

Abstract
The channel pore-forming α subunit Kv4.2 is a major constituent of A-type (IA) potassium currents and a key regulator of neuronal membrane excitability. Multiple mechanisms regulate the properties, subcellular targeting, and cell-surface expression of Kv4.2-encoded channels. In the present study, shotgun proteomic analyses of immunoprecipitated mouse brain Kv4.2 channel complexes unexpectedly identified the voltage-gated Na+ channel accessory subunit Navβ1. Voltage-clamp and current-clamp recordings revealed that knockdown of Navβ1 decreases IA densities in isolated cortical neurons and that action potential waveforms are prolonged and repetitive firing is increased in Scn1b-null cortical pyramidal neurons lacking Navβ1. Biochemical and voltage-clamp experiments further demonstrated that Navβ1 interacts with and increases the stability of the heterologously expressed Kv4.2 protein, resulting in greater total and cell-surface Kv4.2 protein expression and in larger Kv4.2-encoded current densities. Together, the results presented here identify Navβ1 as a component of native neuronal Kv4.2-encoded IA channel complexes and a novel regulator of IA channel densities and neuronal excitability.
Introduction
Somatodendritic A-type (IA) voltage-gated K+ channels are key regulators of neuronal excitability, contributing to resting membrane potentials and action potential repolarization and functioning to modulate the frequency of repetitive firing, the current thresholds for action potential generation, and the back-propagation of action potentials into dendrites (Hoffman et al., 1997; Birnbaum et al., 2004; Kim et al., 2005; Yuan et al., 2005). Whole-cell voltage-clamp recordings from neurons obtained from mice (Kv4.2−/−) harboring a targeted disruption of the Kcnd2 (Kv4.2) locus revealed that the K+ channel pore-forming α subunit, Kv4.2, is a major constituent of IA in hippocampal and cortical pyramidal neurons, as well as in dorsal horn neurons of the spinal cord (Chen et al., 2006; Hu et al., 2006; Nerbonne et al., 2008; Norris and Nerbonne, 2010). The functional properties of Kv4.2-encoded channels are regulated by multiple mechanisms, including post-translational modifications and interactions with accessory subunits (Birnbaum et al., 2004). Phosphorylation of the Kv4.2 α subunit by different kinases, for example, modulates the cell-surface expression, densities, and activity-dependent trafficking of Kv4.2-encoded IA channels (Birnbaum et al., 2004; Varga et al., 2004; Hammond et al., 2008). In addition, interaction of Kv4.2 α subunits with accessory subunits, such as the K+ channel interacting proteins (KChIPs) and the dipeptidyl peptidase-like proteins, regulates the subcellular targeting, surface expression, and biophysical properties of heterologously expressed Kv4.2-encoded channels (Nadal et al., 2003; Birnbaum et al., 2004; Rhodes et al., 2004; Jerng et al., 2005; Zagha et al., 2005; Nadin and Pfaffinger, 2010; Norris et al., 2010; Sun et al., 2011).
Experiments in heterologous expression systems have provided valuable insights into the functional effects of a number of putative accessory subunits on the properties of Kv4.2-encoded channels and have indicated that Kv4 channel α subunits function in macromolecular protein complexes (Birnbaum et al., 2004). Little is known, however, about the composition of native neuronal Kv4.2-encoded channels or the roles that the various Kv4 channel accessory subunits play in the regulation of neuronal excitability. The present study identifies the voltage-gated Na+ channel accessory subunit Navβ1 as a component of native neuronal Kv4.2 channel complexes and a key modulator of action potential repolarization and repetitive firing in cortical pyramidal neurons. Navβ1 is a single transmembrane multifunctional protein that, in addition to functioning as a cell adhesion molecule, has been shown to modulate voltage-gated Na+ (Nav) currents and Nav channel cell-surface expression and subcellular localization (Isom et al., 1992; Isom, 2001, 2002; Brackenbury et al., 2008, 2010; Aman et al., 2009; Patino and Isom, 2010; Brackenbury and Isom, 2011). The experiments here identified the presence of Navβ1 in native Kv4.2 channel complexes immunoprecipitated from the mouse brain. Voltage-clamp and current-clamp recordings revealed that acute knockdown of Navβ1 decreases IA densities in isolated cortical neurons. In addition, in vivo loss of Navβ1 impairs action potential repolarization and repetitive firing in cortical pyramidal neurons in slices prepared from animals (Scn1b−/−) lacking Navβ1. Biochemical and voltage-clamp experiments further demonstrated that Navβ1 functions to stabilize heterologously expressed Kv4.2 protein, resulting in greater total and cell-surface Kv4.2 protein expression and increased Kv4.2-encoded current densities.
Materials and Methods
All experiments were performed in accordance with the guidelines published in the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experimental protocols were approved by the Animal Care and Use Committee of Washington University School of Medicine. Generation and characterization of the Kv4.2-targeted deletion (Kv4.2−/−) mouse line has been described previously (Guo et al., 2005; Hu et al., 2006; Nerbonne et al., 2008). Scn1b−/− mice were generated from Scn1b+/− heterozygotes (Chen et al., 2004), congenic on the C57BL/6 background, and genotypes were confirmed by PCR screening as described previously. Male and female mice were used in all experiments.
Immunoprecipitation of mouse brain Kv4.2 channel complexes.
For immunoprecipitation (IP) of Kv4.2 channel complexes, flash-frozen brains from adult wild-type (WT) or Kv4.2−/− mice were homogenized in ice-cold lysis buffer containing the following (in mm): 20 HEPES, pH 7.4, 110 potassium acetate, pH 7.4, 1 MgCl2, 150 NaCl, with 0.1 μm CaCl2, complete mini EDTA-free protease inhibitor mixture tablet (Roche), 1 mm Pefabloc (Sigma), 1 μg/ml pepstatin A (Calbiochem), 1 X Halt phosphatase inhibitor mixture (Pierce), and 0.5% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propane-sulfonate hydrate (Sigma). After a 15 min rotation at 4°C, 40 mg of each soluble protein fraction was used for IPs with an anti-Kv4.2 rabbit polyclonal antibody (RbαKv4.2; Millipore Bioscience Research Reagents) cross-linked to protein A-magnetic beads (Invitrogen) using 20 mm dimethyl pimelimidate (Pierce) (Schneider et al., 1982). Following mixing of the protein samples with the antibody-coupled beads for 2 h at 4°C, the beads were collected and washed four times with ice-cold lysis buffer. Protein complexes were eluted from the beads in 2% Rapigest (Waters) in 100 mm Tris, pH 8.5, at 60°C for 5 min.
Mass spectrometric analyses.
Immunoprecipitated protein samples were reduced, alkylated, trypsinized, and analyzed using shotgun proteomics by Multidimensional Protein Identification Technology (MudPIT) as described previously (Link et al., 1999; Washburn et al., 2001; Arnett et al., 2008; Marionneau et al., 2009). Briefly, a fritless microcapillary (100 μm inner diameter) column was packed sequentially as follows: 9 cm of 5 μm C18 reverse-phase (Synergi 4 μ Hydro RP80a; Phenomenex), 3 cm of 5 μm strong cation exchange (Partisphere SCX; Whatman), and 2 cm of C18 reverse-phase packing material. The trypsin-digested samples were loaded directly onto the triphasic column, equilibrated in 0.1% formic acid and 2% acetonitrile. The column was placed in line with a nanoESI-LTQ linear ion trap mass spectrometer (Thermo Scientific), and an automated six-cycle multidimensional chromatographic separation was performed using buffer A (0.1% formic acid, 5% acetonitrile), buffer B (0.1% formic acid, 80% acetonitrile), and buffer C (0.1% formic acid, 5% acetonitrile, and 500 mm ammonium acetate) at a flow rate of 300 nl/min. The first cycle was a 20 min isocratic flow of buffer B. Cycles 2–6 consisted of 3 min of buffer A, 2 min of buffer C, and 5 min of buffer A, followed by a 60 min linear gradient to 60% buffer B. Cycles 2–6 used 15, 30, 50, 70, and 100% of buffer C, respectively. During the linear gradient, eluting peptides were analyzed by one full mass spectrometric (MS) scan (200–2000 m/z), followed by (five) MS/MS scans on the five most abundant ions detected in the full MS scan while operating under dynamic exclusion.
The program extractms2, developed and provided by Jimmy Eng and John R. Yates III (The Scripps Research Institute, La Jolla, CA), was used to generate the ASCII peak list and identify +1 or multiply charged precursor ions from unprocessed MS data files. Tandem spectra were searched with no protease specificity using SEQUEST-PVM (Sadygov et al., 2002) against the RefSeq mouse protein database (released May 2005) containing 28,818 entries. For multiply charged precursor ions (z ≥ +2), an independent search was performed on both the +2 and +3 mass of the parent ion. Data were processed and organized using the BIGCAT software analysis suite (McAfee et al., 2006). A weighted scoring matrix was used to select the most likely charge state of multiply charged precursor ions (Link et al., 1999; McAfee et al., 2006). From the database search, tryptic peptide sequences with SEQUEST cross-correlation scores (Cn) ≥1.5 for +1 ions, ≥2 for +2 ions, and ≥2 for +3 ions were considered significant and used to create the list of identified proteins. To compare the relative abundances of the proteins identified by MS analyses, protein abundance factors (Powell et al., 2004) were calculated for each identified protein by normalizing the total number of nonredundant spectra that correlated significantly with each open reading frame to the molecular weight of the cognate protein (×104).
