

Abstract
An important factor that defines the toxicity of elements such as cadmium(II), mercury(II), and lead(II) with biological macromolecules is metal ion exchange dynamics. Intriguingly, little is known about the fundamental rates and mechanisms of metal ion exchange into proteins, especially helical bundles. Herein, we investigate the exchange kinetics of cadmium(II) using de novo designed three-stranded coiled coil peptides that contain metal complexing cysteine thiolates as a model for the incorporation of this ion into trimeric, parallel helical bundles. Peptides were designed containing both single cadmium(II) binding site, GrandL12AL16C [Grand=AcG-(LKALEEK)5-GNH2], GrandL26AL30C, and GrandL26AE28QL30C, as well as GrandL12AL16CL26AL30C with two cadmium(II) binding sites. The binding of cadmium(II) to any of these sites is of high affinity (KA > 3×107 M−1). Using 113Cd NMR spectroscopy, cadmium(II) binding to these designed peptides was monitored. While the cadmium(II) binding is in extreme slow exchange without showing any chemical shift changes, incremental line broadening for the bound 113cadmium(II) signal is observed when excess 113cadmium(II) is titrated into the peptides. Most dramatically, for one site, L26AL30C, all 113cadmium(II) NMR signals disappear once a 1.7:1 ratio of cadmium(II)/(peptide)3 is reached. The observed processes are not compatible with simple “free-bound” two-site exchange kinetics at any time regime. The experimental results can, however, be simulated in detail with a multi-site binding model, which features additional cadmium(II) binding site(s) which, once occupied, perturb the primary binding site. This model is expanded into differential equations for five-site NMR chemical exchange. The numerical integration of these equations exhibits progressive loss of the primary site NMR signal without a chemical shift change and with limited line broadening, in good agreement with the observed experimental data. The mathematical model is interpreted in molecular terms as representing binding of excess cadmium(II) to surface Glu residues located at the helical interfaces. In the absence of cadmium(II), the Glu residues stabilize the three-helical structure though salt bridge interactions with surface Lys residues. We hypothesize that cadmium(II) interferes with these surface ion pairs, destabilizing the helical structure, and perturbing the primary cadmium(II) binding site. This hypothesis is supported by the observation that the cadmium(II)-excess line broadening is attenuated in GrandL26AE28QL30C where a surface Glu(28), close to the metal binding site, was changed to Gln. The external binding site may function as an entry pathway for cadmium(II) to find its internal binding site following a molecular rearrangement which may serve as a basis for our understanding of metal complexation, transport and exchange in complex native systems containing α-helical bundles.
Introduction
Metal ion transport and homeostasis are critical cellular processes required for the maintenance of cellular health. One may distinguish two important classes of metal ion control. The first category addresses the proper handling of essential elements (e.g., Fe, Ni, Cu) in order to localize these metals in the appropriate protein, while minimizing the potential deleterious effects of the free ion. The second group contains the (usually) purely toxic ions (e.g., mercury(II), cadmium(II), arsenic(III) or lead(II)) which a cell encounters and which must be sequestered and detoxified before they are released back into the cellular environment. Nature often employs different strategies for uptake and transport of different essential metal ions. Some proteins such as albumin (binds Ca2+, Na+, K+) or calmodulin (binds Ca2+) sequester metals directly without any assistance by other proteins.1,2 On the other hand, intracellular trafficking of transition metals such as copper and nickel occurs by the assistance of metallochaperone proteins, that are part of the cellular machinery which ensure that the proper metal ion is delivered to appropriate targets in a safe manner.3–7 Remarkably, little is known about the fundamental rates and exchange pathways that control both the uptake and transport of metal ions by proteins into different cellular environments or different protein structure types.
Lower organisms such as bacteria, have evolved a detoxification mechanism to cope with heavy metals such as mercury(II), cadmium(II), arsenic(III) and lead(II) by using different repressor proteins such as MerR,8,9 CadC/CmtR,10–13 ArsR/SmtB,14 and pbrR.15,16 In the absence of heavy metals these proteins bind to DNA, blocking the transcription of the genes that code for proteins that extrude heavy metals from cells. Upon metal binding the repressor protein either twists the DNA (MerR) or dissociates from DNA (CadC/CmtR, ArsR/SmtB) after undergoing a conformational change which then allow transcription to occur. In all of these cases, the proteins display exclusive Cys ligation around the metal centers, and in the SmtB class utilize helical domains for metal interaction. A fundamental understanding of heavy metal biochemistry requires a complete description of the metal’s first coordination sphere and the corresponding physical properties that such a site confers. The rates for metal incorporation and removal are essential parts for proper functioning of metalloregulatory and metallochaperone proteins. However, complete understanding of the mechanistic pathways in which metals insert into, and are released from, metalloproteins are not well documented.6,7,17–22 More specifically, the exchange rates for metal ion insertion into metalloregulatory proteins are not known and are of fundamental interest. Unfortunately, such information cannot be acquired by the study of small molecule systems and native proteins are often too complex to identify individual process critical for metal sequestration clearly.
Using a series of de novo parallel three-stranded coiled coils we have designed homoleptic thiol-rich binding sites for heavy metals such as cadmium(II), mercury(II), lead(II), and arsenic(III) at the hydrophobic core of these polypeptides that has helped us clarify the heavy metal biochemistry of different metalloregulatory proteins. Following pioneering work by DeGrado and co-workers,23 the de novo polypeptide systems are designed based on the general sequence Ac-G(LKALEEK)xG-NH2, where x is the number of heptad repeats.24 Residues in positions 1 and 4 of the heptad form well defined hydrophobic cores at the interior of the polypeptides. These peptides self-assemble in aqueous solution to form three-stranded coiled coils [Ac-G(LKALEEK)xG-NH2]3, above pH 5.5.25 These peptides provide us with simpler constructs for understanding more complicated native proteins.. The successes using this strategy include the first spectroscopic and structural models24,26–28 for mercury(II) binding to MerR, arsenic(III) complexation29,30 by ArsR and trigonal31 cadmium(II) for CmtR. We have also demonstrated how to control the coordination geometry of metal ions such as cadmium(II) at the peptide interior. Using a combination of 113cadmium NMR and 111mcadmium PAC32 (Perturbed Angular Correlation) spectroscopies it was shown that the peptide TriL16C [TRI = Ac-G(LKALEEK)4G-NH2] bound cadmium(II) as a mixture of 3-coordinate trigonal planar CdS3 and 4-coordinate pseudotetrahedral CdS3O (O = exogenous water molecule) species.33 Removal of steric bulk directly above the metal binding site by replacing a Leu to an Ala led to the isolation of pure 4-coordinate CdS3O species in the peptide TriL12A16C.31,34 Pure 3-coordinate CdS3O geometry was achieved by increasing steric bulk in the first and second coordination sphere of the metal ion that blocked the access of the exogenous water molecule to the metal site, both employing unnatural amino acids. In the first strategy, metal binding Cys residues were replaced by bulky penicillamine (Pen = geminal dimethyl cysteine) yielding the peptide TriL16Pen.31 In the second strategy, chirality of the Leu above the Cys site was modified by replacing L-Leu to D-Leu that caused the Leu side chains to orient towards the metal binding site (towards C-terminus of the bundle) which resulted in exclusive CdS3 geometry in TriL12LDL16C.35 Using this knowledge, heterochromic constructs GrandL16PenL26AL30C,36 and GrandL12LDL16CL26AL30C35 [Grand = Ac-G(LKALEEK)5G-NH2] were designed subsequently, each peptide containing both 3- and 4-coordinate sites which showed site-selective molecular recognition of cadmium(II) for the 4-coordinate CdS3O geometry. Recently we have shown that depending on the topological position of the metal binding site (middle of the helix vs. helical terminus) the physical properties of cadmium(II) can be fine-tuned by the protein matrix even though the metal is bound as “identical” 4-coordinate CdS3O geometry at both the binding sites in the peptide GrandL12AL16CL26AL30C.37
In this study we have investigated the exchange dynamics associated with cadmium(II) binding to various derivatives of GRAND series of peptides. Figure 1 shows a model of the three-stranded coiled coil. Clarification of the rules that we elucidate for encapsulation of heavy metals such as cadmium(II), and the factors that influence these rates, should serve as the basis for our general understanding heavy metal complexation and exchange processes in more complex systems such as metallochaperones, and metalloregulatory proteins such as CmtR and CadC which belong to the ArsR/SmtB metal sensing family which sense cadmium(II) and lead(II).10,11 Previous kinetic studies propose that cadmium(II) insertion into three-stranded coiled coils follows a “Breathing Mechanism”.38 The assembly is thought to provide an opening for the metal by separation of two of the helices at the helical interface, in a slow and partially rate-determining step. Next, cadmium(II) binds to the Cys residues at the interior of the coiled coil as a fast step with a concomitant loss of exogenous water molecule(s) bound to the metal ion. Subsequently, salt bridges that help stabilize the aggregate are reformed yielding the final metallated species. Here we are able to describe the breathing model in more detail. The insights are based on 113Cd NMR studies using excess cadmium(II) as compared to the primary binding site. The data is only compatible with a multi-site exchange process that involves (several) surface sites that bind cadmium(II) and destabilize the three-stranded coiled coil structure. From this work, we also infer that surface glutamate residues play a crucial role in assisting the insertion of the metal ion from the helical interface to the primary metal binding site at the hydrophobic interior.
