

Abstract
Ankyrin-B (AnkB) loss-of-function may cause ventricular arrhythmias and sudden cardiac death in humans. Cardiac myocytes from AnkB heterozygous mice (AnkB+/−) show reduced expression and altered localization of Na/Ca exchanger (NCX) and Na/K-ATPase (NKA), key players in regulating [Na]i and [Ca]i. Here we investigate how AnkB reduction affects cardiac [Na]i, [Ca]i and SR Ca release. We found reduced NCX and NKA transport function but unaltered [Na]i and diastolic [Ca]i in myocytes from AnkB+/− vs. wild-type (WT) mice. Ca transients, SR Ca content and fractional SR Ca release were larger in AnkB+/− myocytes. The frequency of spontaneous, diastolic Ca sparks (CaSpF) was significantly higher in intact myocytes from AnkB+/− vs. WT myocytes (with and without isoproterenol), even when normalized for SR Ca load. However, total ryanodine receptor (RyR)-mediated SR Ca leak (tetracaine-sensitive) was not different between groups. Thus, in AnkB+/− mice SR Ca leak is biased towards more Ca sparks (vs. smaller release events), suggesting more coordinated openings of RyRs in a cluster. This is due to local cytosolic RyR regulation, rather than intrinsic RyR differences, since CaSpF was similar in saponin-permeabilized myocytes from WT and AnkB+/− mice. The more coordinated RyRs openings resulted in an increased propensity of pro-arrhythmic Ca waves in AnkB+/− myocytes. In conclusion, AnkB reduction alters cardiac Na and Ca transport and enhances the coupled RyR openings, resulting in more frequent Ca sparks and waves although the total SR Ca leak is unaffected. This could enhance the propensity for triggered arrhythmias in AnkB+/− mice.
Keywords: ankyrin-B, Na/K-ATPase, Na/Ca exchanger, intracellular Na, Ca sparks, SR Ca leak
1. Introduction
Ankyrin-B (AnkB) is a multivalent “adaptor” protein that targets and tethers select membrane proteins to the cytoskeleton. AnkB deficiency emerged as an important pro-arrhythmic factor a few years ago, when it was found that AnkB loss-of-function mutations generate complex cardiac phenotypes in humans. These phenotypes may include bradycardia, atrial fibrillation, conduction defects, stress- or exercise-induced ventricular arrhythmias and sudden cardiac death [1–5].
The Na/Ca exchanger (NCX) is the main pathway for Ca extrusion in cardiac myocytes. It exchanges one Ca ion for three Na ions and is a major contributor to Na influx during the cardiac cycle. To maintain low [Na]i and thus facilitate Ca extrusion through NCX, Na is pumped out of the cells by the Na/K-ATPase (NKA). Therefore, NCX and NKA are essential in the regulation of cardiac [Na]i, [Ca]i and contractility [6]. AnkB directly associates with NCX and NKA in vivo and in vitro, and is required for the membrane targeting and stability of both NCX and NKA in cardiac myocytes [1–7,10]. Notably, one AnkB mutant (E1425G) that is linked to complex cardiac arrhythmias in humans exhibits a 60–70% loss of association with NCX and NKA [11]. In fact, the clinical severity of human AnkB loss-of-function variants has been directly linked with the inability to target NCX and NKA to the cardiac myocyte sarcolemma [3]. Collectively, these data support a major role of altered NCX and NKA membrane expression and function in ankyrin-B-associated cardiac disease.
While ankyrin-B−/− mice die shortly after birth, ankyrin-B+/− mice are viable and largely reproduce the human cardiac ankyrin-B syndrome [1]. Cardiac myocytes from AnkB+/− mice show reduced NCX and NKA protein expression (by ~20%) but no change in the mRNA levels [1]. Immunofluorescence staining indicates that this reduction occurs predominantly at the T-tubules [1]. In contrast, the protein expression and distribution of SR Ca-ATPase, ryanodine receptors (RyRs), L-type Ca channels, plasma membrane Ca-ATPase, Na channels and K channels or associated subunits are unaltered in AnkB+/− mice [1]. Voltage-clamped cardiac myocytes from AnkB+/− mice exhibit elevated Ca transients and show early and delayed afterdepolarizations and extrasystoles during β-adrenergic stimulation [1]. Action potential duration, inward Ca current and diastolic Ca are not significantly altered.
The mechanistic basis for the increased Ca transients and arrhythmogenic events in AnkB+/− mice is not elucidated. Here we investigated how the coordinated loss of NCX and NKA at the T-tubules alters cardiac myocyte [Na]i, spontaneous SR Ca release and Ca waves in AnkB+/− mice.
2. Materials and methods
2.1 Cardiac myocytes isolation; Ca transients and fractional SR Ca release measurements
Ventricular myocytes were isolated from mice heterozygous for ankyrin-B null allele (AnkB+/−) and wild-type (WT) littermates by perfusion with 0.8 mg/ml collagenase on a Langendorff apparatus, as previously described [12]. All animal protocols were approved by the animal welfare committee at the University of California Davis. 14 AnkB+/− and 14 WT mice were used for this study. All measurements were at room temperature (21–25 °C).
For Ca transient measurements, myocytes were loaded with Fluo-4 AM (10 μM for 40 min) and excited at 480 nm. Fluorescence was recorded at 535±20 nm. Using an in vivo calibration, we determined that the intracellular Fluo-4 concentration in Fluo-AM loaded myocytes is 25–30 μM (see Supplemental Methods and Supplemental Fig. 1). The fractional release of Ca from the SR during the twitch was calculated as the ratio between the amplitude of twitch Ca transient and the amplitude of Ca transient evoked by 10 mM caffeine.
