leupeptin (all 8 μg/ml; pH 7.4) and incubated at 4°C for 30 min. Homogenates were centrifuged at 1,500 × g at 4°C for 15 min, and supernatants were stored at −20°C until assays were performed. Human and murine TGF-β was measured by enzyme-linked immunosorbent assay (ELISA) according to the instructions of the manufacturer (both R&D systems, Minneapolis, MN). IL-1β, IL-6, IL-17, keratinocyte chemoattractant (KC), monocyte chemoattractant protein 1 (MCP-1) and tumor necrosis factor alpha (TNF-α) were determined by ELISA (BD Biosciences, San Jose, CA). For Western blot analysis, lung homogenates were prepared as described above, with addition of 1 mM sodium orthovanadate (Sigma-Aldrich, St. Louis, MO). Samples were separated by SDS-PAGE and transferred onto activated polyvinylidene difluoride (PVDF) membranes (Millipore, Etten Leur, The Netherlands). Membranes were blocked with either 5% (wt/vol) nonfat dry milk in Tris-buffered saline containing 0.1% Tween (TBS-T) (actin) or 5% bovine serum albumin (Sigma) in TBS-T (Smad2/3, phospho-Smad2/3). Blots were probed with primary antibody (1:1,000; BD Transduction Laboratories for Smad2/3; Santa Cruz Biotechnology, Santa Cruz, CA, for phospho-Smad2/3 and actin), followed by incubation with a horseradish peroxidase (HRP)-conjugated secondary antibody. HRP activity was visualized with ECL reagent (Amersham Pharmacia Biotech, Roosendaal, The Netherlands) using a Lumi-Imager (Roche Diagnostics, Almere, The Netherlands). Aspartate transaminase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine were determined with commercially available kits (Sigma) using a Hitachi analyzer (Boehringer Mannheim, Mannheim, Germany).
Statistical analysis.
Values are expressed as means ± standard error of the mean (SEM). Differences between groups were analyzed by Mann-Whitney U test. Wilcoxon matched pairs test was used for analysis of continuous variables. For survival analysis, Kaplan-Meier analysis followed by log rank test was performed. These analyses were performed using GraphPad Prism version 5.01, GraphPad Software (San Diego, CA). Values of P of <0.05 were considered statistically significant.
RESULTS
Increased TGF-β expression in patients with severe melioidosis.
To obtain insight into TGF-β expression during melioidosis, we first quantified TGF-β levels in plasma from 33 patients with culture-proven B. pseudomallei infection and 30 local healthy controls. TGF-β expression was significantly elevated in patients with melioidosis (5,223 pg/ml ± 909.2) compared to controls (3,144 pg/ml ± 433.8; P < 0.05) (Fig. 1). In five patients from whom blood could be obtained after recovery (14 days after admission), TGF-β levels showed complete normalization (Fig. 1). The mortality rate in this cohort of patients was 44%. It has been previously reported that TGF-β levels at admission are associated with poor outcome in patients with pneumonia-induced sepsis (29). However, in our cohort of septic patients, elevation of plasma TGF-β level was not associated with an adverse outcome: on admission, patients who went on to die did not have higher TGF-β concentrations than those who survived (data not shown).
Fig 1.

TGF-β levels are elevated in melioidosis patients. (A) On admission, TGF-β levels in plasma of patients with culture-proven melioidosis (n = 33) were elevated compared to healthy controls (n = 30). (B) Plasma levels of TGF-β decreased in melioidosis patients 2 weeks after successful antibiotic treatment (n = 6). *, P < 0.05.
Elevated TGF-β levels in lung tissue during experimental pneumonia-derived melioidosis.
A large number of patients with melioidosis present with pneumonia with bacterial dissemination to distant organs (3, 10, 22). Considering that it is not feasible to study TGF-β expression at tissue level in patients, we made use of our well-established murine model of experimental melioidosis in which mice are intranasally infected with a lethal dose of B. pseudomallei (24, 26, 28). At 72 h postinfection, TGF-β concentrations measured in the pulmonary compartment were significantly elevated compared to baseline (P < 0.001) (Fig. 2), which is in full agreement with the observed increase in TGF-β level in patients.
Fig 2.