Plasmids.
The mouse Kv4.2, KChIP2, Navβ1, and Kv2.1 cDNAs were purchased from Open Biosystems and the sequences were verified. The pCMV-Script plasmid was purchased from Stratagene. The enhanced yellow fluorescent protein (EYFP)-C-terminally tagged Navβ1 construct was generated by subcloning Navβ1 from the pCMV-SPORT6 into the pEYFP-N1 vector (Clontech). The Myc-N-terminally tagged TASK1 was generated by cloning the coding region of mouse TASK1 into the pCMV-Tag3B (Myc-tagged) vector (Stratagene). The Myc-TASK1 coding sequence was subcloned into the α-MHC vector (Gulick and Robbins, 2009) at the SalI site. Plasmids expressing short hairpin RNA (shRNA) sequences targeting Scn1b (Navβ1) were obtained from the Genome Sequencing Center at Washington University School of Medicine. The nontargeted shRNA control was obtained from Sigma (Mission shRNA). The Navβ1 targeted shRNA sequences used were as follows: CTCTCTCACCAGCCTTCAATT, GCCATTACATCCGAGAGCAAA, GAGGAATTTGTCAAGATCCTA, CGACTACGAATGTCACGTCTA, and CGTCTCCTCTTCTTTGATAAT. The nontargeted shRNA sequence used was CAACAAGATGAAGAGCACCAA. Each of the (five targeted and one nontargeted) shRNA sequences was provided in a pLKO.1-puro expression vector. The coding sequence of the Puromycin-resistance gene was replaced with the sequence coding for the red fluorescent protein tdTomato to allow transfected cells to be identified under epifluorescence illumination.
Cell culture and transient transfections.
Human embryonic kidney 293 (HEK-293) cells were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin, in 37°C, 5% CO2:95% air incubator. Cells were transiently transfected with 0.5 μg of the Kv4.2 plasmid alone or with Navβ1 and/or KChIP2 at 80–90% confluence using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Experiments were also performed with Kv2.1 or TASK1 coexpressed with Navβ1. The relative amounts of the cDNA constructs used for the transfections were 1:2 for Kv4.2:Navβ1, 1:1 for Kv4.2:KChIP2, 1:1:2 for Kv4.2:KChIP2:Navβ1, 1:2 for Kv2.1:Navβ1, and 1:2 for TASK1:Navβ1. The absolute amounts of the various constructs were calculated and pCMV-Script plasmid was used as a filler plasmid to keep the total DNA constant at 2 μg in each transfection.
Coimmunoprecipitation of heterologously expressed proteins.
The EYFP-tagged Navβ1 construct was used in coimmunoprecipitation experiments. Twenty-four hours after transfections, HEK-293 cells were washed twice with PBS and lysed in lysis buffer (as described above). For IPs, soluble protein fractions were collected and incubated with magnetic beads coupled to a rabbit polyclonal anti-Kv4.2 antibody (RbαKv4.2; Millipore Bioscience Research Reagents) or to a rabbit polyclonal anti-enhanced green fluorescent protein (EGFP) antibody (RbαEGFP; Millipore Bioscience Research Reagents). After a 2 h incubation at 4°C, beads were washed four times in lysis buffer, and protein complexes were eluted with 1X SDS sample buffer at 60°C for 5 min. Protein eluates from IPs were fractionated by gel electrophoresis. Western blot analyses were performed as described previously (Marionneau et al., 2008). The mouse monoclonal anti-Kv4.2 and anti-KChIP2 antibodies were developed by and obtained from the University of California, Davis/NIH NeuroMab Facility (supported by NIH Grant U24NS050606 and maintained by the University of California, Davis). A mouse monoclonal anti-EGFP antibody was purchased from Millipore Bioscience Research Reagents. Goat anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies were purchased from Pierce.
Cycloheximide treatment.
To evaluate the stability of total and cell-surface Kv4.2 protein, transfected HEK-293 cells were treated with the protein translation inhibitor, cycloheximide (Calbiochem) at 100 μg/ml in DMEM at 37°C for varying times (0, 30, 60, 120, and 480 min). Cell-surface biotinylation assays were then performed as described below. Kv4.2 protein expression (total or cell surface) in cycloheximide-treated cells is expressed as the percentage of Kv4.2 protein expression (total or cell surface) in untreated cells.
Cell-surface biotinylation and endocytosis assays.
Surface biotinylation of HEK-293 cells was completed as described previously (Marionneau et al., 2008). Briefly, cells were incubated with the cleavable EZ-Link Sulfo-NHS-SS-Biotin (0.5 mg/ml) (Pierce) in ice-cold PBS, pH 7.4, for 30 min at 4°C. Free biotin was quenched with Tris-saline (10 mm Tris, pH 7.4, 120 mm NaCl), and detergent-soluble cell lysates were prepared. Biotinylated cell-surface proteins were affinity-purified using NeutrAvidin-conjugated agarose beads (Pierce), and purified cell-surface proteins were analyzed by Western blot (as described above). Mouse monoclonal anti-transferrin receptor (TransR), anti-Kv2.1, and anti-myc antibodies were purchased from Invitrogen, the University of California, Davis/NIH NeuroMab Facility, and Millipore, respectively. Bands corresponding to Kv4.2, Kv2.1, and Myc-TASK1 were normalized to bands corresponding to TransR from the same sample. Kv4.2 protein expression (total or cell surface) is expressed relative to Kv4.2 protein expression (total or cell surface) in cells transfected with Kv4.2 only.
To assay endocytosis, cells were biotinylated (pulse) and washed with Tris-saline solution as described above. Cells were then returned to culture medium (chase) at 37°C for varying times (0, 15, 30, and 60 min). At the end of each chase time, cells were rinsed with ice-cold PBS and incubated with the impermeable reducing agent sodium 2-mercaptoethanesulfonate (100 mm, in 50 mm Tris, pH 8.6, 100 mm NaCl, 2.5 mm CaCl2) at 4°C for 15 min to remove the biotin remaining at the cell surface. This procedure was repeated twice, and cells were then incubated with 5 mg/ml iodoacetamide in PBS at 4°C for 15 min to modify free SH groups. Detergent-soluble cell lysates were prepared, and biotinylated proteins were purified using NeutrAvidin-conjugated agarose beads (Pierce). For each chase time, nonreduced samples were used to estimate the degradation of biotinylated proteins or spontaneous debiotinylation. Reduced samples at 0 min (t0 min, reduced) were used to evaluate background (usually ≤10%). Results are expressed as the percentage of biotinylated surface proteins that were endocytosed at each time point, i.e., %Fraction Endocytosed (at x min), calculated as (timexmin, reduced − time0 min, reduced)/timexmin, nonreduced × 100.
Electrophysiological recordings from HEK-293 cells.
Whole-cell Kv currents were recorded at room temperature from transiently transfected HEK-293 cells using an Axopatch-1B amplifier (Molecular Devices), as described previously (Li et al., 2005). Voltage-clamp paradigms were controlled using the pClamp 9 software package (Molecular Devices) interfaced to the electrophysiological equipment using a Digidata 1322A A/D converter (Molecular Devices). Data were acquired at 10 kHz, and current signals were filtered on-line at 5 kHz before digitization and storage. Recording pipettes contained the following (in mm): 115 KCl, 15 KOH, 10 EGTA, 10 HEPES, and glucose 5 (pH 7.2, 295–310 mosM). Pipette resistances were 1.8–2.8 MΩ when filled with the pipette solution. The bath solution contained the following (in mm): 140 NaCl, 4 KCl, 1 CaCl2, 2 MgCl2, 10 HEPES, and glucose 5 (pH 7.4, 295–310 mosM). After establishing the whole-cell configuration, ±10 mV steps from a holding potential (HP) of −70 mV were applied to allow measurements of whole-cell membrane capacitances and input resistances. Whole-cell membrane capacitances and series resistances were routinely compensated (80%) electronically. The voltage errors resulting from the uncompensated series resistances were always ≤6 mV and were not corrected. Only data obtained from cells with input resistances >200 MΩ and capacitive transients well described by single exponentials were analyzed. Kv currents were evoked by 400 ms depolarizing voltage steps to potentials between −40 and +30 mV from an HP of −70 mV; voltage steps were presented in 10 mV increments at 15 s intervals.
Electrophysiological data were compiled and analyzed using Clampfit 9 (Molecular Devices) and Excel (Microsoft). Whole-cell membrane capacitances were calculated by integrating the area under the capacitive transients evoked during the ±10 mV voltage steps from the HP, before compensation. Peak currents at each voltage step were defined as the maximal Kv current amplitudes. For each cell, current amplitudes were normalized to the whole-cell membrane capacitance, and current densities (pA/pF) are reported.
Scn1b (Navβ1) shRNA screening.