2.2 Diastolic [Ca]i measurements
Myocytes were loaded with 10 μM Fura-2-AM for 45 min. Fura-2 was excited alternatively at 340±4 and 380±4 nm and emitted fluorescence was recorded at 510±20 nm. After subtracting the background, the fluorescence ratio (R=F340/F380) was converted to [Ca]i using the expression [Ca]i=Kdβ(R−Rmin)/(Rmax−R), where Kd=236 nM [13] and Rmin, Rmax, and β were determined experimentally.
2.3 Intracellular Na concentration ([Na]i) and Na/K-ATPase function measurements
Myocytes were incubated with SBFI-AM (10 μM for 90 min). Dual excitation measurements (at 340 and 380 nm) were performed and the F340/F380 ratio was calculated after background subtraction. F340/F380 was converted to [Na]i by a three-point calibration at the end of each experiment [14].
The Na/K-ATPase (NKA) function was assessed in intact myocytes as the [Na]i-dependence of the pump-mediated Na extrusion [14]. Myocytes were Na-loaded by inhibiting NKA in K-free solution. [Na]i decline was then measured upon pump reactivation in a 0Na/4 mM K solution. Since cell volume does not change with this protocol [14], [Na]i decline reflects Na efflux. The rate of [Na]i decline (−d[Na]i/dt) was plotted vs. [Na]i and fitted with the Hill equation: −d[Na]i/dt = Vmax/(1+(K0.5/[Na]i)nHill), where Vmax is the maximum Na extrusion rate, K0.5 is the [Na]i for the half-maximal activation of the pump and nHill is the Hill coefficient.
2.4 Ca spark recordings in intact cardiac myocyte
Ca sparks were recorded with a laser-scanning confocal microscope as we previously reported [15]. Fluo-4-loaded myocytes were excited with the 488 nm line of an argon laser. Fluorescence was collected through a 515–530 nm band-pass filter. Longitudinal line scans were acquired at 3 ms intervals.
To monitor Ca sparks, myocytes were paced at different frequencies (0.5, 1 or 2 Hz) until Ca transients reached steady-state. Stimulation was then stopped and spontaneous Ca sparks were recorded in a 0Na/0Ca Tyrode’s solution (with Li substituted for Na and 10 mM EGTA), in order to inhibit both the Na/Ca exchanger and L-type Ca channels. SR Ca load is maintained for at least 5 min in this 0Na/0Ca solution [16]. Finally, SR Ca content was assessed from the amplitude of the Ca transient generated by rapid application of 10 mM caffeine. Because NCX is blocked in 0Na/0Ca solution, occurrence of spontaneous Ca waves does not significantly affect SR Ca content because most (>95%) of Ca released during a wave is taken back into the SR. F/F0 ratios, where F0 is the mean fluorescence intensity measured after stopping the pacing, were converted to [Ca]i using the diastolic [Ca]i values measured as described above (85 nM, see Results section) and assuming that Fluo-4 Kd is 1100 nM [17]. SR Ca content was calculated as: [Ca]SRT (μmol/Lcytosol)=Bmax/(1+(Km/[Ca]i))+Bmin, where Bmax=244 μmol/Lcytosol, Bmin=−28 μmol/Lcytosol and Km=673 nM [6]. Ca sparks were detected, counted and characterized using IDL software (Research Systems, Inc., Boulder, CO, USA) and confirmed visually as described previously [15].
2.5 Ca spark measurements in permeabilized cardiac myocytes
Myocytes were permeabilized with 50 μg/mL saponin for 1 min in internal solution. Free [Ca] and [Mg] were adjusted at 50 nM and 1 mM, respectively, using Maxchelator and the EGTA concentration was 0.5 mM. Cells were then washed and incubated with Fluo-4 pentapotassium salt (10 μM) for 10 min. Ca sparks were measured in quiescent cells using line-scan recordings and analyzed using the IDL software as described above.
2.6 SR Ca leak measurements
Fluo-4-loaded myocytes were pre-conditioned with three different protocols: i) steady-state pacing at 1 Hz in 1 mM Ca, ii) pacing for two beats following a previous caffeine, and iii) steady-state pacing at 1 Hz in 4 mM Ca. This was done to attain various levels of SR Ca content. The perfusate was then switched to 0Na/0Ca solution and 1 mM tetracaine was added after reaching a stable [Ca]i [18]. Tetracaine, a RyR inhibitor, blocks the SR Ca leak and thus results in a decline in resting [Ca]i. The magnitude of this decline reflects the magnitude of the SR Ca leak. Tetracaine was then removed and SR Ca content was assessed with caffeine.
2.7 Statistical analysis
The data are presented as mean ± standard error. Normal distribution was verified using the D’Agostino normality test in GraphPad Prism. Means were considered statistically different when P<0.05.
3. Results
3.1 Elevated Ca transients and SR Ca content in myocytes from AnkB+/− vs. WT mice
Myocytes from WT and AnkB+/− mice were field-stimulated at 1 Hz until Ca transients reached steady-state (Fig. 1A). Pacing was then stopped for 10 sec, followed by rapid application of 10 mM caffeine to empty the SR of Ca and thus measure its content. Ca transients and SR Ca content were significantly larger in myocytes from AnkB+/− vs. WT mice (by 47% and 13%, respectively; Fig. 1B). In addition, there was a 30% increase in the fractional release of Ca from the SR during the twitch (Fig. 1C). The decay of twitch Ca transients, which is indicative of SERCA function, was not significantly different in AnkB+/− vs. WT mice (Fig. 1D). This is in agreement with previous reports that SERCA (and phospholamban) protein expression and distribution are not altered in AnkB+/− mice [1]. The slightly (not significant) smaller decay time in AnkB+/− mice (Fig. 1D) is most likely due to the larger Ca transients (Supplemental Fig. 2) [19].
Figure 1. Larger Ca transients and SR Ca content and reduced NCX function in myocytes from AnkB+/− mice.