Figure 1.

A model of the parallel three-stranded coiled coil peptides investigated in this study. The model is derived based on the X-ray structure of a related peptide CSL9C (CS = Coil Ser) (PDB ID: 3LJM).48 Peptide backbone is represented as gray cartoon, amino acid side chains that are modified yielding different derivatives of GRAND peptides (GrandL12AL16C, GrandL26AL30C, GrandL12AL16CL26AL30C, and GrandL26AE28QL30C) used in this study are shown as ball and stick representations. N→C terminus corresponds to top→bottom in this view parallel to the helical axis.
Materials and Methods
a) Peptide Synthesis and Purification
The GRAND peptides (see Table 1 for sequence nomenclature) were synthesized on an Applied Biosystems 433A peptide synthesizer using standard Fmoc protocols,39 and purified and characterized as described previously.40 Our initial 113cadmium(II) NMR titration experiments with GrandL12AL16C, where the CdS3O metal binding site is located in the middle of the helical scaffold, suggested that an exchange process was occurring when excess metal was titrated. Next, experiments were performed with GrandL26AL30C, to see whether the metal site being located proximal to the C-terminus end of the bundle would show different exchange behavior than the L12AL16C site located in the middle of the helix. Next, the peptide GrandL12AL16CL26AL30C was studied to see whether the same sites (L12AL16C, L26AL30C) would show similar exchange properties, as was observed in individual peptides, when incorporated into a single peptide. The results from these experiments led us to hypothesize that there are additional surface binding sites (Glu) that are contributing to the exchange process. To test this hypothesis, we prepared GrandL26AE28QL30C, where a surface Glu residue close to the primary metal binding site was changed to a non-metal binding Gln. Finally, experiments were performed with the GrandL16PenL19IL23PenL26I peptide to see whether the 3-coordinate CdS3 sites also undergo exchange broadening like the 4-coordinate CdS3O sites under similar experimental conditions.
Table 1.
Peptide sequences used in this study.
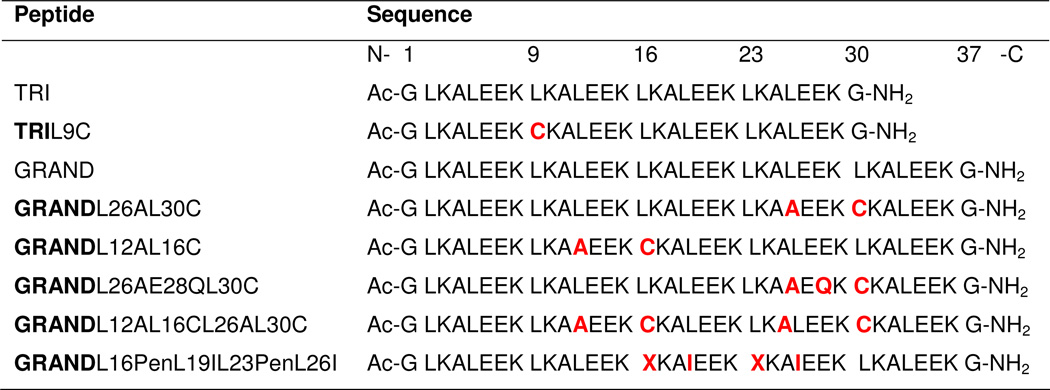 |
 = Penicillamine (Pen). Residues in red represent changes from the parent TRI or GRAND sequence. Sequences are shown as N- to C-terminus from left to right.
= Penicillamine (Pen). Residues in red represent changes from the parent TRI or GRAND sequence. Sequences are shown as N- to C-terminus from left to right.
b) UV-Vis Spectroscopy
UV-Vis spectroscopy was used to determine the stoichiometry and pH dependence of cadmium(II) binding to the peptide GrandL26AE28QL30C. All solutions were purged with argon before use to minimize the chances of oxidation of the peptides and formation of disulfide bonds. Fresh stock solutions of the purified peptides were prepared for each experiment in doubly distilled water and their concentrations determined by quantization of the Cys thiol groups using a known assay with 4,4’-dipyridyl disulphide.41
c) Metal Binding Titrations
cadmium(II) into peptide titrations were performed at room temperature on a Cary 100 Bio UV-Vis spectrometer using a 1-cm quartz cuvette. Aliquots of 8.932 mM CdCl2 stock solution were added into a 3-mL solution containing 60 µM peptide and 50 mM TRIS buffer at pH 8.5. In each case, the difference spectra were obtained by subtracting the background spectrum of the peptide in the absence of metal (60 µM peptide and 50 mM TRIS buffer at pH 8.5). The difference molar extinction coefficients (Δε) are calculated based on the total metal concentration.
d) pH Titrations
UV-Vis pH titrations were carried out at room temperature on an Ocean Optics SD 2000 fiber optic spectrometer and the pH measured using a mini-glass combination pH electrode (Hamilton Biotrode) coupled to a Fisher Accumet digital pH meter model 805 MP. pH titrations were performed by adding small aliquots of concentrated solution of KOH to unbuffered solutions containing CdCl2 (20 µM) and peptide (60 µM), and monitoring the change in absorbance at 235 nm as a function of pH. Equilibration time was always allowed before reading the final pH. In all cases, reverse titrations were carried out by adding small aliquots of concentrated solution of HCl to verify the reversibility of the process. The UV-Vis pH titration curve of the peptide GrandL26AE28QL30C was analyzed using the model and the fitting equation published previously for the simultaneous release of two Cys thiol protons upon formation of tristhiolato cadmium(II) species (CdS3)− at high pH from a monothiolato cadmium(II) complex (CdS(SH)2)+ that exists at low pH.42,43
e) 113Cd NMR Spectroscopy
All the spectra were collected at room temperature on a Varian Inova 500 spectrometer (110.92 MHz for 113Cd) equipped with a 5 mm broadband probe. 113Cd NMR spectra were externally referenced to a 0.1 M Cd(ClO4)2 solution in D2O. A spectral width of 847 ppm (93,897 Hz) was sampled using a 5.0 µs 90° pulse and 0.05 second acquisition time with no delay between scans. Samples were prepared under a flow of argon by dissolving 30 – 35 mg of the lyophilized and degassed peptides in 450 – 500 µL 15% D2O/H2O solution. The peptide concentrations were determined by using the assay with 4,4’-dipyridyl disulphide,41 and the concentrations range from 9 to 18 mM peptide, which corresponds to 3 – 6 mM three-stranded coiled coil. The final samples were prepared by the addition of the appropriate amount of 250 mM 113Cd(NO3)2 solution (prepared from 95% isotopically enriched 113CdO obtained from Oak Ridge National Laboratory) and the adjustment of the pH with KOH or HCl solutions. The pH value was measured both before and after the experiment. An argon atmosphere was maintained when possible but the samples came in contact with air during addition of 113Cd(NO3)2, pH adjustment and acquisition. The data were analyzed using the software MestRe-C.44 All free induction decays (FIDs) were zero filled to double the original points and were processed by application of 100 Hz line broadening prior to Fourier transformation.
