

Abstract
Long QT syndrome is characterized by prolongation of the corrected QT (QTc) interval on the surface electrocardiogram and is associated with precipitation of torsade de pointes (TdP), a polymorphic ventricular tachycardia that may cause sudden death. Acquired long QT syndrome describes pathologic excessive prolongation of the QT interval, upon exposure to an environmental stressor, with reversion back to normal following removal of the stressor. The most common environmental stressor in acquired long QT syndrome is drug therapy. Acquired long QT syndrome is an important issue for clinicians and a significant public health problem concerning the large number of drugs with this adverse effect with a potentially fatal outcome, the large number of patients exposed to these drugs, and our inability to predict the risk for a given individual. In this paper, we focus on mechanisms underlying QT prolongation, risk factors for torsades de pointes and describe the short- and long-term treatment of acquired long QT syndrome.
1. Introduction
Acquired long QT syndrome is a disorder of cardiac repolarization most often due to specific drugs, hypokalemia, or hypomagnesemia that may precipitate torsade de pointes and cause sudden cardiac death. Selzer and Wray first reported QT prolongation and ventricular fibrillation as a response to quinidine in 1964 [1]. Two years later, Dessertenne [2] described torsades de pointes, a polymorphic ventricular tachycardia where QRS complexes twist around an isoelectric line in a sinusoidal fashion in an elderly woman with complete atrioventricular block and syncopal attacks (Figure 1). Torsade de pointes is usually self-limited but may degenerate into ventricular fibrillation. The incidence of acquired long QT syndrome is difficult to be estimated. Although the chances of provoking torsades de pointes by a noncardiac medication are generally lower than antiarrhythmic medications, a number of noncardiovascular drugs have been recently withdrawn from market because of unexpected sudden cardiac deaths associated with prolongation of QT interval and torsades de pointes [3]. The frequency of drug-induced long QT syndrome and our inability to predict the risk for a given individual, makes long QT syndrome an important issue for clinicians. This paper focuses on mechanisms underlying QT prolongation, risk factors for torsades de pointes and describes the short- and long-term treatment of acquired long QT syndrome.
Figure 1.
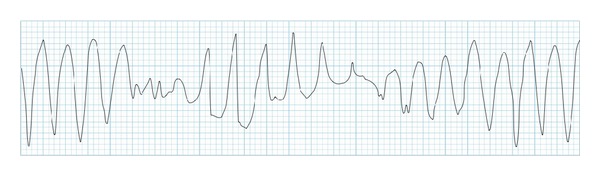
Torsades de pointes.
2. QT Interval Measurement
QT interval on the surface electrocardiogram describes the manifestation of ventricular depolarization and repolarization. It is measured from the beginning of QRS complex to T wave termination and averaged over 3 to 5 beats in a single lead. Longest QT intervals are usually measured in precordial leads and V3 or V4 leads appear more reliable for assessing QT prolongation [4]. Prominent U waves should be included in the measurement if they merge into the T wave. QT interval is influenced by heart rate. The RR interval preceding the QT interval should be measured for rate correction. Several formulae have been proposed for heart rate correction of the QT interval. The most commonly used formulae are Fridericia's cube root formula (QTc = QT/RR1/3) and Bazett's square root formula (QTc = QT/RR1/2). Although there is no consensus on best QTc method, Bazett's formula is considered the gold standard, even though it may overestimate QT prolongation [5]. In general, QT prolongation is considered when the QTc interval is greater than 440 ms, but arrhythmias are most often associated with values of 500 ms or more. QTc interval is longer in adult women because of a relative shortening of the QTc interval in men during adolescence [6]. Intervals of 440 to 460 milliseconds in men and 440 to 470 milliseconds in women are considered borderline [7]. QT intervals may also vary due to ECG acquisition technique, electrolyte imbalance, sympathovagal activity, intra- and interobserver variability, and diurnal variation which can be up to 75–100 ms [8, 9]. It is important to notice that for every individual there is a different relation between the QT interval and the heart rate, and even though rate-correction formulae are useful clinically, they may not be accurate enough, especially when assessing the minor changes of the QT interval induced by drugs.
3. Mechanisms of Drug-Induced QT Prolongation
QT interval on the surface electrocardiogram represents the summation of action potentials in ventricular myocytes. QT prolongation entails action potential prolongation, that results from an increase in inward current (e.g., through sodium or calcium channels) or a decrease in outward current (e.g., through potassium channels). Myocardial repolarization is primarily mediated by efflux of potassium ions. Two subtypes of the delayed rectifier potassium current, IKr (rapid) and IKs (slow), are predominantly responsible for repolarization. The two currents have different activation, inactivation, and deactivation characteristics, different sensitivities to blocking drugs [10–12], different rate, and catecholamine sensitivity [13, 14] and were later found to be the result of expression of different genes [15, 16].
The hallmark mechanism of acquired LQTS and TdP is the blockade of IKr by specific drugs [17]. IKr current proteins are encoded by the human ether-a-go-go-related gene HERG (now termed KCNH2) [18]. Two structural characteristics account for the unusual susceptibility of IKr channels to various drugs [19]. First, the presence of aromatic aminoacids (Tyr652 and Phe656) with side chains directed towards the large central cavity of the pore region provides high-affinity binding sites for many of compounds. These amino acids are not present in most other potassium channels, and mutation of KCNH2 at these sites to other amino acids reduces binding affinity of several drugs. Second, while most potassium channels contain two proline residues in the helix that forms part of the pore that restrict access to the drug binding site, these two prolines are absent in KCNH2. Mutation of these residues to the conserved Pro-Val-Pro results in reduced drug binding [20].
