

Abstract
Enhanced renin-angiotensin-aldosterone system (RAAS) activation contributes to proteinuria and chronic kidney disease by increasing glomerular and tubulointerstitial oxidative stress, promotion of fibrosis. Renin activation is the rate limiting step in angiotensin (Ang II) and aldosterone generation, and recent work suggests direct renin inhibition improves proteinuria comparable to that seen with Ang type 1 receptor (AT1R) blockade. This is important as, even with contemporary use of AT1R blockade, the burden of kidney disease remains high. Thereby, we sought to determine if combination direct renin inhibition with AT1R blockade in vivo, via greater attenuation of kidney oxidative stress, would attenuate glomerular and proximal tubule injury to a greater extent than either intervention alone. We utilized the transgenic Ren2 rat with increased tissue RAS activity and higher serum levels of aldosterone, which manifests hypertension and proteinuria. Ren2 rats were treated with renin inhibition (aliskiren), AT1R blockade (valsartan), the combination (aliskiren+valsartan), or vehicle for 21 days. Compared to Sprague-Dawley controls, Ren2 rats displayed increased systolic pressure (SBP), circulating aldosterone, proteinuria and greater urine levels of the proximal tubule protein excretory marker beta-N-acetylglucosaminidase (β-NAG). These functional and biochemical alterations were accompanied by increases in kidney tissue NADPH oxidase subunit Rac1 and 3-nitrotyrosine (3-NT) content as well as fibronectin and collagen type III. These findings occurred in conjunction with reductions in the podocyte-specific protein podocin as well as the proximal tubule-specific megalin. Further, in transgenic animals there was increased tubulointerstitial fibrosis on light microscopy as well as ultrastructural findings of glomerular podocyte foot-process effacement and reduced tubular apical endosomal/lysosomal activity. Combination therapy led to greater reductions in SBP and serum aldosterone, but did not result in greater improvement in markers of glomerular and tubular injury (ie. β-NAG) compared to either intervention alone. Further, combination therapy did not improve markers of oxidative stress and podocyte and proximal tubule integrity in this transgenic model of RAAS-mediated kidney damage despite greater reductions in serum aldosterone and BP levels.
Keywords: Aldosterone, Combination, Renin inhibition, AT1R blockade, Podocyte, β-NAG, Oxidative Stress
Introduction
The renin-angiotensin-aldosterone system (RAAS) plays a key role in regulating normal cardiovascular and kidney function [1]. However, inappropriate activation of the RAAS contributes to maladaptive kidney tissue remodeling through promotion of oxidative stress and inflammation, processes that contribute to the progressive disease as determined by proteinuria and tubulointerstitial fibrosis [2,3]. Anti-hypertensive agents that inhibit the actions of angiotensin II (Ang II), such as of the Ang type 1 receptor (AT1R) and direct renin inhibition (DRI), have been shown to reduce proteinuria as well as slow the decline in glomerular filtration function and progression of kidney disease [5–7]. However, prevalent and incident rates for chronic kidney disease (CKD) and progression to end-stage renal disease are increasing despite widespread use of AT1R blockade and increasing use of DRI [8,9]. It is of note then, these strategies only attenuate progression on survival analysis across trials and only partially reduce proteinuria in preclinical models, suggesting there remains a significant amount of residual risk for progression of CKD [8,9].
Independent of Ang II, elevations in aldosterone directly contributes to kidney tissue remodeling and albuminuria processes that are associated with tubulointerstitial fibrosis. In the context of elevations in systolic blood pressure, a subset of patients treated with AT1R blockade exhibit a phenomenon known as “aldosterone breakthrough” [10,11]. Aldosterone breakthrough is most likely due to the activities of non-ACE enzymes that cleave Ang I to Ang II which eventually lead to increased Ang II that induces the synthesis and release of aldosterone following activation of adrenal AT2R [11]. As renin activation is a pivotal step in generation of Ang II and downstream aldosterone, renin inhibition alone or in combination with AT1R blockade is an attractive therapeutic target to prevent glomerular/tubular injury through targeting greater reductions in aldosterone [12,13].
In this regard, work done in a model of enhanced RAAS activation the transgenic TG(mRen2)27 rat (Ren2) suggest that adrenal glomerulosa production of aldosterone is, in part, renin dependent and that direct renin inhibition may regulate aldosterone production directly through actions in the zona glomerulosa [14,15]. Inappropriate activation of mineralcorticoid receptor (MR) signaling pathways, in the absence of salt deprivation, further promotes progressive proteinuria and renal tubulointerstitial fibrosis [2,7]. MR-dependent changes in kidney function are thought to largely occur through non-genomic actions including generation of oxidative stress. Data from human studies suggest direct renin inhibition may reduce aldosterone to a greater extent than AT1R blockade [6,14–20]. Accordingly, we hypothesized that combination therapy with in vivo direct renin inhibition and AT1R blockade would attenuate proteinuria and tubulointerstitial fibrosis to a greater extent than either intervention alone, in part, through reductions in aldosterone as well as attenuation of kidney tissue oxidative stress.
Due to species specificity for only human and mouse renin, aliskiren cannot be studied effectively in conventional rat models [16,21,22]. To circumvent this issue, we utilized the transgenic Ren2, which harbors both the native Ren1 and the murine renin transgene, with increased tissue Ang II and circulating aldosterone and manifests hypertension, proteinuria and tubulointerstitial fibrosis [14,15,23–27]. Thereby, use of the Ren2 rat allows for investigation of the specific role that combination of direct renin inhibition with AT1R blockade compared to the individual interventions on kidney injury.
Methods
Animals and treatments
All animal procedures were approved by the University of Missouri animal care and use committees and housed in accordance with NIH guidelines. Transgenic TG(mRen2)27 (Ren2) rats (6–9 weeks of age) and age-matched Sprague-Dawley (SD) littermates were randomly assigned to sham-treated (R2-C and SD-C, respectively; n=5 each), aliskiren-treated (R2-A; n=6 each) at 50mg/kg/day, valsartan treated (R2-V; n=5) at 30mg/kg/day, or a combination of aliskiren and valsartan (R2-A+V; n=6) in saline via intraperitoneal injection for 21 days. Aliskiren was provided by Novartis research laboratories and prepared fresh daily in sterile 0.9% normal saline. Dosing was based on previous studies in Ren2 rats [14,16].
