

Abstract
Growing evidence indicates that glia pathology and amino-acid neurotransmitter system abnormalities contribute to the pathophysiology and possibly the pathogenesis of major depressive disorder. This study investigates changes in glial function occurring in the rat prefrontal cortex (PFC) after chronic unpredictable stress (CUS), a rodent model of depression. Furthermore, we analyzed the effects of riluzole, a Food and Drug Administration-approved drug for the treatment of amyotrophic laterosclerosis, known to modulate glutamate release and facilate glutamate uptake, on CUS-induced glial dysfunction and depressive-like behaviors. We provide the first experimental evidence that chronic stress impairs cortical glial function. Animals exposed to CUS and showing behavioral deficits in sucrose preference and active avoidance exhibited significant decreases in 13C-acetate metabolism reflecting glial cell metabolism, and glial fibrillary associated protein (GFAP) mRNA expression in the PFC. The cellular, metabolic and behavioral alterations induced by CUS were reversed and/or blocked by chronic treatment with the glutamate-modulating drug riluzole. The beneficial effects of riluzole on CUS-induced anhedonia and helplessness demonstrate the antidepressant action of riluzole in rodents. Riluzole treatment also reversed CUS-induced reductions in glial metabolism and GFAP mRNA expression. Our results are consistent with recent open-label clinical trials showing the drug's effect in mood and anxiety disorders. This study provides further validation of hypothesis that glial dysfunction and disrupted amino-acid neurotransmission contribute to the pathophysiology of depression and that modulation of glutamate metabolism, uptake and/or release represent viable targets for antidepressant drug development.
Keywords: antidepressant, stress, glia, glutamate, glutamate transporter, metabolism
Introduction
Major depressive disorder (MDD) is a common and disabling illness affecting a rising percentage of the world's population. At present, nearly all of the medications available for the treatment of MDD have been developed out of the monoaminergic deficit hypothesis of depression that arose in the mid 1960s.1 Although this approach has led to great advances in our ability to treat MDD, the limitations of our current armamentarium of antidepressant drugs are becoming increasingly evident.2 This realization presents a strong impetus to search beyond the monoaminergic systems for a better understanding of the pathophysiological mechanisms underlying MDD, and to consider these newly identified mechanisms in the search for novel antidepressant medications.
Growing evidence indicates that glial elements are involved in the neuropathology of several neuropsychiatric illnesses including MDD.3 Post-mortem studies of tissues from patients with MDD describe a reduced number and an altered morphology of glial cells in several brain regions, in particular the prefrontal cortex (PFC).3–7 Other post-mortem studies demonstrating altered expression of glial fibrillary associated protein (GFAP), glial-specific excitatory amino-acid transporters (EAATs) and glutamine synthetase (GS) in tissue from individuals with MDD8,9 suggest that the glial cell abnormalities include changes in astrocytic cell function. Recent studies provide evidence that stress exposure may be related to some of the reported glial cell pathology by demonstrating that animals exposed to chronic stress have a decreased glial density in the hippocampus10 and a reduced production of glial cells in the adult hippocampus and PFC.11–13
Stress-related effects on glutamate (Glu) neuro-transmission are increasingly implicated in the pathophysiology and pathogenesis of MDD,3,14–16 and there is now strong evidence that marked abnormalities exist within the glutamatergic and GABAergic amino-acid neurotransmitter (AANt) systems of individuals with MDD.17 In this context, the role of glial cells in the regulation of the AANt system function18,19 through the clearance of Glu from the extracellular space via the EAATs20 and recycling of Glu through the glutamate/glutamine (Glu/Gln) cycle21 seems especially relevant to the pathophysiological processes underlying mood disorders.17 Specifically, impaired glial cell function would be predicted to lead to increased glutamatergic activation, especially at extrasynaptic sites that have been shown to promote neurotoxic-like effects.22,23 Recent evidences showing NMDA antagonists to produce rapid antidepressant responses have further fueled interest in this model.24 The hypothesis that increased levels of extracellular Glu could be related to depressive-like behaviors, and that increased Glu uptake could have protective, antistress or antidepressant-like effects is supported by a recent study demonstrating that ceftriaxone, a β-lactam agent known to increase EAAT expression,25 has significant antidepressant-like properties in the tail suspension and forced swim tests (FSTs).26
Riluzole, a drug currently used to slow the progression of amyotrophic lateral sclerosis, is believed to alter Glu neurotransmission by both decreasing presynaptic release and facilitating glial cell Glu uptake.27–31 Interestingly, riluzole has also been shown to be effective in open-label studies in several psychiatric disorders including treatment-resistant MDD.31 The goal of the current study is to test two related hypotheses: (1) chronic stress leads to impaired astrocytic function and reduced AANt cycling, resulting in AANt perturbations that are associated with characteristic stress-induced behaviors, and (2) riluzole, a drug that facilitates Glu uptake will attenuate the effects of stress on AANt cycling, and reverse or attenuate the effects of stress on tests of anhedonia and despair.
Materials and methods
Animals
Male Sprague–Dawley rats (Charles River) were housed in wire bottom cages under a 12-h light/12-h dark cycle at constant temperature (25 °C) and humidity with free access to food and water except when animals were subjected to light disturbance or deprivation stressors during the chronic unpredictable stress (CUS) procedure. Animals weighed between 250 and 300 g at the time of the first stressor. Experiments began after at least 1 week of habituation to the housing conditions. Animal use procedures were in accordance with the Yale University Care and Use of laboratory animals guidelines.
Chronic unpredictable stress procedure
CUS is an experimental procedure in which animals are exposed to a variable sequence of mild and unpredictable stressors. Our CUS procedure was successfully used in the laboratory to produce behavioral changes as well as alteration of glial cell proliferation in the PFC.12 The CUS animals were subjected to exactly the same sequence of 12 stressors (2 per day for 35 days) described in Banasr et al.12: cage rotation, light on, light off, cold stress, isolation, crowding, cold swim stress, food and water deprivation, wet bedding, stroboscope, cage tilt and odor exposure. To simulate a realistic course of antidepressant intervention, animals were first exposed to CUS for 15 days, and then were administered with riluzole (4 mg/kg, i.p) daily for 21 days with continued CUS procedure. The dose used in this study was similar to Risterucci et al.32 and was shown to block behavioral deficit induced by PFC ibotenate-induced excitotoxic lesion. On days 15 and 35 of the study, animals were tested in the sucrose preference test (SPT). On day 28, animals were tested in the active avoidance test (AAT). All animals were tested for locomotor activity on days 15, 28 and 35.