Ikr blockade causes a delay in phase 3 rapid repolarization of the action potential (Figure 2), which is reflected by QT prolongation. Prolonged repolarization can cause early afterdepolarizations (EADs) due to activation of inward depolarizing currents (L-type calcium channels or sodium-calcium exchange current) [21], that appear as depolarizing oscillations in membrane voltage during phases 2 and 3 of the action potential (Figure 3). EADs that reach threshold voltage can cause a ventricular extrasystole preceded by a long QT interval on the surface ECG. On the other hand, dispersion of refractoriness due to heterogeneity in ventricular repolarization can create zones of unidirectional block. Repetitive extrasystoles, unidirectional block and zones of slow conduction can lead to reentry and TdP [22]. Torsades de pointes is usually preceded by a short-long-short ECG sequence (Figure 4) [23]. In this case one or more premature ventricular complexes are followed by a compensatory pause. The subsequent sinus beat may have an especially long QT and deformities of T or U waves. This sinus beat is followed by another premature ventricular complex that precipitates torsades de pointes [24].
Figure 2.
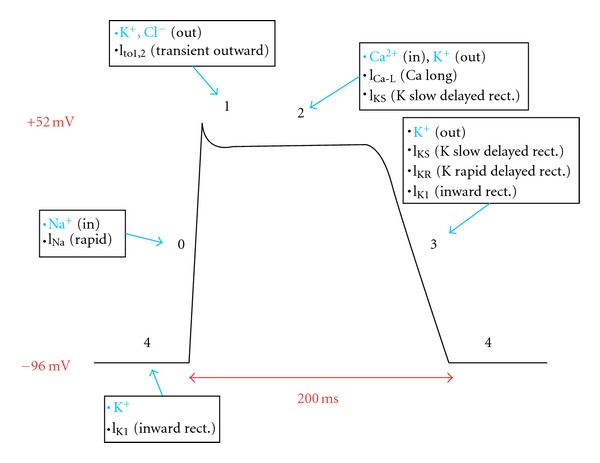
Myocardial action potential. Phase 0 rapid depolarization is mediated by sodium entry into cells. Phase 1 and 3 repolarization results from potassium efflux from cells. Balanced slow calcium entry and potassium exit cause the plateau in phase 2. Potassium reenters and sodium exits cells during phase 4 recovery.
Figure 3.
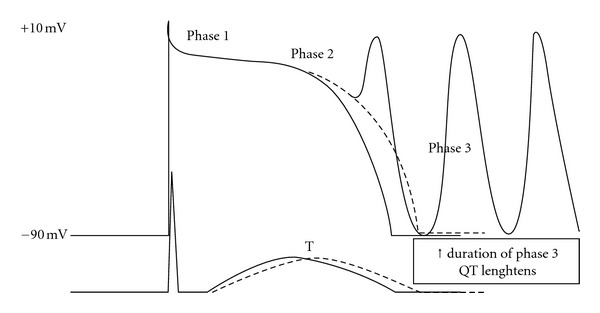
Multiple early afterdepolarizations (EADs) from progressively more negative transmembrane potential.
Figure 4.
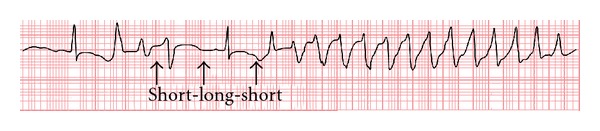
Short-long-short sequence preceding TdP.
Several other ECG variables besides QT interval have been proposed to be predictors of TdP. QT dispersion, which represents the difference between maximum and minimum QT intervals, was supposed to be a more direct measure of spatial heterogeneity of repolarization [25], but proved to be a disappointing tool, because it is mostly dependent on T wave morphology [26]. An increasing number of basic and clinical studies suggest that the interval from the peak to the end of the electrocardiographic T wave (Tp-e) corresponds to transmural dispersion of repolarization [27–29]. Prolonged QTc interval and Tpeak-Tend was found to correlate with increased risk for torsades de pointes during acquired bradyarrhythmias [30]. The Tp-e/QT ratio serves as a more sensitive index of arrhythmogenesis as it provides an estimate of dispersion of repolarization relative to total duration of repolarization. Thereby, it eliminates the confounding effects of variability of heart rate and interindividual variation of QT interval [31]. Outlined evidence clearly suggests the applicability of Tp-e/QT ratio as a potentially important index of arrhythmogenesis, even though direct validation of Tp-e interval as a body surface index of transmural dispersion of repolarization is still lacking [32]. More recent studies have also provided guidelines for the estimation of transmural dispersion of repolarization in the case of more complex T waves, including negative, biphasic, and triphasic T waves [33]. In such cases, the interval from the nadir of the first component of the T wave to the end of the T wave was shown to provide an electrocardiographic approximation of transmural dispersion of repolarization. T-wave alternans or a change in amplitude or polarity of the T-wave on alternating beats has been observed in LQTS as a precursor to TdP [34]. T-wave alternans is thought to result from alternation of the M-cell APD, leading to exaggeration of transmural dispersion of repolarization during alternate beats, and thus the potential for development of TdP [35].
