

Abstract
Background:
Rathke's cleft cyst (RCC) is a lesion derived from maldeveloped remnants of a dorsal invagination of the stomodeal ectoderm (Rathke's pouch). Although commonly found on autopsy, these lesions rarely become symptomatic during an individual's lifetime. When symptoms occur, they most often include headaches, visual disturbances, and/or varying degrees of hypopituitarism. The natural history remains unclear. The current standard of care includes surgical drainage and biopsy of the cyst wall or surgical resection of symptomatic lesions; however, debate exists regarding the management of asymptomatic lesions. Rare reports of spontaneously resolving RCC can be found in the literature.
Case Description:
We describe the management of a case of RCC in an 8½-year-old girl who presented with a history of growth deceleration since 4 years of age and near-growth arrest since 7 years of age. Her parents also described a tendency towards polydipsia since she was 2 years of age. Endocrine evaluation revealed growth hormone deficiency, central hypothyroidism, and diabetes insipidus, but normal cortisol secretion. The patient experienced no symptoms characteristic of intracranial or sellar mass. Neurologic examination was normal; formal ophthalmologic examination revealed no deficits. The magnetic resonance imaging (MRI) was consistent with RCC. The patient was treated medically for her hormone deficiencies. Over the next year, her sellar mass spontaneously involuted. Twenty-seven months after her initial presentation to our clinic, imaging revealed no sellar mass; the patient remained on hormone replacement therapy.
Conclusion:
Although the natural history of RCC requires further study, observation with serial MRI may be an acceptable management strategy in the absence of debilitating symptoms.
Keywords: Pituitary cyst, Rathke's cleft cyst, spontaneous involution, transsphenoidal surgery
INTRODUCTION
A Rathke's cleft cyst (RCC) is derived from remnants of a maldeveloped Rathke's pouch, which is a depression in the roof of the developing oral cavity that gives rise to the adenohypophysis.[8,20,22] The failure of Rathke's pouch to regress following formation of the adenohypophysis and neurohypophysis may result in a cyst that is filled with proteinaceous fluid and lined by ciliated or non-ciliated, cuboidal or columnar epithelium, mucous-producing goblet cells, and cells of the anterior pituitary gland.[9,24] RCCs are relatively common, appearing in up to 10–22% of normal autopsy cases but are rarely symptomatic.[8,15,27,30,33] With the advent and more common use of high-resolution magnetic resonance imaging (MRI) techniques for the evaluation of neurologic conditions, these lesions are more frequently being diagnosed in asymptomatic patients. When symptoms are present, they may include headache, visual loss (due to suprasellar extension and compression of the optic apparatus), and/or endocrinopathies (due to disruption of the hypothalamic–pituitary axis),[1,22] although cases of RCCs manifesting as pituitary apoplexy secondary to intracyst hemorrhage have been reported.[21]
The natural history of RCCs is poorly understood, but high rates of recurrence after surgical resection have been reported.[1,3,15,16,18] Surgical drainage of the cyst and biopsy of the cyst wall or surgical cyst excision, most frequently through a transsphenoidal approach, are considered for symptomatic patients.[3,15,18,20] However, instances of cyst reduction associated with corticosteroid replacement therapy have been reported.[8,17] Recently, Amhaz et al.[2] described nine cases of spontaneously resolving RCCs. In their series, only one patient was receiving steroid replacement therapy (with intranasal fluticasone). Although the phenomenon of a spontaneously involuting RCC rarely has been reported previously, the mechanism by which it occurs remains speculative.[12,17,19,20,25,29] In addition, these reports have been in adolescents (with the youngest patient being 14 years old) and adults.
In this report, we describe a case of a spontaneous involuting sellar lesion, a RCC, in an 8½-year-old girl who had normal cortisol secretion and therefore did not receive corticosteroid replacement therapy. Rather, this patient's lesion regressed in the presence of levothyroxine and desmopressin (DDAVP), and, later, growth hormone replacement therapy, which she received for treatment of central hypothyroidism, diabetes insipidus, and growth hormone deficiency, respectively. We report what is to our knowledge the first documented case of a spontaneously involuting RCC in a patient as young as the patient in this report.