Sucrose preference test
The SPT was performed on days 15 and 35 and consisted in 48 h exposition to a palatable sucrose solution (1%; Sigma, St Louis, MO, USA), followed by 4 h of water deprivation and a 1-h exposure to two identical bottles, one filled with sucrose solution and the other with water. This procedure was adapted from Willner33 and has been previously used successfully in our lab.12 Sucrose and water consumption was determined by measuring the change in volume of fluid consumed. Sucrose preference was defined as the ratio of the volume of sucrose vs water consumed during the 1-h test.
Active avoidance test
The active avoidance procedure was performed in one learned helplessness chamber of custom-built shuttle boxes (Med Associates). After a 5 min habituation, animals received 30 randomized escapable footshocks at an intensity of 0.65 mA. In each shock trial, the gate separating the two halves of the shuttle box opened 5 s before shock onset and remained open for the duration of the shock. The average intertrial interval was 60 s (range 20–100 s). The first 5 trials required one crossing to terminate footshock (FR1) and the remaining 25 trials required two crossings (FR-2). Results are expressed as number of escape failures, that is, the number of times that the animal did not terminate the footshock.
In situ hybridization
After all animals were tested in behavior, a subset was killed by decapitation 24 h after the last stressor and/ or the last drug injection for in situ hybridization and the other subset was used for the nuclear magnetic resonance (NMR) study. Brains were removed, rapidly frozen on dry ice and stored at 80 °C. Cryocut sections (14 μm thickness) containing prelimbic cortex (4.2–2.2 mm from bregma) were subjected to in situ hybridization analysis as previously described.34 Briefly, radiolabeled (35S) riboprobes were generated by in vitro transcription (Megashortscript; Ambion) from PCR product amplified from whole brain using gene-specific primers. The GFAP, GLT-1, GLAST, GS probes were verified by sequencing and previously used in our laboratory.34 The optical density was quantified within a 500 μm2 box in the prelimbic cortex using NIH ImageJ software. Results are expressed as mean±s.e.m. percentage from control (home cage animals injected with saline for 21 days).
[2-13C]acetate infusion and sample preparation
To determine whether CUS and/or chronic riluzole treatment induce changes in glial metabolism, we used a similar approach to Chowdhury et al.35 based on infusion of [2-13C]acetate followed by 1H-[13C]-NMR ex vivo. At 24 h after the last stressor or/and injection, rats were anesthetized with urethane (1.5 g kg−1, i.p.) and 30 min later, [2-13C]acetate (99 atom%; Cambridge Isotopes, 2 M, pH 7) was infused through a tail vein. The solution was delivered by variable rate infusion (from 0–15 s at a rate of 3.12 ml min−1 kg−1 followed by 436 ml min−1 kg−1 from 15 s to 4 min, at 249 μl min−1 kg−1 from 4 to 8 min and 125 μl min−1 kg−1 from 8 to 15 min). Immediately after infusion, rats were killed using a directed microwave pulse (5 kW, Model TMW 6402C; Muromachi Microwave Fixation System) to the head to quickly arrest metabolism allowing brain tissue to be removed without post-mortem changes. PFC was then dissected, frozen in N2 liquid and stored at −80 °C until concentrations of the major amino acids (Glu, GABA and Gln) in these tissue samples as well as their 13C enrichments were determined using 1H-[13C]-NMR spectroscopy. Blood was sampled from the heart at the end of experiment for determination of the isotopic fractional enrichments and concentrations of the infused substrate. After centrifugation, collected blood was frozen in liquid N2, and stored at −80 °C for subsequent analysis.
PFC metabolites were extracted from frozen tissue (130–150 mg) as described previously.36 Briefly, tissue was ground with 0.1 mol l−1 HCl in methanol (1:2, wt/vol). A known quantity of [2-13C] glycine (0.5 μmol) was added as an internal concentration reference. The tissue powder was homogenized with ice-cold ethanol (1:6, wt/vol; 60% ethanol) then clarified by centrifugation (20 000 g). The supernatant was lyophilized and resuspended in 500μl of a phosphate-buffered (100 mmol l−1, pH 7) deuterium oxide (Cambridge Isotope Laboratories) solution containing 0.5 mmol l−1 3-trimethylsilyl[2,2,3,3-D4]-propionate (TSP), which served as a chemical shift reference.
Frozen blood plasma samples were thawed for direct measurement of glucose (Beckman Glucose Analyzer II, Model 6517; Beckman Instruments Inc.). The remaining plasma was mixed with deuterium oxide and passed through a centrifugal filter (10 kDa cutoff, Nanosep, Centrifugal Devices; Pall Life Science). Samples were loaded into 5 mm NMR tubes along with known quantities of TSP and formate, the latter serving as a concentration standard.
Nuclear magnetic resonance analysis of cortical extracts
Fully relaxed 1H-[13C]-NMR spectra of plasma and cortical extracts were acquired at 11.7 T (1H resonance frequency of 500.13 MHz) using a Bruker AVANCE spectrometer (Bruker Instruments). 1H-[13C]-NMR spectra were acquired as two subspectra—one involving broadband 13C inversion pulses applied in alternate scan blocks, whereas 13C-decoupling was applied in both. Subtraction of the scans obtained with 13C inversion (12C–13C) from those without inversion (12C+13C) gave the difference spectrum (2×13C), containing only 13C-coupled 1H resonances at twice the true intensity. The total carbon isotope composition was given by the 12C+13C subspectrum. The 13C atom percentage enrichment was calculated as the ratio, 13C/(12C+13C)×100, followed by subtraction of 1.1% to remove 13C arising from natural abundance.35 Absolute concentrations of metabolites were determined relative to [2-13C]glycine, added during tissue extraction as an internal concentration reference. The isotopic 13C enrichments of Glu-C4, C3, GABA-C2, Gln-C4, succinate (Suc)-C2+C3, aspartate (Asp)-C3, alanine (Ala)-C3 and lactate (Lac)-C3 were calculated from the ratio of the areas of these resonances in the 1H-[13C]-NMR difference spectrum (2×13C only) and the nonedited spectrum (12C+13C).