Abnormal, giant T-U waves and a slow QRS upstroke separate TdP initiation in LQTS patients from PVCs in other heart disease and from other PVCs in LQTS patients. Abnormal T-U waves support the notion that EADs are the trigger for TdP in LQTS. If found, they may be an indicator for imminent risk of TdP [36]. Short-term variability of QT intervals (as measured from 30 consecutive beats) is increased in patients with a history of drug-induced long QT syndrome, suggesting that it could prove to be a useful noninvasive, easily obtainable parameter aiding the identification of the patient at risk for potentially life-threatening arrhythmia in the context of drugs with QT prolonging potential [37].
Although evaluating the effect of a new drug on the QTc interval is important, conclusions on the potential clinical risk of TdP associated with its use, based solely on its ability to prolong the QTc interval, might turn out to be highly flawed. Tpeak-Tend measurement and Tp-e/QT ratio, giant T-U waves, slow QRS upstroke, and short-term variability of QT intervals tend to be useful clinical variables to predict risk of TdP.
4. Risk Factors
Multiple clinical risk factors (Table 1) are often present in an individual case. These factors provide a starting point for basic research into underlying mechanisms at the genetic, molecular and cellular level. The occurrence of drug-induced LQTS is unpredictable in any given individual, but a common observation is that most patients have at least one identifiable risk factor in addition to drug exposure [38].
Table 1.
Risk factors for drug-induced torsade de pointes.
| Female sex |
| Hypokalemia |
| Bradycardia |
| Recent conversion from atrial fibrillation |
| Congestive heart failure |
| Left ventricular hypertrophy |
| High drug concentrations |
| Rapid rate of intravenous infusion with a QT-prolonging drug |
| Base-line QT prolongation |
| Subclinical long QT syndrome |
| Ion-channel polymorphisms |
| Severe hypomagnesemia |
A female preponderance has been consistently observed in multiple studies, with TdP occurring two to three times more commonly in women than in men [39]. These clinical observations, coupled with the finding that the QT shortens after puberty in males but not female [40], suggest that sex hormones modulate repolarization. Testosterone, by increasing IKr, shortens QTc and has been implicated as the major factor lowering risk of TdP in males [41].
Hypokalemia is another common risk factor in drug-induced LQTS. Low extracellular potassium paradoxically reduces IKr by enhanced inactivation [42] or exaggerated competitive block by sodium [43]. As a result, hypokalemia prolongs the QT interval. However, the fact that low extracellular potassium increases IKr blockade by drug, is of most importance in clinical practice [44]. Correction of extracellular potassium to the high normal range can shorten QT interval and associated morphological abnormalities [45, 46].
Pauses, usually after an ectopic beat, precipitate drug-induced TdP. It is presumed that pause generates the dispersion of many electrophysiological properties, notably repolarization times, that underlie torsades de pointes [47]. In Holter recordings of patients with drug-induced TdP an increase in underlying sinus heart rate was reported in the minutes prior to an event [48]. This finding suggests that a pause in the setting of heightened sympathetic activation and long QT intervals may be especially arrhythmogenic.
The period shortly after conversion of atrial fibrillation is characterized by increased risk of torsades de pointes. Studies using QT/RR plots during atrial fibrillation have shown rate-independent QT prolongation after conversion to sinus rhythm [49]. Dofetilide causes only minor QT prolongation during atrial fibrillation, but significantly more QT prolongation when given to the same patients after cardioversion to sinus rhythm [50]. Congestive heart failure [51] and left ventricular hypertrophy are other high-risk situations for drug-induced torsades de pointes, but further investigation is needed on molecular and cellular mechanisms.
For the majority of drugs (with the exception of class IA drugs), risk increases with higher drug concentrations. Class IA drugs (quinidine, disopyramide, and procainamide) block outward potassium currents and inward sodium currents. Sodium current blockade increases as serum levels increase, but potassium current blockade predominates at low serum levels. Therefore, TdP frequently occurs at low or subtherapeutic serum levels [52]. Administration of more than one drug that prolong repolarization increases the risk of drug-induced LQTS, but in most cases the mechanism of increased risk is due to drug-drug interactions altering metabolism, rather than simple additive effects on IKr. Cytochrome P450 superfamily of proteins is responsible for the metabolism of most of the drugs by liver and CYP3A4 is the predominant cytochrome P450. Coadministration of drugs which are substrates for CYP3A4 and/or IKr blockers results in further QT prolongation. Drugs that prolong QT interval and inhibitors of CYP3A4 are shown in Tables 2 and 3. Amiodarone rarely causes torsades de pointes despite significant QT prolongation. Amiodarone blocks IKr without reverse use dependence and prolongs action potential duration in a homogenous manner, thus reducing heterogeneity of refractoriness and making myocardium less susceptible to reentry. Additional electrophysiologic effects that explain its safety include noncompetitive β antagonism and inward L-calcium channel blockade which may reduce EADs [53]. The incidence of torsades de pointes at currently used doses is <1% [54, 55] while with sotalol the incidence ranges from 0.8 to 3.8% [56] and with ibutilide from 3.6 to 8.3% [57, 58]. Verapamil, even though a potent IKr blocker, rarely causes torsades de pointes [59]. Verapamil reduces EADs by blocking inward calcium current [60], reduces transmural dispersion of refractoriness, and shortens QT interval and the incidence of TdP in a model of acquired LQTS from combined IKs and IKr block [61].
Table 2.