Systolic blood pressure (SBP), Aldosterone, and Urine Measures
Restraint conditioning was initiated before blood pressure measurements were performed as previously described. SBP was measured in triplicate on separate occasions throughout the day using the tail-cuff method (Harvard Systems, Student Oscillometric Recorder) prior to initiation of treatment and on days 19 or 20 prior to sacrifice at 21 days [14,24–26]. Serum aldosterone was measured at the end of the treatment period via by radioimmunoassay using a double antibody assay at the Vanderbilt Hormone & Analytic Service Core Laboratory at the Vanderbilt Diabetes Research and Training Center. Blood samples were taken prior to sacrifice via tail vein phlebotomy.
Both creatinine and protein concentrations in urine were analyzed on an automated clinical chemistry analyzer (Olympus AU680) using commercial assays [14,24–26]. Urine beta-N-acetylglucosaminidase (β-NAG) was determined by colorimetric assay (Roche Diagnostics, Indianapolis, IN) [26]. Creatinine was determined using an automated Jaffe reaction assay and urine protein using an automated colorimetric assay. The chemistry instrument was calibrated and proper controls performed prior to analysis.
3-Nitrotyrosine (3-NT) immunostaining
3-NT was quantified as previously described [24–26]. Briefly, tissue sections were incubated overnight with 1:200 primary rabbit polyclonal anti-nitrotyrosine antibody (Millipore; Billerca, MA; Cat #AB5411). Sections were then washed and incubated 30 min with secondary antibodies, biotinylated link, and streptavidin-HRP. After several rinses with distilled water, diaminobenzidine was applied for 12 min, and sections were again rinsed and stained with hematoxylin for 45 sec, rehydrated, and mounted with a permanent media. The slides were viewed under a bright field (Nikon 50i) microscope and 40X images captured with a snapcf camera.
Western Blots
Kidney protein was quantified using BCA assay (Fisher – Thermo Scientific, Pierce BCA Protein Assay Cat#; 23225). Laemmli buffer was added to the lysates and equal amounts were loaded onto Criterion gels 7.5%. Components of Lamelli buffer include the following: 1.5 g SDS, 3.75 mL 1 M Tris pH 6.8, 0.015 g bromophenol blue, 1.16 g DTT, 3.75 mL H2O, and 7.5 mL Glycerol. Blots were blocked in 1%BSA in 1X tris buffered saline with Tween 20 for 1hr. TBST was prepared from 10X stock consisting of 80 g NaCl, 20 g KCl, 300 g Tris and brought up to a volume of 10 L; 100 mL of the 10X TBS in 900 mL DI water and then add 1 mL Tween 20 (Fisher Scientific BP337500). and incubated overnight at 4°C with rabbit monoclonal anti-fibronectin antibody (Epitomics Inc, CA; Cat #1573-1) or rabbit polyclonal anti-megalin (Santa Cruz, CA; Cat # SC16478). Bands were visualized with ECL on a Biorad Phosphorimager and quantified with Image Lab software (Biorad, Hercules CA). For megalin, the NuPage large protein analysis system was used (Invitrogen, Grand Island, NY). Briefly, lysates were prepared for loading on to Novex 3–8% Tris-Acetate gels using sample buffer and reducing agent supplied with the kit. Proteins were transferred for ~18hrs at 15V at 4°C. Blots were blocked as above and anti-megalin antibody was added for overnight incubation at 4°C.
Immunohistochemistry
Harvested kidney tissue was prepared as previously described [14,24–26]. Briefly, rehydrated paraffin embedded sections were blocked in 5% BSA, 5% donkey serum and 0.01% sodium azide in HEPES buffer for 4 hours in a humidity chamber. Following a brief rinse sections were incubated with 1:50 goat anti-megalin, 1:200 mouse monoclonal Rac1 (Millipore; Cat #05389), and 1:100 rabbit polyclonal podocin (Santa Cruz, CA; Cat #SC21009) antibodies in 10-fold diluted blocking agent overnight. After washing, sections were incubated for four hours with 1:300 Alexa-fluor donkey anti-goat 647 (Invitrogen, Grand Island NY; Cat #A21447) the slides were examined under a bi-photon confocal microscope, and the images were captured with LSM imaging system. Signal intensities were analyzed with MetaVue.
Light Microscopic Analysis for Tubulointerstitial Fibrosis
Five μm thick paraffin sections were mounted on glass slides and stained with Verhoeff van Gieson (VVG) stain, which stains collagen fibers pink, to evaluate interstitial fibrosis, as previously described [24–26]. The relative amount of collagen within 10 representative regions of interest was determined with the aid of MetaVue Software and an average value recorded for each kidney sample and expressed as arbitrary units. Samples from five rats from each of the five treatment groups were analyzed.
Ultrastructural observations with transmission electron microscopy (TEM)
Kidney cortical tissue was thinly sliced, placed immediately in primary TEM fixative, and prepared as previously described [24–26]. A JOEL 1400 TEM microscope (JOEL Ltd, Tokyo, Japan) was utilized to view all samples.
Statistical analysis
This investigation was powered based on prior sensitivity and variability measurements of albuminuria to achieve a significance of p<0.05 with a power of 0.8 [14,24–26]. All values are expressed as the mean ± standard error. Statistical analyses were performed in SPSS 13.0 (SPSS Inc., Chicago, IL) using ANOVA with Fisher's LSD for post hoc multiple comparisons.
Results
Combination therapy lowers SBP and Aldosterone more than individual therapy, but not proteinuria or β-NAG in the transgenic Ren2 rat
Consistent with prior observations in the Ren2 [14,24–27], there were increases in SBP in the Ren2 compared to SD controls (p<0.05) that was improved with administration of both aliskiren and valsartan in Ren2 animals (Fig 1A; each p<0.05). There were greater reductions in SBP in the combination treated Ren2 compared to treatment with either aliskiren or valsartan alone (each p<0.05).
Figure 1. Experimental measures at end of treatment period in the Ren2 rat.