Nuclear magnetic resonance analysis of blood samples
The concentration and 13C isotopic enrichment of plasma [2-13C]acetate (2-CH3, 1.9 p.p.m.) and total acetate concentration were determined using 1H-NMR without 13C decoupling using a repetition time of 20 s. Formate was added to the sample as a concentration standard. The isotopic 13C enrichment of acetate-C2 (1.9 p.p.m.) was calculated by dividing the areas of the 13C satellites with the total area (12C+13C) of their respective resonances.36
Statistical analysis
Statistical differences were determined by analysis of the variance (ANOVA, StatView 5) followed by Newman–Keuls post hoc analysis. Repeated-measures ANOVAs were used to analyze the effect of the treatments when the same behavioral test was used at different time points for the same individual. This analysis was followed by Dunnett's post hoc analysis. The F-values and group and experimental degrees of freedom are included in the legends of the figures. For experiments with two groups, Student's t-test was used. The level of statistical significance was set at P < 0.05 using two-tailed tests.
Results
CUS-induced effects on depressive-like behaviors are reversed by chronic riluzole treatment
In this study, we used a well-documented animal model of depression, CUS that results in multiple behavioral effects, many of which are relevant to those observed clinically, most notably anhedonia.33 Moreover, these changes are reversed after chronic, but not acute antidepressant treatment, a time-dependent effect that is poorly modeled by other behavioral paradigms (for example, learned helplessness or FST). The CUS procedure used in this study was the same procedure reported previously by our lab and consists of 12 different randomized stressors, 2 per day for 35 days.12 Home cage control (CTR) and CUS animals were subjected to the SPT, which is used as an index of the hedonic-like state of animals on days 15 and 35 (Figure 1; experimental design).
Figure 1.

Timeline of experimental procedures. Rats are handled daily (home cage control, CTR) or subjected to the chronic unpredictable stress (CUS) procedure for 35 days. Animals are administered saline or riluzole (4 mg/kg) for the 21 last days of the experiment. The efficacy of CUS or riluzole on behavioral performances of the animals in locomotor activity, active avoidance and sucrose preferences test are measured.
We found that CUS induces a significant decrease in the ratio of sucrose vs water consumed after 15 days of stress compared to CTR animals (Figure 2a; n = 16 per group; t-test, P < 0.05). No difference between groups was observed in total fluid consumption for the 1 h test. In addition, no changes in spontaneous locomotor activity were found at this time point (Supplementary Table S1). To simulate a realistic course of antidepressant intervention, animals were then split into two experimental groups (based on their SPT performances, n = 8 per group) and injected with riluzole (4 mg kg−1) or saline i.p. daily for 21 days with continued CUS procedure (Figure 1; experimental design). Animals were tested in the AAT, index of helplessness or despair,37 on day 28 and retested in SPT on day 35.
Figure 2.
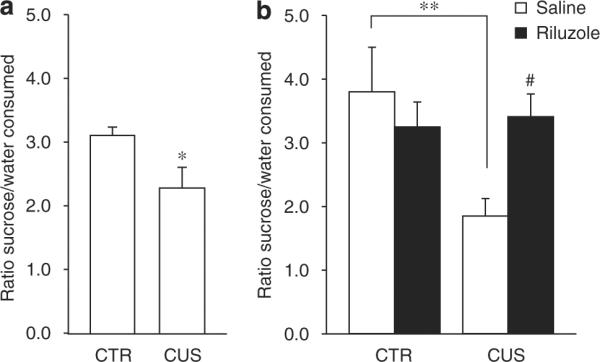
Effects of chronic unpredictable stress (CUS) and riluzole in the sucrose preference test. (a) On day 15, CUS animals showed a significant decrease in sucrose preference when compared to home cage control (CTR) animals (t30 = 2.36, P = 0.025). (b) Sucrose preference was decreased by 35 days CUS exposure (F1,28 = 3.42, P < 0.01) and was reversed by chronic riluzole treatment (F1,28 = 5.258, P < 0.05). Error bars represent s.e.m. *P < 0.05, **P < 0.01 compared to CTR and #P < 0.05 compared to CUS, two-way analysis of the variance (ANOVA), Student–Newman–Keuls post hoc analysis.
CUS animals injected with saline (CUS+SAL) showed a higher number of escape failures than CTR animals injected with saline (CTR+SAL) (Figure 3; n = 8 per group; two-way ANOVA, post hoc Student–Newman–Keuls, P < 0.01). Although behavioral performances in AAT of CTR animals injected with riluzole (CTR+RIL) and CUS animals treated with riluzole (CUS+RIL) were not different from that of CTR+SAL group, the number of escape failures was significantly lower in CUS+RIL group compared to CUS+SAL group (Figure 3a; two-way ANOVA, post hoc Student–Newman–Keuls, P < 0.001). Latency to escape was also increased in CUS+SAL animals compared with the other groups. This effect was significant from the second FR-2 block of five escapable shocks (footshocks 10–15) to the end of the test. CUS+RIL animals showed performances in AAT significantly different from CUS+SAL as soon as the first FR-2 block of five escapable shocks (Figure 3b; two-way repeated-measures ANOVA, post hoc Dunnett's, P < 0.01). These results demonstrate that CUS induces helplessness in the AAT and that riluzole prevents this effect.
Figure 3.
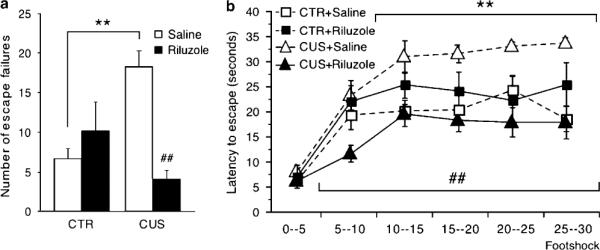
Effects of chronic unpredictable stress (CUS) and riluzole in the active avoidance test. (a) CUS animals showed a significant increase of escape failures (F1,28 = 7.798, P < 0.01) and this effect was reversed by chronic riluzole treatment (F1,28 = 19.05, P < 0.001). Error bars represent s.e.m. **P < 0.01 compared to home cage control (CTR) and ##P < 0.01 compared to CUS, two-way analysis of the variance (ANOVA) and Student–Newman–Keuls post hoc analysis. (b) Latency to escape was significantly increased in CUS animals (F1,28 = 7.79, P < 0.01) starting from the second FR-2 block of five escapable shocks (footshocks 10–15) to the end of the test (F1,168 = 27.37, P < 0.01). Riluzole-treated animals showed performances in this test significantly different from CUS (F1,28 = 16.45, P < 0.01) as soon as the first FR-2 block of five escapable shocks (F1,168 = 45.57, P < 0.01). Error bars represent s.e.m. **P < 0.01 compared to CTR and ##P < 0.01 compared to CUS, Two-way repeated-measures ANOVA followed by Dunnett's post hoc analysis.