f) Line Fitting Analysis of the 113Cd NMR Spectra
Line fitting analysis of the 113Cd NMR peaks was performed using the software MestRe-C by first defining the region of the spectra to be analyzed followed by selecting the peak of interest. Fittings were performed by keeping the chemical shift and the intensity of the peaks fixed while varying the line width (LW) of the peak during the iterative fitting process. The ratio of Lorentzian/Gaussian functions for each peak were kept constant at 1 (L/G = 1) during the fitting analysis.
g) Two-Site Exchange Kinetics Modeling
The fractions of the species as a function of total cadmium(II) and total protein concentration were obtained from numerical integrations of simple binding kinetics equations. While this simple case has an analytical solution (the well-known quadratic equation), we used numerical integration because it was needed for the subsequent more complicated binding simulations and served as a test-bed. The equations used were:
| [Eq 1] |
with the starting conditions
| [Eq 2] |
The integrations were allowed to reach equilibrium (tkin = ∞). The details of chemical shift and line width changes of two-site chemical exchange in the slow/intermediate regime, were obtained from numerical integrations of the Bloch-McConnell (BMC) equations45 which are valid for all time regimes without any approximations:
| [Eq 3] |
The variables in the BMC equations are related to the variables in the kinetic equations [1] as follows:
| [Eq 4] |
The FIDS were computed until the signal had decayed to < 0.01 of the starting signal. NMR spectra such as shown in Figure 3 were obtained by Fourier transformation of the calculated FIDs.
Figure 3.

Results of two-site NMR titration simulations performed using equation 3 in Materials and Methods. The “bound” cadmium(II) signal is shown at 25000 Hz and the “free” cadmium(II) signal is shown at −25000 Hz (δν = 450 ppm at 111 MHz). Both signals have a R2 of 1000 s−1 (linewidth = 318 Hz). The parameters chosen for these simulations are:
A. kon=3×109M−1s−1, koff=100 s−1 (KA=3×107 M−1). From bottom to top, the traces correspond to [Cd]total/[P] total = 0.25, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75 and 2.00 with [P] total = 4 mM (trimer).
B. An enlargement of the “bound” cadmium(II) signal obtained from simulations in (A).
C. An enlargement of the “bound” cadmium(II) signal of a simulation with kon=3×1010M−1s−1, koff=1000 s−1 (KA=3×107 M−1). Other parameters are the same as in (A).
D. An enlargement of the “bound” cadmium(II) signal of a simulation with kon=3×1010M−1s−1, koff=10,000 s−1 (KA=3×106 M−1). Other parameters are the same as in (A).
h) Five-Site Exchange Kinetics Modeling
A model for the binding of cadmium(II) to one of the sites of the Grand peptide is shown in Figure 4. In order to obtain quantitative estimations of the behavior of the scheme, we solve for the fractions of species at several different total cadmium(II) concentrations by numerical integration of the kinetic equations (5) until equilibrium is reached.
| Eq 5 |
Figure 4.

A cartoon representation of the model containing two metal binding sites. The scheme has 5 thermodynamic “species”, P, Q, R, S and free metal, Cd. The scheme has 5 NMR species α, β, γ, δ and ε. The direct communication between species Q and R (“Cd internalization”) is important for the kinetics of metal binding but is thermodynamically irrelevant. In the cartoons, cadmium(II) is represented as a green circle, Cys-SH groups as yellow circles, Glu-COO− groups as red circles and Lys NH3+ groups as blue circles.
The initial free cadmium(II) and free protein concentrations correspond to the known total cadmium(II) and total protein concentration, respectively, with all other initial concentrations set to zero:
| [Eq 6] |
These kinetic calculations were carried out for a variety of starting conditions. An example is shown in Figure S9 of the SI. The obtained steady-state (equilibrium) concentrations as a function of total concentrations describe the sought-after description of the thermodynamics of the scheme as shown in Figure 5. The scheme was then extended to incorporate NMR line-broadening effects due to kinetic effects. For the 113Cd signal, the scheme in Figure 4 corresponds to a 5-site exchange. Free cadmium(II) in solution is designated as “site” α. Other possible sites where cadmium(II) can bind are to the site β in species Q, to site γ in species R, or to sites δ or ε in species S. The relevant kinetic parameters are summarized in Figure 6. The effects of exchange kinetics on NMR relaxation is described by Bloch-McConnell equations,45 which for a generic 5-site exchange scheme are:
| Eq 7 |
where are the complex coherences of species j, ωj the angular NMR frequencies in the different sites j, and the the intrinsic transverse relaxation rates of sites j. This generic scheme was adapted to the thermodynamic/kinetic scheme in Figure 6 by the following substitutions for the non-NMR parameters in the kinetic matrix:
| Eq 8 |
where the concentrations of the species Cd, P, Q, R and S were taken from the endpoints of the integrations of Eq 5. The equilibrium concentrations obtained from the computations with Eq 5 were used as starting conditions:
| [Eq 9] |
Figure 5.

Equilibrium concentrations of different species obtained after 5 seconds of kinetic simulations as a function of total metal concentration. This simulation time is 50 times longer than it is needed to reach equilibrium (see Figure S9). [Cd]free is red; [P] is cyan; [Q] is black; [R] is green; [S] is purple. Simulation conditions: Total protein concentration 4 mM; kPQ= kRS=kint=3×105 M−1s−1; kQP=kSR= 0.01 s−1; kPR=kQS=kext=107 M−1s−1; kRP=kSQ=103 s−1, kRQ= 103 s−1, kQR= 0.333 s−1. (Corresponding to the equilibrium association constants KPQ=3×107 M−1 and KPR=104 M−1).
Figure 6.

Five-site NMR exchange parameters for the metal in the scheme of Figure 4. In the cartoons, cadmium(II) is a green circle, Cys-SH groups are yellow circles, Glu-COO− groups are red circles and Lys NH3+ groups are blue circles. The relevant exchange rates are related to the kinetic constants in Figure 4 as indicated. (Also see Equations 8 and 9 and Tables S1 and S2 of the SI)
Equations [8] were solved for different equilibrium conditions (obtained from Eq 5) by numerical integration to obtain the different coherences as a function of time, i.e. to obtain the FIDS of all species. The FIDS were co-added, zero-filled, and transformed using a complex Fourier transform to obtain the spectra as shown in Figure 7. All programs were written in Fortran-90, compiled using GNU-compilers and executed using a 2.4 GHz Intel Core 2 Duo Apple MacbookPro running MacOS10.6.8.
Figure 7.

Simulation results of the 5-site exchange NMR spectra according to the model represented in Figure 6 and using equations 8 and 9. Site α (free cadmium(II)) resonates at −25000 Hz; site β (the “primary” buried site in species Q) at +25000 Hz; site γ (the surface site in species R) at −15000 Hz (concentration too low to be visible); site δ (the buried site in species S) at 0 Hz; site ε (the surface site in species S) at −10000 Hz. All sites have R2 = 1000 s−1. Site-site exchange rates were derived from the rates in Figure 4, as indicated in Figure 6 with values as listed in the legend of Figure 5 and tables S1 and S2 of the SI.
A. Total protein concentration = 4 mM (trimer); total ligand concentration = 9.0 mM.
B. Simulation results obtained for different ratios of [Cd]total/[P] total. Only the signal for site β (the buried site in species Q) at +25000 Hz is shown. From bottom to top the traces are obtained using the following ratio: [Cd]total/[P] total = 0.25, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75 and 2.00 with [P] total = 4 mM (trimer).