Drugs implicated in torsades de pointes.
| (1) Antiarrhythmic medications |
| Class IA (Quinidine, Procainamide, and Disopyramide) |
| Class III (Dofetilide, Ibutilide, Sotalol, and Amiodarone) |
| Class IV (Verapamil) |
| (2) Promotility medications |
| Cisapride |
| (3) Antimicrobial medications |
| Macrolides |
| Erythromycin |
| Clarithromycin |
| Fluoroquinolones |
| Antiprotozoals |
| Pentamidine |
| Antimalarials |
| Chloroquine |
| (4) Antipsychotic medications |
| Phenothiazine neuroleptics |
| Thioridazine |
| Chlorpromazine |
| Butyrophenone neuroleptics |
| Haloperidol |
| (5) Miscellaneous medications |
| Arsenic trioxide |
| Methadone |
Table 3.
Inhibitors of CYP3A4.
| (1) Antihypertensive medications |
| Dihydralazine |
| Diltiazem |
| Verapamil |
| (2) Antidepressant and anxiolytic medications |
| Fluoxetine |
| Midazolam |
| (3) Antimicrobial medications |
| Macrolides |
| Clarithromycin |
| Erythromycin |
| Isoniazid |
| HIV agents |
| (4) Endocrine medications |
| Contraceptives |
| Ethinylestradiol |
| Antiprogesterone agent |
| Estrogen receptor modulators |
| Tamoxifen |
| (5) Food and herbal constituents |
| Bergamottin (grapefruit juice) |
| Glabridin (licorice) |
Subclinical mutations or polymorphisms in congenital LQTS genes have been described as a risk factor for the drug-associated form [38]. Patients with subclinical congenital LQTS may develop TdP after exposure to a QT prolonging agent [62]. In approximately hundred patients with drug-induced form of LQTS, congenital LQTS disease genes were identified in a percentage of 5–10% and their mutations classified them as having the congenital syndrome [63]. Identification of these cases emphasizes the increasing recognition of incomplete penetrance. As a result, many patients with the congenital syndrome have normal baseline ECGs, but they may be at increased risk for torsades during drug challenge [64, 65]. First-degree relatives of patients with drug-induced TdP exhibit greater abnormalities of cardiac repolarization in comparison with first-degree relatives of patients tolerating QT-prolonging antiarrhythmic therapy [66].
This variability in the risk of torsade de pointes involves defining the molecular mechanisms that control the duration of action potential and the QT interval in the normal heart and in diseases such as the congenital long QT syndrome or heart failure. The term repolarization reserve has been proposed as a unifying framework for analysis of risk factors and their clinical mechanisms [67]. Repolarization reserve characterizes the capacity of the myocardium to affect orderly and provide rapid repolarization through normal mechanisms. In normal hearts, repolarization is accomplished by multiple and redundant mechanisms. The presence of a single risk factor is usually insufficient to elicit a LQTS phenotype. Multiple subclinical lesions are necessary in the repolarization process before superimposition of an IKr-blocking drug can produce marked action potential prolongation and torsades de pointes.
Genetic modulation of repolarization reserve is thought to contribute to interindividual differences in susceptibility to QT-prolonging drugs. Genetic variations in KCNH2, KCNE, KCNE2, and ANKB have been identified in some patients with drug-induced torsades de pointes [68, 69]. The gene in which mutations seem most common is KCNQ1, whose expression results in IKs [62, 63]. Studies in human myocytes and in computational models have implicated variability in IKs amplitude as a major contributor to variability in response to IKr block (i.e., to repolarization reserve) [70, 71]. IKs amplitude is readily increased by interventions such as adrenergic stimulation [72] or endothelin [73], but also by compensatory increase through posttranscriptional upregulation of underlying units of IKs, likely mediated by microRNA changes due to sustained reductions of IKr [74]. The way in which the above-mentioned and other mechanisms might contribute to IKs regulation during challenge with an IKr blocker remains a broad area for investigation, both at the clinical level and at the molecular level.
Current studies investigate the relationship of common variants within the human genome termed polymorphisms and variable risk for torsades de pointes. The most compelling example to date is a single nucleotide polymorphism (SNP), common in African Americans, that results in substitution of a tyrosine for serine at position 1103 of the cardiac sodium channel. The SCN5A variant S1103Y points to the potential role of ethnicity as a genetic determinant of repolarization reserve [75]. This is further confirmed by the unique distribution of certain ion channel variants across different ethnic groups [76–78]. Pharmacogenetics additionally may determine arrhythmia risk in patients with acquired LQTS. Genetically determined reduced activity of cytochrome P450 enzyme CYP3A4 may decrease efficient metabolism of the QT prolonging drugs thioridazine, erythromycin, and terfenadine [79].
5. Treatment
The cornerstone of the management of acquired LQTS includes the identification and discontinuation of any precipitating drug and the aggressive correction of any metabolic abnormalities, such as hypokalemia or hypomagnesemia. Most of the episodes of torsade de pointes are short-lived and terminate spontaneously. However, prolonged episodes result in hemodynamic compromise and require immediate cardioversion.
Short-term treatment of the syndrome focuses on prevention of recurrence of torsade de pointes and includes administration of intravenous magnesium sulfate and temporary transvenous cardiac pacing. Intravenous isoproterenol is rarely needed. Important step in the management of acquired LQTS is withdrawal of offending agents and correction of electrolyte abnormalities [80]. The effectiveness of lidocaine, phenytoin, or atropine even though reported to be beneficial is uncertain [81].