(A) Ca transient and SR Ca content measurements - representative traces in a WT and an AnkB+/− myocyte. Myocytes were paced at 1 Hz until Ca transients reached steady-state, then pacing was stopped for 10 sec, followed by application of 10 mM caffeine. Mean values for Ca transient amplitude and SR Ca content (18 cells, 5 animals for both WT and AnkB+/− mice) (B), SR fractional release (18 cells, 5 mice for both WT and AnkB+/− mice) (C), decay time of the twitch Ca transient (18 cells, 5 mice for both WT and AnkB+/− mice) (D) and decay time of the caffeine-induced Ca transient (9 cells, 5 mice for both WT and AnkB+/− mice) (E). (F) Mean diastolic [Ca]i in myocytes from WT and AnkB+/− mice (n=10, 4 mice for both WT and AnkB+/− mice).
3.2 Reduced NCX and NKA function but unaltered [Na]i and diastolic [Ca]i in AnkB+/− myocytes
Cardiac myocytes from AnkB+/− mice show modestly (~20%) reduced NCX and NKA protein expression and even more dramatic loss of apparent localization at T-tubules [1]. This is expected to result in decreased NCX and NKA function. To test this, we determined NCX function from the decay of the caffeine-induced Ca transient, as this is mediated mostly by NCX [6] (Fig. 1E). The decay was 40% slower in myocytes from AnkB+/− mice (time constant τ=7.4±0.8 sec vs. 5.2±0.6 sec in WT; Fig. 1E), indicating that NCX function is significantly reduced compared to WT mice.
NKA function was determined in intact myocytes by measuring the [Na]i-dependence of the NKA-mediated Na efflux. Myocytes were Na-loaded by blocking NKA in K-free solution (Fig. 2A). Then NKA was reactivated by re-admission of 4 mM [K]o (and removal of extracellular Na) and we monitored the ensuing decline in [Na]i. This was then numerically differentiated and the rate of [Na]i decline (−d[Na]i/dt) was plotted as a function of [Na]i (Fig 2B). The curves were fit with a Hill expression to determine Vmax, K0.5 and the Hill coefficient. We found that Vmax was significantly reduced by 22% (5.0±0.5 vs. 6.4±0.4 mM/min in WT) whereas the K0.5 and Hill coefficient were unchanged in myocytes from AnkB+/− mice (Fig. 2B). Thus, NKA function is also reduced in AnkB+/− vs. WT mice.
Figure 2. NKA function is reduced but [Na]i is unchanged in myocytes from AnkB+/− mice.

(A) Measurement of NKA-mediated Na efflux in intact myocytes from WT and AnkB+/− mice - representative trace in an AnkB+/− myocyte. NT – normal Tyrode’s solution; (B) The rate of [Na]i decline (−d[Na]i/dt) was calculated by numerical differentiation, plotted as a function of [Na]i and fitted with Hill equation to determine Vmax, K0.5 and the Hill coefficient. Mean data in myocytes from WT (8 cells, 6 mice) and AnkB+/− mice (10 cells, 5 mice) indicate that K0.5 is not changed whereas Vmax is significantly lower in AnkB+/− myocytes (Inset). (C) [Na]i measurements at rest and during pacing (2Hz) in the absence and in the presence (2Hz+ISO) of 1 μM isoproterenol in a WT myocyte. (D) Mean [Na]i in myocytes from WT (12 cells, 7 mice) and AnkB+/− mice (14 cells, 6 mice) at rest and during pacing with and without isoproterenol.
NKA is the main pathway for Na extrusion and NCX is the main route for Ca efflux in cardiac myocytes. Since both NCX and NKA activities are reduced, we might expect that both [Na]i and diastolic [Ca]i are increased in myocytes from AnkB+/− mice. To test this, we measured [Na]i in myocytes from WT and AnkB+/− mice using SBFI (Fig. 2C). Neither resting [Na]i, nor steady-state [Na]i during field-stimulation at 2 Hz were altered in AnkB+/− vs. WT mice (Fig. 2D). We also added 1 μM isoproterenol and again [Na]i was not significantly different between groups (Fig. 2D). Thus, [Na]i is remarkably unaltered in AnkB+/− mice despite reduced NKA expression and function. This might be attributable to a concomitant decrease in Na influx in AnkB+/− vs. WT myocytes. NCX is the dominant pathway for Na influx in cardiac myocytes [20] and NCX function is decreased by ~40% in AnkB+/− mice (Fig. 1E), which may explain the absence of [Na]i elevation. Thus, larger Ca transients and SR Ca content in myocytes from AnkB+/− mice are not due to alterations in global [Na]i.
We also measured diastolic [Ca]i with Fura-2 and found no detectable difference between WT and AnkB+/− mice (85±6 vs. 87±5 nM, respectively; Fig. 1F). While this was slightly surprising based on the larger Ca transients and SR Ca load in AnkB+/− myocytes, it is consistent with the unaltered [Na]i.
3.3 Elevated Ca spark frequency in intact myocytes from AnkB+/− mice through a mechanism independent of the SR Ca content
The higher fractional SR Ca release during twitch Ca transients stimulated us to investigate why SR Ca release is enhanced in myocytes from AnkB+/− mice vs. WT, and how this could increase the propensity for triggered ventricular arrhythmias in AnkB+/− mice. To test this, we recorded Ca sparks and waves in intact myocytes from AnkB+/− and WT mice. Myocytes were paced for 5 min at 0.5, 1 or 2 Hz to reach various SR Ca loads. Pacing was then stopped and diastolic Ca sparks were measured for 10–12 seconds in 0Na/0Ca solution (Fig. 3A). The 0Na/0Ca solution was used to block both NCX (and thus prevent SR Ca unloading) and SR Ca release induced by stochastic openings of L-type Ca channels. Finally, the SR Ca content was measured by rapid application of 10 mM caffeine. Representative confocal line-scan images are shown in Fig. 3A and selected areas are shown in Fig. 3B. The AnkB+/− myocyte has a higher number of Ca sparks compared to WT and exhibits a spontaneous Ca wave. Ca spark frequency was significantly higher in myocytes from AnkB+/− compared to WT mice for all pre-conditioning pacing frequencies (Fig. 3C). Histograms showing the distribution of Ca spark frequency are presented in Supplemental Fig. 3. All other Ca spark characteristics (amplitude, duration and width) were not significantly different in AnkB+/− vs. WT mice (Supplemental Table 1).