Results
A) UV-Vis Spectroscopy
Results from UV-Vis spectroscopy can be found in the Supporting Information.
B) 113Cd NMR Spectroscopy
The cadmium(II) binding to the peptide GrandL12AL16C, which has been shown to bind one cadmium(II) ion with a 4-coordinate CdS3O geometry,36 was investigated by 113Cd NMR. The NMR spectrum shows a resonance at 572 ppm in the presence of 1.0 equivalent 113cadmium(II) (Figure 2A). When 1.5 equivalents of 113cadmium(II) were added, the peak was slightly broadened, with no change in the chemical shift, indicating slow/intermediate exchange. The resonance further broadened and decreased in intensity in the presence of 2.0 equivalents of 113cadmium(II) (Figure S3 of the SI shows fittings of the NMR spectra used to obtain the linewidths). However, tn the presence of excess cadmium(II) no additional resonance was observed which could be attributed to the free 113cadmium(II), expected to appear in the up-field region between 0–100 ppm (see Figure S4 of the SI for the full chemical shift scale).46
Figure 2.

113Cd NMR spectra of solutions containing A) 3.2 mM (GrandL12AL16C)3 (the metal binding site located almost in the middle of the peptide) at pH 8.5 in the presence of 1.0, 1.5 and 2.0 equivalents of 113Cd(NO3)2, B) 4.0 mM (GrandL26AL30C)3 (the metal site located almost in the middle of the peptide) at pH 8.5 in the presence of 0.2 to 1.7 equivalents of 113Cd(NO3)2, and C) 3.2 mM (GrandL12AL16CL26AL30C)3 (two metal binding sites, located in the middle and at the helical terminus of the peptide, respectively) at pH 8.5 with different equivalents of added 113Cd(NO3)2. The most downfield peak marked with a star in C) is an impurity most likely in the form of a peptide of shorter length that is produced during automated peptide synthesis and is inseparable by HPLC.
After the initial observations with the GrandL12AL16C peptide, a careful stepwise titration was performed with the peptide GrandL26AL30C, which also binds one cadmium(II) ion in CdS3O geometry.36 Addition of 0.2 to 0.8 equivalents of 113cadmium(II) to a solution containing 4 mM (GrandL26AL30C)3 at pH 8.5 led to a gradual increase in the intensity of the 113Cd NMR resonance with a chemical shift of 587 ppm, while no line broadening of the peak was observed (Figure 2B). When 1.0 equivalent of 113cadmium(II) was added, the peak slightly decreased in intensity, but no broadening was observed. With the gradual addition of more than 1.0 equivalent of 113cadmium(II), the resonance at 587 ppm decreased in intensity and subsequently broadened. In the presence of 1.7 equivalents of 113cadmium(II), the resonance had broadened beyond detection (Figure 2B). The linewidths as measured from the spectra (see Figure S5 of the SI) are listed in Table 2. The resonance at 587 ppm did not shift during the course of 113cadmium(II) addition, showing that exchange is occurring on the slow/intermediate exchange regime. Again, in the presence of excess cadmium(II) no additional resonance was observed which could be attributed to the free 113cadmium(II) (Figure S6 of the SI shows the full chemical shift scale). The comparison of these two results shows that the metal binding sites in the GrandL12AL16C and GrandL26AL30C peptides have different properties.
Table 2.
113Cd NMR frequencies and line widths for the bound 113cadmium(II) signal. Base frequency: 111 MHz (500 MHz 1H). The shifts were externally referenced to a 0.1 M Cd(CIO4)2 solution in D2O.
| Peptide | Eq. of added Cd | LW(Hz) |
|---|---|---|
| GrandL12AL16C | 1.0 | 309 |
| 1.5 | 393 | |
| 2.0 | 608 | |
| GrandL26AL30C | 0.2 | 368 |
| 0.4 | 420 | |
| 0.6 | 377 | |
| 0.8 | 411 | |
| 1.0 | 422 | |
| 1.1 | 470 | |
| 1.2 | 538 | |
| 1.3 | 570 | |
| 1.4 | 606 | |
| 1.5 | 634 | |
| GrandL12AL16CL26AL30C | 1 | 308 (572 ppm site) |
| 422 (589 ppm site) | ||
| 1.5 | 287 (572 ppm site) | |
| 391 (589 ppm site) | ||
| 2 | 303 (572 ppm site) | |
| 381 (589 ppm site) | ||
| 2.5 | 343 (572 ppm site) | |
| 472 (589 ppm site) | ||
| 3.0 | 348 (572 ppm site) | |
| GrandL26AE28QL30C | 1.0 | 378 |
| 1.1 | 407 | |
| 1.2 | 401 | |
| 1.3 | 413 | |
| 1.4 | 518 | |
| 1.5 | 524 | |
| 2.0 | 737 | |
We next investigated by NMR the peptide GrandL12AL16CL26AL30C to assess whether these two binding sites when incorporated into a single peptide have different properties. Furthermore, we examined whether placing two metal binding sites in the same peptide (GrandL12AL16CL26AL30C) alters the properties of the metal bound states when compared to the peptides containing a single metal binding site (GrandL12AL16C and GrandL26AL30C). GrandL12AL16CL26AL30C binds cadmium(II) ions as 4-coordinate pseudotetrahedral CdS3O species at both metal binding sites (16 and 30 positions).37 Gradual addition of 0.5 equivalent of 113cadmium(II) to a solution containing 3.2 mM (GrandL12AL16CL26AL30C)3 at pH 8.5 led to an increase in intensity of the 113Cd NMR resonances at 572 and 589 ppm up to 2.0 equivalents of 113cadmium(II) (Figure 2C) (the most downfield peak marked with a star is an impurity from a peptide of shorter length that is produced during automated peptide synthesis and is inseparable by HPLC). The signals at 572 ppm and 589 ppm correspond to chemical shifts of cadmium(II) in the L12AL16C and L26AL30C sites, respectively.37 In the presence of 2.0 equivalents (stoichiometric) of 113cadmium(II), the two peaks gained full intensity. Adding an additional 0.5 equivalent of 113cadmium(II) (2.5 equivalents total) resulted in a significant decrease in intensity and broadening of the peak at 589 ppm. This observation is similar to the results obtained for the single site peptide GrandL26AL30C. The peak at 572 ppm underwent only a slight decrease in intensity and increase in broadening in the presence of 2.5 equivalents of 113cadmium(II). None of the peaks shifted their respective resonance frequencies, indicating that the process is occurring on the slow exchange regime of NMR time scale. When 3.0 equivalents of 113cadmium(II) were added, the peak at 589 ppm had broadened beyond detection. The 572 ppm peak, on the other hand, was still present and did not broaden or lose further intensity. The linewidths as measured from the spectra (see Figure S7 of the SI) are listed in Table 2. Again, in the presence of excess cadmium(II) no additional resonance was observed which could be attributed to the free 113cadmium(II). These results show that the two metal binding sites in GrandL12AL16CL26AL30C have different properties from each other, but that each site appears to behave similarly to that observed for the single site peptides (e.g., L12AL16C sites in GrandL12AL16CL26AL30C and GrandL12AL16C behave the same). This later observation demonstrates that adding a second metal binding site (at least 14 residues away) to either GrandL12AL16C or GrandL26AL30C does not cause significant perturbation to the metal ion bound at the original site.