Intravenous magnesium is the agent of choice for immediate treatment of torsade de pointes irrespective of the serum magnesium level. 2 g bolus of magnesium sulfate is followed by intravenous infusion of magnesium at a rate of 2–4 mg per minute [82]. The mechanism by which magnesium prevents the recurrences of torsade de points is unclear. Its action is probably mediated through blockage of sodium or calcium currents. The only side effect of intravenous magnesium is flushing during the bolus injection. Administration of potassium is an important adjunct to intravenous magnesium for the short-term prevention of torsade de pointes, especially if the serum potassium level is low. Serum potassium should be maintained in the high normal range. Overdrive transvenous pacing shortens QTc and is highly effective in preventing recurrences of torsades de pointes [83], especially when they are precipitated by a pause or bradycardia. Short-term pacing rates of 90 to 110 beats/min are recommended. Cardiac pacing prevents pauses and shortens the QTc interval by enhancing the repolarizing potassium currents [84]. Isoproterenol is useful if temporary pacing is unavailable or while preparing for transvenous catheter insertion [85]. Unlike acquired LQTS, isoproterenol is contraindicated in patients with congenital LQTS or ischemic heart disease. Side effects include palpitations and flushes.
Long-term treatment is rarely required. Conditions that predispose to electrolyte imbalance must be corrected. In cases of sick sinus syndrome or atrioventricular block and bradycardia, permanent pacing may be indicated [86].
References
- 1.Selzer A, Wray HW. Quinidine syncope. Paroxysmal ventricular fibrillation occurring during treatment of chronic atrial arrhythmias. Circulation. 1964;30:17–26. doi: 10.1161/01.cir.30.1.17. [DOI] [PubMed] [Google Scholar]
- 2.Dessertenne F. Ventricular tachycardia with 2 variable opposing foci. Archives des Maladies du Coeur et des Vaisseaux. 1966;59(2):263–272. [PubMed] [Google Scholar]
- 3.Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM, Bor DH. Timing of new black box warnings and withdrawals for prescription medications. Journal of the American Medical Association. 2002;287(17):2215–2220. doi: 10.1001/jama.287.17.2215. [DOI] [PubMed] [Google Scholar]
- 4.Sadanaga T, Sadanaga F, Yao H, Fujishima M. An evaluation of ECG leads used to assess QT prolongation. Cardiology. 2006;105(3):149–154. doi: 10.1159/000091227. [DOI] [PubMed] [Google Scholar]
- 5.Indik JH, Pearson EC, Fried K, Woosley RL. Bazett and Fridericia QT correction formulas interfere with measurement of drug-induced changes in QT interval. Heart Rhythm. 2006;3(9):1003–1007. doi: 10.1016/j.hrthm.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 6.Reardon M, Malik M. QT interval change with age in an overtly healthy older population. Clinical Cardiology. 1996;19(12):949–952. doi: 10.1002/clc.4960191209. [DOI] [PubMed] [Google Scholar]
- 7.Surawicz B, Knilans T, Chou T-C. Chou’s Electrocardiography in Clinical Practice. Philadelphia, Pa, USA: W.B. Saunders Company; 2001. [Google Scholar]
- 8.Morganroth J, Brozovich FV, McDonald JT, Jacobs RA. Variability of the QT measurement in healthy men, with implications for selection of an abnormal QT value to predict drug toxicity and proarrhythmia. American Journal of Cardiology. 1991;67(8):774–776. doi: 10.1016/0002-9149(91)90541-r. [DOI] [PubMed] [Google Scholar]
- 9.Molnar J, Zhang F, Weiss J, Ehlert FA, Rosenthal JE. Diurnal pattern of QTc interval: how long is prolonged? Possible relation to circadian triggers of cardiovascular events. Journal of the American College of Cardiology. 1996;27(1):76–83. doi: 10.1016/0735-1097(95)00426-2. [DOI] [PubMed] [Google Scholar]
- 10.Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current: differential sensitivity to block by class III antiarrhythmic agent. Journal of General Physiology. 1990;96(1):195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanguinetti MC, Jurkiewicz NK. Delayed rectifier outward K+ current is composed of two currents in guinea pig atrial cells. American Journal of Physiology—Heart and Circulatory Physiology. 1991;260(2):H393–H399. doi: 10.1152/ajpheart.1991.260.2.H393. [DOI] [PubMed] [Google Scholar]
- 12.Gintant GA. Two components of delayed rectifier current in canine atrium and ventricle: does IKs play a role in the reverse rate dependence of class III agents? Circulation Research. 1996;78(1):26–37. doi: 10.1161/01.res.78.1.26. [DOI] [PubMed] [Google Scholar]
- 13.Sanguinetti MC, Kurkiewicz NK, Scott A, Siegl PKS. Isoproterenol antagonizes prolongation of refractory period by the class III antiarrhythmic agent E-4031 in guinea pig myocytes: mechanism of action. Circulation Research. 1991;68(1):77–84. doi: 10.1161/01.res.68.1.77. [DOI] [PubMed] [Google Scholar]
- 14.Jurkiewicz NK, Sanguinetti MC. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent: Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circulation Research. 1993;72(1):75–83. doi: 10.1161/01.res.72.1.75. [DOI] [PubMed] [Google Scholar]
