

Abstract
Pulmonary toxicity induced by vesicants is associated with oxidative stress. In the present studies we analyzed the role of reactive nitrogen species (RNS) generated via inducible nitric oxide synthase (iNOS) in lung injury and inflammation induced by vesicants using 2-chloroethyl ethyl sulfide (CEES) as a model. C57Bl/6 (WT) and iNOS−/− mice were sacrificed 3 d or 14 d following intratracheal administration of CEES (6 mg/kg) or control. CEES intoxication resulted in transient (3 d) increases in bronchoalveolar lavage (BAL) cell and protein content in WT, but not iNOS−/− mice. This correlated with expression of Ym1, a marker of oxidative stress in alveolar macrophages and epithelial cells. In contrast, in iNOS−/− mice, Ym1 was only observed 14 d post exposure in enlarged alveolar macrophages, suggesting that they are alternatively activated. This is supported by findings that lung tumor necrosis factor and lipocalin Lcn2 expression, mediators involved in tissue repair were also upregulated at this time in iNOS−/− mice. Conversely, CEES-induced increases in the proinflammatory genes, monocyte chemotactic protein-1 and cyclooxygenase-2, were abrogated in iNOS−/− mice. In WT mice, CEES treatment also resulted in increases in total lung resistance and decreases in compliance in response to methacholine, effects blunted by loss of iNOS. These data demonstrate that RNS, generated via iNOS play a role in the pathogenic responses to CEES, augmenting oxidative stress and inflammation and suppressing tissue repair. Elucidating inflammatory mechanisms mediating vesicant-induced lung injury is key to the development of therapeutics to treat mustard poisoning.
Keywords: iNOS, RNS, CEES, Ym1, Lcn2, COX-2, MCP-1, lung function
Introduction
Sulfur mustard is a toxic vesicant that has been used as a chemical warfare agent (Heymann, 2004; Marshall, 1984). The lung is a major target and pulmonary toxicity is the main cause of mortality and long term morbidity following sulfur mustard exposure (Ghanei and Harandi, 2007). Both acute and chronic effects of sulfur mustard inhalation have been described, including pulmonary inflammation and disruption of the alveolar epithelial barrier, as well as bronchitis, and pulmonary fibrosis (Balali-Mood et al., 2011; Ghanei et al., 2008; Weinberger et al., 2011). Toxicity is attributed to the lipophilic nature of sulfur mustard which allows it to rapidly penetrate target tissues and cells and alkylate DNA, resulting in cytotoxicity, apoptosis and autophagy.
Accumulating evidence suggests that cytotoxic mediators released by inflammatory leukocytes play a role in the pathogenesis of lung injury induced by sulfur mustard and related vesicants (Ghabili et al., 2011; Weinberger et al., 2011). Of particular importance are reactive nitrogen species (RNS), including nitric oxide, which is generated by activated macrophages via an inducible form of the enzyme nitric oxide synthase (iNOS) (Laskin et al., 2010). Nitric oxide and its oxidation products are known to damage membrane lipids, proteins and DNA and are involved in the pathogenesis of pulmonary edema, acute lung injury, emphysema and damage to pulmonary surfactants (Ricciardolo et al., 2006; Sugiura and Ichinose, 2011).
Previous studies have demonstrated that iNOS expression is markedly upregulated in alveolar macrophages following acute exposure of animals to sulfur mustard and related vesicants (Malaviya et al., 2010; Sunil et al., 2011a; Sunil et al., 2011b). In the present studies, mice with a targeted disruption of the iNOS gene were used to analyze the role of RNS in vesicant-induced pulmonary toxicity using 2-chloro ethyl sulfide (CEES), a half mustard analog of sulfur mustard, which has been reported to induce similar pathologic effects on the lung (Sunil et al., 2011a; Weinberger et al., 2011). Our findings that iNOS−/− mice are less sensitive to CEES-induced lung inflammation, injury and oxidative stress, and that alterations in lung function are blunted, provide support for the hypothesis that RNS play a key role in the pathogenic pulmonary response to vesicants. Elucidating specific cytotoxic/pro-inflammatory mediators that contribute to the toxicity of vesicants is critical in identifying new targets for the development of effective therapeutics to treat sulfur mustard poisoning.
Materials and methods
Animals and treatments
Female specific pathogen-free C57Bl6/J wild type (WT) and iNOS−/− mice (8–10 weeks) were obtained from the Jackson Laboratories (Bar Harbor, ME). Animals were housed in filter-top, microisolation cages and maintained on food and water ad libitum. All animals received humane care in compliance with the institution’s guidelines, as outlined in the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Animals were anesthetized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg) and then placed on a titling rodent work stand (Hallowell EMC, Pittsfield, MA) in a supine position and restrained using an incisor loop. The tongue was extruded using a cotton tip applicator and the larynx visualized by a hemi-sectioned 3 mm diameter speculum attached to an operating head of an otoscope (Welch Allyn, Skaneateles Falls, NY). PBS containing 12% ethanol or CEES (Sigma-Aldrich, St. Louis, MO; 6 mg/kg diluted in ethanol) was administered via Clay Adams Intramedic PE-10 (I.D 0.76 mm, O.D. 1.22 mm) polyethylene tubing (Becton, Dickinson and Company, Franklin Lakes, NJ) attached to a 271/2 gauge hypodermic needle (0.4 × 13 mm). The tubing was advanced approximately 10 mm past the epiglottis and 0.05 ml of control or CEES instilled into the trachea. The tubing and speculum were withdrawn immediately after instillation. Animals were then removed from the work stand and maintained in a vertical position until normal respiration was observed (less than 1 min). All instillations were performed by David Reimer, D. V. M., Laboratory Animal Services, Rutgers University. Preparation and instillation of CEES, which included the use of double gloves, safety glasses and masks, were performed in a designated room under a chemical hood by personnel who followed Rutgers University Environmental Health and Safety guidelines. CEES was prepared from an 8.5 M stock solution immediately prior to instillation.