Discussion
The aim of this work is to describe at a molecular level how metal ions insert into helical scaffolds using de novo designed three-stranded coiled coil peptides as model systems with the long-term objective of better understanding of metal exchange processes into more complex metalloproteins. The specific questions relating to cadmium(II) exchange that are being addressed in this study are: Does the cadmium(II) initially coordinate to any of the amino acids located at the surface of the peptide? Do the cysteine residues at the designed metal binding site play any role in internalizing the surface bound cadmium(II)? Does the exchange rate depend on the location of the metal binding site within the three-stranded coiled coil interior (middle of the helix vs. towards the helical terminus)? Can one site fine-tune the exchange behavior of a distant second metal site within the same peptide? And, is there any difference in the exchange behavior of the 4-coordinate CdS3O vs. 3-coordinate CdS3 structure?
a) Effect of Peptide Design on the Properties of Cadmium(II) Binding
UV-Vis spectroscopy shows that both peptides GrandL12AL16C and GrandL26AL30C bind cadmium(II) with high affinity (see Figure S1 of the SI) (KA > 3×107 M−1). PAC spectroscopy shows that the cadmium(II) ions are encapsulated in a pseudotetrahedral CdS3O geometry.37,43 While no three-dimensional structure is as of yet available for these complexes, it has been assumed that the peptides are parallel triple-helical bundles in which three cysteines, one from each strand of the peptide, and an internal water molecule, bound along the helical axis, serve as ligands to the metal ion. The NMR spectra in Figure 2A and 2B are in agreement with these results. The chemical shifts at 572 and 587 ppm for GrandL12AL16C and GrandL26AL30C respectively, correspond to a pseudotetrahedral CdS3O environment, and the chemical shifts of the sites are not very different. Moreover, both binding sites show slow exchange characteristics, in agreement with the tight binding seen by UV-Vis spectroscopy. The combined results would, therefore, suggest that the designed cadmium(II) binding sites in GrandL12AL16C and GrandL26AL30C are very similar. However, when excess cadmium(II) is added, the NMR data reveal significant differences between the sites: the bound cadmium(II) resonance for GrandL12AL16C remains visible when excess cadmium(II) is present (Figure 2A), whereas the bound cadmium(II) resonance for GrandL26AL30C disappears when similar amount of excess cadmium(II) is present (Figure 2B). As will be discussed in detail below, these differences can, in rather involved ways, be attributed to differences in the kinetics of cadmium(II) binding and release. Here, however, it suffices to establish that while the binding site affinities and coordination characteristics are independent of the location of the binding site in the peptide, the binding/release kinetics of these sites is not.
The results in Figure 2C indicate that the site-specific similarities and differences of the two single site peptides are retained in the dual-site peptide GrandL12AL16CL26AL30C. First, the 113Cd chemical shifts at 572 and 589 ppm are almost identical to those seen in the peptides with the individual binding sites. Using a combination of 113Cd NMR and 1H-1H NOESY spectroscopies,37 we confirmed that the 589 ppm resonance in GrandL12AL16CL26AL30C indeed belongs to the L26AL30C site and the 572 resonance to the L12AL16C site. The correspondences of the 113Cd shifts in the different peptides strongly suggest that there is no communication between these sites. Secondly, the difference in kinetic behavior of the two sites as seen in the individual single-site peptides is also maintained in GrandL12AL16CL26AL30C: the results in Figure 2C clearly show that site L26AL30C is more dynamic than site L12AL16CL. Combined, these results indicate that the L26AL30C site, which is located close to the C-terminal end of the three-stranded coiled coil, is more susceptible to exchange of cadmium(II) as compared to the L12AL16C site which is located almost in the middle of the helical scaffold. Due to its location, the L26AL30C site is more exposed to the bulk solvent since the α helices are subject to significant fraying.30,47–49 These results, therefore, demonstrate that even though both the L12AL16C and L26AL30C sites in the GrandL12AL16CL26AL30C peptide bind cadmium(II) in “identical” first coordination sphere geometries (4-coordinate CdS3O), the L26AL30C site is kinetically more labile. For GrandL12AL16CL26AL30C, one may also conclude that the sites do not affect each other’s thermodynamic and kinetic properties.
b) Exchange Processes in 113cadmium(II) NMR Spectroscopy: Formulation of Two-Site Exchange Modeling and its Implications
According to solid state NMR results, the aqueous 113cadmium(II) NMR signal is expected to occur around 50 ppm – 100 ppm relative to 113Cd(ClO4)2, depending on counter ions and ionic strength.46 However, in practice, the NMR signal of 113cadmium(II) is difficult to detect in aqueous solution. This is likely due to broadening caused by the sizeable 113cadmium(II) chemical shift anisotropy (CSA) relaxation.50 This may be exacerbated by exchange broadening caused by dynamic rearrangement of the hydration shell. In contrast, all peptides studied show a well-defined 113cadmium(II) resonance around 550 ppm with a 300 Hz linewidth in the presence of substoichiometric amounts of 113cadmium(II) (Figure 2). The properties of these signals suggests a rigid, well-defined structure for bound 113cadmium(II). Indeed, the 4-coordinate pseudotetrahedral CdS3O environment has high symmetry; therefore, a small CSA and CSA broadening are to be expected.
The equilibrium constant KA for the binding of cadmium(II) to the Cys-substituted Grandpeptides is > 3×107 M−1.51 Based on the commonly quoted52 maximum diffusion limited bi-molecular on-rate (kon) of 3×1010 M−1s−1, one expects a maximum off-rate (koff) < 1000 s−1. The predicted maximum off-rate is much smaller than the expected chemical shift difference of 3.5×106 rad/s−1 between bound and free cadmium(II) signal (500 ppm at 111 MHz). In agreement with the predicted slow-exchange regime, no chemical shift change was observed during the titration from substoichiometric to excess equivalents of 113cadmium(II) (Figure 2). However, all spectra show increasing line broadening and decrease in intensity for the “bound” 113cadmium(II) signal when excess equivalents of 113cadmium(II) were added. The least amount of broadening and intensity change was observed for the site L12AL16C, while the largest amount of change was seen for site L26AL30C, both in the single site peptides (GrandL12AL16C, GrandL26AL30C) and in the dual site peptide (GrandL12AL16CL26AL30C). The signal intensity loss and progressive broadening is inconsistent with a two-site slow exchange mechanism. In two-site slow exchange scheme, the intensity of the “bound” resonance should not change when excess free metal is present. In addition, the line width R2 / π of a “bound” resonance (when excess is present) is independent of the ratio metal free/bound and is determined by the intrinsic linewidth at that site, augmented with the constant life-time broadening koff / π:
| [Eq. 10] |
To demonstrate these points, and to explore the limits of the kinetic parameters, two-site chemical exchange simulations were carried out with parameters relevant to the experimental data, using the methods described in the Materials and Methods section. Figure 3A represents a classical slow exchange case. Using a kon of 3×109 M−1s−1 and a koff of 100 s−1, no shifts are observed for the “bound” signal over the titration regime. Also the exchange line broadening, as compared to the intrinsic linewidth of 300 Hz is minimal. In Figures 3C and 3D, the on-rate is increased up to the theoretical limit of 3×1010 M−1s−1, together with a koff of 1000 s−1 or 10,000 s−1, respectively. The simulations now do show significant exchange broadening at suprastoichiometic conditions. However, the broadening is not progressive, while the “bound” signal intensity remains constant (see Table 3). In addition, under these simulation conditions, significant chemical shifts changes for the “bound” signal are being observed, that are not observed in the experimental results.
Table 3.