On day 35, all animals were subjected to a second SPT. We confirmed that 35 days of CUS induce decreases in the ratio of sucrose to water consumed when compared with CTR+SAL12 (Figure 2b; n = 8 per group, two-way ANOVA, post hoc Student–Newman–Keuls, P < 0.01). Although the effect of CUS on sucrose preference was seen as early 15 days, continuation of CUS for 21 more days significantly increases the magnitude of this effect (Figures 2a and b; repeated-measures ANOVA, post hoc Dunnett's, P < 0.05). Finally, we demonstrate that chronic treatment with riluzole significantly reversed the effect of CUS on sucrose preference (Figure 2b; twoway ANOVA, post hoc Student–Newman–Keuls, P < 0.01). Again, total fluid consumption during the 1-h test did not differ between groups. In addition, changes in spontaneous locomotor activity were analyzed on days 28 and 35 in the same animals and did not differ between groups throughout the experiment, demonstrating that the results obtained in AAT and SPT were independent of this experiment bias (Supplementary Table S1). In summary, our results demonstrate that CUS induces anhedonia and helplessness and that these behavioral deficits can be prevented and/or reversed by chronic treatment with riluzole.
We also characterized the antidepressant-like properties of acute and chronic (10 days) treatment with riluzole in the FST, a behavioral test used as a screen to predict antidepressant-like properties of new compounds (Supplementary Figures S1a and b). Although time spent immobile was decreased in both conditions, only chronic treatment with riluzole induced a significant effect (Supplementary Figure S1b; n = 6–7 per group; t-test, P < 0.05). We also analyzed the effects of acute and chronic treatment with riluzole on spontaneous locomotor activity (Supplementary Table S1). Whereas chronic riluzole has no effect, acute riluzole induces a statistically significant decrease in locomotor activity (Supplementary Table S1; n = 6 per group; t-test, P < 0.05). Thus, it is not possible to rule out the fact that an acute antidepressant-like effect of riluzole in the FST was masked by a general decrease in locomotor behavior. In any case, these results show that chronic treatment with riluzole has antidepressant-like properties in CUS-induced anhedonia and helplessness, as well as the FST.
CUS effects on Glu/Gln cycling and GABA synthesis are reversed by chronic riluzole treatment
To determine whether chronic stress or riluzole treatment alters glial metabolism and AANt cycling and/or synthesis, we analyzed the changes in concentrations of amino acids and metabolites occurring in the PFC of CUS animals and/or riluzole-treated animals. For this study, we used a similar approach to Chowdhury et al.35 based on infusion of [2-13C]acetate, which has been shown to be metabolized preferentially in astroglia38 (see Figure 4a), followed by 1H-[13C]-MRS ex vivo. The initial results demonstrate that average concentrations of acetate were similar in plasma samples from all groups (mean±s.e.m.; CTR+SAL = 16.1±1.0 mmoll−1; RIL = 17.3±0.5 mmoll−1; CUS+SAL = 15.9±1.0 mmoll−1 and CUS+RIL = 15.8±0.8 mmoll−1, n = 8 per group). Similar results were found for the percentage 13C enrichments: mean±s.e.m.; CTR+SAL = 91.2±0.5%; CTR+RIL = 92.3±0.8%; CUS+SAL = 92.8±0.4% and CUS+RIL = 93.0±0.7%). In addition, total levels of amino acids and metabolites were determined in the different groups by averaging the respective [2-13C]acetate infusion data; total PFC concentrations of Glu, GABA, Gln, Asp, Suc, Ala, NAA, Cre and Myo-Ins were similar in all animal groups (not shown). These results indicate that changes in PFC levels of the 13C-labeled amino acids found in this study are not related to general metabolic alterations or changes in total amino-acid concentrations. We did not detect β-hydroxybutyrate35 in the blood or brain extract spectra of any group, thus indicating the treatments did not induce ketosis in the animals.
Figure 4.

Effects of chronic unpredictable stress (CUS) and riluzole on 13C concentrations and -enrichment of Glu-C4, Gln-C4 and GABA-C2 after 13C-acetate infusion. (a) Schematic depiction of the major metabolic pathways of 13C isotopic label flow from the astroglial substrate, [2-13C]acetate. After transport into the brain from blood, [2-13C]acetate is metabolized in the mitochondria to acetyl-CoA (labeled at C2, Ac2CoA) and enters the astroglial TCA cycle (TCAa) as citrate labeled at C4 after condensation with oxaloacetate (oaa). Further metabolism along the cycle labels α-ketoglutarate (α-KG) at C4, which can then exchange with the astroglial glutamate pool (small pool glutamate) transferring 13C label to C4 (Glu4). With time the 13C label traverses the complete TCA cycle, labeling the other carbon positions, e.g., C3, C2, C1. The initial rate of label trapping at Gln4 is related to astroglial TCA cycle flux, whereas Glu4 and Gaba2 are related to the glutamate/GABA/glutamine cycle fluxes. Astroglia precursors (mainly Gln4) are released from the astroglia, transferring the label to neurons for synthesis of Glu4 and Gaba2. Only flux pathways labeling Gln4, Glu4, Gaba2 from [2-13C] acetate are shown. The pyruvate carboxylation pathway (anaplerosis) in astroglia is depicted by the dashed arrow. The continuous metabolism of unlabeled glucose in neurons and astroglia through pyruvate dehydrogenase (pyr → acetyl-CoA) serves as a constant dilution flux. Definitions: (subscripts) a, astroglia; n, neuron; acetyl-CoA, acetyl-coenzyme A; suc, succinate; TCAn, neuronal TCA cycle. (b) 13C concentration of Glu-C4 was significantly decreased by CUS (F1,28 = 2.79, P < 0.05) and this effect was reversed by riluzole treatment (F1,28 = 7.8, P < 0.05). (c) CUS significantly reduced 13C concentration of Gln-C4 (F1,28 = 13.71, P < 0.001) and riluzole did not significantly reverse this effect. (d) 13C concentration of GABA-C2 was significantly decreased by CUS (F1,28 = 4.63, P < 0.05) and this effect was reversed by riluzole treatment (F1,28 = 5.39, P < 0.01).