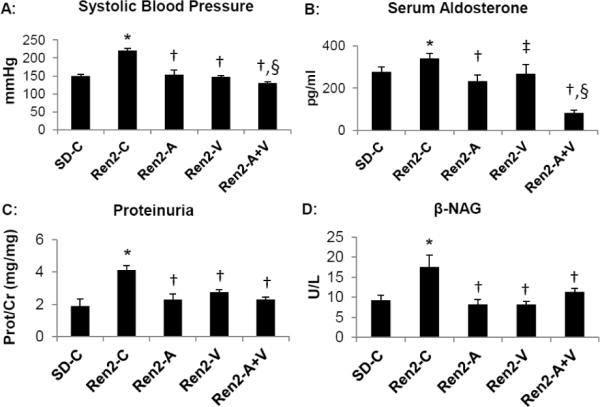
A) Systolic blood pressure measured by tail cuff. B) Serum aldosterone determined by radioimmunoassay. C) Proteinuria determined by urine protein to creatinine ratio. D) N-acetyl-beta-glucosaminidase (β-NAG) determined by colorimetric assay. Values presented as mean ± standard error. *, p<0.05 when Ren2 controls (Ren2-C) are compared to age-matched Sprague-Dawley controls (SD-C); †, p<0.05 when Ren2 rats treated with either aliskiren (Ren2-A), valsartan (Ren2-V), or combination (Ren2-A+V) are compared to age-matched Ren2-C; ‡, p<0.1 when Ren2-V are compared to Ren2-C; and §, p<0.05 when combination treated Ren2 rats are compared to all other groups.
Excessive MR activation leads to both glomerular and tubular injury independent of Ang II actions [28,29]. In this context, we measured circulating aldosterone. Consistent with previous work there increases in serum aldosterone levels in the Ren2 compared to SD controls (Fig 1B; p<0.05)) [30]. Similar to our observation with SBP, administration of aliskiren reduced serum aldosterone (p<0.05) along with a trend with valsartan (p<0.1). There were greater reductions in the combination treated Ren2 compared to treatment with either aliskiren or valsartan alone (each p<0.05).
SBP and aldosterone are important modifiable factors in mediating progressive kidney disease as measured by proteinuria. In this context, there were increases in proteinuria in Ren2 transgenic compared to SD controls (p<0.05), findings that were prevented over three weeks with administration of aliskiren, valsartan, and the combination in Ren2 animals (each p<0.05) (Fig 2A).
Figure 2. Semi-quantitative analysis of Oxidative Stress in the Ren2.

A) Representative images of immunohistochemistry analysis of kidney cortical tissue NADPH oxidase subunit Rac1 and B) 3-nitrotyrosine (3-NT), as a marker for lipid peroxidative and oxidative stress, with corresponding average gray-scale intensities below. Values presented as mean ± standard error. *, p<0.05 when Ren2 controls (Ren2-C) are compared to age-matched Sprague-Dawley controls (SD-C); †, p<0.05 when Ren2 rats treated with either aliskiren (Ren2-A), valsartan (Ren2-V), or combination (Ren2-A+V) are compared to age-matched Ren2-C. Scale bar = 50 μm.
Proteinuria is traditionally thought to be derived from glomerular origins with alterations in basement membrane and the podocyte slit-pore diaphragm [31]. However, recent work support impairments in proximal tubule reabsorption of protein, as well as a glomerular contribution to early development of proteinuria [32]. Injury to the proximal tubule brush border is characterized by release of enzymes such as β-NAG a glycosidase found in proximal tubular epithelial cell lysosomes [32]. In young Ren2 rats there were parallel increases in β-NAG and proteinuria compared to SD controls (p<0.05) improved with aliskiren, valsartan, and the combination treatment (Fig 1D; each p<0.05).
Combination adds little to improvements in oxidative stress in the transgenic Ren2 rat
The NADPH oxidase enzyme complex is an important regulator of RAAS-dependent generation of oxidant stress in the kidney. Consistent with this notion, there were increases in cortical tissue proximal tubule intensity of subunit Rac1 in the Ren2 rat compared to SD controls (p<0.05). These collective findings were improved to a similar extent with aliskiren, valsartan or combination treatment (each p<0.05). Immunostaining for 3-nitrotyrosine (3-NT), as a marker for peroxynitrite (ONOO−) formation, was similarly increased in the Ren2 when compared to SD renal cortical tissue (p<0.05; Fig 2). 3-NT represents increased oxido- and nitroso- lipid products, not simply reactive oxygen (ROS) formation. Similar to Rac1, 3-NT content was improved to a similar extent with aliskiren, valsartan or combination treatment (each p<0.05).
Combination does not improve proximal tubule protein handling in the transgenic Ren2 rat
Oxidant injury to the proximal tubule cell is associated with loss of the brush border endocytotic receptor megalin that is responsible for proximal tubule cell reabsorption of protein. To this point, the increases in proteinuria and β-NAG observed in Ren2 controls, was associated with a parallel reduction in megalin (Fig 3A and B, p<0.05). Treatment with both aliskiren and valsartan increased megalin to a similar extent in the Ren2 (each p<0.05). However, combination aliskiren and valsartan had no impact on megalin expression in the Ren2 (p>0.05 when compared to Ren2 controls and p<0.05 when compared to aliskiren and valsartan treatment). Ultrastructural analysis of the S-1 segment of the proximal tubule utilizing TEM corroborated the megalin findings (Fig 3C). Representative images demonstrated very little endosomal lysosomal activity (megalin dependent process) in the Ren2-C as compared to SD-C, findings were improved with both aliskiren and valsartan treatment. However there were no qualitative differences between the three treatment groups.
Figure 3. Proximal tubule protein handling in the Ren2.

A) Representative images from immunohistochemistry analysis of megalin with corresponding measures to the below. Scale bar = 50 μm. B) Western blot analysis of megalin. Values presented as mean ± standard error. *, p<0.05 when Ren2 controls (Ren2-C) are compared to age-matched Sprague-Dawley controls (SD-C); †, p<0.05 when Ren2 rats treated with either aliskiren (Ren2-A), valsartan (Ren2-V), or combination (Ren2-A+V) are compared to age-matched Ren2-C; §, p<0.05 when combination treated Ren2 rats are compared to aliskiren or valsartan treatment. C) Representative images from ultrastructural analysis utilizing transmission electron microscopy for the S-1 region of the PTC. Images depict from Ren2-C (top right panel) demonstrate very little endosomal lysosomal activity with decreased clathrin coated pits, endosomes, and apical vacuoles within the endosomal regions as compared to SD-C (top left panel). Note that Ren2-A (bottom left panel), Ren2-V (bottom middle panel), and Ren2-A+V (bottom right panel) restore these decreased entities. Scale bar = 0.2 μm.