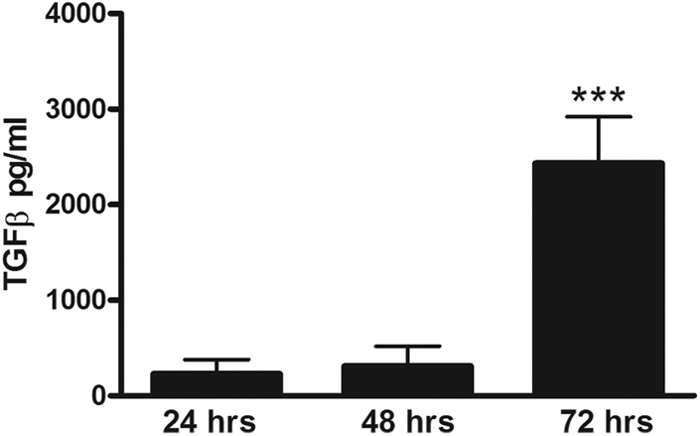
Increased expression of TGF-β in lung tissue of wild-type mice infected with B. pseudomallei. Elevated pulmonary TGF-β levels were observed in mice (n = 8) 72 h after intranasal infection with 7.5 × 102 CFU of B. pseudomallei compared to 24 h after infection. ***, P < 0.001.
Anti-TGF-β treatment results in decreased lung Smad2 phosphorylation and a diminished inflammatory response.
To acquire further understanding of the function of TGF-β during melioidosis, we treated wild-type mice inoculated with 7.5 × 102 CFU of B. pseudomallei with a neutralizing anti-TGF-β antibody or control IgG antibody. We first evaluated whether the anti-TGF-β antibody was successful in inhibiting Smad signaling, which is the major pathway activated by TGF-β. Therefore, we measured total and phosphorylated Smad2 and Smad3 expression using Western blot analysis and found that mice treated with anti-TGF-β showed decreased Smad2 phosphorylation. These data indicate inhibition of the Smad pathway (Fig. 3). To determine the effect of the inhibition of TGF-β and Smad2/3 signaling on inflammation, we first measured cytokine levels in whole lung and spleen homogenates harvested from both the anti-TGF-β-treated and control mice at 72 h postinfection, with a lethal dose of B. pseudomallei. Despite decreased MCP-1 levels in lung homogenates of anti-TGF-β-treated mice, we could not demonstrate significantly lower levels of IL-1β, IL-6, and TNF-α in both the lungs and spleens of these mice (Table 1). No difference in IL-17, a TGF-β driven cytokine, was found in both organs. In addition, we evaluated the effect of TGF-β suppression on organ damage as reflected in kidney and liver function tests. A marked decrease in AST (88.0 U/liter ± 65.8 versus 414.3 U/liter ± 369.2) and BUN (5.7 ± 3.2 versus 18.4 ± 14.8) levels was seen in mice treated with anti-TGF-β, demonstrating less organ damage in this group (P < 0.05) (Fig. 4).
Fig 3.

Decreased expression of phosphorylated Smad2 in mice receiving anti-TGF-β treatment. TGF-β signaling was evaluated by quantifying the total levels of Smad2 and Smad3 and their phosphorylation in lung tissue by Western blot 48 h after intranasal infection with B. pseudomallei. The left 4 lanes show the expression of Smad2 (A) and Smad3 (B) in lung tissue of control mice and the right 4 lanes of mice that received anti-TGF-β treatment (2 × 200 μg TGFβ antibody). The bar graph shows the ratio of expressed phosphorylated Smad/total Smad corrected for actin loading levels. (C) A significant decrease in p-Smad2 is demonstrated in mice that were treated with anti-TGF-β antibody. *, P < 0.05.
Table 1.
Cytokine profile during experimental melioidosis in mice treated with control antibody or anti-TGF-βa
| Cytokine/organ | Cytokine level (pg/ml) |
|
|---|---|---|
| Control antibody | Anti-TGF-β | |
| Lungs | ||
| IL-1β | 12,416 ± 1,626 | 9,036 ± 1,540 |
| IL-6 | 14,954 ± 5,050 | 5,037 ± 2,066 |
| IL-17 | 192.9 ± 14.92 | 183 ± 17.86 |
| KC | 18,777 ± 3,996 | 10,524 ± 2,741 |
| MCP-1 | 22,490 ± 3,105* | 13,095 ± 3,049 |
| TNF-α | 4,859 ± 671 | 3,392 ± 628.5 |
| Spleen | ||
| IL-1β | 710 ± 176.6 | 827 ± 79.42 |
| IL-6 | 920.2 ± 453.8 | 111.4 ± 47.73 |
| IL-17 | 62.59 ± 4.21 | 84.58 ± 16.4 |
| KC | 7,300 ± 1,840 | 3,061 ± 514.5 |
| MCP-1 | 3,502 ± 1,824 | 831.8 ± 434.9 |
| TNF-α | 239.7 ± 134.5 | 180.7 ± 92.83 |
Mice (n = 8/group) were inoculated with 7.5 × 102 CFU of B. pseudomallei intranasally and treated with anti-TGF-β antibody or control IgG antibody. After 72 h, mice were sacrificed, lungs and spleens were removed and homogenized, and cytokines were measured. Data are expressed as mean ± SEM.