Sample collection
Animals were euthanized 3 d or 14 d after treatment by intraperitoneal injection of Nembutal (200 mg/kg). PBS (1 ml) was instilled and withdrawn into the lungs three times through a cannula in the trachea. BAL was collected by slowly withdrawing the fluid. BAL fluid was centrifuged (300 × g, 8 min), supernatants collected, aliquoted, and stored in diethyl triamine pentaacetic acid (DTPA, 5 mM) at −80°C until analysis. Cell pellets were resuspended in 1 ml PBS and viable cells (10 μl) counted on a hemocytometer using trypan blue dye exclusion. For differential analysis, cytospin preparations were fixed in methanol and stained with Giemsa (Labchem Inc., Pittsburgh, PA). A total of 300 cells were counted by light microscopy.
Measurement of BAL protein
Total protein was quantified in cell-free BAL using a BCA Protein Assay kit (Pierce Biotechnologies Inc., Rockford, IL) with bovine serum albumin as the standard. Samples (25 μl) from 6 mice per treatment group were analyzed in triplicate. Samples were evaluated at 560 nm on a Vmax MAXline™ microplate reader (Molecular Devices, Sunnyvale, CA).
Real time quantitative PCR
Total mRNA was extracted from tissue samples using an RNeasy Mini kit (Qiagen, Valencia, CA). mRNA was reverse-transcribed using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. Standard curves were generated using serial dilutions from pooled cDNA samples. Real time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on a 7900HT thermocycler. All PCR primers were generated using Primer Express 3.0 software (Applied Biosystems). Samples from 3 animals per treatment group were pooled and analyzed in triplicate. Data were normalized to GAPDH mRNA expression. Forward and reverse primer sequences were tumor necrosis factor (TNF)α: AAA TTC GAG TGA CAA GCC CTA, CCC TTG AAG AGA ACC TGG GAG TAG; monocyte chemotactic protein (MCP)-1: TTG AAT GTG AAG TTG ACC CGT AA, GCT TGA GGT TGT GGA AAA G; cyclooxygenase (COX)- 2: CAT TCT TGC CCA GCA CTT CAC, GAC CAG GCA CCA GAC CAA AGA C; and GAPDH: TGA AGC AGG CAT CTG AGG G, CGA AGG TGG AAG AGT GGG AG
Immunohistochemistry
Tissue sections were deparaffinized with xylene (4 min, × 2) followed by decreasing concentrations of ethanol (100% - 50%) and finally, water. After antigen retrieval using citrate buffer (10.2 mM sodium citrate, 0.05% Tween 20, pH 6.0) and quenching of endogenous peroxidase with 3% H2O2 for 15 min, sections were incubated for 1–4 h at room temperature with 5–100% goat serum to block nonspecific binding. This was followed by overnight incubation at 4°C with rabbit IgG or rabbit polyclonal anti-COX-2 (1:400, Abcam, Cambridge, MA), or anti-Ym1 antibodies (1:200, Stem Cell, Vancouver, Canada). Sections were then incubated with biotinylated secondary antibody (Vector Labs, Burlingame, CA) for 30 min at room temperature. Binding was visualized using a Peroxidase Substrate Kit DAB (Vector Labs). Random sections from three mice per treatment group were analyzed for each antibody.
Western blot analysis
BAL protein samples (2 μg) were fractionated on 4–15% Tris-glycine gels (Bio Rad, Hercules, CA) and transferred to polyvinylidene fluoride membranes (PVDF, 0.45 μm pore size; Millipore, Billerica, MA). Non-specific binding was blocked by incubation of the blots for 1 h at room temperature with 5% non-fat dry milk in TBS-T (0.02 M Tris-base, 0.137 M sodium chloride, 0.1 % Tween-20) buffer. Blots were incubated overnight at 4°C with anti-Lcn-2 antibody (Abcam, Cambridge, MA, 1:100). After 4 washes in 0.1% TBS-T buffer, blots were incubated with anti-rabbit HRP-conjugated secondary antibody (1:20,000; Cell Signaling, Danvers, MA) for 1 h at room temperature. Bands were visualized using an ECL Plus Western blotting system (GE Healthcare, Piscataway, NJ).
Measurement of lung mechanics and function
Animals were anesthetized with ketamine (80 mg/kg) and xylazine (10 mg/kg). After 5 min, a tracheotomy was performed using an 18 gauge cannula and the animals attached to a SciReq flexiVent (Montreal, Canada). Baseline lung mechanics and function were assessed as previously described (Sunil et al., 2011a). Animals were then challenged intratracheally with increasing doses of methacholine (0–96 mg/ml) and measurements of lung mechanics and function repeated. Data were analyzed using flexiVent software version 5.2.
Statistical analysis
All experiments were repeated at least 3 times. Data were analyzed by two-way ANOVA followed by post hoc analysis (Bonferroni’s method for multiple comparison); a p value of <0.05 was considered statistically significant.
Results
Effects of loss of iNOS on CEES-induced lung injury, inflammation and oxidative stress
Initially we assessed the effects of loss of iNOS on BAL protein and cell number, markers of alveolar epithelial cell injury and lung inflammation (Bhalla, 1999). Exposure of WT mice to CEES resulted in transient increases in BAL protein and cell content which were observed 3 d post exposure (Fig. 1). By 14 d, protein and cell number were at control levels. In contrast, in iNOS−/− mice, CEES had no effect on BAL protein or cell number at 3 d, and levels of both markers were reduced to below control at 14 d post exposure. In both WT and iNOS−/− mice treated with control, the majority of cells (>99%) recovered in BAL were macrophages, with a small percentage (<1%) neutrophils. After administration of CEES, a transient increase in neutrophils was observed at 3 d (3–5%).
