

Abstract
Notch is an ancient transmembrane receptor with critical roles in cell-fate choices. While the “canonical” Notch pathway and its core members are well established -- involving ligand-induced cleavage of Notch for transcriptional regulation -- it has been unclear whether Notch can also function independent of ligand and transcription (‘non-canonically’) through a common mechanism. Recent studies suggest that Notch can non-canonically exert its biological functions by posttranslationally targeting Wnt/β-catenin signaling, an important cellular and developmental regulator. The non-canonical Notch pathway appears to be highly conserved from flies to mammals. Here, we discuss the emerging conserved mechanism and role of ligand/transcription-independent Notch signaling in cell and developmental biology.
Keywords: Notch, Wnt/β-Catenin, Numb, DAPT, Stem and Progenitor Cells, Cancer
Canonical vs. Non-Canonical Notch Signaling
Nearly a century ago, the name Notch was given to an allele found to cause notched fly wings; since this time, the gene encoding the transmembrane protein Notch has been extensively investigated for its function and mechanisms [1–4]. The investigation led to the identification of key members of Notch signaling including ligands, proteases, and transcriptional co-factors, forming the dogma of the canonical Notch signal transduction pathway (Box 1). While Notch mediates a number of biological processes through the canonical pathway, a ligand- or transcription-independent (non-canonical) function of Notch has also been reported [5–29]. However, due to lack of mechanistic understandings, it was unknown whether the non-canonical function represented a general role for Notch. Over the past several years, multiple laboratories have reported a novel non-canonical role for Notch[5, 15, 17, 30]: antagonizing Wnt/β-catenin signaling (Box 2) -- a critical regulator of development and disease -- independent of Notch ligand-dependent cleavage or nuclear localization. Given the considerable reciprocal involvement of Notch and Wnt/β-catenin signaling in fundamental cellular processes like expansion and differentiation, understanding the non-canonical role of Notch could provide invaluable insights into regenerative medicine and disease therapeutics. This review will summarize our current understanding of non-canonical Notch biology, and discuss in detail the emerging role and mechanism of non-canonical Notch regulation of Wnt/β-catenin signaling.
Box 1. Canonical vs. Non-Canonical Notch Signaling.
Notch is an evolutionarily conserved single-pass transmembrane receptor that affects numerous cell fate decisions through short-range cell-cell interactions. Notch protein (cLIN-12 and cGLP-1 in Caenorhabditis elegans, Notch in Drosophila, Notch1–4 in mammals) consists of the extracellular domain (NECD) with 29–36 epidermal growth factor (EGF) repeats for ligand binding, the transmembrane domain (TM), and the intracellular domain (NICD) with transcriptional activity [1, 77]. The canonical Notch pathway initiates when Notch ligands – transmembrane proteins characterized by three motifs: DSL (Delta, Serrate, LAG-2), DOS (Delta and OSM-11 like) and EGF repeats – bind to the EGF repeats 11–12 and 24–29 of NECD from adjacent cells (Figure Ia). The ligand-NECD interaction allows members of the α-secretase/metalloprotease family (ADAM10/Kuzmanian, ADAM17/TACE) to shed NECD, leading to sequential cytoplasmic cleavage of NICD by γ-secretase – a multi-subunit protease complex composed of presenilin (PS), nicastrin (NCT), Aph-1, Pen-2 and others [78–80]. The resulting NICD translocates to the nucleus, where the RAM domain of NICD interacts with the DNA-binding transcription factor CSL (CBF1/RBPjk in vertebrates, Suppressor of Hairless in Drosophila, Lag-1 in C. elegans). NICD functions as a co-activator for CSL, Mastermind-like proteins (Mastermind in drosophila, MAML1 in mammals, Lag-3 in C. elegans) and other cofactors such as CBP/p300 to transcriptionally activate Notch target genes [81–83]. In the absence of NICD, CSL functions as a sequence-specific repressor [84]. In addition to the RAM domain, NICD consists of an Ankyrin repeat domain, which is involved in protein interactions, a transactivation domain and a PEST domain rich in proline, glutamate, serine and threonine residues. Non-canonical Notch signaling is CSL-independent and can be either ligand-dependent or independent (Figure Ib). Although some genes are affected by non-canonical Notch function, in most cases the mediators of non-canonical Notch signaling are unknown (summarized in Table 1). The most well studied and conserved effect of non-canonical Notch function is regulation of Wnt/β-catenin signaling:;Notch binds and titrate levels of the obligate Wnt-signaling component active β-catenin. Therefore, active β-catenin activity may serve as a useful readout for non-canonical Notch signals. Currently, at least in mammals, there is no simple genetic approach or tool available to test non-canonical Notch function in vivo; testing likely requires combinatorial deletion/overexpression of Notch members including Notch, NICD, CSL, Mastermind, Ligands, and Presenilin.