, P < 0.05.
Fig 4.

Less organ damage in mice receiving anti-TGF-β treatment. Levels of (A) BUN, (B) creatinine, (C) AST, and (D) ALT were measured in B. pseudomallei-infected mice (n = 8/group) that were treated with anti-TGF-β antibody and their controls at 72 h as parameters for organ damage. Expression of AST and BUN were decreased, demonstrating less organ damage. *, P < 0.05. Data shown from single independent experiments.
TGF-β facilitates bacterial growth.
To determine whether TGF-β impacts on bacterial growth during melioidosis, we determined bacterial loads in lung homogenate (the primary site of infection), blood, and spleen (to evaluate dissemination of the bacterium to distant sites) from B. pseudomallei-infected mice treated with anti-TGF-β or control antibody. Five out of seven blood cultures were positive in the control group compared to 1 out of 7 in the anti-TGF-β-treated group (data not shown). In addition, bacterial growth was significantly decreased in the lungs and spleens of mice that were administered anti-TGF-β antibody, showing a protective effect of TGF-β inhibition (P < 0.05) (Fig. 5).
Fig 5.

Anti-TGF-β treatment results in diminished bacterial growth during experimental melioidosis. Mice (n = 8/group) were treated with anti-TGF-β antibody or control IgG antibody and inoculated with 7.5 × 102 CFU of B. pseudomallei intranasally. Bacterial numbers were quantified 72 h postinfection. Less growth was seen in the lungs (A) and spleens (B) of mice treated with anti-TGF-β antibody. *, P < 0.05. Data shown from single independent experiments.
No beneficial effect on survival of anti-TGF-β treatment.
The experiments above demonstrated a protective effect of anti-TGF-β treatment on inflammation and bacterial growth during experimental melioidosis. Given that TGF-β is upregulated in patients with severe melioidosis and that anti-TGF-β treatment has anti-inflammatory effects and leads to less bacterial growth in the lungs and spleen, we next assessed the effect of TGF-β inhibition on survival in experimental melioidosis. Mice received anti-TGF-β antibody or control IgG antibody 2 h prior and 48 h after infection with low-dose (3.0 × 102 CFU) or high-dose (7.5 × 102 CFU) B. pseudomallei and were followed until death. No differences in time to death were observed between all groups, demonstrating no effect on survival of anti-TGF-β treatment despite its observed protective effect on inflammation during both low-dose and high-dose infection (shown for 7.5 × 102 CFU B. pseudomallei in Fig. 6).
Fig 6.

No influence on survival of anti-TGF-β treatment during murine melioidosis. Mice (n = 12/group) received either 2× 200 μg control antibody (black squares) or 2× 200 μg anti-TGF-β antibody (white squares) and were infected with 7.5 × 102 CFU, a lethal dose, of B. pseudomallei intranasally, after which they were followed until death. Mortality was assessed every 4 h. No difference in survival was seen.
DISCUSSION
In the present study, we aimed to characterize the expression and function of the pleiotropic cytokine TGF-β in melioidosis, linking observational studies in patients with culture-proven disease with functional studies in mice. Our study demonstrates that patients with severe melioidosis have strongly increased TGF-β levels in plasma. In line, TGF-β expression increased over time in mice intranasally inoculated with B. pseudomallei. To assess the function of TGF-β during melioidosis, we used a depleting TGF-β antibody, which successfully inhibited the major TGF-β signaling Smad pathway. In mice treated with this anti-TGF-β antibody, diminished bacterial outgrowth was seen and less distant organ damage. However, the observed protective effects of anti-TGF-β treatment did not translate into an improved survival in our model of experimental melioidosis.