Concentrations of 13C-labeled amino acids from [2-13C]acetate were found to be altered in PFC samples from CUS- and riluzole-treated animals (Figures 4b–d; Table 1). Indeed, whereas 13C concentrations of Glu-C4, Gln-C4 and GABA-C2 were similar in both CTR+RIL and CTR+SAL groups, the CUS+SAL group showed significant reductions of 13C concentrations of Glu-C4 (Figure 4b; n = 8 per group; two-way ANOVA; post hoc Student–Newman–Keuls, P < 0.05), Gln-C4 (Figure 4c; P < 0.01) and GABA-C2 (Figure 4d; P < 0.05) when compared to CTR+SAL. We found similar reductions in the percentage of 13C enrichments of Glu-C4, and GABA-C2 when comparing CTR+SAL vs CUS+SAL groups (Table 1). These results suggest that CUS decreases glial metabolism and AANt cycling in the rat PFC.
Table 1.
Effects of CUS and riluzole on percentage of 13C enrichment
| Treatment | Glu-C4 | Gln-C4 | GABA-C2 |
|---|---|---|---|
| CTR+Saline | 4.0 ± 0.2 | 13.2 ± 0.2 | 2.6 ± 0.2 |
| CTR+Riluzole | 3.8 ± 0.1 | 12.8 ± 0.1 | 2.7 ± 0.1 |
| CUS+Saline | 3.4 ± 0.1* | 11.5 ± 0.1** | 2.0 ± 0.1* |
| CUS+Riluzole | 4.0 ± 0.2# | 12.6 ± 0.2## | 2.8 ± 0.2## |
Table summarizing the effect of CUS and riluzole on the percentage 13C enrichment of Glu-C4, Gln-C4 and GABA-C2. CUS decreased the percentage of 13C enrichment of Glu-C4 (F1,28 = 3.4, P < 0.05), Gln-C4 (F1,28 = 12.12, P < 0.01) and GABA-C2 (F1,28 = 4.93, P < 0.05). Riluzole significantly reversed the effect of CUS on the percentage of 13C enrichment of Glu-C4 (F1,28 = 7.78, P < 0.05), Gln-C4 (F1,28 = 12.51, P < 0.01) and GABA-C2 (F1,28 = 7.696, P < 0.01).
Error bars represent s.e.m.
P < 0.05,
P < 0.01 compared to CTR
P < 0.05,
P < 0.01 compared to CUS, two-way ANOVA, Student–Newman–Keuls post hoc analysis.
Interestingly, we also found that 13C concentrations of Glu-C4 and GABA-C2 in CUS+RIL animals were significantly different from CUS+SAL animals (Figures 4b–d; two-way ANOVA, post hoc Student–Newman–Keuls, P < 0.05). Although riluzole's effect on Gln-C4 13C concentration in CUS+RIL animals did not reach significance when compared to CUS+SAL, a strong trend was observed, and Glu-C4 13C concentrations were not significantly different from CTR+SAL or CTR+RIL group (Figures 4b–d; two-way ANOVA, post hoc Student–Newman–Keuls, P = 0.073). In addition, chronic riluzole treatment significantly increased the percentage 13C enrichments of all three metabolites in CUS-treated animals (Table 1). This suggests that riluzole reverses the effects of CUS on glial metabolism and AANt cycling.
CUS and riluzole affect expression of glial-specific markers
To determine whether glial alterations are present in our animal model of depression, we investigated the effects of CUS and/or riluzole on expression levels of genes specific to glial cells using in situ hybridization. We analyzed the changes in expression levels of GFAP, the two glial-specific EAATs (GLT-1/EAAT2 and GLAST/ EAAT1) and GS. We found that CUS induces a significant 20% decrease in GFAP mRNA expression and no change in expression level of the other markers (Figures 5a–c; CTR+SAL: n=4, CUS+SAL: n = 5; two-way ANOVA, post hoc Student–Newman–Keuls, P < 0.05). Although chronic riluzole had no effect on GFAP expression in nonstressed animals, this treatment reversed the effect of CUS (Figures 5a and b; RIL = 4, CUS+SAL: n = 5, CUS+RIL: n = 5; two-way ANOVA, post hoc Student–Newman–Keuls, P < 0.05). This decrease in GFAP expression suggests that the morphology and/or architecture of astrocytes may be altered by CUS and that riluzole treatment blocks these changes.
Figure 5.
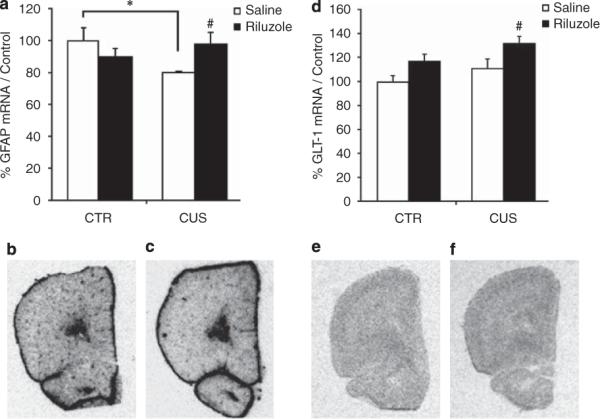
Effects of chronic unpredictable stress (CUS) and riluzole on mRNA levels of glial-specific markers. (a) CUS showed a significant decrease of mRNA levels of glial fibrillary associated protein (GFAP) compared to home cage control (CTR) animals (F1,16 = 4.52, P < 0.05). CUS animals treated with riluzole were significantly different from CUS animals treated with saline and not different from CTR animals treated with saline or riluzole (F1,16 = 5.7, P < 0.05). Representative autograph of effect of CUS (c) on GFAP mRNA expression compared to CTR (b). (d) Although CUS has no effect on mRNA level of GLT-1, chronic riluzole treatment significantly increased GLT-1 transcript (F1,16 = 4.9, P < 0.05). Representative autograph of effect of CUS (e) on GLT-1 mRNA expression compared to a CUS animal treated with riluzole (f). Error bars represent s.e.m. *P < 0.05 compared to CTR and #P < 0.05 compared to CUS, two-way analysis of the variance (ANOVA), Student–Newman–Keuls post hoc analysis.
In addition, we found that CUS has no effect on the expression of GLT-1 (Figures 5d and e), GLAST or GS (Table 2). However, chronic riluzole treatment induced a significant ~30% increase in GLT-1 expression in the PFC in the CUS+RIL group (Figures 5d–f; two-way ANOVA, post hoc Student–Newman–Keuls, P < 0.05). No change was observed on the expression of GLAST and GS transcripts following riluzole treatment in stressed and nonstressed animals (Table 2). Increased GLT-1 expression found in this experiment suggests that riluzole treatment may attenuate glutamatergic hyperactivity, classically described after stress.39
Table 2.