Improvements in tubulointerstitial fibrosis in the transgenic Ren2 rat
Progressive kidney disease is characterized morphologically by tubulointerstitial fibrosis. Consistent with previous work in the Ren2 model [14,24–26], there were increases in tubulointerstitial fibrosis in Ren2 compared to SD controls (p<0.05), improved with both aliskiren, valsartan, and combination treatment in the Ren2 (Fig 4A; each p<0.05)). Commensurate with our fibrosis findings, we observed increases in collagen type III and fibronectin in Ren2 compared to SD controls (Fig 4B and C, respectively; each p<0.05). There were improvements with aliskiren, valsartan, and combination treatment in fibronectin (each p<0.05). There were similar reduction in collagen III with aliskiren and valsartan treatment (each p<0.05); however, there was a non-statistical trend with combination treatment.
Figure 4. Tubulointerstitial Fibrosis in the Ren2.

A) Verhoeff-Van Gieson (VVG) stain for elastin and collagen with measures of tubulointerstitial fibrosis below. Scale bar = 50 μm. B) Immunohistochemistry analysis of collagen type III. C) Western blot analysis of fibronectin. Values presented as mean ± standard error. *, p<0.05 when Ren2 controls (Ren2-C) are compared to age-matched Sprague-Dawley controls (SD-C); †, p<0.05 when Ren2 rats treated with either aliskiren (Ren2-A), valsartan (Ren2-V), or combination (Ren2-A+V) are compared to age-matched Ren2-C. Scale bar = 50 μm. D) Representative images from ultrastructural analysis of transmission electron microscopy for the S-1 region of the proximal tubule. Images depict depicts loss of basal polarity with elongated canalicular plasma membrane infoldings and mitochondria, and basement membrane (BM) thickening in the basal region of the R2-C (middle panel) compared to SD-C (left panel), findings improved only with combination Ren2-A+V (right panel) and not in Ren2-A and Ren2-V as depicted in Figure 2C. Scale bar = 0.2 μm
Early stage proximal tubule epithelial cell promotion of tubulointerstitial fibrosis is characterized by ultrastructural loss of basal polarity and basement membrane thickening. Consistent with reductions in apical endosomal findings in the Ren2 (Fig 4D), there were additional reductions in basilar lysosomes with parallel loss in basal polarity compared to SD controls. The loss of polarity was manifested by ultrastructural remodeling consisting of mitochondrial fragmentation, which is characterized by loss of elongation and spherical enlargement with mitochondrial matrix abnormalities. Further, there was basement membrane thickening and loss of elongated canalicular plasma membrane infoldings characterized by electron dense finger-like protrusions along the canaliculi in Ren2 renal tissue. Treatment with combination aliskiren and valsartan improved this loss of basal polarity but not aliskiren or valsartan alone in Ren2 rats.
Improvements in glomerular filtration barrier/podocyte injury in the transgenic Ren2 rat
Ang II as well as aldosterone contributes to oxidant injury to the glomerular filtration barrier characterized by increases in the podocyte slit-pore diaphragm. Podocin is a lipid raft-associated protein that plays a critical role in recruiting nephrin and structural organization of the slit diaphragm. Consistent with prior observations of podocyte injury-associated increases in albuminuria, there was decreased expression of the podocyte-specific protein podocin in Ren2 cortical tissue when compared to SD controls (Fig 5A; p<0.05), improved with aliskiren, valsartan or combination treatment in the Ren2 (each, p<0.05).
Figure 5. Podocyte Foot Process Integrity in the Ren2.
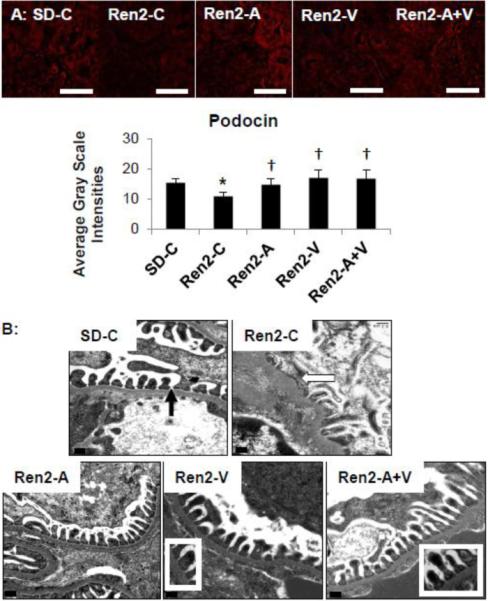
A) Representative images of immunohistochemistry analysis of the podocyte specific protein podocin with corresponding measures below. Values presented as mean ± standard error. *, p<0.05 when Ren2 controls (Ren2-C) are compared to age-matched Sprague-Dawley controls (SD-C); †, p<0.05 when Ren2 rats treated with either aliskiren (Ren2-A), valsartan (Ren2-V), or combination (Ren2-A+V) are compared to age-matched Ren2-C. Scale bar = 50 μm. B) Representative images from ultrastructural analysis of transmission electron microscopy for the glomerular filtration barrier on transmission electron microscopy. Images depict Ren2-C (top right panel) displays loss of glomerular filtration barrier integrity with podocyte foot-process effacement and loss of the slit-pore diaphragm (arrows) compared to SD-C (top left panel). Note that Ren2-A (bottom left panel), Ren2-V (bottom middle panel), and Ren2-A+V (bottom right panel) restore these findings (inserts). Scale bar = 0.2 μm
Ultrastructural analysis of the glomerular filtration barrier was consistent with the podocin findings. The Ren2 gomerular tissue displayed increases in basement membrane thickness with additional podocyte foot process effacement and loss of the slit-pore integrity compared to SD controls. These observed ultrastructural abnormalities were corrected to a similar extent with aliskiren, valsartan or combination treatment in the Ren2 kidney.