To our knowledge, this is the first report on the role of TGF-β in melioidosis. Earlier studies on TGF-β expression during severe bacterial infection are conflicting: in patients with sepsis, both up- and downregulation of TGF-β have been described (11, 21, 29). In patients with Chagas' disease, caused by the parasite Trypanosoma cruzi, elevated levels of TGF-β are seen (18). Of note, high levels of circulating TGF-β during sepsis caused by community-acquired pneumonia have been shown to correlate with higher APACHE II scores and mortality (29). Our study demonstrates that severe melioidosis is characterized by high levels of TGF-β as well, although we could not demonstrate a relation between TGF-β levels and mortality in our cohort of patients.
In our study TGF-β inhibition led to less bacterial growth and decreased organ damage. The mechanism in which TGF-β inhibition exerts its protective effects during melioidosis remains to be clarified. TGF-β has been shown in previous studies to control both the innate and adaptive immune system by its anti-inflammatory properties (4). Yet, TGF-β can also induce differentiation of T helper cells into proinflammatory IL-17, producing T lymphocytes (Th17 cells) (17, 30). Surprisingly, decreased IL-17 levels have been found in diabetic melioidosis patients. To assess whether the protective effect of anti-TGF-β treatment was caused by inhibition of Th17 cells, we measured IL-17 levels in the lungs, spleen, and plasma by ELISA. However, there was no difference in IL-17 production in mice treated with the anti-TGF-β antibody and their controls (Table 1).
The function of TGF-β in immune responses depends heavily on its surroundings, for example, on circulating cytokines and chemokines and differentiation state of target cells. Although TGF-β might be upregulated in the event of invading pathogens, this does not always seem to contribute to efficient bacterial clearance. It has been described that TGF-β expression is upregulated and exploited by some pathogens, such as T. cruzi and Francisella tularensis, to facilitate entry, replication, and persistence in the host. In these instances, elevated TGF-β expression might suppress the killing activity of macrophages, thereby enhancing the intracellular proliferation of the microorganisms and facilitating virulence (9). The facultative intracellular bacterium B. pseudomallei might use a similar mechanism to remain virulent after host invasion in order to try to escape from the immune response.
Our study has several limitations. We used a selective TGF-β antibody instead of TGF-β KO mice, resulting in incomplete suppression of TGF-β activity. As mentioned above, TGF-β KO mice die shortly after birth due to major inflammatory disease. We made use of TGF-β blocking antibodies; however, it should be noted that, currently, no valid bioactivity assay exists to measure TGF-β activity. Nevertheless, the selective anti-TGF-β antibody was successful in inhibiting the Smad pathway largely. It has to be noted, however, that TGF-β can activate Smad-independent pathways as well (30).
In conclusion, TGF-β levels are strongly increased during severe melioidosis. Inhibition of TGF-β with a selective TGF-β antibody has a protective effect: inflammation was reduced, and less bacterial growth and distant organ damage were seen. However, this protective effect did not improve survival. Although it is clear that TGF-β is involved in the pathogenesis of melioidosis, its role in host defense against B. pseudomallei seems to be limited.
ACKNOWLEDGMENTS
We are grateful to Marieke ten Brink and Joost Daalhuisen for expert technical assistance.