- 15.Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 16.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and IsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 17.Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. The Journal of Clinical Investigation. 2005;115:2025–2032. doi: 10.1172/JCI25539. A comprehensive review of molecular and cellular mechanisms underlying acquired LQTS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 19.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. Expert review of the role of KCNH2 in arrhythmias, in both congenital and drug induced LQTS. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. Physicochemical Features of the hERG Channel Drug Binding Site. Journal of Biological Chemistry. 2004;279(11):10120–10127. doi: 10.1074/jbc.M310683200. [DOI] [PubMed] [Google Scholar]
- 21.Roden DM, Lazzara R, Rosen M, Schwartz PJ, Towbin J, Michael Vincent G. Multiple mechanisms in the long-QT syndrome: current knowledge, gaps, and future directions. Circulation. 1996;94(8):1996–2012. doi: 10.1161/01.cir.94.8.1996. [DOI] [PubMed] [Google Scholar]
- 22.Antzelevitch C. Role of transmural dispersion of repolarization in the genesis of drug-induced torsades de pointes. Heart Rhythm. 2005;2(11):S9–S15. doi: 10.1016/j.hrthm.2004.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89(11):1363–1372. doi: 10.1136/heart.89.11.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swerdlow CD, Martin DJ, Kass RM, et al. The zone of vulnerability to T wave shocks in humans. Journal of Cardiovascular Electrophysiology. 1997;8(2):145–154. doi: 10.1111/j.1540-8167.1997.tb00776.x. [DOI] [PubMed] [Google Scholar]
- 25.Hii JTY, Wyse DG, Gillis AM, Duff HJ, Solylo MA, Mitchell LB. Precordial QT interval dispersion as a marker of torsade de pointes: disparate effects of class Ia antiarrhythmic drugs and amiodarone. Circulation. 1992;86(5):1376–1382. doi: 10.1161/01.cir.86.5.1376. [DOI] [PubMed] [Google Scholar]
- 26.Shah RR. Drug-induced QT dispersion: does it predict the risk of torsade de pointes? Journal of Electrocardiology. 2005;38(1):10–18. doi: 10.1016/j.jelectrocard.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Antzelevitch C. Tpeak-Tend interval as an index of transmural dispersion of repolarization. European Journal of Clinical Investigation. 2001;31(7):555–557. doi: 10.1046/j.1365-2362.2001.00849.x. [DOI] [PubMed] [Google Scholar]
- 28.Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. American Journal of Physiology—Heart and Circulatory Physiology. 2007;293(4):H2024–H2038. doi: 10.1152/ajpheart.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm. 2007;4(7):964–972. doi: 10.1016/j.hrthm.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topilski I, Rogowski O, Rosso R, et al. The morphology of the QT interval predicts torsade de pointes during acquired bradyarrhythmias. Journal of the American College of Cardiology. 2007;49(3):320–328. doi: 10.1016/j.jacc.2006.08.058. [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi M, Shimizu M, Ino H, et al. T wave peak-to-end interval and QT dispersion in acquired long QT syndrome: a new index for arrhythmogenicity. Clinical Science. 2003;105(6):671–676. doi: 10.1042/CS20030010. [DOI] [PubMed] [Google Scholar]
- 32.Xia Y, Liang Y, Kongstad O, et al. In vivo validation of the coincidence of the peak and end of the T wave with full repolarization of the epicardium and endocardium in swine. Heart Rhythm. 2005;2(2):162–169. doi: 10.1016/j.hrthm.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Emori T, Antzelevitch C. Cellular basis for complex T waves and arrhythmic activity following combined IKr and IKs block. Journal of Cardiovascular Electrophysiology. 2001;12(12):1369–1378. doi: 10.1046/j.1540-8167.2001.01369.x. [DOI] [PubMed] [Google Scholar]
- 34.Grabowski M, Karpinski G, Filipiak KJ, Opolski G. Images in cardiovascular medicine. Drug-induced long-QT syndrome with macroscopic T-wave alternans. Circulation. 2004;110(18):459–460. doi: 10.1161/01.CIR.0000147541.49575.15. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu W, Antzelevitch C. Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation. 1999;99(11):1499–1507. doi: 10.1161/01.cir.99.11.1499. [DOI] [PubMed] [Google Scholar]
- 36.Kirchhof P, Franz MR, Bardai A, Wilde AM. Giant T-U waves precede torsades de pointes in long QT syndrome. A systematic electrocardiographic analysis in patients with acquired and congenital QT prolongation. Journal of the American College of Cardiology. 2009;54(2):143–149. doi: 10.1016/j.jacc.2009.03.043. [DOI] [PubMed] [Google Scholar]
- 37.Hinterseer M, Thomsen MB, Beckmann BM, et al. Beat-to-beat variability of QT intervals is increased in patients with drug-induced long-QT syndrome: a case control pilot study. European Heart Journal. 2008;29(2):185–190. doi: 10.1093/eurheartj/ehm586. [DOI] [PubMed] [Google Scholar]
- 38.Roden DM, Woosley RL, Primm RK. Incidence and clinical feature of the quinidine-associated long QT syndrome: implications for patient care. American Heart Journal. 1986;111(6):1088–1093. doi: 10.1016/0002-8703(86)90010-4. [DOI] [PubMed] [Google Scholar]
- 39.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. Journal of the American Medical Association. 1993;270(21):2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 40.Rautaharju P, Zhou SH, Wong S, et al. Sex differences in the evolution of the electrocardiographic QT interval with age. Canadian Journal of Cardiology. 1992;8(7):690–695. [PubMed] [Google Scholar]