Figure 3. Increased Ca spark frequency in intact myocytes from AnkB+/− mice, independent of the larger SR Ca content.

(A) Representative examples of line-scan measurements of spontaneous Ca sparks in myocytes from WT and AnkB+/− mice. Myocytes were paced for 5 minutes at 1 Hz, then pacing was stopped for Ca spark measurements in 0Na/0Ca solution. SR Ca content was measured at the end by rapid application of 10 mM caffeine. (B) Expanded scale for the areas marked with a white box in panel A. (C) Mean Ca spark frequency in myocytes from WT and AnkB+/− mice pre-conditioned by pacing at 0.5, 1 and 2 Hz. (D) SR Ca content in myocytes from WT and AnkB+/− mice paced at 0.5, 1 and 2 Hz. (E) Ca spark frequency normalized to the SR Ca content for myocytes pre-conditioned by pacing at 0.5, 1 and 2 Hz. For each pacing frequency, n=10 cells from 4–6 mice for both groups. (F) Ca spark frequency in myocytes from WT mice under control conditions (Ctl; 7 cells, 3 mice) and in the presence of 5 μM ouabain (9 cells, 3 mice). Myocytes were pre-conditioned by pacing at 1 Hz. *P<0.05, **P<0.01.
Spontaneous SR Ca release increases with increasing SR Ca load [18] and SR Ca content was higher in myocytes from AnkB+/− mice (Fig. 3D). However, Ca spark frequency was still significantly increased in AnkB+/− myocyte even when normalized to the SR Ca content (Fig. 3E). Although Ca spark frequency depends non-linearly on the SR load, this strong effect suggests that AnkB reduction may enhance spark-mediated diastolic SR Ca release both by increasing SR Ca load and by sensitizing RyRs.
To determine whether the reduction in NCX and NKA activity in AnkB+/− mice may play a role in the elevated Ca spark frequency, we repeated Ca spark measurements in WT myocytes in the presence of 5 μM ouabain. At this concentration, ouabain practically inhibits all ouabain-sensitive NKA-α2, which is concentrated in the T-tubules, and has only a minor effect on NKA-α1, resulting in a reduction in total NKA activity by about 20–25% [21]. This is comparable to the reduction in NKA expression [1] and function (see above) observed in myocytes from AnkB+/− mice. NKA inhibition will also reduce Ca extrusion through NCX because of elevated [Na]i (locally and/or bulk cytosolic), thus mimicking to some extent the situation in AnkB+/− mice. We reported previously that in mouse myocytes 5 μM ouabain significantly increases Ca transient amplitude (by ~25%) and moderately raises [Na]i (by ~2 mM), without significantly affecting the SR Ca content [22]. Ouabain (5 μM) increased Ca spark frequency from 0.32±0.08 to 0.99±0.23 sparks/(100·μm·s) (Fig. 3F) without significantly affecting the SR Ca content (132±10 μmol/Lcytosol in the absence vs. 135±10 μmol/Lcytosol in the presence of ouabain). Of note, ouabain application only partially replicates the AnkB+/− mice because in these mice the expression of NKA-α1 isoform and NCX are also reduced [1].
In both humans with AnkB loss-of-function mutations and AnkB+/− mice, ventricular arrhythmias occur preponderantly during stress or exercise [1]. To mimic this, we measured spontaneous Ca sparks in myocytes stimulated with 1 μM isoproterenol (ISO; Fig. 4). With ISO, Ca spark frequency increased in both groups but was significantly higher in myocytes from AnkB+/− vs. WT mice for all three pacing rates (Fig. 4C). However, with ISO the SR Ca load was similar in AnkB+/− and WT mice under all circumstances (Fig. 4D). This demonstrates that the enhanced spark-mediated SR Ca release in AnkB+/− vs. WT mice during activation of the β-adrenergic receptors with ISO is mostly independent of SR Ca load, and may reflect enhanced sensitivity of SR Ca release.
Figure 4. Elevated Ca spark frequency but similar SR Ca content in intact myocytes from WT and AnkB+/− mice in the presence of isoproterenol.

(A) Representative example of Ca sparks measurements in the presence of 1 μM ISO in an AnkB+/− myocyte. (B) Expanded scale for the areas marked with a white box in panel A. (C) Ca spark frequency in myocytes from WT and AnkB+/− mice pre-conditioned by pacing at 0.5, 1 and 2 Hz in the presence of 1 μM ISO. (D) SR Ca content in myocytes from WT and AnkB+/− mice paced at 0.5, 1 and 2 Hz in the presence of 1 μM ISO. For each pacing frequency, n=10 cells from 4–6 mice for both groups. *P<0.05, **P<0.01.