Chinese Hamster Ovary (CHO) cells were maintained in Ham's F-12 Medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin, in 37°C, 5% CO2:95% air incubator. Cells were transfected at ∼70% confluence using Lipofectamine 2000 (Invitrogen) according to the directions from the manufacturer. Briefly, cells were cotransfected with EYFP-tagged Navβ1 and each of the shRNAs (five Scn1b (Navβ1) targeted and one nontargeted) at a 1:1 ratio. Cells were incubated in the transfection mix for 8 h at 37°C and then lysed in lysis buffer (as described above) 48 h after transfection. Protein lysates were analyzed by Western blot (as described above).
Isolation, maintenance, and transfection of cortical neurons.
Neurons were isolated from the primary visual cortices of postnatal day 6–8 C57BL/6 WT mice using previously described methods (Locke and Nerbonne, 1997; Nerbonne et al., 2008; Norris and Nerbonne, 2010; Norris et al., 2010). Briefly, mice were anesthetized with isoflurane, decapitated, and the brains were rapidly removed. The posterior cortices were dissected, minced, and incubated at 37°C in Neurobasal medium (Invitrogen) containing papain (20 U/ml) (Worthington Biochemicals) under 95% O2:5% CO2 for 90 min. Isolated cortical neurons were obtained by trituration and subsequent centrifugation (at 500 g for 15 min) through a bovine serum albumin gradient. Dissociated cells were resuspended in Neurobasal medium and plated on previously prepared monolayers of (rat) neocortical astrocytes (Locke and Nerbonne, 1997). Neurons were transfected with (10 nm) of the Scn1b targeted shRNA or the nontargeted shRNA within 5 h of plating using PepMute siRNA transfection reagent (SignaGen Laboratories) according to the directions from the manufacturer. After incubation with the transfection solution for 5 h at 37°C, the medium was replaced with fresh Neurobasal medium. Neuronal/glial cultures were maintained in 95% O2:5% CO2 incubator at 37°C.
Preparation of acute cortical (and hippocampal) slices.
Brain slices were prepared from the primary visual cortices (or hippocampi) of C57BL/6 WT and Scn1b−/− mice at postnatal day (P) 11–12 using standard procedures (Davie et al., 2006). This age was selected primarily because the Scn1b−/− mice die prematurely, beginning at P13 (Chen et al., 2004). For experiments, mice were decapitated and the brains were rapidly removed and placed in ice-cold, oxygenated artificial cerebrospinal fluid (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 25 dextrose (∼310 mosM), saturated with 95% O2:5% CO2. Coronal slices (350 μm) containing the primary visual cortex (or hippocampus) were cut on a Leica VT1000 S vibrating blade microtome (Leica Microsystems). Slices were incubated in ACSF for at least 30 min before transfer to the recording chamber.
Electrophysiological recordings from cortical (and hippocampal) pyramidal neurons.
Whole-cell voltage-clamp recordings were obtained from tdTomato-expressing cortical neurons 24–72 h following transfection with the nontargeted, or Scn1b-targeted, shRNA construct. Whole-cell current-clamp recordings were obtained from visually identified layer 5 pyramidal neurons in cortical slices (or from CA1 pyramidal neurons in hippocampal slices) using differential interference contrast with infrared microscopy. All recordings were obtained at room temperature (22–24°C). Data were collected using a Multiclamp 700B patch-clamp amplifier interfaced with a Digidata 1332 and the pCLAMP 9 software (Molecular Devices) to a Gateway computer. In all experiments, tip potentials were zeroed before membrane-pipette seals were formed; pipette capacitances and series resistances were compensated electronically by ∼90%. Signals were acquired at 20–50 kHz and filtered at 10 kHz before digitization and storage. These data acquisition parameters fully capture the action potential parameters measured. For voltage-clamp recordings, the bath solution contained the following (in mm): 140 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 5 glucose, 0.001 tetrodotoxin, and 0.1 CdCl2 (pH 7.4, ∼300 mosM). For the current-clamp experiments, slices were perfused continually with ACSF (see above) saturated with 95% O2:5% CO2. The recording pipette solution for voltage-clamp recordings contained the following (in mm): 130 KCl, 10 HEPES, 10 glucose, 0.83 CaCl2, and 2.6 BAPTA (pH 7.4, 300 mosM), and 3 MgATP and 0.5 NaGTP were added the day of recording. Recording pipettes for the current-clamp experiments contained the following (in mm): 120 potassium methyl sulfate, 20 KCl, 10 HEPES, 0.2 EGTA, 8 NaCl, 4 Mg-ATP, 0.3 Tris-GTP, and phosphocreatine 14 (pH 7.25, ∼300 mosM). All reagents were from Sigma unless otherwise noted.
The rapidly activating and rapidly inactivating Kv current, IA, was isolated using a two-step voltage protocol as previously described (Norris and Nerbonne, 2010). Briefly, whole-cell Kv currents were first evoked in response to 4 s depolarizing voltage steps to potentials between −40 and +40 mV (in 10 mV increments) from a holding potential of −70 mV. A prepulse paradigm that included a brief (60 ms) step to −10 mV before the 4 s depolarizing voltage steps to potentials between −40 and +40 mV (in 10 mV increments) was then used. Off-line subtraction of the currents evoked after the prepulse from the currents evoked without the prepulse was performed to isolate IA. Single action potentials and action potential trains were elicited from the resting membrane potential in response to brief (5 ms) and prolonged (500 ms) depolarizing current injections of variable amplitudes. All current-clamp recordings were obtained within 5 min after achieving the whole-cell configuration.
Data analyses.
Data were compiled and analyzed using ClampFit (Molecular Devices), Microsoft Excel, and Prism (GraphPad Software). For the voltage-clamp experiments, only data from cells with input resistances >300 MΩ and access resistances <20 MΩ were included in the analyses. Capacitive currents, elicited by short (25 ms) voltage steps (±10 mV) from the holding potential (−70 mV), were measured in each cell. Only cells with capacitive transients well described by a single exponential (consistent with a single electrical compartment) were analyzed further. The whole-cell membrane capacitance (Cm) of each cell was calculated by dividing the integral of capacitive transient by the membrane voltage. Input resistances were calculated from the steady-state currents elicited by the same ±10 mV steps (from the holding potential). For each cell, the series resistance was calculated by dividing the time constant of the decay of the capacitive transient (fit by a single exponential) by the Cm. IA amplitudes in individual cells were determined by subtracting the currents evoked after the prepulse from the currents evoked without the prepulse (see voltage-clamp protocols described above). Current amplitudes, measured in individual cells, were normalized for differences in cell size (whole-cell Cm), and current densities (pA/pF) are reported.
All current-clamp recordings were obtained from cells with overshooting action potentials and with stable resting membrane potentials ≤−55 mV. Input resistances (Rin) were determined from the change in membrane potential produced by a 20 pA hyperpolarizing current injection from the resting potential. The current threshold for action potential generation was defined as the minimal current injection, applied (for 5 ms) from the resting membrane potential, required to evoke a single action potential. The properties (amplitudes, thresholds, widths at half-maximum, and decay times) of individual action potentials were determined off-line using Mini Analysis (version 6.0; Synaptosoft). In each cell, action potential amplitude was measured as the voltage difference between the resting membrane potential and the peak of the action potential. The voltage threshold (Vthr) for action potential generation in each cell was determined from the third derivative of the variation in the membrane voltage as a function of time (dV/dt) during the rising phase of the action potential. Differentiated traces were filtered with a digital Gaussian filter and smoothed by 30 points to determine Vthr (Synaptosoft). The width at half-maximum of the action potential in each cell was determined from measurement of the duration of the action potential when the membrane voltage had returned from the peak halfway back to the resting membrane potential. Action potential decay times in each cell were determined as the time required for the membrane voltage to decrease from 90% to 37% of the peak amplitude.
Statistics.
Results are expressed as means ± SEM. Statistical analyses were performed using the (unpaired) Student's t test, the two-way ANOVA, or the Kolmogorov–Smirnov test. Student's t tests and two-way ANOVA were performed and frequency histograms were generated using Prism (version 4.0; GraphPad Software).
Results
Navβ1 is identified in mouse brain Kv4.2 channel complexes
As illustrated in the Western blot in Figure 1A, the Kv4.2 protein was readily immunoprecipitated from adult WT mouse brain, but not from Kv4.2−/− brains. IPs were digested in-solution with trypsin, and the resulting tryptic peptides were analyzed using MudPIT (Fig. 1B). Consistent with previous reports (Marionneau et al., 2009, 2011), the MS analyses unambiguously identified the three Kv4 α subunits, Kv4.2, Kv4.3, and Kv4.1, as well as several previously described Kv4 accessory subunits: KChIP2, KChIP3, and KChIP4 (Rhodes et al., 2004; Jerng et al., 2005; Marionneau et al., 2009, 2011) as well as DPP6 and DPP10 (Nadal et al., 2003; Jerng et al., 2005; Zagha et al., 2005; Marionneau et al., 2009, 2011). Importantly, none of these proteins were identified in the RbαKv4.2 IPs from Kv4.2−/− brains.
Figure 1.