Figure 1.

Effects of loss of iNOS on BAL protein and cell number. BAL was collected 3 d or 14 d after treatment of WT or iNOS−/− mice with control (PBS) or CEES (6 mg/kg). Left: Cell-free supernatants were analyzed in triplicate for protein using a BCA protein assay kit. Right: Viable cells were enumerated by trypan blue due exclusion. Each bar is the average ± SE (n = 6 mice). *Significantly different (p ≤ 0.05) from control; #Significantly different (p ≤ 0.05) from WT.
We next analyzed the effects of CEES on lung expression of Ym1, a marker of oxidative stress and alternative macrophage activation (Gordon and Martinez, 2010; Zhang et al., 2009). Treatment of WT mice with CEES resulted in a transient increase in Ym1 expression which was observed in alveolar macrophages and epithelial cells after 3 d (Fig. 2). By 14 d, Ym1 expression was significantly reduced and only evident at low levels in alveolar macrophages. Whereas CEES administration had no effect on Ym1 expression in iNOS−/− mice 3 d post exposure, after 14 d, intense staining was evident in alveolar macrophages; Ym1 positive macrophages also appeared significantly enlarged in lungs from iNOS−/− mice relative to WT mice.
Figure 2.

Effects of loss of iNOS on CEES-induced Ym1 protein expression. Lungs sections, prepared 3 d and 14 d after treatment of WT and iNOS−/− mice with control (PBS) or CEES were stained with antibody to Ym1. Binding was visualized using a peroxidase DAB substrate kit. One representative section of the alveolar region from 3 separate experiments is shown (Original magnification, × 1000).
Effects of loss of iNOS on CEES-induced inflammatory gene expression
In further studies we analyzed expression of TNFα, MCP-1 and COX-2, inflammatory proteins implicated in pulmonary injury (Laskin et al., 2011; Weinberger et al., 2011). Treatment of WT mice with CEES had no significant effect on TNFα mRNA expression at 3 d or 14 d post exposure (Fig. 3). In contrast, in iNOS−/− mice an increase in TNFα was noted at 14 d post exposure, with no effect at 3 d. Increased expression of MCP-1 and COX-2 mRNA was also noted in WT mice 3 d post CEES exposure (Fig. 3). These effects were transient, and by 14 d levels were at or below control. COX-2 protein also increased in Type II alveolar epithelial cells 3 d after CEES administration to WT mice, a response which persisted for at least 14 d (Fig. 4). Loss of iNOS resulted in a delayed upregulation of COX-2 protein which was noted 14 d post CEES exposure (Fig. 4). This was correlated with a significant decrease in COX-2 mRNA expression at 3 d post CEES exposure in iNOS−/− mice, but no significant effect at 14 d. Loss of iNOS also blunted the effects of CEES on MCP-1 mRNA expression.
Figure 3.

Effects of loss of iNOS on CEES-induced inflammatory gene expression. Lung tissue was collected 3 d or 14 d after treatment of WT or iNOS−/− mice with control (PBS) or CEES. RNA was extracted from the tissue, pooled and analyzed in triplicate by real time PCR for TNFα, MCP-1 and COX-2 gene expression. Data were normalized to GAPDH and presented as fold change relative to control. Each bar is the average ± SE (n = 3 mice). *Significantly different (p ≤ 0.05) from control; #Significantly different (p ≤ 0.05) from WT.
Figure 4.

Effects of loss of iNOS on CEES-induced COX-2 protein expression. Lungs sections prepared 3 d and 14 d after treatment of WT and iNOS−/− mice with control (PBS) or CEES were stained with antibody to COX-2. Binding was visualized using a peroxidase DAB substrate kit. One representative section of the alveolar region from 3 separate experiments is shown (Original magnification, × 1000).
Effects of loss of iNOS on CEES-induced Lcn2 expression
Lcn2 is an acute phase protein thought to be important in restoring homeostasis after pulmonary injury (Borkham-Kamphorst et al., 2011; Playford et al., 2006; Roudkenar et al., 2008). Low levels of Lcn2 were detected in BAL from both WT and iNOS−/− mice treated with control (Fig. 5). CEES administration resulted in increased Lcn2 in both mouse strains at 3 d. Whereas in WT mice this was transient, in iNOS−/− mice, Lcn2 remained significantly elevated for at least 14 d post exposure (Fig. 5).
Figure 5.
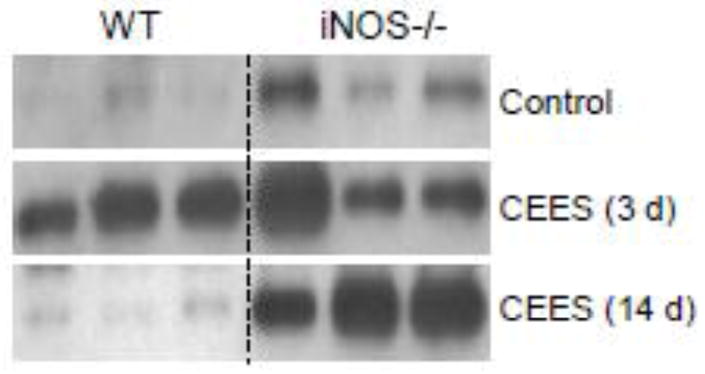
Effects of CEES on BAL Lcn2 protein levels. BAL was collected 3 d or 14 d after treatment of WT and iNOS−/− mice with control (PBS) or CEES. Samples (2 μg) were analyzed by western blotting for Lcn2 protein. Each lane represents one animal.