3.4 Total RyR-mediated SR Ca leak is unaltered in AnkB+/− mice
Ca sparks account for only part of RyR-mediated diastolic SR Ca leak [23–25]. The non-spark SR Ca release (i.e. release that causes such small, narrow or brief fluorescent signals that they are below the threshold criteria for Ca sparks) may be due to either alternative anatomical arrangement of RyRs or specific channel gating properties [26]. We measured the total RyR-mediated diastolic SR Ca leak as the decline in diastolic [Ca]i upon blocking RyRs with 1 mM tetracaine (Fig. 5A) [18]. To vary the SR Ca load, myocytes were paced to steady-state at 1 Hz in 1 or 4 mM Ca or paced for two beats following a previous caffeine application before measuring the SR leak. For the range of SR Ca contents obtained with these pre-conditioning protocols, the total RyR-mediated SR Ca leak was similar in myocytes from WT and AnkB+/− mice (Fig. 5B). Taken together with the larger Ca spark frequency in AnkB+/− myocytes, these data indicate that, at a given SR Ca content, RyR function is biased towards producing more Ca sparks at the expense of smaller, non-spark mediated releases in AnkB+/− mice.
Figure 5. Similar RyR-mediated diastolic SR Ca leak in myocytes from WT and AnkB+/− mice.

(A) Representative example of SR Ca leak measurements. Each myocyte was pre-conditioned by steady-state pacing at 1 Hz in 1 and 4 mM external Ca and by pacing for only two beats following a previous caffeine application. The perfusate was then switched to 0Na/0Ca solution and 1 mM tetracaine was added. Tetracaine blocks the RyR-mediated SR Ca leak, which resulted in a resting [Ca]i decline. Diastolic [Ca]i returned to its initial value after removing tetracaine. SR Ca content was assessed at the end with caffeine. (B) RyR-mediated diastolic SR Ca leak, measured as the decline in diastolic [Ca]i upon tetracaine application, vs. SR Ca content in myocytes from WT and AnkB+/− mice. Mean data from 24 (5 mice) WT and 21 (5 mice) AnkB+/− myocytes.
IP3 receptor expression is also reduced in AnkB+/− myocytes [1] and this could in principle contribute to the differences in the SR Ca release between WT and AnkB+/− myocytes. However, in WT myocytes, without IP3-inducing agonists, the IP3 receptor-mediated SR Ca leak is negligible (Supplemental Fig. 4), whether or not RyRs are simultaneously blocked with tetracaine.
3.5 Permeabilization equalizes Ca spark frequency in myocytes from AnkB+/− and WT mice
The SR Ca load-independent component of the higher Ca spark frequency in AnkB+/− myocytes may be due to intrinsic differences in RyRs organization (protein expression is unaltered [1]) or local cytosolic RyR regulation, resulting in increased function. To investigate this, we measured Ca sparks in saponin-permeabilized myocytes, as this allows better control of the intracellular environment. After permeabilization and 10 min stabilization in 50 nM free Ca internal solution, we recorded Ca sparks and measured the SR Ca content (Fig. 6). Representative examples of line-scan images in permeabilized WT and AnkB+/− myocytes are shown in Fig. 6A. After permeabilization, both Ca spark frequency (3.8±0.3 vs. 4.1±0.3 sparks/100·μm−1·s−1 in WT and AnkB+/− mice, respectively; Fig. 6B) and SR Ca content (107±5 μmol/Lcytosol in WT and 102±6 in μmol/Lcytosol AnkB+/− mice; Fig. 6C) were similar in myocytes from WT and AnkB+/− mice. This result rules out major intrinsic differences in RyR organization as a contributor to the enhanced SR Ca release in AnkB+/− myocytes, but leaves open the possibility of altered cytosolic RyR regulation.
Figure 6. Ca spark measurements in saponin-permeabilized myocytes.

(A) Representative linescan images in WT and AnkB+/− myocytes. Ca spark-frequency (B) and SR Ca content (C) in permeabilized WT and AnkB+/− myocytes. Free [Ca]i=50 nM. n=12 cells (3 mice) for both WT and AnkB+/− mice.
3.6 Increased propensity for Ca waves in AnkB+/− vs. WT myocytes
Arrhythmia-triggering delayed afterdepolarizations are generated by the transient inward current activated by spontaneous Ca waves. We found a significantly larger propensity for Ca wave occurrence in myocytes from AnkB+/− vs. WT mice, both under control conditions (9 out of 45 AnkB+/− myocytes displayed waves vs. 0 out of 45 for WT; Fig. 7A) and in the presence of ISO (28 out of 53 AnkB+/− myocytes with waves vs. 15 out of 50 for WT; Fig. 7B). The propagation velocity of Ca waves was similar in myocytes from WT and AnkB+/− mice (62±6 vs. 69±6 μm·s−1).
Figure 7. Increased incidence of Ca waves in myocytes from AnkB+/− vs. WT mice both under control conditions.
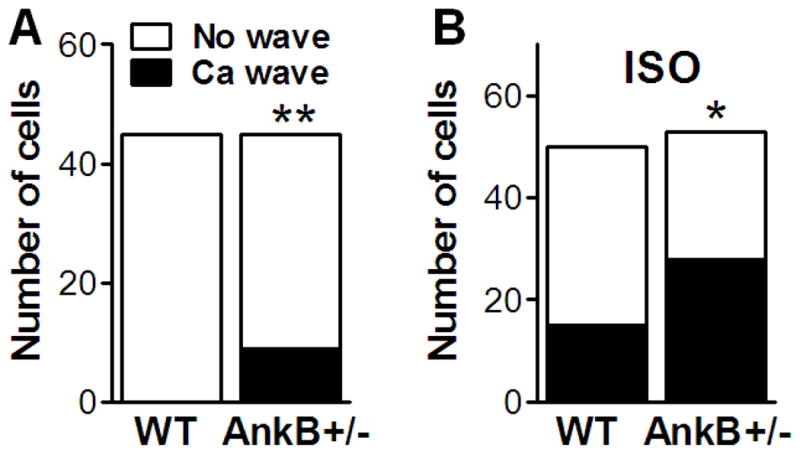
(A) and in the presence of 1 μM ISO (B). Shown is the number of cells that exhibited Ca waves for each group.