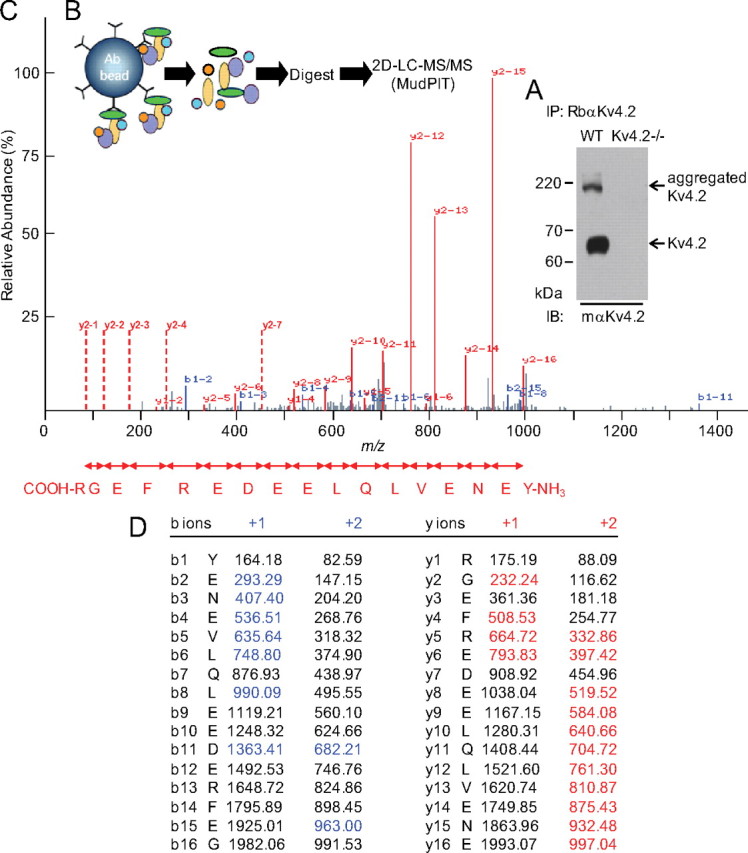
Identification of Navβ1 in mouse brain Kv4.2 channel complexes. A, A representative Western blot of fractionated proteins immunoprecipitated (IP) from adult WT and Kv4.2−/− mouse brains with a rabbit polyclonal anti-Kv4.2 antibody (RbαKv4.2), probed (IB) with a mouse monoclonal anti-Kv4.2 antibody (mαKv4.2). Although clearly evident in the RbαKv4.2-IP from the WT mouse brain, no Kv4.2 protein is identified in the Kv4.2−/− IP. Similar results were obtained in 4 independent experiments. The strategy used to isolate Kv4.2 complexes is diagrammed in (B). IPs were digested with trypsin and were analyzed by MudPIT. C, A representative MS/MS spectrum of a Navβ1 tryptic peptide is shown. The observed fragment ions matching the calculated m/z values of the C- (y-ions) or the N- (b-ions) termini of the peptide are in red and blue, respectively. Vertical red dashed lines indicate the position of unobserved doubly charged y-ions. The amino acid sequence derived from the m/z differences of the doubly charged y-ion series is given in the carboxyl-to-amino-terminal direction. D, The corresponding exact masses of detected fragment ions are indicated in red (y-ions) and blue (b-ions).
Unexpectedly, the MS analyses also revealed that the voltage-gated Na+ channel accessory subunit, Navβ1, coimmunoprecipitates with Kv4.2 from WT mouse brain. Navβ1 was not identified, however, in the (control) immunoprecipitated samples from Kv4.2−/− brain. A Navβ1 tryptic peptide was identified in both the +2 and +3 charged state. The calculated protein abundance factor (see Materials and Methods) for Navβ1 was 0.8, which compares with protein abundance factors in the range of 1.4 to 5.6 calculated for the various KChIP and DPP proteins (Marionneau et al., 2011). As validation of the Navβ1 peptide identification, a representative MS/MS spectrum of the +3 charged peptide, together with the amino acid sequence matching the spectral data (Fig. 1C) and the corresponding masses of the identified fragmented b- and y-ions, (Fig. 1D) are presented in Figure 1.
Navβ1 coimmunoprecipitates with Kv4.2
To provide an independent validation of the association of Navβ1 with Kv4.2, coimmunoprecipitation experiments were completed on lysates of HEK-293 cells transiently cotransfected with cDNA constructs encoding mouse Kv4.2 and an EYFP-tagged mouse Navβ1 (Navβ1-EYFP). Parallel control experiments were performed on cells transiently transfected with Kv4.2 and KChIP2, Kv4.2 and EYFP, or Navβ1-EYFP alone. As illustrated in Figure 2A, Navβ1-EYFP coimmunoprecipitates with Kv4.2, as does KChIP2 (Rhodes et al., 2004). Control experiments revealed that EYFP does not coimmunoprecipitate with Kv4.2 and that Navβ1-EYFP is not immunoprecipitated with the RbαKv4.2 antibody in the absence of Kv4.2 (Fig. 2A). Parallel experiments with an antibody against EGFP (to immunoprecipitate the EYFP-tagged Navβ1) demonstrated that Kv4.2 coimmunoprecipitates with Navβ1-EYFP (Fig. 2B). In addition, Navβ1-EYFP coimmunoprecipitated with Kv4.2 when KChIP2, Navβ1-EYFP, and Kv4.2 were coexpressed (Fig. 3A). Importantly, however, KChIP2 and Navβ1-EYFP do not coimmunoprecipitate in the absence of Kv4.2 (Fig. 3B), indicating no direct interactions between the KChIP2 and Navβ1 proteins.
Figure 2.

Coimmunoprecipitation of heterologously expressed Kv4.2 and Navβ1. Cell lysates were prepared 24 h following transfection of HEK-293 cells with cDNA constructs encoding Navβ1-EYFP alone or Kv4.2 with EYFP, KChIP2, or Navβ1-EYFP. A, IPs were performed with a RbαKv4.2 antibody. Western blot analyses of the lysates (left) probed with monoclonal anti-Kv4.2, anti-KChIP2, or anti-EGFP antibodies confirmed robust protein expression of all constructs. Following IP with RbαKv4.2, Western blots (right) revealed that Navβ1-EYFP, like KChIP2, coimmunoprecipitates with Kv4.2 whereas EYFP does not. B, IPs performed with a RbαEGFP antibody revealed that Kv4.2 coimmunoprecipitates with Navβ1-EYFP but not with EYFP.
Figure 3.

Navβ1 coimmunoprecipitates with Kv4.2 in the presence (and absence) of KChIP2. Twenty-four hours following transfection of HEK-293 cells with cDNA constructs encoding Kv4.2, KChIP2, and/or Navβ1-EYFP, cell lysates were prepared. A, IPs were performed with a RbαKv4.2 antibody. Robust protein expression of all constructs was confirmed by Western blot analyses of the lysates (left) probed with a monoclonal anti-Kv4.2, anti-KChIP2, or anti-EGFP antibody. Following IP with RbαKv4.2, Western blots (right) revealed that Navβ1-EYFP coimmunoprecipitates with Kv4.2 in the presence (and absence) of KChIP2. B, In contrast, Navβ1-EYFP does not coimmunoprecipitate with KChIP2 in the absence of Kv4.2.
Navβ1 increases Kv4.2-encoded current densities
To explore the functional consequences of the interaction between Kv4.2 and Navβ1, whole-cell voltage-clamp recordings were obtained from HEK-293 cells expressing Kv4.2 alone or in combination with Navβ1. As illustrated in Figure 4, Kv4.2-encoded current densities in cells coexpressing Kv4.2 and Navβ1 were significantly (p < 0.01) higher than in cells expressing Kv4.2 alone. Consistent with previous reports (An et al., 2000; Bähring et al., 2001; Foeger et al., 2010), peak Kv4.2-encoded current densities were also significantly (p < 0.001) higher in cells coexpressing Kv4.2 and KChIP2 (Fig. 4). Interestingly, in cells expressing Kv4.2 with both KChIP2 and Navβ1, peak current densities were significantly (p < 0.05) higher than in cells coexpressing Kv4.2 with either Navβ1 or KChIP2 (Fig. 4). In contrast with KChIP2 (An et al., 2000), however, Navβ1 coexpression did not measurably affect the kinetics or the voltage-dependent properties of Kv4.2-encoded currents (not illustrated).
Figure 4.

Navβ1 increases heterologously expressed Kv4.2 current densities. A, Representative whole-cell, voltage-gated K+ currents recorded from transiently transfected HEK-293 cells. Currents were evoked in response to 400 ms voltage steps to potentials between −40 and +30 mV from a holding potential of −70 mV. B, Mean ± SEM peak current densities in cells transfected with Kv4.2 (black), Kv4.2 + Navβ1 (red), Kv4.2 + KChIP2 (blue), and Kv4.2 + KChIP2 + Navβ1 (purple) are plotted as a function of test potential. Peak current densities measured in cells expressing Kv4.2 alone (n = 18) are significantly (two-way ANOVA) different from those measured in cells expressing Kv4.2 + Navβ1 (‡p < 0.01, n = 20) or Kv4.2 + KChIP2 (*p < 0.001, n = 11). In addition, peak current densities in cells coexpressing Kv4.2 + KChIP2 + Navβ1 (n = 10) are significantly (two-way ANOVA) different from those measured in cells expressing only Kv4.2 + Navβ1 (*p < 0.001, n = 20) or Kv4.2 + KChIP2 (†p < 0.05, n = 11).