CASE REPORT
History
This patient is an 8½-year-old girl who came to medical attention because of growth deceleration. Her growth curve revealed a linear growth at the 50th percentile until 4 years of age. Since that time, she had subtle, but steady growth deceleration, reaching the 10th percentile by 7 years of age. Since the age of 7 years, she had experienced almost no growth. Additionally, her parents described a tendency towards polydipsia, with a preference toward water beginning approximately at the age of 2 years. This was associated with polyurea, with urine that was described by the parents as light in color. There were no reports of headaches, nausea, vomiting, irritability, symptoms consistent with seizure, numbness/tingling/weakness of the extremities, visual disturbances, or other focal neurologic deficits.
Examination
On the initial examination, the patient was awake and alert, interactive, with no focal findings elicited on neurologic examination and normal visual field testing to confrontation. A formal ophthalmologic examination was within normal limits.
An endocrine work-up conducted at this time revealed hypopituitarism with central hypothyroidism (Free T4 level, 0.1 ng/dL; thyroid-stimulating hormone level, 1.71 mIU/L), diabetes insipidus (urine specific gravity, 1.005; sodium level, 141 mmol/L), and growth hormone deficiency, as determined by an IGF-1 level of <25 ng/mL (reference range 97–352 ng/mL). Cortisol secretion was determined to be intact by cosyntropin stimulation test (cortisol levels of 11.5, 17.9, 15.1, and 24.5 mcg/dL at 0, 20, 30, and 60 min, respectively, after the injection of 10 mcg of cosyntropin). The patient was started on levothyroxine (12.5 mcg daily) and DDAVP (0.1 mg daily).
An MRI study of the brain demonstrated a hypointense cystic lesion in the middle of the pituitary gland (17.1 × 13.9 × 11.4 mm3) with peripheral enhancement [Figure 1a–c]. The lesion was mostly isointense compared to brain on T1-weighted MRI and hyperintense to brain on T2-weighted imaging, with a concentric hypointense region surrounding the lesion that was a representative of hemosiderin. There was a small intracystic nodule that was hyperintense on T1 - weighted imaging and hypointense on T2-weighted imaging. There was superior displacement of the pituitary without compression of the optic apparatus. A computed tomographic (CT) scan of the head obtained at this time showed an enlarged sella turcica with no evidence of abnormal calcification. On the basis of her symptoms and radiographic findings, the differential diagnosis at this time included RCC, pars intermedia cyst, or (less likely) an early craniopharyngioma or macroadenoma. Due to the peripheral hemosiderin identified on the T2 images, the possibility of Sheehan's-like syndrome or hemorrhage into a RCC was considered. The possibility of undergoing endoscopic transsphenoidal cyst fenestration and cyst wall biopsy was discussed with the patient's family. However, given that the optic apparatus was intact and owing to the poor likelihood of resolution of the endocrinopathies in response to surgery, the patient's family requested close observation and follow-up.
Figure 1.
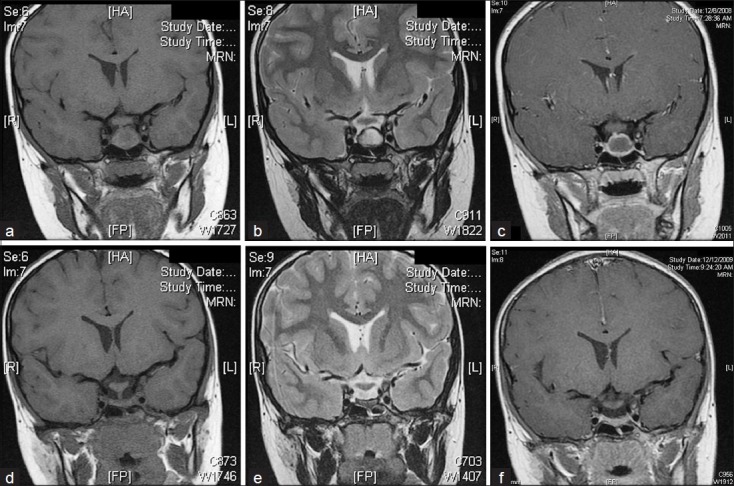
(a–c) Coronal MR image showing a cystic lesion in the middle of the pituitary gland, measuring 17.1 × 13.9 × 11.4 mm3. (a) On T1-weighted imaging, the lesion is mostly isointense to brain, with a small hyperintense intracystic nodule. (b) On T2-weighted imaging, the lesion is hyperintense to brain, with the intracystic nodule being hypointense. There is a concentric hypointense region surrounding the lesion. (c) Contrast-enhanced T1-weighted imaging shows peripheral enhancement. (d–f) Approximately 1 year later, the size of the lesion is dramatically decreased. (d) This T1-weighted image shows a flattened gland that is approximately 3 mm in height. (e) T2-weighted image similarly demonstrates a flattened gland with no definite mass. A hypointense rim surrounds the gland. (f) T1-weighted image with contrast demonstrates no abnormal enhancement.