Effects of CUS and riluzole on EAAT-1 and GS transcripts (% from CTR)
| Genes | CTR + Saline | CTR + Riluzole | CUS + Saline | CUS + Riluzole |
|---|---|---|---|---|
| EAAT-1/GLAST | 100 ± 7 | 101 ± 7 | 99 ± 11 | 98 ± 7 |
| Glutamine synthetase | 100 ± 7 | 100 ± 5 | 102 ± 5 | 104 ± 4 |
Discussion
This study provides the first experimental evidence of impaired cortical glial function in an animal model of depression. The results demonstrate that animals exposed to CUS exhibit behavioral deficits in tests measuring helplessness and anhedonia, as well as decreases in 13C-acetate metabolism and GFAP mRNA expression in the PFC. The cellular and behavioral alterations induced by CUS were reversed or blocked by chronic treatment with the Glu-modulating drug riluzole. The beneficial effects of riluzole on CUS-induced anhedonia and helplessness clearly define chronic riluzole as an antidepressant treatment in this model. Chronic riluzole was also able to reverse the CUS-induced decrease in 13C-acetate metabolism and GFAP expression. These effects were associated with a selective increase in the expression of the glial-specific EAAT, GLT-1. Together, our results suggest that chronic stress alters glial cell function and that the cellular mechanism of riluzole antidepressant action may involve the attenuation of glial dysfunction and an enhancement of extrasynaptic Glu clearance.
Glial impairment in an animal model of depression
Stress-based animal models are a leading approach for analyzing cellular and molecular mechanisms underlying the pathophysiology of depression. These models have also been of tremendous utility in testing antidepressant properties of new compounds. CUS is a dependable depression model with high face, predictive and construct validity.33 CUS simulates realistic conditions for human depression and results in multiple behavioral changes similar to those observed clinically. In this study, we confirmed that our CUS procedure decreases sucrose preference,12 a measure of anhedonia in rodents33 and we show that the same procedure induces helplessness, demonstrated by increased escape failures in AAT. Knowing that anhedonia and helplessness are major symptoms of human depression (Diagnostic and Statistical Manual of Mental Disorders, 4th edn), these results provide further evidence of the validity of CUS and support the hypothesis that cellular changes found in this model may also occur in patients affected by stress-related illnesses.
At the cellular level, we found that animals subjected to CUS show decreased GFAP mRNA expression in the PFC. Decreased GFAP immunostaining and a reduced number of GFAP-positive cells were recently reported after chronic psychosocial stress in the hippocampus, another major limbic brain region involved in depression.10 As two previous studies,11,12 failed to demonstrate any effects of stress on the proliferation of GFAP expressing cells, it does not appear the findings are a result of decreased astrocyte proliferation, but rather an effect on mature cortical astrocytes. Although hippocampal glial loss was not found in post-mortem tissue from depressed patients,40 studies have consistently reported a decreased number and altered morphology of glial cells in several regions of the frontal cortex.3 This glial pathology has been associated with decreased GFAP immunostaining.41 As GFAP is a key cytoskeletal protein of astrocytes, this could be related to observed morphological alteration of cortical astrocytes. Whether the glial morphological changes found in the human brain or in this study are associated with alteration of glial function or behavioral deficits remain unknown.
Abnormal increases in Glu efflux have been reported in the PFC in response to stress and, corticosterone treatment,39 and elevated levels of Glu have been recently found in PFC tissue of depressed subjects.42 This increase in Glu is postulated to contribute to the morphological alterations observed in limbic regions in MDD and after stress.16 In addition to the important role of astrocytes in nurturing neurons, they are actively involved in maintaining synaptic transmission.43 Glu clearance is believed to be a major function of EAATs located on glial cells. Although we did not find altered expression of GLAST or GLT-1 mRNA in CUS animals, we cannot rule out the possibility of decreased protein levels or transporter activity. Indeed, there are reports suggesting loss of GFAP can decrease EAAT activity through alterations of GLT-1 trafficking at the membrane in the cortex and hippocampus.44 Alterations of EAAT activity and glial metabolism are consistent with the results obtained in our 13C-acetate analysis. Because glial cells are the main cell type responsible for uptake and metabolism of 13C-acetate in the brain,38 the lower 13C labeling of glial (Gln-C4) and neuronal (Glu-C4 and GABA-C2) products of AANt cycling in PFC extracts indicates that glial metabolism is decreased in animals subjected to CUS. The AANt cycling is also dependent on GS, a glial-specific enzyme responsible for the recycling of neuronally released Glu and GABA into glutamine.21,45 Even though we did not see changes in GLT-1, GLAST or GS mRNA expression after CUS, the results obtained in the 13C-acetate indicate that Glu flux through these enzyme/transporters is reduced in astrocytes. In support of this, a previous study has shown that glial-mediated Glu uptake is reduced after chronic stress in rodents.46 In addition, recent work carried out in our lab demonstrated that local infusion of a gliotoxin in the PFC resulting in decreases in GLT-1, GS and the number of astrocytes was sufficient to induce a depressive-like phenotype similar to CUS (Banasr and Duman 2007, 37th Annual Meeting of the Society for Neurosciences, 383.2). Together, our results along with data from the literature suggest that dysregulation of the primary functions of astrocytes in the PFC is associated with stress and depression and may contribute to the expression of the depressive-like symptoms.