Effects of loss of iNOS on CEES-induced alterations in lung function
To analyze the role of RNS in CEES-induced alterations in lung function, WT and iNOS−/− mice were examined by a series of perturbations including single frequency and broadband forced oscillation, and pressure volume loops. While no major changes in baseline lung resistance or compliance were observed in either WT or iNOS−/− mice at 3 d post CEES exposure, a significant increase in central airway resistance was observed in iNOS−/− mice at this time (Table 1). Additionally, significant decreases in central airway resistance, as well as total lung compliance and static lung compliance, were detected in iNOS−/−, but not in WT mice, at 14 d post CEES exposure (Table 1).
Table 1.
Effects of CEES on baseline lung function. WT and iNOS−/− mice were exposed to control (PBS) or CEES (6 mg/kg). Lung function was assessed at 3 d and 14 d later. Each value represents the mean ± SE (n = 3–7 mice).
| WT | iNOS−/− | ||||
|---|---|---|---|---|---|
| Control | CEES | Control | CEES | ||
| Resistance | 0.99 ± 0.127 | 0.81 ± 0.016 | 0.94 ±0.094 | 1.16 ± 0.309 | 3 d |
| Central Airway Resistance | 0.36 ± 0.073 | 0.29 ± 0.071 | 0.47 ± 0.074 | 0.66± 0.148# | |
| Compliance | 0.03 ± 0.002 | 0.03± 0.003 | 0.02 ± 0.002 | 0.03± 0.009 | |
| Static Compliance | 0.07 ± 0.004 | 0.06 ± 0.011 | 0.07 ± 0.004 | 0.06 ± 0.007 | |
| Resistance | 0.97 ± 0.078 | 1.05 ± 0.225 | 0.65 ± 0.033# | 0.94± 0.178 | 14 d |
| Central Airway Resistance | 0.31±0.059 | 0.33± 0.078 | 0.33 ± 0.020 | 0.22 ± 0.038* | |
| Compliance | 0.03 ± 0.001 | 0.03± 0.003 | 0.04 ± 0.004 | 0.03 ± 0.004* | |
| Static Compliance | 0.07 ± 0.002 | 0.07 ± 0.003 | 0.09 ± 0.003 | 0.08 ± 0.005* | |
Significantly different (p ≤ 0.05) from control
Significantly different (p ≤ 0.05) from WT.
Upon challenge with methacholine, dose-related increases in lung resistance and central airway resistance were observed in WT animals, with concomitant decreases in total and static compliance (Fig. 6). Loss of iNOS increased the sensitivity of the mice to methacholine; however, this was only noted 3 d post exposure. Treatment of WT mice with CEES resulted in enhanced sensitivity to methacholine with respect to total lung resistance and compliance at 3 d post exposure, but had little effect on the responsiveness of central airway resistance and static compliance to methacholine. At 14 d post exposure the only significant effect observed in WT mice was hyper-responsiveness in central airway resistance. In iNOS−/− mice treated with CEES, resistance and compliance in response to methacholine were reduced relative to both control exposure, and to WT mice (Fig. 6). At 14 d, while compliance remained attenuated in iNOS−/− mice, central airway resistance was similar to WT animals.
Figure 6.

Effects of CEES on lung responsiveness to methacholine. Total lung resistance, central airway resistance, total lung compliance, and static compliance were evaluated in response to increasing doses of methacholine following exposure of WT and iNOS−/− mice to control (PBS) or CEES. Values were normalized and expressed as percentage change from baseline. Each point is the average ± SE (n = 6 mice). *Significantly different (p ≤ 0.05) from control.
Discussion
Nitric oxide synthases consist of a family of enzymes that catalyze the generation of nitric oxide from L-arginine (Laskin et al., 2010). Whereas nitric oxide produced via constitutive isoforms of NOS are important in cell signaling, the maintenance of vascular and airway tone, angiogenesis, peristalsis and nervous system activity, the inducible isoform is involved in inflammatory responses and has been implicated in tissue injury induced by a variety of pulmonary toxicants [reviewed in (Laskin et al., 2011)]. In the present studies, we investigated the contribution of RNS generated via iNOS to the pulmonary toxicity of CEES, a half-mustard analog of sulfur mustard, using mice lacking iNOS. In accord with previous findings with vesicants (Anderson et al., 2000; Malaviya et al., 2010), CEES was found to induce a transient increase in BAL protein and cell content in WT mice, confirming the acute toxicity of this vesicant (Sunil et al., 2011a). In contrast, in iNOS−/− mice, CEES had no effect on BAL protein or cell number 3 d post-exposure, and by 14 d, levels were below control. These data suggest that loss of iNOS stabilizes the alveolar epithelium and protects against lung injury. BAL protein and cell number were found to be increased in control animals at 14 d when compared to 3 d. The reason for this increase is not clear. Aging has been reported to cause changes in alveolar epithelial permeability (Meyer et al., 1996; Tankersley et al., 2003), and this may contribute to the effects. It is also possible that there were some delayed consequences of the intratracheal instillation procedure. This may also account for decreases in baseline lung resistance between day 3 and day 14 in iNOS−/− mice.