Biochemical experiments revealed that total Kv4.2 protein expression in HEK-293 cells coexpressing Kv4.2 and Navβ1 was significantly (p < 0.001) higher than in cells expressing Kv4.2 alone (Fig. 5A,B). Similar results were obtained with KChIP2 coexpression, although total Kv4.2 coexpression in cells expressing KChIP2 was significantly (p < 0.001) higher than in cells coexpressing Navβ1 (Fig. 5A,B). Cell-surface Kv4.2 expression was also significantly higher in cells coexpressing KChIP2 (p < 0.001) or Navβ1 (p < 0.01), compared with cells expressing Kv4.2 alone (Fig. 5A,B). For KChIP2, the relative increase in cell-surface Kv4.2 expression was significantly (p < 0.001) higher than the increase in total Kv4.2 protein, whereas total and cell-surface Kv4.2 were increased similarly in cells coexpressing Navβ1 (Fig. 5A,B). Consistent with the electrophysiological data (Fig. 4), total and cell-surface Kv4.2 expression was significantly (p < 0.001) higher in cells expressing Kv4.2 with both Navβ1 and KChIP2 compared with cells expressing Kv4.2 with either Navβ1 or KChIP2 (Fig. 5A,B). In contrast with the effects on total and cell-surface Kv4.2 protein levels, coexpression of Navβ1 did not measurably affect the expression levels of either the voltage-gated K+ channel α subunit, Kv2.1, or the two-pore potassium channel subunit, TASK1 (Fig. 5C), revealing that the effects of Navβ1 are subunit-specific (see Discussion).
Figure 5.

Coexpression with Navβ1 increases total and cell-surface Kv4.2 protein expression. A, Representative Western blots of total (left) and cell surface (right) Kv4.2 from HEK-293 cells transiently transfected with cDNA constructs encoding Kv4.2 alone or with KChIP2 and/or Navβ1. Samples were probed in parallel with the anti-transferrin receptor (TransR) antibody. B, Mean ± SEM total and cell-surface Kv4.2 protein expression in HEK-293 cells transiently transfected with Kv4.2 alone (n = 8–20), Kv4.2 + Navβ1 (n = 8–20), Kv4.2 + KChIP2 (n = 14–18), and Kv4.2 + KChIP2 + Navβ1 (n = 14–16). Expression of Kv4.2 in each sample was first normalized to the TransR protein in the same blot and then expressed relative to Kv4.2 protein expression (total or cell surface) in cells transfected with Kv4.2 alone. Relative (mean ± SEM) Kv4.2 total and cell-surface expression was significantly (t test) higher in cells expressing Kv4.2 + KChIP2 (*p < 0.001) or Kv4.2 + Navβ1 (*p < 0.001; ‡p < 0.01) than in cells with Kv4.2 alone. Kv4.2 total and cell-surface expression, however, was significantly (#p < 0.001) higher in cells expressing Kv4.2 + KChIP2 than in cells expressing Kv4.2 + Navβ1. In addition, relative (mean ± SEM) cell-surface Kv4.2 expression was significantly (§p < 0.001) higher than total Kv4.2 expression in cells coexpressing Kv4.2 with KChIP2 but not with Navβ1. Cells coexpressing Kv4.2 with both KChIP2 and Navβ1 had significantly (¶p < 0.001) higher mean ± SEM total and cell-surface Kv4.2 protein expression levels than cells coexpressing Kv4.2 with only KChIP2 or Navβ1. C, In contrast, coexpression of Navβ1 did not affect the total expression of either Kv2.1 (n = 2) or TASK1 (n = 2).
Acute knockdown of Navβ1 decreases IA densities in cortical neurons
To test directly the hypothesis that Navβ1 regulates native neuronal Kv4-encoded IA, an shRNA-mediated RNA interference approach was used to allow acute knockdown of the expression of Navβ1 in cortical neurons. To identify shRNA sequences that effectively reduce the expression of the Navβ1 protein, five shRNA sequences targeting Scn1b (Navβ1) were screened in CHO cells expressing Navβ1-EYFP (see Materials and Methods). As illustrated in the Western blot in Figure 6A, Navβ1 was readily detected in cells coexpressing Navβ1-EYFP and the (control) nontargeted shRNA. In contrast, Navβ1 protein levels were undetectable in cells coexpressing Navβ1-EYFP and one of the Scn1b-targeted shRNA sequences (CGTCTCCTCTTCTTTGATAAT) (Fig. 6A). Similar experiments on the other four shRNAs targeting Navβ1 revealed different degrees of knockdown and the sequence used in Figure 6A was selected for use in neurons. Plasmids encoding this Scn1b-targeted, or the nontargeted, shRNA sequence, together with the red fluorescent protein, tdTomato, were used in subsequent experiments in isolated cortical neurons. Within ∼24 h of transfection, tdTomato expression was readily detected in cortical neurons under epifluorescence illumination.
Figure 6.

shRNA-mediated knockdown of Navβ1 decreases IA densities in cortical neurons. A, Specific shRNA sequences targeting Scn1b (Navβ1) were screened in CHO cells. Navβ1-EYFP was coexpressed with either a control (nontargeted) shRNA or with the Scn1b-targeted shRNA. Protein lysates were prepared from transfected cells and analyzed by Western blot using an anti-EGFP antibody. Blots were also probed with an anti-GAPDH antibody to verify equal loading of proteins. Navβ1 is robustly expressed in cells expressing the nontargeted shRNA but is undetectable in cells transfected with the Scn1b shRNA. B, Representative whole-cell Kv currents, recorded in response to voltage steps ranging from −40 mV to +40 mV in 10 mV increments from a holding potential of −70 mV in cortical neurons expressing the nontargeted shRNA (Ba) or the Scn1b targeted shRNA (Ca) revealed marked differences in peak, but not steady-state currents. Bb–Cb, In each cell, recordings were also obtained using the same depolarizing steps, preceded by a brief prepulse to −10 mV to inactivate IA. The voltage protocols are illustrated in the insets. Currents recorded with the prepulse (b) were subtracted offline from the control records (a) in individual cells to isolated IA (a-b). The subtracted records are also shown on an expanded time scale. D, Mean ± SEM IA densities are plotted as a function of test potential. *Values in neurons expressing the Scn1b shRNA are significantly (*p < 0.001) different from those in neurons expressing the nontargeted shRNA.
Whole-cell Kv currents, evoked in response to voltage steps to potentials ranging from −40 mV to +40 mV (in 10 mV increments) from a holding potential of −70 mV, were examined in tdTomato-positive cortical neurons expressing the nontargeted, or the Scn1b-targeted, shRNA (Fig. 6Ba,Ca). In each cell, outward Kv currents evoked at the same test potentials were also recorded following a brief prepulse to −10 mV (Fig. 6Bb,Cb) to inactivate IA (Norris and Nerbonne, 2010). Off-line subtraction of the recordings with the prepulse from the recordings without the prepulse allowed the isolation of IA (Fig. 6Ba,Bb,Ca,Cb). Analyses of these subtracted records revealed that the mean ± SEM IA density is significantly (p < 0.001) reduced in cells expressing the Scn1b-targeted shRNA, compared with cells expressing the nontargeted shRNA (Fig. 6D). The kinetics and voltage-dependent properties of IA were indistinguishable, however, in cells expressing the nontargeted, and the Scn1b-targeted, shRNAs (data not shown). In addition, further analyses revealed that the slowly inactivating (delayed rectifier) and steady-state outward K+ currents were not measurably different in cells expressing the Scn1b-targeted, and the nontargeted, shRNAs (Fig. 6), suggesting that the effects of acute knockdown of Navβ1 are specific to IA among the Kv currents (See Discussion).
Loss of Navβ1 prolongs action potentials and increases repetitive firing in cortical pyramidal neurons
To explore the hypothesis that the regulation of Kv4.2-encoded IA channels by Navβ1 plays a role in the regulation of the excitability of cortical neurons, the effects of the targeted deletion of Scn1b (Navβ1) on action potential waveforms and repetitive firing properties were examined. Whole-cell current-clamp recordings were obtained from layer 5 pyramidal neurons in acute slices prepared from the cortices of WT and Scn1b−/− mice (Fig. 7A). Resting membrane potentials (Vm) and input resistances (Rin) were similar in WT and Scn1b−/− layer 5 pyramidal neurons (Fig. 7B). In addition, analyses of single action potentials, elicited by brief (5 ms) depolarizing current injections (Fig. 7A), revealed that mean ± SEM voltage (Vthr) and current (Ithr) thresholds for action potential generation, as well as mean ± SEM action potential amplitudes (APA), are also indistinguishable in WT and Scn1b−/− neurons (Fig. 7B). The mean ± SEM action potential decay time (2.00 ± 0.05 ms) and widths (2.57 ± 0.06 ms) at half-maximum measured in Scn1b−/− neurons, however, were significantly (p < 0.01) longer than the mean ± SEM values (decay time: 1.78 ± 0.04 ms; width at half-maximum: 2.30 ± 0.05 ms) in WT neurons (Fig. 7C).
Figure 7.