Riluzole: an antidepressant that acts on glia
In this study we demonstrate that the cellular and behavioral alterations induced by CUS are reversed and/or blocked by chronic treatment with riluzole. The antidepressant actions of riluzole were only evident after chronic treatment, including in the FST, which is typically responsive to acute antidepressants. Although acute riluzole induced a trend in decreasing time spent immobile in the FST this effect was not significant and possibly due to the sedative properties of riluzole.31 This sedative-like effect was confirmed in a parallel group in which acute riluzole decreased locomotor activity, however the locomotor suppressant effects of riluzole were not seen with chronic treatment. In CUS animals, chronic riluzole treatment reversed helplessness in the AAT and anhedonia in SPT. The efficacy of riluzole in these three behavioral tests clearly defines riluzole as an antidepressant and further validates the findings of several open-label clinical studies demonstrating riluzole's efficacy in the treatment of mood and anxiety disorders.31
At a cellular level, chronic riluzole treatment increased GLT-1 mRNA expression in the PFC. The increased GLT-1 expression may provide a mechanism contributing to the increased Glu uptake that is observed following riluzole treatment.27–30 Through the enhanced uptake of riluzole via EAATs it is possible that riluzole helps to prevent the over-stimulation of extrasynaptic Glu receptors, a process believed to result in an excitotoxic cascade leading to a variety of pathological consequences.23 Chronic riluzole treatment also prevented or reversed the CUS-induced decrease in GFAP expression suggesting that the drug may reverse or block the glial morphological changes (atrophy or loss) found in the PFC of individuals with MDD. This effect would be consistent with riluzole's reported protective effects on glial cells against Glu toxicity.47 These results support the hypothesis that enhancement of Glu uptake is a new cellular mechanism for antidepressant action. This is consistent with another recent study in which ceftriaxone, which also increases Glu transport,25 was reported to have antidepressant properties in FST, tail suspension and novelty suppressed feeding.26
Finally, chronic riluzole also blocks the CUS-induced decrease in glial metabolism as the reductions in 13C labeling Glu-C4, Gln-C4 and GABA-C2 found after chronic stress were reversed by riluzole. The beneficial effect of riluzole on glial metabolism and glial function supports the hypothesis that a glioprotective drug may have antidepressant properties, and that glial dysfunction in the PFC contributes to the expression of the symptoms of MDD.13,48 It is important to note that glioprotective properties have also been reported with typical antidepressant agents. Fluoxetine treatment prevented glial loss in the hippocampus of rats subjected to chronic psychosocial stress,10 and reversed the decrease in glial cell proliferation observed in the PFC after chronic stress.11,12 Preliminary data from our laboratory (data not shown) also suggest that chronic fluoxetine may increase GLT-1 mRNA expression in CUS-treated animals.
These glial changes are likely associated with changes in neuronal function in the PFC. Many studies show alterations in the morphology of PFC neurons after stress49 similar to those observed in post-mortem tissue of depressed patients.3 Questions still remain regarding whether these neuronal changes are the primary cause or a consequence, or occur in parallel to the glial dysfunction.50 However, the highly consistent finding of glial loss and atrophy even in younger individuals with MDD suggests that glial dysfunction may precede neuronal atrophy.3 If so, then antidepressants that protect against glial loss, insure glial function and normalize Glu and GABA metabolism and neurotransmission will be of tremendous help in the development of antidepressants with a new mechanism of action.
In conclusion, this series of studies demonstrate that CUS causes changes in glial cell AANt metabolism, the expression of GFAP the major intermediate filament protein in mature astrocytes, and produces a depressive-like phenotype. These findings are consistent with a growing number of studies showing significant glial cell pathology and markedly abnormal AANt concentrations in individuals with MDD. Furthermore, we show that riluzole, a drug known to modulate Glu neurotransmission and facilitate glial Glu clearance, reverses several of the cellular, physiological and behavioral consequences of CUS. This result is consistent with evidence that riluzole and other Glu-modulating agents are efficacious in treating severe treatment-resistant MDD. In sum, although these findings do not conclusively identify a patho-physiological mechanism relating stress, glial pathology and behavior, they do support the general hypothesis that glial cell pathology and AANt abnormalities contribute the pathophysiology of MDD, and that this pathological process can be targeted in the development of novel antidepressant medications.
Supplementary Material
{kind=link}
Acknowledgments
We thank Dr Jane Taylor and Dr Angus Narin for the use of their locomotor activity apparatus and microwave fixation system. We also thank Richard Trinko and Xiaoxian Ma for their technical assistance with the acetate infusion studies, and Mathew Girgenti for his technical assistance with the in situ hybridization studies. This study was supported by NARSAD (2007 NARSAD Forster Bam Investigator GS), K02 (MH076222, GS), VA CT Research Enhancement Award Program (REAP) research center (GS and RSD), R01 (MH25642, RSD), R01 (MH45481, RSD), R01 (DK027121, KB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Drs James CK Lai, Fahmeed Hyder and Robin A de Graaf for helpful discussions and Dr Prajna P Siddiqui for performing the brain tissue extracts.
Footnotes
Author Contributions: Mounira Banasr was responsible for all animal treatments and behavioral analyses. She also contributed significantly to the study design and writing of the paper. Golam Chowdhury was responsible for conducting and analyzing the ex vivo 13C-NMR studies and contributed in the overall study design and preparation of the paper. Rose Twillinger and Dr Samuel Newton performed the in situ hybridization studies. Ronald S Duman contributed to design and analysis related to the behavioral and cellular components of the study, as well as the preparation of the paper. Kevin Behar contributed to design and analysis related to the NMR components of the study as well as the preparation of the paper. Gerard Sanacora provided the study's conception. He also coordinated all study activities and contributed significantly to the preparation of the paper.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Schildkaut JJ. The catacholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- 2.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- 3.Rajkowska G, Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets. 2007;6:219–233. doi: 10.2174/187152707780619326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cerebral Cortex. 2002;12:386–394. doi: 10.1093/cercor/12.4.386. [DOI] [PubMed] [Google Scholar]
- 5.Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- 6.Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res. 2004;67:269–275. doi: 10.1016/S0920-9964(03)00181-6. [DOI] [PubMed] [Google Scholar]