W. J. Wiersinga is supported by a VENI grant from the Netherlands Organization of Scientific Research.
Footnotes
Published ahead of print 13 February 2012
REFERENCES
- 1. Cheng AC, Currie BJ. 2005. Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18:383–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Currie BJ. 2003. Melioidosis: an important cause of pneumonia in residents of and travellers returned from endemic regions. Eur. Respir. J. 22:542–550 [DOI] [PubMed] [Google Scholar]
- 3. Currie BJ, Ward L, Cheng AC. 2010. The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl. Trop. Dis. 4:e900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. 2010. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 10:554–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koh GC, et al. 2011. Glyburide is anti-inflammatory and associated with reduced mortality in melioidosis. Clin. Infect. Dis. 52:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kulkarni AB, Karlsson S. 1993. Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am. J. Pathol. 143:3–9 [PMC free article] [PubMed] [Google Scholar]
- 7. Letterio JJ, Roberts AB. 1998. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 16:137–161 [DOI] [PubMed] [Google Scholar]
- 8. Levy MM, et al. 2003. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit. Care Med. 31:1250–1256 [DOI] [PubMed] [Google Scholar]
- 9. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. 2006. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 24:99–146 [DOI] [PubMed] [Google Scholar]
- 10. Limmathurotsakul D, Chaowagul W, Day NP, Peacock SJ. 2009. Patterns of organ involvement in recurrent melioidosis. Am. J. Trop. Med. Hyg. 81:335–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marie C, Cavaillon JM, Losser MR. 1996. Elevated levels of circulating transforming growth factor-beta 1 in patients with the sepsis syndrome. Ann. Intern. Med. 125:520–521 [DOI] [PubMed] [Google Scholar]
- 12. McCartney-Francis N, Jin W, Wahl SM. 2004. Aberrant Toll receptor expression and endotoxin hypersensitivity in mice lacking a functional TGF-beta 1 signaling pathway. J. Immunol. 172:3814–3821 [DOI] [PubMed] [Google Scholar]
- 13. McCartney-Francis NL, Wahl SM. 2002. Dysregulation of IFN-gamma signaling pathways in the absence of TGF-beta 1. J. Immunol. 169:5941–5947 [DOI] [PubMed] [Google Scholar]
- 14. Shull MM, et al. 1992. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359:693–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simpson AJ. 2001. Melioidosis: a clinical model for gram-negative sepsis. J. Med. Microbiol. 50:657–658 [DOI] [PubMed] [Google Scholar]
- 16. Smits HH, et al. 2009. Cholera toxin B suppresses allergic inflammation through induction of secretory IgA. Mucosal Immunol. 2:331–339 [DOI] [PubMed] [Google Scholar]
- 17. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. 2006. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 24:179–189 [DOI] [PubMed] [Google Scholar]
- 18. Waghabi MC, et al. 2009. Pharmacological inhibition of transforming growth factor beta signaling decreases infection and prevents heart damage in acute Chagas' disease. Antimicrob. Agents Chemother. 53:4694–4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walsh KP, Brady MT, Finlay CM, Boon L, Mills KH. 2009. Infection with a helminth parasite attenuates autoimmunity through TGF-beta-mediated suppression of Th17 and Th1 responses. J. Immunol. 183:1577–1586 [DOI] [PubMed] [Google Scholar]
- 20. West TE, Ernst RK, Jansson-Hutson MJ, Skerrett SJ. 2008. Activation of Toll-like receptors by Burkholderia pseudomallei. BMC Immunol. 9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. White M, et al. 2010. Transforming growth factor beta-1 and interleukin-17 gene transcription in peripheral blood mononuclear cells and the human response to infection. Cytokine 50:322–327 [DOI] [PubMed] [Google Scholar]
- 22. Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. 2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4:272–282 [DOI] [PubMed] [Google Scholar]
- 23. Wiersinga WJ, et al. 2007. Endogenous interleukin-18 improves the early antimicrobial host response in severe melioidosis. Infect. Immun. 75:3739–3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wiersinga WJ, et al. 2007. Toll-like receptor 2 impairs host defense in gram-negative sepsis caused by Burkholderia pseudomallei (Melioidosis). PLoS Med. 4:e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wiersinga WJ, Wieland CW, Roelofs JJ, van der Poll T. 2008. MyD88 dependent signaling contributes to protective host defense against Burkholderia pseudomallei. PLoS One 3:e3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wiersinga WJ, de Vos AF, de Beer R, R B, Wieland CW, Roelofs JJ, Woods DE, van der Poll T. 2008. Inflammation patterns induced by different Burkholderia species in mice. Cell Microbiol. 10:81–87 [DOI] [PubMed] [Google Scholar]
- 27. Wiersinga WJ, van der Poll T. 2009. Immunity to Burkholderia pseudomallei. Curr. Opin. Infect. Dis. 22:102–108 [DOI] [PubMed] [Google Scholar]
- 28. Wiersinga WJ, et al. 2010. Urokinase receptor is necessary for bacterial defense against pneumonia-derived septic melioidosis by facilitating phagocytosis. J. Immunol. 184:3079–3086 [DOI] [PubMed] [Google Scholar]
- 29. Wu HP, et al. 2009. Plasma transforming growth factor-beta1 level in patients with severe community-acquired pneumonia and association with disease severity. J. Formos. Med. Assoc. 108:20–27 [DOI] [PubMed] [Google Scholar]
- 30. Yoshimura A, Wakabayashi Y, Mori T. 2010. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 147:781–792 [DOI] [PMC free article] [PubMed] [Google Scholar]