Discussion
Results from this investigation support the concept that combination therapy with direct renin inhibition and an AT1R blockade contribute to greater reductions in SBP as well as aldosterone levels compared to either intervention alone in conditions of RAAS activation such as the transgenic Ren2 rat overexpressing the murine renin transgene. Both individual treatment strategies substantially reduced proteinuria and urine levels of the proximal tubular derived β-NAG in the transgenic rat. Each of the treatments also lessened renal cortical measures of oxidative stress and histological evidence of glomerular and tubular injury in this rat model overexpressing the mouse renin gene [33–35]. However, combination therapy provided little additional benefit compared to either intervention alone in improving proteinuria or glomerular and proximal tubule injury in this transgenic model. Thereby our data highlight the importance of Ang II-dependent changes in blood pressure on markers of podocyte and proximal tubule integrity in this transgenic model.
The finding that combination AT1R blockade and direct renin inhibition led to greater reductions in serum aldosterone levels in the Ren2 is novel, and corroborates an observation from one human cohort that combination of aliskiren and valsartan led to greater reductions in urinary aldosterone excretion compared to either intervention alone in sodium replete subjects [36]. The premise that inhibition of renin activation in combination with blockade of the AT1R would lead to a more effective reduction of adrenal production of aldosterone has potential relevance as a therapeutic strategy. This is especially pertinent when considering the demonstrated importance of enhanced local renin activation in adrenal aldosterone generation in this model of enhanced tissue renin expression [15,34,37. Indeed, previous work in this transgenic model support the notion that the adrenal gland has a high concentration of mouse and rat renin content, both (pro)renin and active renin, in the adrenal zona glomerulosa [15]. Further, it has been observed that aldosterone production is highly dependent on tissue renin activity, wherein inhibition of renin with a monocolonal antibody and CP71632 led to reductions in activity and aldosterone [15]. Another report indicated that Ren2 adrenal renin activity is stimulated by cAMP, calcium, and Ang II, suggesting an activated paracrine adrenal RAAS as well as diminished negative feedback by Ang II on renin-dependent aldosterone generation [38].
Increased tissue renin expression is associated with enhanced oxidative stress in the heart, kidneys and adrenal in the Ren2 rat [14,34,35,39]. In this regard, there is evidence that Ang II promotes aldosterone production through a NADPH oxidase and mitochondrial dependent generation of reactive oxygen species [40]. Oxidant stress in this transgenic model is driven by AT1R activation of the enzyme complex NADPH oxidase and mitochondrial uncoupling [14, 39]. Thereby, the observed reductions in circulating aldosterone with renin inhibition as well as in combination with AT1R blockade are likely driven by renin inhibition of renin-dependent adrenal aldosterone production that is unique to this model in conjunction with blockade of AT1R-dependent aldosterone production.
Mineralocorticoid receptor signaling also contributes substantially to kidney tissue injury through increased generation of reactive oxygen species. Our finding that direct renin inhibition and AT1R blockade led to improvements in both proximal tubule and podocyte injury is consistent with prior observations in this model [14,24–26]. We hypothesized the greater reductions in aldosterone with combination therapy would contribute to a greater protection from oxidant stress-induced glomerular and proximal tubule injury. However, our finding that combination therapy did not provide an additional benefit differs from two other reports [41–43].
Recent work from a rat model of ureteral obstruction as well as two mouse models including an endothelial nitric oxide (eNOS) deficient model [41] and the db/db [42] support that combination aliskiren and valsartan led to greater reductions in proteinuria as well as markers of glomerular and proximal tubule injury compared to either intervention alone. Findings from the the eNOS−/− model that manifest hypertension and the db/db mouse, a model of progressive insulin resistance and beta cell dysfunction suggest that combination of aliskiren and valsartan at lower doses than used in our study led to greater reductions in BP as well as improvement in proteinuria in both models that were related to improvements in glomerulosclerosis, NADPH oxidase-dependent oxidant stress, and the podocyte-specific markers [41]. However, in the rat model of unilateral ureteral obstruction (UUO) that manifests significant tubulointerstitial fibrosis, there was no additional benefit with combination on BP or proteinuria but had a greater impact on scores of tubulointerstitial injury compared to either intervention alone [43]. The discordant data between the three groups would suggest our findings are very model and blood pressure-dependent rather than due to success or failure of the combination strategy.
In this study conducted in a rodent model of RAAS activation the data supports the notion that RAAS activation of NADPH oxidase contributes to ROS formation in kidney tissue injury as evidenced by increases tubulointerstitial fibrosis. Our finding of greater increases in the NADPH oxidase subunit Rac1 and peroxynitrite (ONOO−) as measured by 3-NT content in the Ren2 occur contemporaneous with increases in fibronectin, collagen III, and light microscopy findings of tubulointerstitial fibrosis support oxidant stress as a factor in eliciting kidney tissue injury. Our additional finding that AT1R blockade and direct renin inhibition attenuates these findings is consistent with previous reports in the Ren2 model [14], and the finding that combination led to comparable improvements in markers of fibrosis contrast with findings from the UUO model further supporting a model-dependent effect. Fibrosis in the UUO model is driven by increases in inflammation and infiltration of the interstitium by monocytes/macrophages that have been shown to possess the (pro)renin receptor [42] and the mineralcorticoid receptor [43] and thereby be more susceptible to the combination of renin inhibition and AT1R blockade with downstream reductions in aldosterone. Previous reports in the UUO model suggest blockade of the MR attenuates progression of fibrosis [43] while a previous report in the Ren2 suggest that blood pressure reductions may be paramount in improving markers of glomerular injury rather than specific actions of renin inhibition or AT1R blockade [14].
Our additional finding that glomerular filtration barrier/podocyte and proximal tubule injury occurred in parallel in young Ren2 rats (aged 6–9 weeks) further support a proximal tubule contribution to early stages in the development of proteinuria and not just confined to alterations in the filtration barrier [32,43]. This is evidenced by observed increases in the proximal tubule marker β-NAG in parallel with reductions in megalin along with ultrastructural observations consistent with reductions in endosomal/lysosomal activity support impairments in proximal tubule reabsorption of protein in the Ren2. The proximal tubule findings occurred temporally related to reductions in the podocyte-specific protein podocin and ultrastructural changes consistent with podocyte foot-process effacement with increases in the NADPH oxidase subunit Rac1 and 3-NT content.