Follow-up evaluations
At the 3-month follow-up evaluation, the patient had no new complaints. Her examination remained stable and non-focal with normal visual fields. She continued to take levothyroxine and DDAVP. An MRI study demonstrated a cystic pituitary mass with an enhancement pattern similar to previous studies but with a decrease in the size of the lesion (13.8 × 10.6 × 10.0 mm3) and no compression of the optic apparatus. Given that the patient had normal visual fields and, now, the lesion had regressed, the decision was made to continue monitoring the lesion with serial MRI scans.
At 6-month follow-up, the patient and her family reported no new complaints. Her examination remained stable and non-focal with normal visual fields. She continued to take levothyroxine and DDAVP. An MRI study demonstrated a cystic pituitary mass with a continued decrease in size (13.0 × 9.7 × 10.0 mm3) compared with previous studies. The lesion now had settled in the sella turcica and was distant from the optic chiasm.
At 1-year follow up, the patient had grown 1 inch as a result of the initiation of growth hormone replacement therapy (0.02 mg/kg), which she started 9 months after her initial presentation to our clinic at the recommendation of her endocrinologist. She had no new complaints, and her examination was again stable and nonfocal with normal visual fields. The patient was tolerating all medications, which included levothyroxine, DDAVP, and growth hormone replacement therapy. An MRI study demonstrated a pituitary gland that was almost flat, with a height of 3 mm and only a thin rim of enhancement within the sella turcica [Figure 1d–f]. The optic apparatus remained free of any compression.
At the time this report was written (27 months after her initial presentation to our clinic), the patient remained asymptomatic and demonstrated no recurrence on MRI. She remained on levothyroxine, DDAVP, and growth hormone replacement therapy.
DISCUSSION
RCCs are a relatively common intracranial lesion but are rarely symptomatic. They most commonly occur in women (female-to-male ratio of 2:1), with a mean age at presentation of 38 years old.[32] Symptoms, when they do occur, may include headache, visual disturbances and/or endocrinopathies attributable to hypopituitarism. Interestingly, only the presence of visual disturbances has been correlated with the size of the lesion, whereas the existence of chronic inflammation and lesions demonstrating T1 hyperintensity has been correlated with hypopituitarism.[9,15,20] Furthermore, T1-hyperintense lesions tend to manifest clinically at a smaller size compared with those lesions that are T1-hypointense.[25,33]
Radiographic features of RCC have been reported by many authors and appear to be quite variable, thus hindering the rendering of a definitive diagnosis radiographically.[5,23,31,33] This variability also is maintained in the pediatric population.[7] It has been suggested that the MRI intensity of the intracystic contents is dependent on protein concentration; higher protein concentrations result in increasing T1 intensity and decreasing T2 intensity.[10] Although RCCs are generally regarded to be sellar lesions, they often extend into the suprasellar region, thereby assuming a dumbbell shape.[31] Although mural calcifications on a CT scan are typically considered characteristic of craniopharyngiomas, Shin et al.[28] report that 13% of RCCs demonstrate this finding.
Our patient exhibited a lesion that was T1-isointense and T2-hyperintense, suggesting not only moderate protein and mucopolysaccharide content but also significant water content. In addition, the lesion contained a non-enhancing intracystic nodule that was T1 hyperintense and T2 hypointense relative to the remaining intracystic contents; intracystic nodules with these imaging patterns have been described as characteristic of RCCs.[5] This intensity pattern of intracystic nodules is in contrast to the intracystic nodules of craniopharyngiomas, which tend to be strongly enhancing and T1 hypointense and T2 hyperintense relative to the remaining intracystic contents.