Action potentials are prolonged in layer 5 cortical pyramidal neurons lacking Navβ1. A, Representative action potential waveforms evoked by brief (5 ms) current injections in WT (left) and Scn1b−/− (right) neurons are illustrated. B, Average passive and active membrane properties in WT (n = 33) and Scn1b−/− (n = 41) neurons are summarized. Data are presented as means ± SEM; Vm = resting membrane potential; Vthr = voltage threshold for action potential generation; APA = action potential amplitude; Rin = input resistance; and Ithr = current required to evoke a single action potential. C, Mean ± SEM action potential decay time and width at half-maximum are significantly (*p < 0.001; ‡p < 0.01, t test) different in WT (n = 33) and Scn1b−/− (n = 41) neurons.
Repetitive firing, elicited directly from the resting membrane potential in response to prolonged (500 ms) depolarizing current injections of varying amplitudes, was also examined in WT and Scn1b−/− layer 5 pyramidal neurons in acute cortical slices (Fig. 8). The numbers of action potentials elicited by prolonged depolarizing current injections increased as a function of the current injection amplitude in both WT and Scn1b−/− cortical pyramidal neurons (Figs. 8A,B). At each injected current amplitude, however, the mean ± SEM number of action potentials evoked in Scn1b−/− neurons was significantly (p < 0.01) higher than in WT neurons (Fig. 8B). In addition, the mean ± SEM current threshold required to evoke repetitive firing was significantly (p < 0.01) lower in Scn1b−/− (102.4 ± 7.4 pA) than in WT (142.9 ± 10 pA) layer 5 cortical pyramidal neurons (Fig. 8C).
Figure 8.

Repetitive firing is increased in layer 5 cortical pyramidal neurons lacking Navβ1. A, Voltage recordings from WT (top) and Scn1b−/− (bottom) neurons in response to prolonged (500 ms) depolarizing current injections of varying amplitudes are illustrated. B, Mean ± SEM numbers of action potentials evoked during 500 ms current injections are plotted as a function of the amplitude of the current injection. At all amplitudes, the mean ± SEM number of action potentials is significantly (‡p < 0.01, two-way ANOVA) higher in Scn1b−/− (n = 41) than in WT (n = 34) neurons. C, The mean ± SEM current thresholds required to evoked repetitive firing during a prolonged (500 ms) current injection in WT (n = 34) and Scn1b−/− (n = 41) cortical pyramidal neurons are significantly (‡p < 0.01, t test) different.
Navβ1 increases the stability of Kv4.2
To determine directly if Navβ1 affects the stability of the Kv4.2 protein, cells expressing Kv4.2 alone or in combination with Navβ1 were treated with the protein synthesis inhibitor cycloheximide for various times (30, 60, 90, 120, and 480 min). With de novo protein synthesis blocked, the stability of pre-existing total and cell-surface Kv4.2 was assessed by Western blot and by cell-surface biotinylation, followed by Western blot, analyses. Total and cell-surface Kv4.2 protein expression levels in cycloheximide-treated cells at different time points were measured and expressed as the percentage of total or cell-surface Kv4.2 protein in untreated cells. As illustrated in Figure 9A and B, when Kv4.2 was expressed alone, total Kv4.2 protein expression was significantly (p < 0.001) reduced (by ∼65%) after 30 min of cycloheximide treatment; no further reductions in Kv4.2 were observed at longer times. When Navβ1 was coexpressed with Kv4.2, however, the initial reduction in Kv4.2 (∼50%) was significantly (p < 0.01) less than when Kv4.2 was expressed alone (Fig. 9B). In addition, further reductions in mean ± SEM total Kv4.2 protein were evident after 60 and 120 min in cycloheximide. At 60 min, the Kv4.2 protein was significantly (p < 0.01) higher in cells coexpressing Navβ1 than in cells expressing Kv4.2 alone, whereas, at 120 min, the mean ± SEM fraction of Kv4.2 remaining in cells transfected with and without Navβ1 were not significantly different (Fig. 9B). Cell-surface Kv4.2 protein expression in cells expressing Kv4.2 alone or in combination with Navβ1 was not significantly altered by cycloheximide treatment over the same time period (Fig. 9A,B). There are, therefore, (at least) two cellular pools of Kv4.2: a cell-surface pool that does not appear to turnover measurably in 2 h and an intracellular pool that turns over rapidly. The simplest interpretation of the results in Figure 9 is that coexpression of Navβ1 increases the total expression of Kv4.2 by stabilizing the intracellular pool of Kv4.2.
Figure 9.

Navβ1 increases Kv4.2 protein stability. HEK-293 cells were transiently transfected with Kv4.2 alone or Kv4.2 with Navβ1. Total and cell-surface Kv4.2 expression following exposure to the protein synthesis inhibitor, cycloheximide (A), and in endocytosis assays (C) was examined. A, Representative Western blots of total (left) and cell-surface (right) Kv4.2 protein in extracts prepared 30, 60, and 120 min following exposure to cycloheximide (100 μg/ml). B, For quantification of Kv4.2 expression, protein bands after cycloheximide were measured and normalized to the Kv4.2 protein levels in untreated cells; mean ± SEM values (n = 3–4) are plotted as a function of treatment time. The reduction in total Kv4.2 expression is significantly (*p < 0.01, two-way ANOVA) less in cells coexpressing Kv4.2 and Navβ1 than in cells expressing Kv4.2 alone. In contrast, cell-surface Kv4.2 protein expression in cells expressing Kv4.2 alone or with Navβ1 is not measurably affected by cycloheximide treatment. C, Representative Western blots illustrating total biotinylated Kv4.2 and transferrin receptor (TransR) in nonreduced samples, as well as the proportion of endocytosed Kv4.2 and TransR (in reduced samples). D, Quantification of endocytosed Kv4.2 and TransR proteins. Data are expressed in percentage (%) of endocytosed proteins as means ± SEM (n = 2–10). No significant differences in Kv4.2 cell-surface stability or turnover rates were observed with Navβ1 coexpression.
Pulse-chase experiments were also performed to examine the turnover rate of Kv4.2 at the cell surface. HEK-293 cells expressing Kv4.2 alone or in combination with Navβ1 were first biotinylated at 4°C (pulse), and then returned at 37°C (chase) for various times (0, 15, 30, and 60 min) to allow endocytosis from, and recycling to, the cell surface. After each chase time, cells were treated with a nonpermeable reducing agent to eliminate the biotin on channels remaining at the cell surface and allow independent analysis of endocytosed channels (reduced samples). Control cells that had not been treated (nonreduced samples) were examined in parallel. Consistent with findings in the cycloheximide experiments (Figs. 9A,B), no reduction in total (nonreduced) biotinylated Kv4.2 was observed at any of the chase time points tested in extracts from cells expressing Kv4.2 alone or in combination with Navβ1 (Fig. 9C). Analysis of the reduced samples, however, revealed that ∼20, 30, and 40% of the biotinylated Kv4.2 protein is endocytosed after 15, 30, and 60 min, respectively (Figs. 9C,D). At each chase time, the proportion of endocytosed Kv4.2 protein in cells expressing Kv4.2 alone or in combination with Navβ1 was not significantly different (Figs. 9C,D). Control experiments revealed that, consistent with previous observations (Sheff et al., 2002; Foeger et al., 2010), the turnover rate of the transferrin receptor is rapid, with virtually all of the biotinylated transferrin receptors endocytosed after 15 min (Figs. 9C,D). By comparison, the cell-surface turnover rate of Kv4.2 channels is slow and, in addition, is not measurably affected by Navβ1.
Discussion
The results presented here demonstrate a physiological role for the voltage-gated Na+ channel accessory subunit, Navβ1, in the functioning of neuronal Kv4.2-encoded IA channels. Shotgun proteomic analysis led to the identification of Navβ1 in native mouse brain Kv4.2 channel complexes, and subsequent biochemical and electrophysiological studies in heterologous cells demonstrated a functional role for Navβ1 in regulating Kv4.2 channel protein stability and current densities. Experiments using shRNA-mediated RNA interference to knock-down Navβ1 in isolated cortical neurons further revealed that Navβ1 also regulates IA channels in these cells. In addition, electrophysiological recordings from layer 5 cortical pyramidal neurons in the in vitro slice preparation revealed that in vivo loss of Navβ1 prolongs action potentials and increases repetitive firing in cortical pyramidal neurons, consistent with a role for Navβ1 in the regulation of native neuronal Kv4.2-encoded IA channels.
Navβ1 is a component of neuronal Kv4.2 channel complexes
Results of numerous previous studies suggest that neuronal Kv4.2 channels likely function in macromolecular protein complexes comprising four pore-forming α subunits, together with accessory KChIPx and DPPx subunits, as well as other regulatory/modulatory proteins (An et al., 2000; Nadal et al., 2003; Birnbaum et al., 2004; Jerng et al., 2004; Rhodes et al., 2004; Jerng et al., 2005; Zagha et al., 2005; Kim et al., 2008; Maffie and Rudy, 2008; Norris et al., 2010; Sun et al., 2011). The identification of Navβ1 in native mouse brain Kv4.2 channel complexes, however, was unexpected. Although not quantitative, the results of the MudPIT experiments suggest that the relative abundance of Navβ1 in Kv4.2 channel complexes is lower than the relative abundances of the KChIPx or the DPPx proteins, with a protein abundance factor value of 0.8 for Navβ1 compared with values in the range of 1.4 to 5.6 for the KChIPx and DPPx proteins. The association of Navβ1 with Kv4.2 may, therefore, only occur in particular neurons or in specific neuronal compartments. Alternatively, the relatively lower protein abundance factor (0.8) for Navβ1, compared with the KChIPx and DPPx proteins, could suggest that Navβ1 interacts weakly or only transiently with Kv4.2 channels. It is also possible that the interaction between Kv4.2 and Navβ1 is indirect, requiring an intermediary scaffolding protein.