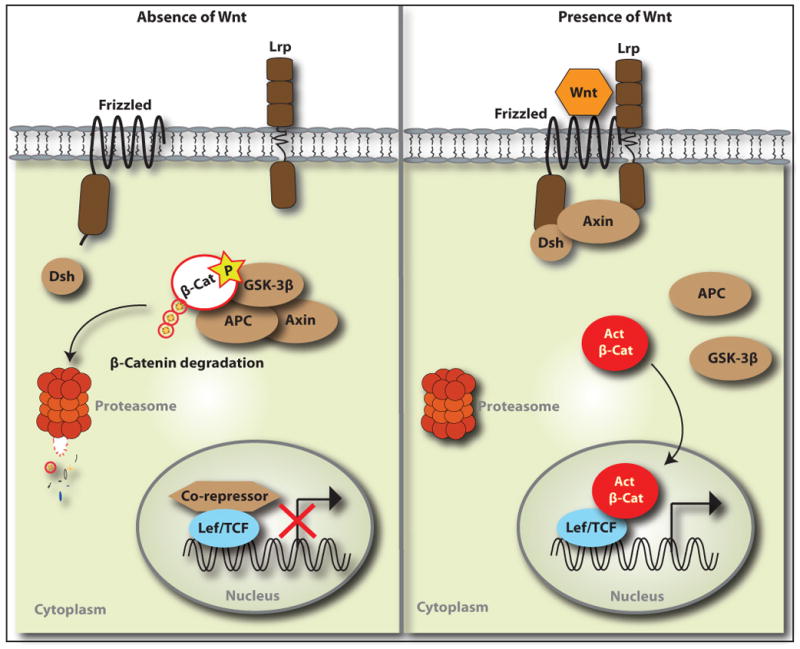
Box 2. Wnt/β-Catenin Signal Transduction Pathway [50, 85, 86].
The Wnt signaling pathway is a conserved cascade that regulates a number of critical developmental and stem cell processes (Figure II). The central signaling component is β-catenin, an obligatory transcriptional mediator. Wnt/β-catenin signaling is initiated when the secreted glycoprotein Wnt binds to the cognate receptor complex of Frizzled and Lrp. This interaction activates the cytoplasmic protein Dishevelled, which stabilizes β-catenin by inhibiting the kinase activity of the destruction complex of adenomatous polyposis coli (APC), axin, and glycogen synthase kinase-3β (GSK3β). Active (unphosphorylated at Ser37/Thr41) β-catenin translocates to the nucleus, where it binds to the TCF/lymphoid enhancer factor (LEF) transcription factors to activate Wnt target genes. In the absence of Wnt, the destruction complex phosphorylates the N-terminal of β-catenin to lead to ubiquitin-mediated proteolytic degradation.