4. Discussion
4.1 Altered [Na]i does not contribute to elevated SR Ca content in cardiac myocytes from AnkB+/− mice
We found larger SR Ca content in myocytes from AnkB+/− vs. WT mice. Prior studies proposed that AnkB+/− myocytes likely have higher [Na]i and [Ca]i, due to reduced NKA and NCX expression, and that these effects would elevate SR Ca load and Ca transient amplitude [1]. Here we found that while the function of both NCX and NKA is reduced in myocytes from AnkB+/− mice, as expected from their lower protein expression and altered localization, [Na]i is not altered (either at rest or during pacing). In agreement with this, diastolic [Ca]i was also similar in myocytes from WT and AnkB+/− mice. Thus, the larger SR Ca content in AnkB+/− myocytes is not caused by elevated [Na]i. Obviously, here we measured global [Na]i and diastolic [Ca]i and this result does not rule out differences in the local, submembrane [Na]i and [Ca]i between WT and AnkB+/− mice (see below).
SR Ca content is determined by the balance of Ca fluxes across both sarcolemma and SR membrane. Mohler et al. have shown that expression, localization and function (Ca current) of L-type Ca channels is not altered in myocytes from AnkB+/− mice [1]. The protein expression of both SR Ca-ATPase and phospholamban is similar in myocytes from WT and AnkB+/− mice [1] and we found that the decay of twitch Ca transients is also unchanged. These results suggest that the SR Ca-ATPase activity is comparable. We also found that the total diastolic SR Ca leak is not significantly altered. Thus, the decreased NCX function seems to be a primary cause for elevated SR Ca content in AnkB+/− myocytes. That is, during [Ca]i decline the SR Ca-ATPase competes better with NCX in AnkB+/− myocytes, biasing the steady-state SR Ca content higher. Further studies are needed to elucidate whether reduced NCX function is solely the result of reduced NCX protein expression and localization at the T-tubules, or that interaction with AnkB directly modulates NCX activity, as is the case for other cytoskeletal proteins such as actin [27] and protein 4.1R [28]. The extent of NCX functional inhibition (~40%) is greater than the ~20% decrease in expression [1], leaving this as a possibility.
4.2 Diastolic SR Ca leak is biased towards Ca sparks vs. smaller release events in myocytes from AnkB+/− mice
We found higher Ca spark frequency in intact myocytes from AnkB+/− vs. WT mice, both under control conditions and in the presence of isoproterenol. Under control conditions, the higher Ca spark frequency is in part due to elevated SR Ca content. However, Ca spark frequency is still much larger in AnkB+/− myocytes even after normalization to the SR Ca content. With isoproterenol, SR Ca load is similar in AnkB+/− and WT myocytes. Thus, factors independent of the SR Ca content essentially contribute to elevate Ca spark frequency in AnkB+/− mice. Ca spark frequency was similar in myocytes from WT and AnkB+/− mice after equalizing the cytosolic environment upon myocyte permeabilization with saponin. This indicates that the higher Ca spark frequency in intact AnkB+/− myocytes is caused by different RyR regulation at the cytoplasmic side, rather than being due to intrinsic differences in RyR expression and/or cluster organization in AnkB+/− vs. WT mice. For example, it is possible that reduced peri-junctional NCX function (due to reduced peri-junctional NCX expression and reduced NKA function) would allow local diastolic [Ca]i (in or near the junctional cleft) to be higher in AnkB+/− vs. WT myocytes, despite unaltered global diastolic [Ca]i. While a role for NCX (and NKA) in regulating cleft [Ca]i is controversial, there is evidence that they are partly located at the junctions [29] and may affect cleft vs. global [Ca]i [30–32]. Our data indicating that acute inhibition of the T-tubular localized NKA-α2 (20–25% of the total NKA) in WT myocytes significantly increases Ca spark frequency despite unaltered SR Ca content support this hypothesis. Elevated cleft [Ca]i may increase Ca spark frequency either directly, by sensitizing RyRs, or indirectly through activation of CaMKII and consequent RyR phosphorylation at S2814, the CaMKII-specific phosphorylation site [33]. AnkB is also essential for the targeting of the regulatory subunit B56α of protein phosphatase 2A (PP2A) [34]. Reduced B56α expression in AnkB+/− myocytes may also enhance basal RyR phosphorylation by CaMKII, and thus may contribute to the enhanced propensity for Ca sparks. Further studies are needed to critically investigate these possibilities.
IP3 receptor expression is also reduced in AnkB+/− myocytes [1], but their role in excitation-contraction coupling in ventricular myocytes is controversial. While studies have implicated IP3 receptors in altering Ca transients when Gαq-coupled agonists are present [35,36], most studies found no significant role of IP3 receptor-dependent Ca release in the absence of neurohumoral agents that activate IP3 production. Consistent with this, we found that in the absence of IP3-inducing agonists, the IP3 receptor-mediated SR Ca leak is negligible in WT myocytes. This suggests that the reduced IP3 receptor expression in AnkB+/− mice does not significantly contribute to the differences in the SR Ca release between WT and AnkB+/− ventricular myocytes.
Ca sparks account for only a fraction of the total RyR-mediated diastolic SR Ca leak [23–25]. Despite the higher Ca spark frequency in AnkB+/− mice, the total RyR-mediated SR Ca leak, measured as the tetracaine-induced decline in diastolic [Ca]i, was comparable in WT and AnkB+/− mice (at similar SR Ca contents). Thus, Ca sparks account for a larger fraction of the total RyR-mediated diastolic SR Ca leak in AnkB+/− vs. WT mice (at comparable SR Ca loads), indicating that in AnkB+/− mice RyR function is biased towards producing more Ca sparks at the expense of smaller, non-spark Ca release events. The non-spark RyR-mediated SR Ca leak is caused by either activation of isolated unclustered RyRs (rogue RyRs) [23] or by spontaneous opening of a single RyR in a release unit without activation of the remaining RyRs in the cluster [26,37]. An increase in the ratio of clustered/rogue RyRs or alterations in RyR cluster organization in AnkB+/− mice are ruled out by our finding that Ca spark frequency is similar in saponin-permeabilized myocytes from WT and AnkB+/− mice. Thus, the bias towards more Ca sparks in intact AnkB+/− myocytes may be caused by more coordinated openings of RyRs in a cluster due to the local ionic environment in intact cells.