In various animals and in man, glomerular abnormalities have been emphasized in the etiology of proteinuria. These abnormalities include reductions in podocyte-specific proteins (podocin) and podocyte foot process effacement/reduction in slit-pore diaphragm integrity on ultrastructure analysis. However, recent work from several groups suggest that at early stages of metabolic kidney disease, we need to also consider proximal tubular contributions from impairments in either the retrieval or degradation process in the proximal tubule [38]. Our finding that urine levels of β-NAG, a specific urine marker for injury to the PTC brush border, was increased in parallel with reductions in megalin expression and ultrastructural observations of reduced endosomal activity support impaired proximal tubule reabsorption of protein.
Megalin is a chaperone protein involved in the retrieval mechanism of albumin reabsorption directly as a receptor and indirectly by affecting the expression of cubilin, which is co-expressed with megalin in the brush border and the endocytic apparatus. Recent data suggest the RAAS may disrupt the retrieval mechanism in the PTC and highlight the impact Ang II has on disruption of cytoskeletal organization [44]. In this regard, AT1AR-dependent signaling reduces megalin expression in cultured proximal tubule cells, and that blockade of the AT1R improves megalin expression and lysosomal degradation of albumin [44,45]. Our finding that both renin inhibition and AT1R blockade improved megalin as well as β-NAG further refine this relationship between Ang II-dependent generation of oxidative stress and proximal tubule handling of protein reabsorption.
Recent work also suggests that aldosterone may regulate proximal tubule brush border NHE3 transport through an epidermal growth factor-receptor (EGFR)-dependent mechanism [47]. Moreover, in this same study antagonism of MR led to reductions in EGFR expression. Interestingly, extracellular domain of megalin contains a single EGF-like repeat [48] and is proposed to bind to the EGFR and mediate downstream cytokine signaling. This suggests the possibility the reductions in aldosterone in the combination arm may explain, in part, the lack of benefit on improved megalin expression compared to the individual component. While it is not known whether the MR binds directly to the EGFR promoter as potential mechanism in megalin expression, our finding is novel and warrants future more mechanistic investigation in culture models. Further, the finding that combination therapy had no impact on megalin yet improved β-NAG further suggest an Ang II-dominant role in regulation of proximal tubule protein handling and may suggest that AT1R effects predominate over MR effects in improving megalin integrity.
Our transgenic model which harbors both the native Ren1 and the murine renin transgene with increased tissue Ang II and circulating aldosterone was chosen for the elicit purpose of interrogating the RAAS. Results from this investigation support that combination DRI with AT1R blockade contributes to greater reductions circulating aldosterone but highlight the importance of Ang II-dependent changes in blood pressure on markers of podocyte and proximal tubule integrity in this transgenic model of Ang II-dependent elevations in blood pressure. Our findings merit future work in other physiological relevant models that display activation of the RAAS such as diabetic or nephrectomized models.
Highlights
We present data on a relevant translational therapeutic strategy for the treatment of kidney disease by comparing the combination of direct renin inhibition with blockade of the angiotensin type 1 receptor (AT1R) to individual components in a transgenic model of RAAS-dependent kidney damage the TG(mRen2)27 rat.
Combination therapy contributed to greater reductions in circulating aldosterone and systolic blood pressure compared to individual components. However, did not improve markers of oxidative stress and podocyte and proximal tubule integrity.
Our data suggest AT1R effects predominate over MR effects in improving markers of kidney injury in this model.
Acknowledgements
The authors would like to acknowledge Brenda Hunter for editing this manuscript. The authors would also like to acknowledge the Electron Microscopic Core Center at the University of Missouri, Columbia, Missouri for their excellent help and tissue preparation of animal samples for viewing. Lastly, the authors would like to acknowledge Charles E Wiedmeyer, DVM, PhD for performing the proteinuria and β-NAG measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1).Nistala R, Whaley-Connell A, Sowers JR. Redox control of renal function and hypertension. Antioxid Redox Signal. 2008;10(12):2047–89. doi: 10.1089/ars.2008.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Jaimes EA, Hua P, Tian RX, Raij L. Human glomerular endothelium: interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am J Physiol Renal Physiol. 2010;298(1):F125–32. doi: 10.1152/ajprenal.00248.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Fujii H, Kono K, Nakai K, Goto S, Komaba H, Hamada Y, Shinohara M, Kitazawa R, Kitazawa S, Fukagawa M. Oxidative and nitrosative stress and progression of diabetic nephropathy in type 2 diabetes. Am J Nephrol. 2010;31(4):342–352. doi: 10.1159/000297290. [DOI] [PubMed] [Google Scholar]
- 4).Rossing P, Hommel E, Smidt UM, Parving HH. Reduction in albuminuria predicts a beneficial effect on diminishing the progression of human diabetic nephropathy during antihypertensive treatment. Diabetologia. 1994;37(5):511–6. doi: 10.1007/s001250050140. [DOI] [PubMed] [Google Scholar]
- 5).Kalaitzidis R, Bakris GL. Effects of angiotensin II receptor blockers on diabetic nephropathy. J Hypertens. 2009;27(5):S15–21. doi: 10.1097/01.hjh.0000357904.71080.7d. [DOI] [PubMed] [Google Scholar]