At one time, it was suggested that cyst wall enhancement on MRI was an effective means of differentiating neoplastic cysts from non-neoplastic cysts.[11] However, more recently, such an enhancement has been recognized to occur in non-neoplastic cysts and to represent pericystic inflammation, squamous metaplasia, hemosiderin, cholesterol deposits, and/or a normal, but compressed, pituitary gland.[5,15,20] The enhancement observed in our patient appeared to be contiguous with the infundibulum. Therefore, we regarded this enhancement to represent normal, but compressed, pituitary gland. However, because a surgical procedure and biopsy were not performed in this case, we cannot definitively exclude the presence of any of the previously mentioned possibilities.
Our patient also exhibited a peripheral, pericystic T2-hypointense rim, which could represent calcification, hemorrhage, or chronic inflammation. Given its absence on CT imaging and its persistence over time on MRI, we regarded this finding to be most consistent with chronic inflammation.
Although we considered pituitary apoplexy as a diagnostic possibility, we consider this to be highly unlikely, as this patient had not experienced any of the precipitating conditions traditionally associated with this state.[4] Infarction of the gland is similarly unlikely, as this patient had never experienced any episodes of profound hypotension that would have compromised blood supply to the gland. Consequently, hemorrhage into this patient's RCC is a much more likely possibility.
Recently, Chaiban et al.,[6] coined the term “Rathke cleft cyst apoplexy” to describe “a syndrome in which patients with a Rathke cleft cyst present with a clinical syndrome associated with hemorrhage into the cyst.” They describe this entity to occur in 20% of patients with a RCC in their series and to result in the acute worsening or new-onset of symptoms (e.g., increasing headache, visual changes, cranial nerve palsies, and/or varying degrees of hypopituitarism) caused by the sudden expansion of the pituitary gland. However, our patient's symptoms developed gradually and at no time did we observe any of the clinical findings typically associated with expansion of sellar contents. Therefore, we consider this syndrome to be absent in this case.
Although we recognize that the unequivocal diagnosis of RCCs can only be made by biopsy of the cyst wall, on the basis of the previously described imaging characteristics of this patient's lesion, we regarded her to have a RCC. The natural history of both asymptomatic and symptomatic RCC is poorly defined. However, these cysts are generally considered to be nonexpansile lesions that are stable over time. Aho et al.[1] described 42 asymptomatic patients in whom the lesion was discovered incidentally and who experienced no growth of the cyst over 5 years of follow-up. Further, Sanno et al.[26] reported that only 5.3% of RCCs demonstrated growth during a mean follow-up period of 38.9 months. Symptomatic lesions are treated by surgical drainage and cyst wall biopsy or by surgical resection with expected and predictable improvement of preoperative visual impairment and headache but only occasional resolution of preoperative endocrinopathies.[1,2,15] Pericystic chronic inflammation that extends to the pituitary, causing irreversible damage, has been proposed as the reason for the failure of endocrinopathies to resolve.[1,2]
Surgical drainage or resection of RCC most often is performed via a transsphenoidal approach with the aim of either cyst decompression through fenestration of the cyst wall or aggressive resection of all cyst wall components. Aho et al.[1] demonstrated that aggressive attempts at resection of the entire cyst wall were associated with higher morbidity (e.g., new-onset endocrinopathies requiring lifelong treatment) but without an improvement in the recurrence rate (18% in 5 years). Additional complications of surgical resection are similar to those of transsphenoidal surgery for other pathologies, including meningitis and cerebrospinal fluid leak.[1,14] Given this morbidity, the potential for spontaneous involution and resultant conservative management is desired.