4.3 Higher frequency of Ca waves in AnkB+/− vs. WT myocytes
The propensity for more coordinated openings of RyRs in a cluster causes a larger elevation in local [Ca]i, and this may facilitate activation of a neighboring RyR cluster, leading to more frequent Ca waves. Indeed, we found that a larger fraction of AnkB+/− myocytes (vs. WT) exhibited Ca waves, both in the absence and in the presence of isoproterenol. Diastolic Ca waves generate a transient inward current, carried primarily by NCX [38,39], which induces delayed afterdepolarizations (DADs). If large enough, these DADs can trigger an action potential and lead to an extrasystole. DADs have been reported to occur in myocytes from AnkB+/− following activation of β-adrenergic receptors with isoproterenol [1]. In agreement with this, in both humans heterozygous for AnkB loss-of-function variants and AnkB+/− mice, ventricular arrhythmias occur preponderantly during or just after stress or exercise, when the sympathetic tone is elevated. Our data show that in the presence of isoproterenol, SR Ca load is similar in WT and AnkB+/− mice, yet the AnkB+/− mice exhibit more Ca sparks and waves. Thus, the increased cooperativity in RyR opening is a major cause for the higher propensity for Ca waves, and thus DADs and triggered arrhythmias, in AnkB+/− mice.
4.4 Conclusions
In summary, we have shown that AnkB reduction alters cardiac myocyte Na and Ca transport but [Na]i and diastolic [Ca]i are not affected. For a given SR Ca content, diastolic Ca spark frequency is higher in myocytes from AnkB+/− vs. WT mice, although the total RyR-mediated SR Ca leak is similar. This bias toward more coordinated RyRs openings is likely due to different RyR regulation at the cytoplasmic side and results in an increased propensity for Ca waves, which may contribute to the increased arrhythmogenicity in AnkB+/− mice.
Supplementary Material
Highlights.
Reduced Na & Ca transport & unaltered [Na]i and diastolic [Ca]i in AnkB+/− myocytes
Ca transients, SR Ca load and fractional SR Ca release are larger in AnkB+/− myocytes
Higher Ca spark frequency and unaltered total SR Ca leak in intact AnkB+/− myocytes
The bias towards Ca spark-mediated leak is due to different cytosolic RyR regulation
The more coordinated RyR openings increase the propensity for Ca waves in AnkB+/− mice
Acknowledgments
We thank Khanha Dao for myocyte preparation and Patrick Wright for animal care.
Funding Sources
This work was supported by grants from NIH (grants HL-109501 to SD; HL-30077 and HL-81526 to DMB; HL-084583 and HL-083422 to PJM), American Heart Association (grant 0735084N to SD), Fondation Leducq Award to the Alliance for Calmodulin Kinase Signaling in Heart Disease (DMB, PJM), and The Saving Tiny Hearts Society (PJM).
Abbreviations
- AnkB
ankyrin-B
- AnkB+/−
mice heterozygous for ankyrin-B null allele
- NCX
Na/Ca exchanger
- NKA
Na/K-ATPase
- CaSpF
Ca spark frequency
- RyR
ryanodine receptor
- ISO
isoproterenol
Footnotes
DISCLOSURES
Emmanuel Camors, None; Peter J. Mohler, None; Donald M. Bers, None Sanda Despa, None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosin S, duBell WH, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–39. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 2.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci USA. 2004;101:9137–42. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohler PJ, Le Scouarnec S, Denjoy I, Lowe JS, Guicheney P, Caron L, et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007;115:432–31. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- 4.Le Scouarnec S, Bhasin N, Vieyres C, Hund TJ, Cunha SR, Koval O, et al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci USA. 2008;105:15617–22. doi: 10.1073/pnas.0805500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. 2011;124:1212–22. doi: 10.1161/CIRCULATIONAHA.111.023986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bers DM. Excitation-contraction coupling and cardiac contractile force. 2. Kluwer Academic Publishers; 2001. p. 427. [Google Scholar]
- 7.Li Z, Burke EP, Frank JS, Bennett V, Philipson KD. The cardiac Na+-Ca2+ exchanger binds to the cytoskeletal protein ankyrin. J Biol Chem. 1992;117:337–45. [PubMed] [Google Scholar]
- 8.Cunha SR, Bhasin N, Mohler PJ. Targeting and stability of Na/Ca exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J Biol Chem. 2007;282:4875–83. doi: 10.1074/jbc.M607096200. [DOI] [PubMed] [Google Scholar]
- 9.Devarajan P, Scaramuzzino DA, Morrow JS. Ankyrin binds to two distinct cytoplasmic domains of Na,K-ATPase α subunit. Proc Natl Acad Sci. 1994;91:2965–69. doi: 10.1073/pnas.91.8.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan C, Puschel B, Koob R, Drenckhahn D. Identification of a binding motif for ankyrin on the α-subunit of Na, K-ATPase. J Biol Chem. 1995;270:29971–75. doi: 10.1074/jbc.270.50.29971. [DOI] [PubMed] [Google Scholar]
- 11.Mohler PJ, Davis JQ, Bennet V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeSantiago J, Maier LS, Bers DM. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J Mol Cell Cardiol. 2002;34:975–84. doi: 10.1006/jmcc.2002.2034. [DOI] [PubMed] [Google Scholar]