The finding of Navβ1 in neuronal Kv4.2 channel complexes also raises the interesting possibility that there are macromolecular protein complexes containing both Na+ and K+ channels in cortical pyramidal (as well, perhaps, as in other) neurons. Evidence for channel-channel macromolecular complexes in the brain was provided recently with the demonstration that Ca2+ entry through Cav3-encoded T-type Ca2+ channels regulates Kv4-encoded IA channels in cerebellar stellate neurons and, in addition, that Kv4.2 coimmunoprecipitates with Cav3.2 and Cav3.3 from rat brain and from tsA201 cells (Anderson et al., 2010). In addition, it has been reported that coexpression with Navβ1 increases the densities of heterologously expressed Kv4.3-encoded currents (Deschênes and Tomaselli, 2002), that transient outward K+ currents are reduced in dorsal root ganglion neurons dissociated from Scn1b-null mice (Lopez-Santiago et al., 2011), and that exposure to a small interfering RNA (RNAi) targeting Navβ1 reduces Kv4-encoded transient outward K+ (Ito) currents as well as voltage-gated Na+ currents in neonatal (rat) cardiac myocytes (Deschênes et al., 2008). Voltage-gated Na+ channel α subunits, however, did not coimmunoprecipitate with Kv4.2 in extracts of mouse brains in the experiments here. Similarly, mouse brain Kv4.2 did not coimmunoprecipitate with a pan-specific Nav α subunit antibody (data not shown), suggesting that the association of Kv4.2 with Navβ1 is not mediated through Nav α subunits and is independent of Navβ1-mediated effects on Nav channels.
Navβ1 modulates IA channels and regulates action potential repolarization and repetitive firing in cortical pyramidal neurons
The results of the electrophysiological experiments detailed here revealed that acute knockdown of Scn1b (Navβ1) selectively reduces IA densities in cortical neurons. Importantly, these experiments also revealed that knockdown of Navβ1 does not measurably affect the slowly inactivating (delayed rectifier) and steady-state outward K+ currents in these cells. The experiments presented here further revealed that the in vivo loss of Navβ1 results in prolonged action potentials and increased repetitive firing rates in layer 5 cortical pyramidal neurons. The functional effects of loss of Navβ1 on action potential waveforms and repetitive firing are similar to the previously reported effects of pharmacological suppression of IA and dominant-negative attenuation ofKv4-encoded currents (Locke and Nerbonne, 1997; Hu and Gereau 4th, 2003; Kim et al., 2005; Yuan et al., 2005). The simplest interpretation of these combined results, therefore, is that Navβ1 regulates action potential repolarization and repetitive firing in cortical pyramidal neurons specifically through the modulation of Kv4-encoded IA channels.
Interestingly, mutations in Navβ1 have been identified in patients with generalized epilepsy with febrile seizures plus (GEFS+) as well as in individuals with temporal lobe epilepsy (TLE) and in severe myoclonic epilepsy of infancy (Dravet syndrome) (Wallace et al., 1998; Scheffer et al., 2007; Patino et al., 2009, 2011; Baulac and Baulac, 2010). Previous studies have also shown that Scn1b−/− mice display spontaneous generalized seizures (Chen et al., 2004), although Nav currents were reportedly unaltered, or only mildly affected, in hippocampal neurons isolated from these mice (Chen et al., 2004; Aman et al., 2009; Patino et al., 2009). The current-clamp experiments presented here revealed that the voltage thresholds, as well as the peak amplitudes, of individual action potentials are indistinguishable in WT and Scn1b−/− neurons, suggesting that Nav currents in layer 5 cortical pyramidal neurons are not affected by the loss of Navβ1. The results presented here, therefore, also suggest that decreased IA densities, rather than or in addition to effects on Nav currents, contributes to increased neuronal excitability and epileptogenesis in GEFS+, TLE, and severe myoclonic epilepsy of infancy. Additional experiments will be necessary to explore this hypothesis directly.
The studies presented here also revealed that the phenotypic effects of the deletion of Scn1b are cell-type specific. In contrast with the action potential prolongation and increased repetitive firing rates observed in Scn1b−/− layer 5 cortical pyramidal neurons (Figs. 7, 8), the waveforms of action potentials in WT and Scn1b−/− hippocampal CA1 pyramidal neurons were indistinguishable (data not shown). Similar results were reported previously by Patino et al. (2009). It has, however, also been reported that action potential amplitudes were larger in Scn1b−/− than in WT, hippocampal CA3 pyramidal neurons (Patino et al., 2009), raising the interesting possibility that IA is also decreased in these cells. In addition, although Scn1b−/− layer 5 cortical pyramidal neurons display increased repetitive firing, reduced repetitive firing was observed in Scn1b−/− cerebellar granule neurons (Brackenbury et al., 2010).Together, these results suggest considerable heterogeneity in the molecular composition of IA channels in different cell types, heterogeneity that may underlie the experimental observation that the detailed time- and voltage-dependent properties of IA in different cell types are distinct (Jerng and Pfaffinger, 2008; Maffie and Rudy, 2008).
Navβ1 acts as a molecular chaperone to regulate Kv4.2 protein expression
The results presented here demonstrate that Navβ1 increases the stability of the Kv4.2 protein without measurably affecting cell-surface Kv4.2 channel turnover rates. The time- and voltage-dependent properties of heterologously expressed Kv4.2-encoded currents were indistinguishable in the absence and the presence of Navβ1. Together, these results suggest that Navβ1 acts as a molecular chaperone, stabilizing newly synthesized Kv4.2 protein, which results (by mass action) in greater channel cell-surface expression and larger current amplitudes/densities. Importantly, the experiments here also suggest that stabilization of Kv4.2 protein by Navβ1 is specific because coexpression of Navβ1 did not affect the protein expression levels of other potassium channel pore-forming subunits, including Kv2.1 or TASK1.
These findings further suggest an intriguing model in which multiple Kv channel accessory subunits and regulatory proteins could participate differently in the regulation of Kv4.2 channel expression and functioning. The biochemical results presented here also suggest the interesting hypothesis that Kv4.2 channels are present in two (or more) cellular pools with distinct properties: an intracellular pool, that turns over relatively rapidly and a pool expressed at the cell surface that is more stable and turns over more slowly. The relative roles of the Navβ1 and KChIP subunits, for example, might be different in different cell types or in different subcellular compartments in the same cell. Further studies, focused on defining the molecular mechanisms involved in the dynamic regulation of neuronal Kv4.2 channel expression, trafficking, and functioning in different cell types and subcellular compartments, are needed to explore these hypotheses in detail.
Footnotes
The financial support provided by the Washington University-Pfizer Biomedical Research Program (to J.M.N.), the National Institutes of Health (R01-HL034161 and R21-NS065295 to J.M.N., R01-GM064779 to A.J.L., and R01-NS076752 to L.L.I.), the National Center for Research Resources (NIH P41RR000954 and UL1 RR024992), the NIH Neuroscience Blueprint Center Core Grant (P30-NS057105), the W.M. Keck Foundation, and the Heartland Affiliate of the American Heart Association (to C.M.) is gratefully acknowledged. Y.C. was supported by the Research Training Grant T32-HL007275 and the Individual National Research Service Award F32-NS065581 from the NIH. We also thank Rick Wilson for maintaining and genotyping mice, Rebecca Mellor for technical assistance with molecular biology, and Dr. John R. Yates III (The Scripps Research Institute, La Jolla, CA) for providing the extractms2 program.