- 41.Arya A. Gender-related differences in ventricular repolarization: beyond gonadal steroids. Journal of Cardiovascular Electrophysiology. 2005;16(5):525–527. doi: 10.1111/j.1540-8167.2005.40845.x. [DOI] [PubMed] [Google Scholar]
- 42.Yang T, Snyders DJ, Roden DM. Rapid inactivation determines the rectification and [K+](o) dependence of the rapid component of the delayed rectifier K+ current in cardiac cells. Circulation Research. 1997;80(6):782–789. doi: 10.1161/01.res.80.6.782. [DOI] [PubMed] [Google Scholar]
- 43.Numaguchi H, Johnson JP, Jr., Petersen CI, Balser JR. A sensitive mechanism for cation modulation of potassium current. Nature Neuroscience. 2000;3(5):429–430. doi: 10.1038/74793. [DOI] [PubMed] [Google Scholar]
- 44.Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr: implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93(3):407–411. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]
- 45.Choy AM, Lang CC, Chomsky DM, Rayos GH, Wilson JR, Roden DM. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation. 1997;96(7):2149–2154. doi: 10.1161/01.cir.96.7.2149. [DOI] [PubMed] [Google Scholar]
- 46.Etheridge SP, Compton SJ, Tristani-Firouzi M, Mason JW. A new oral therapy for long QT syndrome: long-term oral potassium improves repolarization in patients with HERG mutations. Journal of the American College of Cardiology. 2003;42(10):1777–1782. doi: 10.1016/j.jacc.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 47.Roden DM, Anderson ME. The pause that refreshes, or does it? Mechanisms in torsades de pointes. Heart. 2000;84(3):235–237. doi: 10.1136/heart.84.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Locati EH, Maison-Blanche P, Dejode P, Cauchemez B, Coumel P. Spontaneous sequences of onset of torsade de pointes in patients with acquired prolonged repolarization: quantitative analysis of Holter recordings. Journal of the American College of Cardiology. 1995;25(7):1564–1575. doi: 10.1016/0735-1097(95)00100-i. [DOI] [PubMed] [Google Scholar]
- 49.Darbar D, Harris P, Hardy A, et al. Marked steepening of QT restitution following cardioversion of atrial fibrillation. Heart Rhythm. 2004;1:p. S192. [Google Scholar]
- 50.Choy AMJ, Darbar D, Dell’Orto S, Roden DM. Exaggerated QT prolongation after cardioversion of atrial fibrillation. Journal of the American College of Cardiology. 1999;34(2):396–401. doi: 10.1016/s0735-1097(99)00226-0. [DOI] [PubMed] [Google Scholar]
- 51.Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. The New England Journal of Medicine. 1999;341(12):857–865. doi: 10.1056/NEJM199909163411201. [DOI] [PubMed] [Google Scholar]
- 52.Wyse KR, Ye V, Campbell TJ. Action potential prolongation exhibits simple dose-dependence for sotalol, but reverse dose-dependence for quinidine and disopyramide: implications for proarrhythmia due to triggered activity. Journal of Cardiovascular Pharmacology. 1993;21(2):316–322. doi: 10.1097/00005344-199302000-00019. [DOI] [PubMed] [Google Scholar]
- 53.Drouin E, Lande G, Charpentier F. Amiodarone reduces transmural heterogeneity of repolarization in the human heart. Journal of the American College of Cardiology. 1998;32(4):1063–1067. doi: 10.1016/s0735-1097(98)00330-1. [DOI] [PubMed] [Google Scholar]
- 54.Hohnloser SH, Klingenheben T, Singh BN. Amiodarone-associated proarrhythmic effects. A review with special reference to torsade de pointes tachycardia. Annals of Internal Medicine. 1994;121(7):529–535. doi: 10.7326/0003-4819-121-7-199410010-00009. [DOI] [PubMed] [Google Scholar]
- 55.Vorperian VR, Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a meta-analysis. Journal of the American College of Cardiology. 1997;30(3):791–801. doi: 10.1016/s0735-1097(97)00220-9. [DOI] [PubMed] [Google Scholar]
- 56.Lazzara R. Antiarrhythmic drugs and torsade de pointes. European Heart Journal. 1993;14:88–92. doi: 10.1093/eurheartj/14.suppl_h.88. [DOI] [PubMed] [Google Scholar]
- 57.Ellenbogen KA, Stambler BS, Wood MA. Efficacy of intravenous inbutilide for rapid termination of atrial fibrillation and atrial flutter: a dose-response study. Journal of the American College of Cardiology. 1996;28(4):130–136. doi: 10.1016/0735-1097(96)00121-0. [DOI] [PubMed] [Google Scholar]
- 58.Stambler BS, Wood MA, Ellenbogen KA, Perry KT, Wakefield LK, VanderLugt JT. Efficacy and safety of repeated intravenous doses of ibutilide for rapid conversion of atrial flutter or fibrillation. Ibutilide Repeat Dose Study Investigators. Circulation. 1996;94(7):1613–1621. doi: 10.1161/01.cir.94.7.1613. [DOI] [PubMed] [Google Scholar]
- 59.Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circulation Research. 1999;84(9):989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]
- 60.Shimizu W, Ohe T, Kurita T, et al. Effects of verapamil and propranolol on early afterdepolarizations and ventricular arrhythmias induced by epinephrine in congenital long QT syndrome. Journal of the American College of Cardiology. 1995;26(5):1299–1309. doi: 10.1016/0735-1097(95)00313-4. [DOI] [PubMed] [Google Scholar]