- 8.Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci USA. 2005;102:15653–15658. doi: 10.1073/pnas.0507901102. E-pub 17 October 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Si X, Miguel-Hidalgo JJ, O'Dwyer G, Stockmeier CA, Rajkowska G. Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology. 2004;29:2088–2096. doi: 10.1038/sj.npp.1300525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czeh B, Simon M, Schmelting B, Hiemke C, Fuchs E. Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology. 2006;31:1616–1626. doi: 10.1038/sj.npp.1300982. [DOI] [PubMed] [Google Scholar]
- 11.Czeh B, Muller-Keuker JI, Rygula R, Abumaria N, Hiemke C, Domenici E, et al. Chronic social stress inhibits cell proliferation in the adult medial prefrontal cortex: hemispheric asymmetry and reversal by fluoxetine treatment. Neuropsychopharmacology. 2007;32:1490–1503. doi: 10.1038/sj.npp.1301275. [DOI] [PubMed] [Google Scholar]
- 12.Banasr M, Valentine GW, Li XY, Gourley SL, Taylor JR, Duman RS. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biol Psychiatry. 2007;62:496–504. doi: 10.1016/j.biopsych.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Banasr M, Duman RS. Regulation of neurogenesis and gliogenesis by stress and antidepressant treatment. CNS Neurol Disord Drug Targets. 2007;6:311–320. doi: 10.2174/187152707783220929. [DOI] [PubMed] [Google Scholar]
- 14.Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, A Gray N, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry. 2003;53:707–742. doi: 10.1016/s0006-3223(03)00117-3. [DOI] [PubMed] [Google Scholar]
- 15.McEwen BS, Magarinos AM. Stress effects on morphology and function of the hippocampus. Ann NY Acad Sci. 1997;821:271–284. doi: 10.1111/j.1749-6632.1997.tb48286.x. [DOI] [PubMed] [Google Scholar]
- 16.Sapolsky RM. The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry. 2000;48:755–765. doi: 10.1016/s0006-3223(00)00971-9. [DOI] [PubMed] [Google Scholar]
- 17.Kugaya A, Sanacora G. Beyond monoamines: glutamatergic function in mood disorders. CNS Spectr. 2005;10:808–819. doi: 10.1017/s1092852900010403. [DOI] [PubMed] [Google Scholar]
- 18.Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res. 1999;57:417–428. [PubMed] [Google Scholar]
- 19.Schousboe A, Waagepetersen HS. Glial modulation of GABAergic and glutamatergic neurotransmission. Curr Top Med Chem. 2006;6:929–934. doi: 10.2174/156802606777323719. [DOI] [PubMed] [Google Scholar]
- 20.Beart PM, O'Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, et al. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soriano FX, Hardingham GE. Compartmentalized NMDA receptor signalling to survival and death. J Physiol. 2007;584:381–387. doi: 10.1113/jphysiol.2007.138875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans. 2006;34:936–938. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 25.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 26.Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2007;61:250–252. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 27.Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 28.Frizzo ME, Dall'Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–128. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–176. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 30.Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pittenger C, Coric V, Banasr M, Bloch M, Krystal JH, Sanacora G. Riluzole in the treatment of mood and anxiety disorders. CNS Drugs. 2008;22:761–786. doi: 10.2165/00023210-200822090-00004. [DOI] [PubMed] [Google Scholar]
- 32.Risterucci C, Coccurello R, Banasr M, Stutzmann JM, Amalric M, Nieoullon A. The metabotropic glutamate receptor subtype 5 antagonist MPEP and the Na(+) channel blocker riluzole show different neuroprotective profiles in reversing behavioral deficits induced by excitotoxic prefrontal cortex lesions. Neuroscience. 2006;137:211–220. doi: 10.1016/j.neuroscience.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 33.Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural–neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52:90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- 34.Newton SS, Girgenti MJ, Collier EF, Duman RS. Electroconvulsive seizure increases adult hippocampal angiogenesis in rats. Eur J Neurosci. 2006;24:819–828. doi: 10.1111/j.1460-9568.2006.04958.x. [DOI] [PubMed] [Google Scholar]
- 35.Chowdhury GM, Gupta M, Gibson KM, Patel AB, Behar KL. Altered cerebral glucose and acetate metabolism in succinic semialdehyde dehydrogenase-deficient mice: evidence for glial dysfunction and reduced glutamate/glutamine cycling. J Neurochem. 2007;103:2077–2091. doi: 10.1111/j.1471-4159.2007.04887.x. [DOI] [PubMed] [Google Scholar]
- 36.Chowdhury GM, Patel AB, Mason GF, Rothman DL, Behar KL. Glutamatergic and GABAergic neurotransmitter cycling and energy metabolism in rat cerebral cortex during postnatal development. J Cereb Blood Flow Metab. 2007;27:1895–1907. doi: 10.1038/sj.jcbfm.9600490. [DOI] [PubMed] [Google Scholar]
- 37.Murua VS, Gomez RA, Andrea ME, Molina VA. Shuttle-box deficits induced by chronic variable stress: reversal by imipramine administration. Pharmacol Biochem Behav. 1991;38:125–130. doi: 10.1016/0091-3057(91)90599-w. [DOI] [PubMed] [Google Scholar]
- 38.Waniewski RA, Martin DL. Astrocytes and synaptosomes transport and metabolize lactate and acetate differently. Neurochem Res. 2004;29:209–217. doi: 10.1023/b:nere.0000010450.21586.a6. [DOI] [PubMed] [Google Scholar]
- 39.Moghaddam B, Jackson M. Effect of stress on prefrontal cortex function. Neurotox Res. 2004;6:73–78. doi: 10.1007/BF03033299. [DOI] [PubMed] [Google Scholar]
- 40.Muller MB, Lucassen PJ, Yassouridis A, Hoogendijk WJ, Holsboer F, Swaab DF. Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur J Neurosci. 2001;14:1603–1612. doi: 10.1046/j.0953-816x.2001.01784.x. [DOI] [PubMed] [Google Scholar]
- 41.Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY, Stockmeier CA, et al. Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biol Psychiatry. 2000;48:861–873. doi: 10.1016/s0006-3223(00)00999-9. [DOI] [PubMed] [Google Scholar]
- 42.Hashimoto K, Sawa A, Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry. 2007;62:1310–1316. doi: 10.1016/j.biopsych.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 43.Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- 44.Hughes EG, Maguire JL, McMinn MT, Scholz RE, Sutherland ML. Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA-induced EAAT2 cell surface trafficking. Brain Res Mol Brain Res. 2004;124:114–123. doi: 10.1016/j.molbrainres.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 45.Sibson NR, Dhankhar A, Mason GF, Behar KL, Rothman DL, Shulman RG. In vivo 13C NMR measurements of cerebral glutamine synthesis as evidence for glutamate-glutamine cycling. Proc Natl Acad Sci USA. 1997;94:2699–2704. doi: 10.1073/pnas.94.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang CH, Huang CC, Hsu KS. Behavioral stress enhances hippocampal CA1 long-term depression through the blockade of the glutamate uptake. J Neurosci. 2005;25:4288–4293. doi: 10.1523/JNEUROSCI.0406-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dagci T, Yilmaz O, Taskiran D, Peker G. Neuroprotective agents: is effective on toxicity in glial cells? Cell Mol Neurobiol. 2007;27:171–177. doi: 10.1007/s10571-006-9082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471–482. doi: 10.1038/sj.npp.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- 50.Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008 Jul 22; doi: 10.1016/j.biopsych.2008.06.008. E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.