- 13.Groden DL, Guan Z, Stokes BT. Determination of Fura-2 dissociation constants following adjustment of the apparent Ca-EGTA association contant for temperature and ionic strength. Cell Calcium. 1991;12:279–87. doi: 10.1016/0143-4160(91)90002-v. [DOI] [PubMed] [Google Scholar]
- 14.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure, but Na/K-pump function is unchanged. Circulation. 2002;105:2543–48. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 15.Brette F, Despa S, Bers DM, Orchard CH. Spatiotemporal characteristics of SR Ca2+ uptake and release in detubulated rat ventricular myocytes. J Mol Cell Cardiol. 2005;39:804–12. doi: 10.1016/j.yjmcc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Bassani RA, Bers DM. Na-Ca exchange is required for rest-decay but not for rest-potentiation of twitches in rabbit and rat ventricular myocytes. J Mol Cell Cardiol. 1994;26:1335–47. doi: 10.1006/jmcc.1994.1152. [DOI] [PubMed] [Google Scholar]
- 17.Harkins AB, Kurebayashi N, Baylor SM. Resting myoplasmic free calcium in frog skeletal muscle fibers measured with fluo-3. Biophys J. 1993;65:865–81. doi: 10.1016/S0006-3495(93)81112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 19.Bers DM, Berlin JR. Kinetics of [Ca]i decline in cardiac myocytes depend on peak [Ca]i. Am J Physiol. 1995;268:C271–C77. doi: 10.1152/ajpcell.1995.268.1.C271. [DOI] [PubMed] [Google Scholar]
- 20.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 21.Berry RG, Despa S, Fuller W, Bers DM, Shattock MJ. Differential distribution and regulation of mouse Na+/K+-ATPase α1 and α2-subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc Res. 2007;73:92–100. doi: 10.1016/j.cardiores.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Despa S, Wu Y, Lingrel JB, Stefani E, Bers DM. Na/K-ATPase α2-Subunit Preferentially Modulates Ca Transients and SR Ca Release in Cardiac Myocytes. Biophys J. 2010;98:201A. [Google Scholar]
- 23.Sobie EA, Guatimosim S, Gómez-Viquez L, Song LS, Hartmann H, Saleet Jafri M, et al. The Ca2+ leak paradox and rogue ryanodine receptors: SR Ca2+ efflux theory and practice. Prog Biophys Mol Biol. 2006;90:172–85. doi: 10.1016/j.pbiomolbio.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, Rios E, et al. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys J. 2010;98:2111–20. doi: 10.1016/j.bpj.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–57. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato D, Bers DM. How does stochastic ryanodine receptor-mediated ca leak fail to initiate a ca spark? Biophys J. 2011;101:2370–79. doi: 10.1016/j.bpj.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Condrescu M, Reeves JP. Actin-dependent regulation of the cardiac Na+/Ca2+ exchanger. Am J Physiol Cell Physiol. 2006;290:C691–701. doi: 10.1152/ajpcell.00232.2005. [DOI] [PubMed] [Google Scholar]
- 28.Stagg MA, Carter E, Sohrabi N, Siedlecka U, Soppa GK, Mead F, et al. Cytoskeletal protein 4. 1R affects repolarization and regulates calcium handling in the heart. Circ Res. 2008;103:855–63. doi: 10.1161/CIRCRESAHA.108.176461. [DOI] [PubMed] [Google Scholar]
- 29.Jayasinghe ID, Cannell MB, Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys J. 2009;97:2664–73. doi: 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su Z, Sugishita K, Ritter M, Li F, Spitzer KW, Barry WH. The sodium pump modulates the influence of INa on [Ca2+]i transients in mouse ventricular myocytes. Biophys J. 2001;80:1230–7. doi: 10.1016/S0006-3495(01)76099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto T, Su Z, Moseley AE, Kadono T, Zhang J, Cougnon M, et al. Relative abundance of α2 Na+ pump isoform influences Na+-Ca2+ exchanger currents and Ca2+ transients in mouse ventricular myocytes. J Mol Cell Cardiol. 2005;39:113–20. doi: 10.1016/j.yjmcc.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD, et al. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J. 2010;99:755–64. doi: 10.1016/j.bpj.2010.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 34.Bhasin N, Cunha SR, Mudannayake M, Gigena MS, Rogers TB, Mohler PJ. Molecular basis for PP2A regulatory subunit B56α targeting in cardiomyocytes. Am J Physiol, Heart Circ Physiol. 2007;293:H109–19. doi: 10.1152/ajpheart.00059.2007. [DOI] [PubMed] [Google Scholar]
- 35.Proven A, Roderick HL, Conway SJ, Berridge MJ, Horton JK, Capper SJ, et al. Inositol 1,4,5-trisphosphate supports the arrhythmogenic action of endothelin-1 on ventricular cardiac myocytes. J Cell Sci. 2006;119:3363–75. doi: 10.1242/jcs.03073. [DOI] [PubMed] [Google Scholar]
- 36.Wihlborg AK, Balogh J, Wang L, Borna C, Dou Y, Joshi BV, et al. Positive inotropic effects by uridine triphosphate (UTP) and uridine diphosphate (UDP) via P2Y2 and P2Y6 receptors on cardiomyocytes and release of UTP in man during myocardial infarction. Circ Res. 2006;98:970–76. doi: 10.1161/01.RES.0000217402.73402.cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipp P, Niggli E. Submicroscopic calcium signals as fundamental events of excitation--contraction coupling in guinea-pig cardiac myocytes. J Physiol. 1996;492:31–38. doi: 10.1113/jphysiol.1996.sp021286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 39.Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na+/Ca2+ exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97(Suppl 1):36–42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.