Simulated NMR linewidth of the “bound resonance” as a function of the ratio ligand/protein for different kinetic models, shown in Figures 3 and 7.
| Model | Two site | Two site | Two site | Five-site |
|---|---|---|---|---|
| KAapp | 3×107 M−1 | 3×107 M−1 | 3×106 M−1 | 3×107 M−1 |
| kon | 3×109 M−1s−1 | 3×1010 M−1s−1 | 3×1010 M−1s−1 | complex |
| kOff | 100 s−1 | 1000 s−1 | 10000 s−1 | complex |
| R2bound | 1000 s−1 | 1000 s−1 | 1000 s−1 | 1000 s−1 |
| δν(free-bound) | 50000 Hz | 50000 Hz | 50000 Hz | 50000 Hz |
| Ratio | δν1/2bound (Hz) | δν1/2bound (Hz) | δν1/2bound (Hz) | δν1/2bound (Hz) |
| 0.25 | 318 | 318 | 318 | 318 |
| 0.5 | 318 | 318 | 318 | 318 |
| 0.75 | 318 | 318 | 318 | 318 |
| 1 | 330 | 500 | 1400 | 318 |
| 1.25 | 340 | 600 | 3000 | 414 |
| 1.5 | 340 | 600 | 3000 | 628 |
| 1.75 | 340 | 600 | 3000 | 1068 |
| 2 | 340 | 600 | 3000 | 2100 |
Because of these simulated results, we conclude that a simple two-site exchange mechanism, can at no choice of parameters, account for the progressive broadening and intensity loss seen in the experimental data. Moreover, the best simulation with a koff of 10,000 s−1 starts to show intermediate exchange-induced chemical shift changes. Most problematic is the fact that the best simulation requires a very fast kon of 3×1010 M−1s−1. Such a bi-molecular on-rate is the very limit of diffusion control. According to collision theory, this fast rate is only reached when the entire surface of a macromolecule is involved in capturing a substrate, followed by surface diffusion which funnels the substrate into an exposed active site.52,53 In comparison, for the present case, it is quite improbable that the collision rate could be the rate-limiting step. After all, the cadmium(II) needs to bind to an interior site located at the center of the three-helix bundle. Much more likely, one expects that a much slower transient unfolding or breathing process, which would allow access of cadmium(II) to the interior, is rate limiting.
c) Formulation of a Multi-Site Exchange Model
At this point one can conclude that the NMR titration data is incompatible with a simple two-site exchange scheme because at supra-stoichiometric ratios the signal corresponding to the “bound” state loses intensity and it broadens progressively. Several thermodynamic models can be proposed that can account for the reduction of the NMR signals at supra-stoichiometric ratios. A satisfactory model is high-affinity metal binding in the primary binding site in species Q, followed by low-affinity binding in an auxiliary site which changes the properties of the primary binding site as shown in Figure 4. If we choose the equilibrium binding constant KPQ=kPQ/kQP in Figure 4 to be larger than KPR=kPR/kRP then species Q will disappear when the cadmium(II) concentration is increased in excess of stoichiometry with a subsequent increase in the population of the species S. In this model, one will observe the disappearance of the signal with NMR properties β when the metal concentration is increased in excess of stoichiometry. Reasons why the signal of site δ of species S may be undetectable will be discussed below. Invoking this multi-site exchange scheme, the concentrations of the different species in Figure 4 were computed as a function of the ratio of metal/protein concentrations using numerical methods as described in the Material and Methods. With a choice of the equilibrium association constants KPQ= KRS=3×107 M−1 (primary binding site), and KPR= KQS=104 M−1 (secondary binding site), the profile as shown in Figure 5 is obtained. The results from these simulations show that this scheme allows for disappearance of the species Q, because it is being displaced by the species S. Hence, this multi-site exchange scheme can account for the hallmark effect observed in the experimental titrations: the bound cadmium(II) signal loses intensity when excess cadmium(II) is present.
In order to also account for the progressive increase in linewidth of the “bound” resonance (see Table 2), the “species” scheme of Figure 4 must be translated into a “site” scheme for the cadmium(II) ion as shown in Figure 6. The scheme in Figure 4 corresponds to a 5-site chemical exchange scheme. According to this scheme cadmium(II) can be free (denoted as site α), bound to site β in species Q, bound to site γ in species R, or bound to sites δ or ε in species S. Potentially, all 5 sites have different NMR parameters. That is, the primary metal binding site β may change in frequency and linewidth if excess metal binds to the auxillary site in species S (i.e. sites β and δ are different for NMR). The exchange rates between the sites are related to the kinetic constants in Figure 4 and the concentrations of the different species [P],[R],[Q], [S] and free [Cd] as indicated in Figure 6 and equations 7 and 8 in the Materials and Methods section. In the simulations, it was assumed that no direct exchange can occur between the internal and external sites δ and ε in species S because the sites are occupied. It was also assumed that the internal site β cannot directly interchange with the external site ε and that the external site γ cannot directly interchange with the internal site δ, because these processes would require coordinated rearrangement and binding/release of two cadmium(II) ions. The possibility that external cadmium(II) in species R (site γ) can become internalized in species Q (site β) was allowed in the computations. While this internalization process does not change the thermodynamics (all is balanced in Carnot cycles), or the NMR linewidths (see SI), it forms a realistic pathway for the binding of cadmium(II) to the internal site (discussed below).
The detailed-balanced equilibrium kinetic site-site exchange parameters were inserted into five-site chemical exchange Bloch-McConnell equations shown in Equations 7 and 8 in the Materials and Methods. The equations were numerically integrated with the equilibrium concentrations of the different species as starting conditions, listed in Tables S1 and S2 of the SI. The parameters chosen correspond to slow exchange for all sites. The obtained FIDs were Fourier transformed yielding the spectra shown in Figure 7. Figure 7A corresponds to a slight excess of cadmium(II). The resonances for all sites are seen, except for site γ in species R which is too small in intensity. Figure 7B shows a zoom in on the “bound” signal (site β in species Q) as a function of the cadmium(II)/peptide ratio. The simulated spectra correspond very closely to the experimental NMR data for site L26AL30C, both in the single (GrandL26AL30C) and dual site (GrandL12AL16CL26AL30C) peptides (Figures 2B and 2C). As is documented in Table 3, the linewidth of the “bound” resonance increases as the excess cadmium(II) is increased. This progressive broadening, consistent with the experimental data, is caused by increasing lifetime broadening of site β in species Q, because it is converted to site δ in species S, with an increasing rate depending on the concentration of free cadmium(II):
| [Eq 11] |
It thus appears that the simple model of Figure 4, representing five-site metal exchange, can account for both the decrease in intensity of the bound signal as well as progressive line broadening when excess free cadmium(II) is present.
Why the other lines are not observed in the experiment may be understood as follows. As we already know, the signal of 113cadmium(II) ion in water is broadened beyond detection. Hence, it may be expected that the cadmium(II) signals of the solvent exposed sites γ and ε are also broadened away. The cadmium(II) signal of internal site δ in structure S is invisible because we associate this structure with a destabilized bis-liganded internal cadmium(II). Such a destabilization likely causes excessive exchange broadening of the signal. Moreover, 113Cd is also known to the have a large chemical shift anisotropy, which can reach to 100 ppm in asymmetric environments such as this site.50 The associated line broadening effects, especially at higher magnetic fields, would further exacerbate the broadening of cadmium(II) in the site δ in species S. While the allosteric/kinetic/NMR model contains many parameters, only kint, (the bimolecular on-rate to the internal site) and kRP (the off-rate of the external site) can be freely chosen – with all other parameters being restrained by the overall experimental affinity and by the observed experimental line-broadening.
d) Nature of the Additional Cadmium(II) Binding Site(s)
The three-stranded coiled-coil peptides have been designed as repeating heptads LKALEEK, with a hydrophobic core formed by residues in positions 1, and 4. The Glu residues located at the 5th position are involved in inter-helical electrostatic interactions with the Lys residues located at the 7th position. These electrostatic interactions are essential for the formation of stable tri-helical bundles. From our previous studies we know that the peptides containing 4 heptads or more (TRI, GRAND and Coil Ser) exist as parallel three-stranded coiled coils even in the absence of metals at any pH higher than 5.5.25,30,47,48 Species Q in Figure 4 represents the situation where cadmium(II) is bound to three cysteines at the interior of the three-stranded coiled coils under stoichiometric conditions. The analysis of the NMR data leads to the prediction that cadmium(II) must also bind elsewhere, and that this external binding affects the cadmium(II) bound to three cysteines at the interior. It is quite reasonable to hypothesize that the Glu residues can also bind to cadmium(II), and that this binding interferes with the overall stability of the peptide and consequently with the NMR signal of the cadmium(II) bound to the internal site.