The spontaneous involution of RCCs is rare, with only 7 other reports in the literature.[2,12,17,19,20,25,29] However, none of these instances occurred in a patient as young as the patient presented in this report. The mechanism by which this phenomenon occurs remains speculative. Saeki et al.[25] theorize that bleeding, which may or may not be detectable on MRI, followed by subsequent resorption of blood products causes transient fluctuations in the cyst size. Another proposed mechanism is that increased intracystic pressure may damage mucous-producing goblet cells in the cyst wall, causing a reduction in secretion with resultant decrease in the cyst size.[13] Others have suggested that repeated cyst rupture ultimately leads to shrinkage of the lesion.[19] Finally, the reduction of pericystic inflammation, attributed to glucocorticoid administration, may also promote symptom resolution and cyst size reduction.[8,17]
The role of medical therapy in the treatment of RCCs has not been studied. Although exogenous glucocorticoids may promote cyst size reduction, their presence is clearly not necessary, as several of the reported cases of spontaneous involution occurred in their absence.[2,12,20,25,29] Our patient did not receive glucocorticoid replacement therapy. However, she did receive levothyroxine and DDAVP and experienced a subsequent decrease in the size of the cyst. The size of the cyst continued to diminish after growth hormone replacement therapy was initiated approximately 9 months after initial diagnosis of the lesion. Previous reports have not described spontaneous cyst involution in the presence of or in response to these medical therapies. Theoretically, these exogenously administered hormones could augment the negative feedback of the hypothalamic–pituitary axis and thereby suppress functioning anterior pituitary cells of the cyst wall, with resultant hypoplasia of these cells. However, we do not feel that this purely speculative and theoretical mechanism explains the spontaneous involution observed in this patient.
The effect of medical treatment on the growth and/or involution of RCC remains unclear. As mentioned previously, many reports describe spontaneous involution in the absence of any medical therapy. We are unable to assign causation of the spontaneous involution described in this report to the medical therapy our patient received. Nevertheless, considering the poor response of endocrinopathies to surgery, we propose that conservative medical treatment of these hormonal perturbations and observation with serial MRI scans may be an acceptable management of RCCs that spare the optic apparatus. Given the potential to avoid surgical morbidity, further studies into the role of medical therapy and conservative management of these lesions are warranted.
CONCLUSIONS
Although the natural history of RCC requires further study, observation with serial MRI studies may be an acceptable management strategy in the absence of debilitating symptoms. Given that preoperative endocrinopathies rarely resolve following surgical resection of a RCC, medical therapy and observation with serial MRI may be an acceptable alternative for patients presenting with symptoms not attributable to compression of nearby structures (e.g., visual disturbances). The role of medical therapy in promoting spontaneous involution of RCCs remains unclear.
ACKNOWLEDGMENTS
We thank Ronald Alberico, MD (Roswell Park Cancer Institute), for assistance with interpretation of the magnetic resonance images, Mark Burkard, RN (University at Buffalo Neurosurgery), for assistance with gathering clinical information, Paul H. Dressel, BFA (University at Buffalo Neurosurgery), for preparation of the illustrations, and Debra J. Zimmer, AAS CMA-A (University at Buffalo Neurosurgery), for editorial assistance.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2012/3/1/42/94925
Contributor Information
Stephan A. Munich, Email: stephan.munich@gmail.com.
Jody Leonardo, Email: jleonardo@ubns.com.