Oxidative stress plays a key role in vesicant-induced lung injury and is thought to be a primary event triggering the inflammatory cascade (Ghanei and Harandi, 2011; Naghii, 2002; Weinberger et al., 2011). Markers of oxidative stress, such as superoxide dismutase, glutathione, 8-hydroxyguanosine and 4-hydroxynonenal are increased in the respiratory tract after exposure of animals to vesicants including CEES (Elsayed and Omaye, 2004; Sunil et al., 2011a; Ucar et al., 2007). Ym1 is a chitinase-like protein reported to be upregulated in response to oxidative stress (Zhang et al., 2009). Following CEES administration to WT mice we observed a rapid (within 3 d) and transient increase in expression of Ym1 in alveolar macrophages and alveolar epithelial cells, which is in accord with an early induction of oxidative stress by vesicants (Anderson et al., 2000; Ghanei and Harandi, 2011; Naghii, 2002). Our finding that Ym1 was not evident in iNOS−/− mice 3 d post CEES administration indicates that RNS contribute to the oxidative stress response. At 14 d post exposure, Ym1 was expressed by alveolar macrophages in both WT and iNOS−/− mice; this was more prominent in iNOS−/− mice. Ym1 has been described as marker of anti-inflammatory/wound repair macrophages and is associated with Th2 skewing (Menzies et al., 2010; Mosser and Edwards, 2008). The fact that Ym1 expression is markedly increased in lung macrophages in iNOS−/− mice, and that these cells are significantly enlarged relative to WT mice, suggests upregulation of pulmonary repair mechanisms. This may contribute to the reduced sensitivity of iNOS−/− mice to CEES-induced pulmonary toxicity.
TNFα is a macrophage-derived cytokine reported to play a dual role in inflammatory responses to tissue injury. Thus, whereas early release of TNFα promotes inflammation, later in the pathogenic response, it contributes to antioxidant defense and wound repair (Laskin et al., 2011; Saika, 2007). TNFα has previously been reported to be upregulated in alveolar macrophages within 1 h of CEES exposure (Das et al., 2003), which is consistent with its proinflammatory effects. This was, however, transient, as the present studies showed no expression of TNFα 3 d post CEES exposure. In iNOS−/− mice, but not in WT mice, TNFα mRNA increased in the lung 14 d after CEES administration, which coincided with increased numbers of Ym1 positive repair macrophages in the tissue. We speculate that TNFα released by these cells contributes to tissue repair following vesicant exposure. This is supported by recent reports demonstrating that TNFα protects RAW264.7 macrophages against sulfur mustard-induced cytotoxicity (Allon et al., 2010). The fact that increased TNFα was only observed in CEES-treated iNOS−/− mice suggests that in WT mice, the cytotoxic activity of RNS readily overwhelms the protective effects of TNFα.
MCP-1 is a pro-inflammatory chemokine that mediates leukocyte emigration into injured tissues (Chung, 2005; Conti and DiGioacchino, 2001). Although MCP-1 primarily functions as a monocyte chemoattractant, recent evidence suggests that it also contributes to neutrophil accumulation in the lung following injury (Balamayooran et al., 2011). MCP-1 has been shown to play a role in lung pathologies which have been described clinically following vesicant exposure including acute lung injury, bronchitis and chronic obstructive pulmonary disease (COPD) (Chung, 2005; Ghanei and Harandi, 2011; Weinberger et al., 2011). MCP-1 mRNA levels were found to increase in lungs of WT mice 3 d after CEES administration. This was associated with a transient increase in neutrophils in BAL. Similar coordinate increases in BAL neutrophils and MCP-1 levels have been described in humans after sulfur mustard exposure (Emad and Emad, 2007; Ghazanfari et al., 2009), and in mice after CEES administration (Sunil et al., 2011a). Neutrophils are known to participate in both acute and chronic lung injury (Chapman et al., 2009; Chung, 2005). The fact that increases in BAL neutrophil number and lung MCP-1 expression were at control levels in iNOS−/− mice treated with CEES, provide additional support for a role of RNS in initiating vesicant-induced inflammation.
COX-2 is a key enzyme in the biosynthesis of pro-inflammatory prostaglandins and it has been shown to be involved in vesicant-induced dermal toxicity (Wormser et al., 2004). In WT mice, CEES administration resulted in a transient increase in expression of COX-2 mRNA and protein, which is similar to findings in rats treated with sulfur mustard or nitrogen mustard (Malaviya et al., 2010; Sunil et al., 2011b). At 14 d post CEES exposure, COX-2 mRNA decreased in WT mice, while protein levels remained elevated. These findings suggest that at this time, COX-2 is regulated by post translational mechanisms. The observation that COX-2 is suppressed in CEES-treated iNOS−/− mice indicates that RNS regulate early increases in prostaglandin production. Interestingly, COX-2 protein was also upregulated in iNOS−/− mice at 14 d post exposure. Evidence suggests that COX-2-derived eicosanoids such as prostaglandin J2, generated late in inflammatory responses, are important in the resolution of tissue injury (Fukunaga et al., 2005). It is possible that in the absence of RNS, COX-2 derived prostaglandins play a similar protective role in the lung after CEES administration.
Lcn2 is a member of the lipocalin superfamily which functions in regulating inflammatory responses to tissue injury and in maintaining cellular homeostasis (Bahmani et al., 2010; Borkham-Kamphorst et al., 2011). In the lung, Lcn2 is secreted by alveolar macrophages and epithelial cells in response to injury and oxidative stress (Dittrich et al., 2010; Ebrahimi et al., 2010; Sunil et al., 2007; Wu et al., 2010). The present studies show rapid increases in Lcn2 in BAL in both WT and iNOS−/− mice treated with CEES, which is consistent with acute injury and oxidative stress. Whereas this response was transient in WT mice, in iNOS−/− mice Lcn2 remained elevated for at least 14 d. Lcn2 has been reported to protect against oxidative stress by upregulating antioxidants (Bahmani et al., 2010; Roudkenar et al., 2008; Roudkenar et al., 2009). It remains to be determined if Lcn2 similarly functions to protect against CEES-induced lung injury in iNOS−/− mice.
The present studies demonstrate that CEES-induced lung inflammation and alveolar epithelial injury lead to altered lung functioning. However, in WT mice, functional changes were only observed in response to methacholine challenge, indicating that CEES has little effect on the structural integrity of the lung. Interestingly, in iNOS−/− mice, CEES intoxication resulted in increases in central airway resistance 3 d post exposure, and decreases at 14 d. It is possible that the lack of iNOS-derived nitric oxide leads to increased smooth muscle tone in the airway, resulting in increased central airway resistance. The fact that this resistance decreases at 14 d post CEES exposure, with a concomitant loss of compliance, indicates that there is some degree of parenchymal compromise that only occurs at later time points. These findings provide support for the idea that iNOS-derived RNS play a role in maintaining airway tone and in mediating parenchymal changes in the lung.