To test whether Glu residues are involved in cadmium(II) binding, Glu 28 located closest in sequence to the metal binding Cys residue at position 30, was mutated to Gln. The resulting peptide is GrandL26AE28QL30C. Aliquots of 113cadmium(II) were added to a solution containing 3.7 mM (GrandL26AE28QL30C)3 at pH 8.5 and the NMR spectral changes were monitored. In the presence of 1.0 equivalent of 113cadmium(II), a single sharp resonance with a chemical shift of 587 ppm was observed (Figure 8, and Figure S8 of the SI). The 113Cd NMR resonance at 587 ppm broadened slowly with increasing amounts of 113cadmium(II) and was broadened beyond detection in the presence of 2.1 equivalents of 113cadmium(II), which is a significantly larger ratio of cadmium(II)/(peptide)3 than what was needed for the complete broadening of the NMR signal for the GrandL26AL30C peptide. The resonance for the latter peptide was completely broadened beyond detection in the presence of 1.7 equivalents of 113cadmium(II) (Figure 2B). We conclude that the effects seen on the kinetics of the internal cadmium(II) by mutagenesis of a neighboring external Glu, support the hypothesis that the Glu residues are involved in excess cadmium(II) binding.
Figure 8.

113Cd NMR spectra of a solution containing 3.7 mM of the mutant protein (GrandL26AE28QL30C)3 at pH 8.5 with different equivalents of added 113Cd(NO3)2. In this mutant the Glu 28, located close to the primary metal binding Cys site in GrandL26AL30C peptide was replaced by Gln to test the hypothesis whether any surface Glu residue(s) initially coordinate to the cadmium(II) ion. The results from this experiment show that cadmium(II)-induced line broadening is attenuated compared to the GrandL26AL30C peptide, indicating that Glu 28 does play a role in cadmium(II) exchange.
e) Functional Interpretation of the Multi-Site Cadmium(II) Binding
The inferred presence of an external cadmium(II) binding site leads to a model how an external binding site may be essential to the cadmium(II) binding process to the primary site itself. The model has been represented as cartoons for the different states in Figures 4 and 6. In the model, we envision that cadmium(II) is initially coordinated by the Glu residue(s) at the surface (red circles) located closest to the metal binding Cys residues (yellow circles) in species R. As suggested by the data discussed above, this leads to destabilization of the Glu-Lys (blue circle) interaction which in turn would facilitate the sequestration of the metal in the interior. This model is attractive as it can account for relatively fast cadmium(II) binding to the internal binding site as a two-step sequential process involving species R and Q. Figures S9 and S10 of the SI show the time-courses of the simulated binding kinetics according to the model of Figure 4, starting with unbound cadmium(II) and unbound protein. Indeed, one observes that species R becomes rapidly and transiently populated before species Q becomes populated. We believe that this model may also be of relevance for metal binding to naturally occurring proteins.
Although not essential to the sequential binding model, we also propose the nature of this destabilized state which is corroborated by X-ray structures of related metal binding peptides (Coil Ser), where the Cys side chains can adopt multiple conformations partitioned between the helical interior and toward the helical interface..47,48 The Cys sulfur oriented toward the helical interface helps to coordinate the external cadmium(II) ion initially. This situation is symbolized as species R in Figures 4 and 6, where the cadmium(II) is coordinated by Glu and one of the external Cys residues. Next, a rotation of Cys side chains followed by breathing of the helices are thought to internalize the surface bound cadmium(II) to the interior(species S in Figures 4 and 6) where the cadmium(II) at the interior is coordinated by just two Cys thiolates. The latter situation is supported by an X-ray structure of a Coil Ser peptide where one Hg(II) ion is bound to two thiols of Pen (Penicillamine) as linear HgS2 at the interior of the coiled coil, whereas another Hg(II) is present at the helical interface, and coordinated to a Glu and a Pen side chain oriented towards the helical interface (Zastrow, M., Pecoraro, V. L. unpublished results).
f) Comparison of Metal Exchange Between 3- and 4- Coordinate Cadmium(II) Centers
Based on the above discussion we have established that cadmium(II) insertion to peptides as 4-coordinate CdS3O structures are highly intricate in nature involving auxiliary surface residues (Glu) in addition to the primary binding site (Cys) at the polypeptide interior. We have also proposed a mechanism by which the surface bound cadmium(II) can be internalized. As a final note, we investigated the peptide GrandL16PenL19IL23PenL26I, which binds cadmium(II) ions as 3-coordinate CdS3 structures. 37 Figures S11, S12 of the SI show that, unlike the behavior for CdS3O centers, the addition of excess 113cadmium(II) does not cause line broadening to either of the resonances with chemical shifts 681 and 686 ppm. The Pen and Ile residues in the GrandL16PenL19IL23PenL26I peptide provide improved packing conferring higher stability to the coiled coil assembly,37 making the 3-coordinate cadmium(II) sites more rigid and compact. Further, the additional methyl groups on the penicillamine ligand are expected to inhibit, in this very well packed structure, rotation of the sulfur ligand toward the helical interface. In the GrandL12AL16CL26AL30C peptide, on the other hand, each Leu substitution to Cys or Ala results in a loss of free energy of ~ 4 kcal/mole,37 making this an inherently less stable compared to the GrandL16PenL19IL23PenL26I peptide.
Conclusions
In this work we have investigated the exchange of cadmium(II) into de novo designed three-stranded coiled coil peptides that contain both dual and single metal binding sites using 113cadmium(II) spectroscopy and mathematical simulations. Chemical exchange kinetics of cadmium(II) is observed when the metal ion is bound as a 4-coordinate CdS3O structure in pseudotetrahedral geometry. The exchange of cadmium(II) occurs on the slow exchange regime but is incompatible with a simple two-site exchange mechanism. The experimental NMR data are explained by formulating a multi-site binding model which involves coordination of cadmium(II) to surface glutamates, a process which destabilizes the primary cadmium(II) binding site (cysteines) at the interior. The model also provides a general kinetic mechanism for the insertion of metals from the polypeptide surface to the internal binding sites, with likely applications to more complex biological metal-binding proteins. Due to fraying of the coiled coils at the ends, the L26AL30C site, located proximal to the helical terminus, is more susceptible to destabilization by external metal binding as compared to the L12AL16C located almost in the middle of the helix, These differences demonstrate that the dynamics of metal binding and exchange can be significantly different, depending on the location of the metal site within a specified secondary structure. Finally, the 3-coordinate trigonal planar CdS3 structures exchange too slowly to be monitored by 113Cd NMR spectroscopy.
Supplementary Material
Acknowledgement
V.L.P. thanks the National Institutes of Health for support of this research (ES012236). O.I. thanks the Margaret and Herman Sokol Foundation for a Postdoctoral Award. E.R.P.Z. most gratefully thanks the Department of Biological Chemistry of the University of Michigan Medical School for continued support.