- 61.Aiba T, Shimizu W, Inagaki M, et al. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. Journal of the American College of Cardiology. 2005;45(2):300–307. doi: 10.1016/j.jacc.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 62.Napolitano C, Schwartz PJ, Brown AM, et al. Evidence for a cardiac ion channel mutation underlying drug-induced QT prolongation and life-threatening arrhythmias. Journal of Cardiovascular Electrophysiology. 2000;11(6):691–696. doi: 10.1111/j.1540-8167.2000.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 63.Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105(16):1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 64.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome clinical impact. Circulation. 1999;99(4):529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 65.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. The New England Journal of Medicine. 2003;348(19):1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 66.Kannankeril PJ, Roden DM, Norris KJ, Whalen SP, George AL, Murray KT. Genetic susceptibility to acquired long QT syndrome: pharmacologic challenge in first-degree relatives. Heart Rhythm. 2005;2(2):134–140. doi: 10.1016/j.hrthm.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 67.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing and Clinical Electrophysiology. 1998;21(5):1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 68.Paulussen ADC, Gilissen RAHJ, Armstrong M, et al. Genetic variations of HCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. Journal of Molecular Medicine. 2004;82(3):182–188. doi: 10.1007/s00109-003-0522-z. [DOI] [PubMed] [Google Scholar]
- 69.Mank-Seymour AR, Richmond JL, Wood LS, et al. Association of torsades de pointes with novel and known single nucleotide polymorphisms in long QT syndrome genes. American Heart Journal. 2006;152(6):1116–1122. doi: 10.1016/j.ahj.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 70.Jost N, Virag L, Bitay M, et al. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation. 2005;112:1392–1399. doi: 10.1161/CIRCULATIONAHA.105.550111. [DOI] [PubMed] [Google Scholar]
- 71.Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112(10):1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanguinetti MC, Kurkiewicz NK, Scott A, Siegl PKS. Isoproterenol antagonizes prolongation of refractory period by the class III antiarrhythmic agent E-4031 in guinea pig myocytes: mechanism of action. Circulation Research. 1991;68(1):77–84. doi: 10.1161/01.res.68.1.77. [DOI] [PubMed] [Google Scholar]
- 73.Habuchi Y, Tanaka H, Furukawa T, Tsujimura Y, Takahashi H, Yoshimura M. Endothelin enhances delayed potassium current via phospholipase C in guinea pig ventricular myocytes. American Journal of Physiology—Heart and Circulatory Physiology. 1992;262(2):345–354. doi: 10.1152/ajpheart.1992.262.2.H345. [DOI] [PubMed] [Google Scholar]
- 74.Xiao L, Xiao J, Luo X, Lin H, Wang Z, Nattel S. Feedback remodeling of cardiac potassium current expression: a novel potential mechanism for control of repolarization reserve. Circulation. 2008;118(10):983–992. doi: 10.1161/CIRCULATIONAHA.107.758672. [DOI] [PubMed] [Google Scholar]
- 75.Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297(5585):1333–1336. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 76.Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clinic Proceedings. 2003;78(12):1479–1487. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- 77.Koo SH, Ho WF, Lee EJD. Genetic polymorphisms in KCNQ1, HERG, KCNE1 and KCNE2 genes in the Chinese, Malay and Indian populations of Singapore. British Journal of Clinical Pharmacology. 2006;61(3):301–308. doi: 10.1111/j.1365-2125.2005.02545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bezzina CR, Shimizu W, Yang P, et al. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation. 2006;113(3):338–344. doi: 10.1161/CIRCULATIONAHA.105.580811. [DOI] [PubMed] [Google Scholar]
- 79.Fitzgerald PT, Ackerman MJ. Drug-induced torsades de pointes: the evolving role of pharmacogenetics. Heart Rhythm. 2005;2(11):S30–S37. doi: 10.1016/j.hrthm.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 80.Nguyen PT, Scheinman MM, Seger J. Polymorphous ventricular tachycardia: clinical characterization, therapy, and the QT interval. Circulation. 1986;74(2):340–349. doi: 10.1161/01.cir.74.2.340. [DOI] [PubMed] [Google Scholar]
- 81.Vukmir RB, Stein KL. Torsades de pointes therapy with phenytoin. Annals of Emergency Medicine. 1991;20(2):198–200. doi: 10.1016/s0196-0644(05)81223-5. [DOI] [PubMed] [Google Scholar]
- 82.Banai S, Tzivoni D. Drug therapy for torsade de pointes. Journal of Cardiovascular Electrophysiology. 1993;4(2):206–210. doi: 10.1111/j.1540-8167.1993.tb01224.x. [DOI] [PubMed] [Google Scholar]
- 83.Khan IA. Clinical and therapeutic aspects of congenital and acquired long QT syndrome. American Journal of Medicine. 2002;112(1):58–66. doi: 10.1016/s0002-9343(01)01011-7. [DOI] [PubMed] [Google Scholar]
- 84.Damiano BP, Rosen MR. Effects of pacing on triggered activity induced by early afterdepolarizations. Circulation. 1984;69(5):1013–1025. doi: 10.1161/01.cir.69.5.1013. [DOI] [PubMed] [Google Scholar]
- 85.Viskin S. Torsades de pointes. Current Treatment Options in Cardiovascular Medicine. 1999;1:187–195. doi: 10.1007/s11936-999-0022-8. [DOI] [PubMed] [Google Scholar]
- 86.Viskin S. Cardiac pacing in the long QT syndrome: review of available data and practical recommendations. Journal of Cardiovascular Electrophysiology. 2000;11(5):593–600. doi: 10.1111/j.1540-8167.2000.tb00015.x. [DOI] [PubMed] [Google Scholar]