References
- Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci. 2009;29:2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Anderson D, Mehaffey WH, Iftinca M, Rehak R, Engbers JD, Hameed S, Zamponi GW, Turner RW. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat Neurosci. 2010;13:333–337. doi: 10.1038/nn.2493. [DOI] [PubMed] [Google Scholar]
- Arnett DR, Jennings JL, Tabb DL, Link AJ, Weil PA. A proteomics analysis of yeast Mot1p protein-protein associations: insights into mechanism. Mol Cell Proteomics. 2008;7:2090–2106. doi: 10.1074/mcp.M800221-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähring R, Dannenberg J, Peters HC, Leicher T, Pongs O, Isbrandt D. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J Biol Chem. 2001;276:23888–23894. doi: 10.1074/jbc.M101320200. [DOI] [PubMed] [Google Scholar]
- Baulac S, Baulac M. Advances on the genetics of Mendelian idiopathic epilepsies. Clin Lab Med. 2010;30:911–929. doi: 10.1016/j.cll.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol. 2011;2:53. doi: 10.3389/fphar.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Djamgoz MB, Isom LL. An emerging role for voltage-gated Na+ channels in cellular migration: regulation of central nervous system development and potentiation of invasive cancers. Neuroscientist. 2008;14:571–583. doi: 10.1177/1073858408320293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Calhoun JD, Chen C, Miyazaki H, Nukina N, Oyama F, Ranscht B, Isom LL. Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci U S A. 2010;107:2283–2288. doi: 10.1073/pnas.0909434107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA, Isom LL. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. 2004;24:4030–4042. doi: 10.1523/JNEUROSCI.4139-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie JT, Kole MH, Letzkus JJ, Rancz EA, Spruston N, Stuart GJ, Häusser M. Dendritic patch-clamp recording. Nat Protoc. 2006;1:1235–1247. doi: 10.1038/nprot.2006.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes I, Tomaselli GF. Modulation of Kv4.3 current by accessory subunits. FEBS Lett. 2002;528:183–188. doi: 10.1016/s0014-5793(02)03296-9. [DOI] [PubMed] [Google Scholar]
- Deschênes I, Armoundas AA, Jones SP, Tomaselli GF. Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J Mol Cell Cardiol. 2008;45:336–346. doi: 10.1016/j.yjmcc.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foeger NC, Marionneau C, Nerbonne JM. Co-assembly of Kv4 {alpha} subunits with K+ channel-interacting protein 2 stabilizes protein expression and promotes surface retention of channel complexes. J Biol Chem. 2010;285:33413–33422. doi: 10.1074/jbc.M110.145185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick J, Robbins J. Cell-type-specific transgenesis in the mouse. Methods Mol Biol. 2009;561:91–104. doi: 10.1007/978-1-60327-019-9_6. [DOI] [PubMed] [Google Scholar]
- Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM. Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97:1342–1350. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Lin L, Sidorov MS, Wikenheiser AM, Hoffman DA. Protein kinase a mediates activity-dependent Kv4.2 channel trafficking. J Neurosci. 2008;28:7513–7519. doi: 10.1523/JNEUROSCI.1951-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Gereau RW., 4th ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol. 2003;90:1680–1688. doi: 10.1152/jn.00341.2003. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW., 4th The Kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- Isom LL. The role of sodium channels in cell adhesion. Front Biosci. 2002;7:12–23. doi: 10.2741/isom. [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ. Multiple Kv channel-interacting proteins contain an N-terminal transmembrane domain that regulates Kv4 channel trafficking and gating. J Biol Chem. 2008;283:36046–36059. doi: 10.1074/jbc.M806852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, Covarrubias M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci. 2004;27:343–369. doi: 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Kunjilwar K, Pfaffinger PJ. Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J Physiol. 2005;568:767–788. doi: 10.1113/jphysiol.2005.087858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Nadal MS, Clemens AM, Baron M, Jung SC, Misumi Y, Rudy B, Hoffman DA. Kv4 accessory protein DPPX (DPP6) is a critical regulator of membrane excitability in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2008;100:1835–1847. doi: 10.1152/jn.90261.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Guo W, Mellor RL, Nerbonne JM. KChIP2 modulates the cell surface expression of Kv 1.5-encoded K+ channels. J Mol Cell Cardiol. 2005;39:121–132. doi: 10.1016/j.yjmcc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- Locke RE, Nerbonne JM. Role of voltage-gated K+ currents in mediating the regular-spiking phenotype of callosal-projecting rat visual cortical neurons. J Neurophysiol. 1997;78:2321–2335. doi: 10.1152/jn.1997.78.5.2321. [DOI] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Brackenbury WJ, Chen C, Isom LL. Na+ channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J Biol Chem. 2011;286:22913–22923. doi: 10.1074/jbc.M111.242370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffie J, Rudy B. Weighing the evidence for a ternary protein complex mediating A-type K+ currents in neurons. J Physiol. 2008;586:5609–5623. doi: 10.1113/jphysiol.2008.161620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C, Brunet S, Flagg TP, Pilgram TK, Demolombe S, Nerbonne JM. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res. 2008;102:1406–1415. doi: 10.1161/CIRCRESAHA.107.170050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C, LeDuc RD, Rohrs HW, Link AJ, Townsend RR, Nerbonne JM. Proteomic analyses of native brain Kv4.2 channel complexes. Channels. 2009;3:284–294. doi: 10.4161/chan.3.4.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C, Townsend RR, Nerbonne JM. Proteomic analysis highlights the molecular complexities of native Kv4 channel macromolecular complexes. Semin Cell Dev Biol. 2011;22:145–152. doi: 10.1016/j.semcdb.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfee KJ, Duncan DT, Assink M, Link AJ. Analyzing proteomes and protein function using graphical comparative analysis of tandem mass spectrometry results. Mol Cell Proteomics. 2006;5:1497–1513. doi: 10.1074/mcp.T500027-MCP200. [DOI] [PubMed] [Google Scholar]
- Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, Rudy B. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 2003;37:449–461. doi: 10.1016/s0896-6273(02)01185-6. [DOI] [PubMed] [Google Scholar]
- Nadin BM, Pfaffinger PJ. Dipeptidyl peptidase-like protein 6 is required for normal electrophysiological properties of cerebellar granule cells. J Neurosci. 2010;30:8551–8565. doi: 10.1523/JNEUROSCI.5489-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol. 2008;586:1565–1579. doi: 10.1113/jphysiol.2007.146597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris AJ, Nerbonne JM. Molecular dissection of IA in cortical pyramidal neurons reveals three distinct components encoded by Kv4.2, Kv4.3, and Kv1.4 {alpha}-subunits. J Neurosci. 2010;30:5092–5101. doi: 10.1523/JNEUROSCI.5890-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris AJ, Foeger NC, Nerbonne JM. Interdependent roles for accessory KChIP2, KChIP3, and KChIP4 subunits in the generation of Kv4-encoded IA channels in cortical pyramidal neurons. J Neurosci. 2010;30:13644–13655. doi: 10.1523/JNEUROSCI.2487-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA, Isom LL. Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci Lett. 2010;486:53–59. doi: 10.1016/j.neulet.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, O'Malley HA, Gray CB, Miyazaki H, Nukina N, Oyama F, De Jonghe P, Isom LL. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. 2009;29:10764–10778. doi: 10.1523/JNEUROSCI.2475-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA, Brackenbury WJ, Bao Y, Lopez-Santiago LF, O'Malley HA, Chen C, Calhoun JD, Lafrenière RG, Cossette P, Rouleau GA, Isom LL. Voltage-gated Na+ channel beta1B: a secreted cell adhesion molecule involved in human epilepsy. J Neurosci. 2011;31:14577–14591. doi: 10.1523/JNEUROSCI.0361-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DW, Weaver CM, Jennings JL, McAfee KJ, He Y, Weil PA, Link AJ. Cluster analysis of mass spectrometry data reveals a novel component of SAGA. Mol Cell Biol. 2004;24:7249–7259. doi: 10.1128/MCB.24.16.7249-7259.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, Strassle BW, Buchwalder L, Menegola M, Cao J, An WF, Trimmer JS. KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J Neurosci. 2004;24:7903–7915. doi: 10.1523/JNEUROSCI.0776-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadygov RG, Eng J, Durr E, Saraf A, McDonald H, MacCoss MJ, Yates JR., 3rd Code developments to improve the efficiency of automated MS/MS spectra interpretation. J Proteome Res. 2002;1:211–215. doi: 10.1021/pr015514r. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ, Zielinski MA, Xu R, Jackson G, Adams J, Connellan M, Petrou S, Wellard RM, Briellmann RS, Wallace RH, Mulley JC, Berkovic SF. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain. 2007;130:100–109. doi: 10.1093/brain/awl272. [DOI] [PubMed] [Google Scholar]
- Schneider C, Newman RA, Sutherland DR, Asser U, Greaves MF. A one-step purification of membrane proteins using a high efficiency immunomatrix. J Biol Chem. 1982;257:10766–10769. [PubMed] [Google Scholar]
- Sheff D, Pelletier L, O'Connell CB, Warren G, Mellman I. Transferrin receptor recycling in the absence of perinuclear recycling endosomes. J Cell Biol. 2002;156:797–804. doi: 10.1083/jcb.20111048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Maffie JK, Lin L, Petralia RS, Rudy B, Hoffman DA. DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron. 2011;71:1102–1115. doi: 10.1016/j.neuron.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga AW, Yuan LL, Anderson AE, Schrader LA, Wu GY, Gatchel JR, Johnston D, Sweatt JD. Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J Neurosci. 2004;24:3643–3654. doi: 10.1523/JNEUROSCI.0154-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- Yuan W, Burkhalter A, Nerbonne JM. Functional role of the fast transient outward K+ current IA in pyramidal neurons in (rat) primary visual cortex. J Neurosci. 2005;25:9185–9194. doi: 10.1523/JNEUROSCI.2858-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagha E, Ozaita A, Chang SY, Nadal MS, Lin U, Saganich MJ, McCormack T, Akinsanya KO, Qi SY, Rudy B. DPP10 modulates Kv4-mediated A-type potassium channels. J Biol Chem. 2005;280:18853–18861. doi: 10.1074/jbc.M410613200. [DOI] [PubMed] [Google Scholar]