Methacholine is potent muscarinic agonist and as such, is an effective bronchoconstrictor used in animal models to examine hyper-responsiveness of the airway and to assess airway wall stiffness and parenchymal elasticity (Bates and Lauzon, 2007). As previously reported (Leme et al., 2010), total lung resistance and central airway resistance increased in both WT and iNOS−/− mice, while compliance decreased in response to methacholine challenge. CEES administration to WT mice resulted in increased total lung resistance in association with decreased lung compliance 3 d post exposure. These observations suggest that CEES-induced injury and inflammation lead to airflow obstruction and stiffening of the lungs. In contrast, only minimal changes in central airway resistance and static compliance were observed at this early time point, indicating that hyper-responsiveness occurs in the lower airways and results in a loss of parenchymal tethering. Loss of iNOS resulted in a reduced methacholine response 3 d following CEES administration demonstrating that iNOS-derived RNS are involved in the response. At 14 d post treatment, a time when much of the inflammation has resolved in WT mice, the only major functional change was a hyper-responsive central airway resistance, indicative of upper airway constriction. This response was also seen in iNOS−/− mice, but accompanied by elevated lung compliance. This observation is consistent with the apparent increase in reparative processes observed in iNOS−/− animals. Our findings demonstrate that RNS contribute to increases in airway tone and parenchymal damage induced by CEES.
In summary, the present studies demonstrate that lung injury, inflammation and oxidative stress induced by CEES are mediated in part by RNS. These findings are in accord with previous studies in rats treated with nitrogen mustard using anti-oxidants or the iNOS inhibitor aminoguanidine (Korkmaz et al., 2006; Ucar et al., 2007; Yaren et al., 2007), suggesting that it is the absence of iNOS and not differences in biological pathways and immune function that underlie reduce sensitivity of iNOS−/− mice to CEES. iNOS is also important in the pathogenesis of pulmonary diseases observed after vesicant exposure, such as asthma, acute respiratory distress syndrome and COPD (Chen, 2009; Ghanei and Harandi, 2011; Sugiura and Ichinose, 2011). These data suggest that targeting iNOS may be an efficacious approach to mitigating acute vesicant-induced pulmonary injury and long term disease pathogenesis.
Highlights.
Lung injury, inflammation and oxidative stress are induced by the model vesicant CEES
RNS generated via iNOS are important in the CEES-induced pulmonary toxicity
iNOS−/− mice are protected from CEES-induced lung toxicity and altered lung functioning
Acknowledgments
This work was supported by National Institutes of Health, grants AR055073, ES004738, ES005022, CA132624, GM03430, and HL086621. The authors wish to thank David Reimer, D.V.M., Laboratory Animal Services, Rutgers University, for performing animal instillations.
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allon N, Chapman S, Shalem Y, Brandeis R, Weissman BA, Amir A. Lipopolysaccharide induced protection against sulfur mustard cytotoxicity in RAW264.7 cells through generation of TNF-alpha. J Toxicol Sci. 2010;35:345–355. doi: 10.2131/jts.35.345. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Byers SL, Vesely KR. Treatment of sulfur mustard (HD)-induced lung injury. J Appl Toxicol. 2000;20(Suppl 1):S129–132. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat670>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Bahmani P, Halabian R, Rouhbakhsh M, Roushandeh AM, Masroori N, Ebrahimi M, Samadikuchaksaraei A, Shokrgozar MA, Roudkenar MH. Neutrophil gelatinase-associated lipocalin induces the expression of heme oxygenase-1 and superoxide dismutase 1, 2. Cell Stress Chaperones. 2010;15:395–403. doi: 10.1007/s12192-009-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balali-Mood M, Afshari R, Zojaji R, Kahrom H, Kamrani M, Attaran D, Mousavi SR, Zare GA. Delayed toxic effects of sulfur mustard on respiratory tract of Iranian veterans. Hum Exp Toxicol. 2011;30:1141–1149. doi: 10.1177/0960327110389501. [DOI] [PubMed] [Google Scholar]
- Balamayooran G, Batra S, Balamayooran T, Cai S, Jeyaseelan S. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect Immun. 2011;79:2567–2577. doi: 10.1128/IAI.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates JH, Lauzon AM. Parenchymal tethering, airway wall stiffness, and the dynamics of bronchoconstriction. J Appl Physiol. 2007;102:1912–1920. doi: 10.1152/japplphysiol.00980.2006. [DOI] [PubMed] [Google Scholar]
- Bhalla DK. Ozone-induced lung inflammation and mucosal barrier disruption: toxicology, mechanisms, and implications. J Toxicol Environ Health B Crit Rev. 1999;2:31–86. doi: 10.1080/109374099281232. [DOI] [PubMed] [Google Scholar]
- Borkham-Kamphorst E, Drews F, Weiskirchen R. Induction of lipocalin-2 expression in acute and chronic experimental liver injury moderated by pro-inflammatory cytokines interleukin-1beta through nuclear factor-kappaB activation. Liver Int. 2011;31:656–665. doi: 10.1111/j.1478-3231.2011.02495.x. [DOI] [PubMed] [Google Scholar]
- Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther. 2009;121:55–68. doi: 10.1016/j.pharmthera.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Chen HI. From neurogenic pulmonary edema to fat embolism syndrome: a brief review of experimental and clinical investigations of acute lung injury and acute respiratory distress syndrome. Chin J Physiol. 2009;52:339–344. doi: 10.4077/cjp.2009.amh036. [DOI] [PubMed] [Google Scholar]
- Chung KF. Inflammatory mediators in chronic obstructive pulmonary disease. Curr Drug Targets Inflamm Allergy. 2005;4:619–625. doi: 10.2174/156801005774912806. [DOI] [PubMed] [Google Scholar]
- Conti P, DiGioacchino M. MCP-1 and RANTES are mediators of acute and chronic inflammation. Allergy Asthma Proc. 2001;22:133–137. doi: 10.2500/108854101778148737. [DOI] [PubMed] [Google Scholar]
- Das SK, Mukherjee S, Smith MG, Chatterjee D. Prophylactic protection by N-acetylcysteine against the pulmonary injury induced by 2-chloroethyl ethyl sulfide, a mustard analogue. J Biochem Mol Toxicol. 2003;17:177–184. doi: 10.1002/jbt.10076. [DOI] [PubMed] [Google Scholar]
- Dittrich AM, Krokowski M, Meyer HA, Quarcoo D, Avagyan A, Ahrens B, Kube SM, Witzenrath M, Loddenkemper C, Cowland JB, Hamelmann E. Lipocalin2 protects against airway inflammation and hyperresponsiveness in a murine model of allergic airway disease. Clin Exp Allergy. 2010;40:1689–1700. doi: 10.1111/j.1365-2222.2010.03508.x. [DOI] [PubMed] [Google Scholar]
- Ebrahimi M, Roudkenar MH, Imani Fooladi AA, Halabian R, Ghanei M, Kondo H, Nourani MR. Discrepancy between mRNA and protein expression of neutrophil gelatinase-associated lipocalin in bronchial epithelium induced by sulfur mustard. J Biomed Biotechnol. 2010;2010:823131. doi: 10.1155/2010/823131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsayed NM, Omaye ST. Biochemical changes in mouse lung after subcutaneous injection of the sulfur mustard 2-chloroethyl 4-chlorobutyl sulfide. Toxicology. 2004;199:195–206. doi: 10.1016/j.tox.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Emad A, Emad V. Elevated levels of MCP-1, MIP-alpha and MIP-1 beta in the bronchoalveolar lavage (BAL) fluid of patients with mustard gas-induced pulmonary fibrosis. Toxicology. 2007;240:60–69. doi: 10.1016/j.tox.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174:5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- Ghabili K, Agutter PS, Ghanei M, Ansarin K, Panahi Y, Shoja MM. Sulfur mustard toxicity: history, chemistry, pharmacokinetics, and pharmacodynamics. Crit Rev Toxicol. 2011;41:384–403. doi: 10.3109/10408444.2010.541224. [DOI] [PubMed] [Google Scholar]
- Ghanei M, Adibi I, Farhat F, Aslani J. Late respiratory effects of sulfur mustard: how is the early symptoms severity involved? Chron Respir Dis. 2008;5:95–100. doi: 10.1177/1479972307087191. [DOI] [PubMed] [Google Scholar]
- Ghanei M, Harandi AA. Long term consequences from exposure to sulfur mustard: a review. Inhal Toxicol. 2007;19:451–456. doi: 10.1080/08958370601174990. [DOI] [PubMed] [Google Scholar]
- Ghanei M, Harandi AA. Molecular and cellular mechanism of lung injuries due to exposure to sulfur mustard: a review. Inhal Toxicol. 2011;23:363–371. doi: 10.3109/08958378.2011.575413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazanfari T, Yaraee R, Kariminia A, Ebtekar M, Faghihzadeh S, Vaez-Mahdavi MR, Rezaei A, Vojgani M, Soroush MR, Kermani-Jalilvand A, Mohammadi P, Foroutan A, Hassan ZM. Alterations in the serum levels of chemokines 20 years after sulfur mustard exposure: Sardasht-Iran Cohort Study. Int Immunopharmacol. 2009;9:1471–1476. doi: 10.1016/j.intimp.2009.08.022. [DOI] [PubMed] [Google Scholar]
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Heymann WR. Threats of biological and chemical warfare on civilian populations. J Am Acad Dermatol. 2004;51:452–453. doi: 10.1016/j.jaad.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Korkmaz A, Yaren H, Topal T, Oter S. Molecular targets against mustard toxicity: implication of cell surface receptors, peroxynitrite production, and PARP activation. Arch Toxicol. 2006;80:662–670. doi: 10.1007/s00204-006-0089-x. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 2011;51:267–288. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin JD, Heck DE, Laskin DL. Nitric oxide pathways in toxic responses. In: Ballantyne B, Marrs T, Syversen T, editors. General and Applied Toxicology. Wiley-Blackwell; UK: 2010. pp. 425–438. [Google Scholar]