REFERENCES
- 1.Aho CJ, Liu C, Zelman V, Couldwell WT, Weiss MH. Surgical outcomes in 118 patients with Rathke cleft cysts. J Neurosurg. 2005;102:189–93. doi: 10.3171/jns.2005.102.2.0189. [DOI] [PubMed] [Google Scholar]
- 2.Amhaz HH, Chamoun RB, Waguespack SG, Shah K, McCutcheon IE. Spontaneous involution of Rathke cleft cysts: Is it rare or just underreported? J Neurosurg. 2010;112:1327–32. doi: 10.3171/2009.10.JNS091070. [DOI] [PubMed] [Google Scholar]
- 3.Benveniste RJ, King WA, Walsh J, Lee JS, Naidich TP, Post KD. Surgery for Rathke cleft cysts: Technical considerations and outcomes. J Neurosurg. 2004;101:577–84. doi: 10.3171/jns.2004.101.4.0577. [DOI] [PubMed] [Google Scholar]
- 4.Biousse V, Newman NJ, Oyesiku NM. Precipitating factors in pituitary apoplexy. J Neurol Neurosurg Psychiatry. 2001;71:542–5. doi: 10.1136/jnnp.71.4.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byun WM, Kim OL, Kim D. MR imaging findings of Rathke's cleft cysts: Significance of intracystic nodules. AJNR Am J Neuroradiol. 2000;21:485–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Chaiban JT, Abdelmannan D, Cohen M, Selman WR, Arafah BM. Rathke cleft cyst apoplexy: A newly characterized distinct clinical entity. J Neurosurg. 2011;114:318–24. doi: 10.3171/2010.5.JNS091905. [DOI] [PubMed] [Google Scholar]
- 7.Christophe C, Flamant-Durand J, Hanquinet S, Heinrichs C, Raftopoulos C, Sariban E, et al. MRI in seven cases of Rathke's cleft cyst in infants and children. Pediatr Radiol. 1993;23:79–82. doi: 10.1007/BF02012389. [DOI] [PubMed] [Google Scholar]
- 8.Furtado SV, Venkatesh PK, Ghosal N, Hegde AS. Reduction in size of a large Rathke's cleft cyst on treatment with low dose of corticosteroid. Horm Metab Res. 2010;42:227–9. doi: 10.1055/s-0029-1243248. [DOI] [PubMed] [Google Scholar]
- 9.Hama S, Arita K, Nishisaka T, Fukuhara T, Tominaga A, Sugiyama K, et al. Changes in the epithelium of Rathke cleft cyst associated with inflammation. J Neurosurg. 2002;96:209–16. doi: 10.3171/jns.2002.96.2.0209. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi Y, Tachibana O, Muramatsu N, Tsuchiya H, Tada M, Arakawa Y, et al. Rathke cleft cyst: MR and biomedical analysis of cyst content. J Comput Assist Tomogr. 1999;23:34–8. doi: 10.1097/00004728-199901000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Hua F, Asato R, Miki Y, Okumura R, Hashimoto N, Kikuchi H, et al. Differentiation of suprasellar nonneoplastic cysts from cystic neoplasms by Gd-DTPA MRI. J Comput Assist Tomogr. 1992;16:744–9. doi: 10.1097/00004728-199209000-00014. [DOI] [PubMed] [Google Scholar]
- 12.Igarashi T, Saeki N, Yamaura A. Long-term magnetic resonance imaging follow-up of asymptomatic sellar tumors.-- Their natural history and surgical indications. Neurol Med Chir (Tokyo) 1999;39:592–9. doi: 10.2176/nmc.39.592. [DOI] [PubMed] [Google Scholar]
- 13.Ishii T, Yamasaki T, Tanaka J, Tanaka S, Hori T, Muraoka K. Rathke's cleft cyst--Report of three cases. No Shinkei Geka. 1987;15:451–6. [PubMed] [Google Scholar]
- 14.Jho HD. Endoscopic transsphenoidal surgery. J Neurooncol. 2001;54:187–95. doi: 10.1023/a:1012969719503. [DOI] [PubMed] [Google Scholar]
- 15.Kim JE, Kim JH, Kim OL, Paek SH, Kim DG, Chi JG, et al. Surgical treatment of symptomatic Rathke cleft cysts: Clinical features and results with special attention to recurrence. J Neurosurg. 2004;100:33–40. doi: 10.3171/jns.2004.100.1.0033. [DOI] [PubMed] [Google Scholar]
- 16.Laws ER, Kanter AS. Rathke cleft cysts. J Neurosurg. 2004;101:571–2. doi: 10.3171/jns.2004.101.4.0571. [DOI] [PubMed] [Google Scholar]