- 6).Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK, AVOID Study Investigators Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358(23):2433–46. doi: 10.1056/NEJMoa0708379. [DOI] [PubMed] [Google Scholar]
- 7).Epstein M, Williams GH, Weinberger M, Lewin A, Krause S, Mukherjee R, Patni R, Beckerman B. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol. 2006;1(5):940–51. doi: 10.2215/CJN.00240106. [DOI] [PubMed] [Google Scholar]
- 8).Foley RN, Collins AJ. End-stage renal disease in the United States: an update from the United States Renal Data System. J Am Soc Nephrol. 2007;18(10):2644–8. doi: 10.1681/ASN.2007020220. [DOI] [PubMed] [Google Scholar]
- 9).Holtkamp FA, de Zeeuw D, de Graeff PA, Laverman GD, Berl T, Remuzzi G, Packham D, Lewis JB, Parving HH, Lambers Heerspink HJ. Albuminuria and blood pressure, independent targets for cardioprotective therapy in patients with diabetes and nephropathy: a post hoc analysis of the combined RENAAL and IDNT trials. Eur Heart J. 2011;32(12):1493–9. doi: 10.1093/eurheartj/ehr017. [DOI] [PubMed] [Google Scholar]
- 10).Sowers JR, Whaley-Connell A, Epstein M. The emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776–783. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3:486–492. doi: 10.1038/ncpneph0575. [DOI] [PubMed] [Google Scholar]
- 12).Hollenberg NK, Fisher ND, Price DA. Pathways for angiotensin II generation in intact human tissue: evidence from comparative pharmacological interruption of the renin system. Hypertension. 1998;32:387–392. doi: 10.1161/01.hyp.32.3.387. [DOI] [PubMed] [Google Scholar]
- 13).Wiggins KJ, Kelly DJ. Aliskiren: a novel renoprotective agent or simply an alternative to ACE inhibitors? Kidney Int. 2009;76:23–31. doi: 10.1038/ki.2009.105. [DOI] [PubMed] [Google Scholar]
- 14).Whaley-Connell A, Nistala R, Habibi J, Hayden MR, Schneider RI, Johnson MS, Tilmon R, Rehmer N, Ferrario CM, Sowers JR. Comparative effect of direct renin inhibition and AT1R blockade on glomerular filtration barrier injury in the transgenic Ren2 rat. Am J Physiol Renal Physiol. 2010;298(3):F655–61. doi: 10.1152/ajprenal.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Yamaguchi T, Tokita Y, Franco-Saenz R, Mulrow PJ, Peters J, Ganten D. Zonal distribution and regulation of adrenal renin in a transgenic model of hypertension in the rat. Endocrinology. 1992;131(4):1955–62. doi: 10.1210/endo.131.4.1396339. [DOI] [PubMed] [Google Scholar]
- 16).Feldman DL, Jin L, Xuan H, Contrepas A, Zhou Y, Webb RL, Mueller DN, Feldt S, Cumin F, Maniara W, Persohn E, Schuetz H, Jan Danser AH, Nguyen G. Effects of aliskiren on blood pressure, albuminuria, and (pro)renin receptor expression in diabetic TG(mRen-2)27 rats. Hypertension. 2008;52(1):130–6. doi: 10.1161/HYPERTENSIONAHA.107.108845. [DOI] [PubMed] [Google Scholar]
- 17).Nguyen G, Danser AHJ. Prorenin and (pro)renin receptor: a review of available data from in vitro studies and experimental models in rodents. Exp Physiol. 2008;93:557–563. doi: 10.1113/expphysiol.2007.040030. [DOI] [PubMed] [Google Scholar]
- 18).Nguyen G, Delarue F, Berrou J, Rondeau E, Sraer JD. Specific receptor binding of renin on human mesangial cells in culture increases plasminogen activator inhibitor-1 antigen. Kidney Int. 1996;50:1897–1903. doi: 10.1038/ki.1996.511. [DOI] [PubMed] [Google Scholar]
- 19).Kelly DJ, Zhang Y, Moe G, Naik G, Gilbert RE. Aliskiren, a novel renin inhibitor, is renoprotective in a model of advanced diabetic nephropathy in rats. Diabetologia. 2007;50:2398–2404. doi: 10.1007/s00125-007-0795-9. [DOI] [PubMed] [Google Scholar]
- 20).Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Blundell T, Sibanda BL, Pearl L. Three-dimensional structure, specificity and catalytic mechanism of renin. Nature. 1983;304(5923):273–5. doi: 10.1038/304273a0. [DOI] [PubMed] [Google Scholar]
- 22).Lu H, Rateri DL, Feldman DL, RJ, Fukamizu A, Ishida J, Oesterling EG, Cassis LA, Daugherty A. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J Clin Invest. 2008;118(3):984–93. doi: 10.1172/JCI32970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Bohlender J, Menard J, Edling O, Ganten D, Luft FC. Mouse and rat plasma renin concentration and gene expression in (mRen2)27 transgenic rats. Am J Physiol. 1998;274(5 Pt 2):H1450–6. doi: 10.1152/ajpheart.1998.274.5.H1450. [DOI] [PubMed] [Google Scholar]
- 24).Whaley-Connell AT, Chowdhury NA, Hayden MR, Stump CS, Habibi J, Wiedmeyer CE, Gallagher PE, Tallant EA, Cooper SA, Link CD, Ferrario C, Sowers JR. Oxidative stress and glomerular filtration barrier injury: role of the renin-angiotensin system in the Ren2 transgenic rat. Am J Physiol Renal Physiol. 2006;291(6):F1308–14. doi: 10.1152/ajprenal.00167.2006. [DOI] [PubMed] [Google Scholar]
- 25).Whaley-Connell A, Habibi J, Nistala R, Cooper SA, Karuparthi PR, Hayden MR, Rehmer N, DeMarco VG, Andresen BT, Wei Y, Ferrario C, Sowers JR. Attenuation of NADPH oxidase activation and glomerular filtration barrier remodeling with statin treatment. Hypertension. 2008;51(2):474–80. doi: 10.1161/HYPERTENSIONAHA.107.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Whaley-Connell A, Habibi J, Wei Y, Gutweiler A, Jellison J, Wiedmeyer CE, Ferrario CM, Sowers JR. Mineralocorticoid receptor antagonism attenuates glomerular filtration barrier remodeling in the transgenic Ren2 rat. Am J Physiol Renal Physiol. 2009;296(5):F1013–22. doi: 10.1152/ajprenal.90646.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Lee MA, Bohm M, Paul M, Bader M, Ganten U, Ganten D. Physiological characterization of the hypertensive transgenic rat TGR(mREN2)27. Am J Physiol Endocrinol Metab. 1996;270:E919–E929. doi: 10.1152/ajpendo.1996.270.6.E919. [DOI] [PubMed] [Google Scholar]