- Leme AS, Berndt A, Williams LK, Tsaih SW, Szatkiewicz JP, Verdugo R, Paigen B, Shapiro SD. A survey of airway responsiveness in 36 inbred mouse strains facilitates gene mapping studies and identification of quantitative trait loci. Mol Genet Genomics. 2010;283:317–326. doi: 10.1007/s00438-010-0515-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Sunil VR, Cervelli J, Anderson DR, Holmes WW, Conti ML, Gordon RE, Laskin JD, Laskin DL. Inflammatory effects of inhaled sulfur mustard in rat lung. Toxicol Appl Pharmacol. 2010;248:89–99. doi: 10.1016/j.taap.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall E. Iraq’s chemical warfare: case proved: A UN team found mustard and nerve gas bombs on the battlefield; now the challenge is to prevent the war from spreading. Science. 1984;224:130–132. doi: 10.1126/science.224.4645.130. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Henriquez FL, Alexander J, Roberts CW. Sequential expression of macrophage anti-microbial/inflammatory and wound healing markers following innate, alternative and classical activation. Clin Exp Immunol. 2010;160:369–379. doi: 10.1111/j.1365-2249.2009.04086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KC, Ershler W, Rosenthal NS, Lu XG, Peterson K. Immune dysregulation in the aging human lung. Am J Respir Crit Care Med. 1996;153:1072–1079. doi: 10.1164/ajrccm.153.3.8630547. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghii MR. Sulfur mustard intoxication, oxidative stress, and antioxidants. Mil Med. 2002;167:573–575. [PubMed] [Google Scholar]
- Playford RJ, Belo A, Poulsom R, Fitzgerald AJ, Harris K, Pawluczyk I, Ryon J, Darby T, Nilsen-Hamilton M, Ghosh S, Marchbank T. Effects of mouse and human lipocalin homologues 24p3/lcn2 and neutrophil gelatinase-associated lipocalin on gastrointestinal mucosal integrity and repair. Gastroenterology. 2006;131:809–817. doi: 10.1053/j.gastro.2006.05.051. [DOI] [PubMed] [Google Scholar]
- Ricciardolo FL, Di Stefano A, Sabatini F, Folkerts G. Reactive nitrogen species in the respiratory tract. Eur J Pharmacol. 2006;533:240–252. doi: 10.1016/j.ejphar.2005.12.057. [DOI] [PubMed] [Google Scholar]
- Roudkenar MH, Halabian R, Ghasemipour Z, Roushandeh AM, Rouhbakhsh M, Nekogoftar M, Kuwahara Y, Fukumoto M, Shokrgozar MA. Neutrophil gelatinase-associated lipocalin acts as a protective factor against H(2)O(2) toxicity. Arch Med Res. 2008;39:560–566. doi: 10.1016/j.arcmed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Roudkenar MH, Halabian R, Roushandeh AM, Nourani MR, Masroori N, Ebrahimi M, Nikogoftar M, Rouhbakhsh M, Bahmani P, Najafabadi AJ, Shokrgozar MA. Lipocalin 2 regulation by thermal stresses: protective role of Lcn2/NGAL against cold and heat stresses. Exp Cell Res. 2009;315:3140–3151. doi: 10.1016/j.yexcr.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Saika S. Yin and yang in cytokine regulation of corneal wound healing: roles of TNF-alpha. Cornea. 2007;26:S70–74. doi: 10.1097/ICO.0b013e31812f6d14. [DOI] [PubMed] [Google Scholar]
- Sugiura H, Ichinose M. Nitrative stress in inflammatory lung diseases. Nitric Oxide. 2011;25:138–144. doi: 10.1016/j.niox.2011.03.079. [DOI] [PubMed] [Google Scholar]
- Sunil VR, Patel-Vayas K, Shen J, Gow AJ, Laskin JD, Laskin DL. Role of TNFR1 in lung injury and altered lung function induced by the model sulfur mustard vesicant, 2-chloroethyl ethyl sulfide. Toxicol Appl Pharmacol. 2011a;250:245–255. doi: 10.1016/j.taap.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Patel KJ, Nilsen-Hamilton M, Heck DE, Laskin JD, Laskin DL. Acute endotoxemia is associated with upregulation of lipocalin 24p3/Lcn2 in lung and liver. Exp Mol Pathol. 2007;83:177–187. doi: 10.1016/j.yexmp.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Patel KJ, Shen J, Reimer D, Gow AJ, Laskin JD, Laskin DL. Functional and inflammatory alterations in the lung following exposure of rats to nitrogen mustard. Toxicol Appl Pharmacol. 2011b;250:10–18. doi: 10.1016/j.taap.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankersley CG, Shank JA, Flanders SE, Soutiere SE, Rabold R, Mitzner W, Wagner EM. Changes in lung permeability and lung mechanics accompany homeostatic instability in senescent mice. J Appl Physiol. 2003;95:1681–1687. doi: 10.1152/japplphysiol.00190.2003. [DOI] [PubMed] [Google Scholar]
- Ucar M, Korkmaz A, Reiter RJ, Yaren H, Oter S, Kurt B, Topal T. Melatonin alleviates lung damage induced by the chemical warfare agent nitrogen mustard. Toxicol Lett. 2007;173:124–131. doi: 10.1016/j.toxlet.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Weinberger B, Laskin JD, Sunil VR, Sinko PJ, Heck DE, Laskin DL. Sulfur mustard-induced pulmonary injury: therapeutic approaches to mitigating toxicity. Pulm Pharmacol Ther. 2011;24:92–99. doi: 10.1016/j.pupt.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormser U, Langenbach R, Peddada S, Sintov A, Brodsky B, Nyska A. Reduced sulfur mustard-induced skin toxicity in cyclooxygenase-2 knockout and celecoxib-treated mice. Toxicol Appl Pharmacol. 2004;200:40–47. doi: 10.1016/j.taap.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Wu H, Santoni-Rugiu E, Ralfkiaer E, Porse BT, Moser C, Hoiby N, Borregaard N, Cowland JB. Lipocalin 2 is protective against E. coli pneumonia Respir Res. 2010;11:96. doi: 10.1186/1465-9921-11-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaren H, Mollaoglu H, Kurt B, Korkmaz A, Oter S, Topal T, Karayilanoglu T. Lung toxicity of nitrogen mustard may be mediated by nitric oxide and peroxynitrite in rats. Res Vet Sci. 2007;83:116–122. doi: 10.1016/j.rvsc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang M, Kang X, Boontheung P, Li N, Nel AE, Loo JA. Oxidative stress and asthma: proteome analysis of chitinase-like proteins and FIZZ1 in lung tissue and bronchoalveolar lavage fluid. J Proteome Res. 2009;8:1631–1638. doi: 10.1021/pr800685h. [DOI] [PMC free article] [PubMed] [Google Scholar]