- 17.Maruyama H, Iwasaki Y, Tsugita M, Ogami N, Asaba K, Takao T, et al. Rathke's cleft cyst with short-term size changes in response to glucocorticoid replacement. Endocr J. 2008;55:425–8. doi: 10.1507/endocrj.k07e-070. [DOI] [PubMed] [Google Scholar]
- 18.Mukherjee JJ, Islam N, Kaltsas G, Lowe DG, Charlesworth M, Afshar F, et al. Clinical, radiological and pathological features of patients with Rathke's cleft cysts: Tumors that may recur. J Clin Endocrinol Metab. 1997;82:2357–62. doi: 10.1210/jcem.82.7.4043. [DOI] [PubMed] [Google Scholar]
- 19.Nishio S, Morioka T, Suzuki S, Fukui M. Spontaneous regression of a pituitary cyst: Report of two cases. Clin Imaging. 2001;25:15–7. doi: 10.1016/s0899-7071(00)00233-3. [DOI] [PubMed] [Google Scholar]
- 20.Nishioka H, Haraoka J, Izawa H, Ikeda Y. Magnetic resonance imaging, clinical manifestations, and management of Rathke's cleft cyst. Clin Endocrinol (Oxf) 2006;64:184–8. doi: 10.1111/j.1365-2265.2006.02446.x. [DOI] [PubMed] [Google Scholar]
- 21.Onesti ST, Wisniewski T, Post KD. Pituitary hemorrhage into a Rathke's cleft cyst. Neurosurgery. 1990;27:644–6. doi: 10.1097/00006123-199010000-00026. [DOI] [PubMed] [Google Scholar]
- 22.Raper DM, Besser M. Clinical features, management and recurrence of symptomatic Rathke's cleft cyst. J Clin Neurosci. 2009;16:385–9. doi: 10.1016/j.jocn.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 23.Ross DA, Norman D, Wilson CB. Radiologic characteristics and results of surgical management of Rathke's cysts in 43 patients. Neurosurgery. 1992;30:173–9. doi: 10.1227/00006123-199202000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Sade B, Albrecht S, Assimakopoulos P, Vezina JL, Mohr G. Management of Rathke's cleft cysts. Surg Neurol. 2005;63:459–66. doi: 10.1016/j.surneu.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Saeki N, Kubota M, Yamaura A, Ishige N. Fluctuating visual field defects in Rathke's cleft cysts: MRI analysis. J Clin Neurosci. 1999;6:524–7. doi: 10.1016/s0967-5868(99)90018-8. [DOI] [PubMed] [Google Scholar]
- 26.Sanno N, Oyama K, Tahara S, Teramoto A, Kato Y. A survey of pituitary incidentaloma in Japan. Eur J Endocrinol. 2003;149:123–7. doi: 10.1530/eje.0.1490123. [DOI] [PubMed] [Google Scholar]
- 27.Shanklin WM. On the presence of cysts in the human pituitary. Anat Rec (Hoboken) 1949;104:379–407. doi: 10.1002/ar.1091040402. [DOI] [PubMed] [Google Scholar]
- 28.Shin JL, Asa SL, Woodhouse LJ, Smyth HS, Ezzat S. Cystic lesions of the pituitary: Clinicopathological features distinguishing craniopharyngioma, Rathke's cleft cyst, and arachnoid cyst. J Clin Endocrinol Metab. 1999;84:3972–82. doi: 10.1210/jcem.84.11.6114. [DOI] [PubMed] [Google Scholar]
- 29.Simmons JD, Simmons LA. Spontaneous regression of a pituitary cyst. Neuroradiology. 1999;41:27–9. doi: 10.1007/s002340050699. [DOI] [PubMed] [Google Scholar]
- 30.Teramoto A, Hirakawa K, Sanno N, Osamura Y. Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology. 1994;193:161–4. doi: 10.1148/radiology.193.1.8090885. [DOI] [PubMed] [Google Scholar]
- 31.Tominaga JY, Higano S, Takahashi S. Characteristics of Rathke's cleft cyst in MR imaging. Magn Reson Med Sci. 2003;2:1–8. doi: 10.2463/mrms.2.1. [DOI] [PubMed] [Google Scholar]
- 32.Voelker JL, Campbell RL, Muller J. Clinical, radiographic, and pathological features of symptomatic Rathke's cleft cysts. J Neurosurg. 1991;74:535–44. doi: 10.3171/jns.1991.74.4.0535. [DOI] [PubMed] [Google Scholar]
- 33.Wen L, Hu LB, Feng XY, Desai G, Zou LG, Wang WX, et al. Rathke's cleft cyst: Clinicopathological and MRI findings in 22 patients. Clin Radiol. 2010;65:47–55. doi: 10.1016/j.crad.2009.09.010. [DOI] [PubMed] [Google Scholar]