Footnotes
Supporting Information Available
Results from UV-Vis spectroscopy, part of the NMR discussion, parameters for fitting the five-site exchange data, UV-vis titration of cadmium(II) to GrandL26AE28QL30C, GrandL12AL16C, GrandL26AL30C, pH dependence of cadmium(II) binding to GrandL26AE28QL30C, 113Cd NMR spectra of GrandL16PenL19IL23PenL26I in the presence of different equivalents of 113cadmium(II), spectra with full scale of 113Cd NMR chemical shifts for GrandL12AL16C, GrandL26AL30C, simulated binding kinetics of cadmium(II) according to Equations 5, and fittings of 113Cd NMR spectra for GrandL12AL16C, GrandL26AL30C, GrandL12AL16CL26AL30C, GrandL26AE28QL30C, and GrandL16PenL19IL23PenL26I. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Masuoka J, Saltman P. Journal of Biological Chemistry. 1994;269:25557–25561. [PubMed] [Google Scholar]
- 2.Means AR, Dedman JR. Nature. 1980;285:73–77. doi: 10.1038/285073a0. [DOI] [PubMed] [Google Scholar]
- 3.Rosenzweig AC, O'Halloran TV. Curr. Opin. Chem. Biol. 2000;4:140–147. doi: 10.1016/s1367-5931(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 4.Xia W, Li H, Sze K-H, Sun H. Journal of the American Chemical Society. 2009;131:10031–10040. doi: 10.1021/ja900543y. [DOI] [PubMed] [Google Scholar]
- 5.Rosenzweig AC. Accounts of Chemical Research. 2000;34:119–128. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 6.O'Halloran TV, Culotta VC. J. Biol. Chem. 2000;275:25057–25060. doi: 10.1074/jbc.R000006200. [DOI] [PubMed] [Google Scholar]
- 7.Wernimont AK, Huffman DL, Lamb AL, O'Halloran TV, Rosenzweig AC. Nat. Struct. Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 8.Wright JG, Natan MJ, MacDonnell FM, Ralston DM, O'Halloran TV. Prog. Inorg. Chem.:Bioinorg. Chem. 1990;38:323–412. [Google Scholar]
- 9.Wright JG, Tsang H-T, Penner-Hahn JE, O'Halloran TV. J. Am. Chem. Soc. 1990;112:2434–2435. [Google Scholar]
- 10.Ye J, Kandegedara A, Martin P, Rosen BP. J. Bacteriol. 2005;187:4214–4221. doi: 10.1128/JB.187.12.4214-4221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banci L, Bertini I, Cantini F, Ciofi-Baffoni S, Cavet JS, Dennison C, Graham AI, Harvie DR, Robinson NJ. Journal of Biological Chemistry. 2007;282:30181–30188. doi: 10.1074/jbc.M701119200. [DOI] [PubMed] [Google Scholar]
- 12.Busenlehner LS, Weng T-C, Penner-Hahn JE, Giedroc DP. Journal of Molecular Biology. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 13.Busenlehner LS, Cosper NJ, Scott RA, Rosen BP, Wong MD, Giedroc DP. Biochemistry. 2001;40:4426–2236. doi: 10.1021/bi010006g. [DOI] [PubMed] [Google Scholar]
- 14.Shi W, Dong J, Scott RA, Ksenzenko MY, Rosen BP. J. Biol. Chem. 1996;271:9291–9297. doi: 10.1074/jbc.271.16.9291. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Wong M, Stalhandske C, Scott RA, Rosen BP. FASEB JOURNAL. 1999;13:A1464–A1464. [Google Scholar]
- 16.Borremans B, Hobman JL, Provoost A, Brown NL, van der Lelie D. J. Bacteriol. 2001;183:5651–5658. doi: 10.1128/JB.183.19.5651-5658.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC. Biochemistry. 2000;39:14720–14727. doi: 10.1021/bi002207a. [DOI] [PubMed] [Google Scholar]
- 18.Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC. Nat. Struct. Biol. 2001;8:751–755. doi: 10.1038/nsb0901-751. [DOI] [PubMed] [Google Scholar]
- 19.Lamb AL, Wernimont AK, Pufahl RA, O'Halloran TV, Rosenzweig AC. Nat. Struct. Biol. 1999;6:724–729. doi: 10.1038/11489. [DOI] [PubMed] [Google Scholar]
- 20.Huffman DL, O'Halloran TV. J. Biol. Chem. 2000;275:18611–18614. doi: 10.1074/jbc.C000172200. [DOI] [PubMed] [Google Scholar]
- 21.Tosha T, Ng H-L, Bhattasali O, Alber T, Theil EC. Journal of the American Chemical Society. 2010;132:14562–14569. doi: 10.1021/ja105583d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theil EC. Annual Review of Biochemistry. 1987;56:289–315. doi: 10.1146/annurev.bi.56.070187.001445. [DOI] [PubMed] [Google Scholar]
- 23.Bryson JW, Betz SF, Lu ZX, Suich DJ, Zhou HX. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 24.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O'Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. J. Am. Chem. Soc. 1997;119:6195–6196. [Google Scholar]
- 25.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. Journal of Molecular Biology. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 26.Pecoraro VL, Peacock AFA, Iranzo O, Luczkowski M. Bioinorganic Chemistry. American Chemical Society; 2009. pp. 183–197. SE - 112. [Google Scholar]
- 27.Iranzo O, Thulstrup PV, Ryu S-B, Hemmingsen L, Pecoraro VL. Chem. Eur. J. 2007;13:9178–9190. doi: 10.1002/chem.200701208. [DOI] [PubMed] [Google Scholar]
- 28.Dieckmann G, Galang J, DeGrado W, Pecoraro V. Abstracts of papers of the American Chemical Society. 1995;210:552. [Google Scholar]
- 29.Farrer B, McClure C, Penner-Hahn JE, Pecoraro VL. Inorganic Chemistry. 2000;39:5422–5423. doi: 10.1021/ic0010149. [DOI] [PubMed] [Google Scholar]
- 30.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Angew. Chem. Int. Ed. 2006;45:2864–2868. doi: 10.1002/anie.200504548. [DOI] [PubMed] [Google Scholar]
- 32.Hemmingsen L, Narcisz K, Danielsen E. Chemical Reviews. 2004;104:4027–4061. doi: 10.1021/cr030030v. [DOI] [PubMed] [Google Scholar]
- 33.Matzapetakis M, Farrer BT, Weng T-C, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. J. Am. Chem. Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 34.Lee KH, Matzapetakis M, Mitra S, Marsh ENG, Pecoraro VL. J. Am. Chem. Soc. 2004;126:9178–9179. doi: 10.1021/ja048839s. [DOI] [PubMed] [Google Scholar]
- 35.Peacock AFA, Hemmingsen L, Pecoraro VL. Proceedings of the National Academy of Sciences. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iranzo O, Cabello C, Pecoraro VL. Angew. Chem. Int. Ed. 2007;46:6688–6691. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iranzo O, Chakraborty S, Hemmingsen L, Pecoraro VL. Journal of the American Chemical Society. 2011;133:239–251. doi: 10.1021/ja104433n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh D. Chemsitry. Ann Arbor: University of Michigan; 2006. [Google Scholar]
- 39.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. New York: Oxford Univ. Press; 2000. [Google Scholar]
- 40.Farrer BT, Harris NP, Balchus KE, Pecoraro VL. Biochemistry. 2001;40:14696–14705. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 41.Mantle M, Stewart G, Zayas G, King M. Biochem. J. 1990;266:597–604. [PMC free article] [PubMed] [Google Scholar]
- 42.Matzapetakis M, Ghosh D, Weng T-C, Penner-Hahn JE, Pecoraro VL. J. Biol. Inorg. Chem. 2006;11:876–890. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 43.Iranzo O, Lee KH, Jakusch T, Hemmingsen L, Pecoraro VL. Chem. Eur. J. . 2009;15:3761–3772. doi: 10.1002/chem.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cobas C, Cruces J, Sardina FJ. 2.3 ed. Spain: Universidad de Santiago de Compostela; 2000. [Google Scholar]
- 45.Cavanagh JF, W J, Palmer AG, III, Skelton NJ. London: Academic Press; 1996. p. 279. [Google Scholar]
- 46.Summers MF. Coord. Chem. Rev. 1988;86:43–134. [Google Scholar]
- 47.Peacock AFA, Stuckey JA, Pecoraro VL. Angewandte Chemie International Edition. 2009;48:7371–7374. doi: 10.1002/anie.200902166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chakraborty S, Touw DS, Peacock AFA, Stuckey J, Pecoraro VL. Journal of the American Chemical Society. 2010;132:13240. doi: 10.1021/ja101812c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lovejoy B, Choe S, Cascio D, McRorie D, DeGrado W, Eisenberg D. Science. 1993;259:1288–1293. doi: 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- 50.Tinet DFA, Prost R. J. Phys. Chem. 1991;95:8804. [Google Scholar]
- 51.Ghosh D, Lee K-H, Demeler B, Pecoraro VL. Biochemistry. 2005;44:10732–10740. doi: 10.1021/bi0506674. [DOI] [PubMed] [Google Scholar]
- 52.Alberty RA, Hammes GG. J. Phys. Chem. 1958;62:154. [Google Scholar]
- 53.Zhou G, Wong M, Zhou G. Biophysical Chemistry. 1983;18:125. doi: 10.1016/0301-4622(83)85006-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.