- 28).Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: Roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–64. doi: 10.1161/01.HYP.0000255636.11931.a2. [DOI] [PubMed] [Google Scholar]
- 29).Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt sensitive rats and is reversed by aldosterone blocker. Hypertension. 2006;471:1084–93. doi: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- 30).Habibi J, DeMarco VG, Ma L, Pulakat L, Rainey WE, Whaley-Connell AT, Sowers JR. Mineralocorticoid receptor blockade improves diastolic function independent of blood pressure reduction in a transgenic model of RAAS overexpression. Am J Physiol Heart Circ Physiol. 2011;300(4):H1484–91. doi: 10.1152/ajpheart.01000.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Johnstone DB, Holtzman LB. Clinical impact of research on the podocyte slit diaphragm. Nat Clin Pract Nephrol. 2006;2:271–282. doi: 10.1038/ncpneph0180. [DOI] [PubMed] [Google Scholar]
- 32).Russo LM, Sandoval RM, Campos SB, Molitoris BA, Comper WD, Brown D. Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J Am Soc Nephrol. 2009;20(3):489–94. doi: 10.1681/ASN.2008050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Langheinrich M, Lee MA, Böhm M, Pinto YM, Ganten D, Paul M. The hypertensive Ren-2 transgenic rat TGR (mREN2)27 in hypertension research. Characteristics and functional aspects. Am J Hypertens. 1996;9(5):506–12. doi: 10.1016/0895-7061(95)00400-9. [DOI] [PubMed] [Google Scholar]
- 34).Peters J, Ganten D. Adrenal renin expression and its role in Ren2 transgenic rats TGR(mREN2)27. Horm Metab Res. 1998;30(6–7):350–4. doi: 10.1055/s-2007-978897. [DOI] [PubMed] [Google Scholar]
- 35).Böhm M, Lee M, Kreutz R, Kim S, Schinke M, Djavidani B, Wagner J, Kaling M, Wienen W, Bader M, et al. Angiotensin II receptor blockade in TGR(mREN2)27: effects of reninangiotensin-system gene expression and cardiovascular functions. J Hypertens. 1995;13(8):891–9. doi: 10.1097/00004872-199508000-00010. [DOI] [PubMed] [Google Scholar]
- 36).Azizi M, Ménard J, Bissery A, Guyene TT, Bura-Rivière A. Hormonal and hemodynamic effects of aliskiren and valsartan and their combination in sodium-replete normotensive individuals. Clin J Am Soc Nephrol. 2007;2(5):947–55. doi: 10.2215/CJN.00360107. [DOI] [PubMed] [Google Scholar]
- 37.Rocco S, Rebuffat P, Cimolato M, Opocher G, Peters J, Mazzocchi G, Ganten D, Mantero F, Nussdorfer GG. Zona glomerulosa of the adrenal gland in a transgenic strain of rat: a morphologic and functional study. Cell Tissue Res. 1994;278(1):21–8. doi: 10.1007/BF00305774. [DOI] [PubMed] [Google Scholar]
- 38.Peters J, Münter K, Bader M, Hackenthal E, Mullins JJ, Ganten D. Increased adrenal renin in transgenic hypertensive rats, TGR(mREN2)27, and its regulation by cAMP, angiotensin II, and calcium. J Clin Invest. 1993;91(3):742–7. doi: 10.1172/JCI116292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, Ma L, Qazi M, Link D, Karuparthi PR, Stump C, Ferrario C, Sowers JR. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab. 2007;293(1):E355–63. doi: 10.1152/ajpendo.00632.2006. [DOI] [PubMed] [Google Scholar]
- 40.Rajamohan SB, Raghuraman G, Prabhakar NR, Kumar GK. NADPH oxidase-derived H2O2 Contributes to Angiotensin II-induced Aldosterone Synthesis in Human and Rat Adrenal Cortical Cells. Antiox Redox Sign. 2012 doi: 10.1089/ars.2011.4176. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto E, Kataoka K, Dong YF, Nakamura T, Fukuda M, Tokutomi Y, Matsuba S, Nako H, Nakagata N, Kaneko T, Ogawa H, Kim-Mitsuyama S. Aliskiren enhances the protective effects of valsartan against cardiovascular and renal injury in endothelial nitric oxide synthase-deficient mice. Hypertension. 2009;54(3):633–8. doi: 10.1161/HYPERTENSIONAHA.109.133884. [DOI] [PubMed] [Google Scholar]
- 42.Dong YF, Liu L, Lai ZF, Yamamoto E, Kataoka K, Nakamura T, Fukuda M, Tokutomi Y, Nako H, Ogawa H, Kim-Mitsuyama S. Aliskiren enhances protective effects of valsartan against type 2 diabetic nephropathy in mice. J Hypertens. 2010;28(7):1554–65. doi: 10.1097/HJH.0b013e328338bb11. [DOI] [PubMed] [Google Scholar]
- 43.Wu WP, Chang CH, Chiu YT, Ku CL, Wen MC, Shu KH, Wu MJ. A reduction of unilateral ureteral obstruction-induced renal fibrosis by a therapy combining valsartan with aliskiren. Am J Physiol Renal Physiol. 2010;299(5):F929–41. doi: 10.1152/ajprenal.00192.2010. [DOI] [PubMed] [Google Scholar]
- 44.Trachtman H, Weiser AC, Valderrama E, Morgado M, Palmer LS. Prevention of renal fibrosis by spironolactone in mice with complete unilateral ureteral obstruction. J Urol. 2004;172(4 Pt 2):1590–4. doi: 10.1097/01.ju.0000140445.82949.54. [DOI] [PubMed] [Google Scholar]
- 45.Tojo A, Onozato ML, Kurihara H, Sakai T, Goto A, Fujita T. Angiotensin II blockade restores albumin reabsorption in the proximal tubules of diabetic rats. Hypertens Res. 2003;26(5):413–9. doi: 10.1291/hypres.26.413. [DOI] [PubMed] [Google Scholar]
- 46.Hosojima M, Sato H, Yamamoto K, Kaseda R, Soma T, Kobayashi A, Suzuki A, Kabasawa H, Takeyama A, Ikuyama K, Iino N, Nishiyama A, Thekkumkara TJ, Takeda T, Suzuki Y, GejyoF, Saito A. Regulation of megalin expression in cultured proximal tubule cells by angiotensin II type 1A receptor-and insulin mediated signaling cross talk. Endocrinology. 2009;150(2):871–878. doi: 10.1210/en.2008-0886. [DOI] [PubMed] [Google Scholar]
- 47.Drumm K, Kress TR, Gassner B, Krug AW, Gekle M. Aldosterone stimulates activity and surface expression of NHE3 in human primary proximal tubule epithelial cells (RPTEC) Cell Physiol Biochem. 2006;17(1–2):21–8. doi: 10.1159/000091456. [DOI] [PubMed] [Google Scholar]
- 48.Verroust PJ, Birn H, Nielsen R, Kozyraki R, Christensen EI. The tandem endocytic receptors megalin and cubilin are important proteins in renal pathology. Kidney Int. 2002;62(3):745–56. doi: 10.1046/j.1523-1755.2002.00501.x. [DOI] [PubMed] [Google Scholar]