Early Evidence of Non-Canonical Notch Function
Some of the earliest evidence for non-canonical Notch signaling came from in vitro studies, in which increased Notch1 levels inhibited the differentiation of myoblast (C2C12) cells into muscle cells [8–10]. The authors reported that, unlike conventional Notch signaling, the inhibition of myoblast differentiation did not require the CSL interacting domain of Notch1 and was not mediated by CSL or known Notch target genes, suggesting the existence of a CSL-independent Notch pathway [9, 10]. In vivo, Notch loss-of-function studies in Drosophila revealed that Notch exerts its inhibitory effect to select muscle progenitors from the mesoderm even in the absence of ligand and/or CSL [19]. This finding provided compelling evidence that ligand/CSL-independent function of Notch is present and active during development. Since then, ligand/CSL-independent Notch functions have been reported in various systems across species (Table 1). However, in most cases, the key mediators of non-canonical Notch signals are unclear, and the proposed mechanisms appear to vary with context. Could there be a conserved mechanism? While CSL-independent Notch activity may come from interactions of Notch with non-CSL transcription factors in the nucleus [31, 32], this does not explain ligand-independent functions of Notch. Moreover, endogenous Notch protein is mostly detected in the cell membrane and cytoplasm and is rarely observed in the nucleus [33], suggesting Notch may interact in the cytoplasm with other molecules, affecting their function posttranslationally. It is worth noting that the described non-canonical Notch functions have mostly been identified in stem/progenitor cells or embryonic/primordial cells across species, which are capable of expansion and/or differentiation. This suggests that non-canonical Notch signals may play an important role in undifferentiated early cell populations and may interact with conserved cell regulators. Wnt/β-catenin signaling is one such regulator that Notch frequently interacts with throughout development, and we discuss their functional and molecular interactions in the following sections.
Table 1.
Evidence of CSL/ligand-independent Notch signaling
| Species | Cell type | System | Independence | Function | Interacting molecule/signaling (direct or indirect) | References |
|---|---|---|---|---|---|---|
| Human | Stem Cells (hESCs), Cancer | in vitro | Ligand, CSL | Negative regulation of Wnt signaling | Active β-catenin/Wnt signaling | [5] |
| Rodent | Stem Cells (mESCs, NSCs, MSCs), Progenitors (CPCs) | in vivo, in vitro | Ligand, CSL | Negative regulation of Wnt signaling | Active β-catenin/Wnt signaling | [5] |
| T cells | In vitro | CSL | Notch-1 stimulates NF-κB | NF-κB pathway | [28] | |
| Primary embryonic cells | in vitro | PS, Ligand | HES1 activation and MCK inhibition | HES1 and MCK | [6] | |
| Skin progenitors | in vivo | CSL | Leukocytosis, longevity | nd | [7] | |
| Muscle stem cells (C2C12) | in vitro | CSL | Inhibition of Muscle cell differentiation | nd | [8–10] | |
| Fibroblasts (3T3) | in vitro | CSL | Inhibition of E47 | E47 | [11] | |
| CHO cell line | in vitro | CSL | b1 integrin activation | R-Ras | [12] | |
| Avian | Neural crest (stem cells) | in vivo | CSL | Slug expression | Slug | [13, 14] |
| Frog | Embryo | in vivo | CSL | Negative regulation of Wnt signaling | β-catenin/Wnt signaling | [15] |
| Fly | Wing primordium | in vivo | Ligand, CSL | Negative regulation of Wnt signaling | Active β-catenin/Wnt signaling | [16, 17, 27] |
| Muscle progenitors | in vivo | Ligand, CSL | Muscle precursor selection | Wnt signaling | [18, 19] | |
| Neural progenitors | in vivo | Ligand, CSL | Neuronal Cell (MP2) selection | nd | [20] | |
| Blood cells | in vivo | Ligand | Hemocyte survival | Hif-a | [21] | |
| Wing primordium | in vivo, in vitro | CSL | Inhibition of ligand function | Serrate | [22] | |
| Embryo | in vivo | CSL | Dorsal epidermis patterning (closure) | JNK pathway | [23] | |
| Visceral mesoderm progenitors | in vivo | CSL | Inhibition of Wnt signaling | Ubx | [24] | |
| Neural precursors | in vivo | CSL | Repression of neural fate | Wnt signaling | [25, 26] | |
hESC (human Embryonic Stem Cells), mESC (mouse Embryonic Stem Cells), NSCs (Neural Stem Cells), MSCs (Mesenchymal Stem Cells), CPCs (Cardiac Progenitor Cells), PS (presenilin), nd (not determined)
Functional Interaction of Non-Canonical Notch and Wnt/β-Catenin Signaling
Notch exhibits recurrent crosstalk with Wnt/β-catenin signaling in numerous cell types and contexts during development (summarized in Table 1 in [34]). The interaction of Notch and Wnt signaling was first uncovered in the Drosophila wing imaginal disc, where Notch is co-expressed with Wingless (Drosophila Wnt-1) and enforces Wingless signaling [29, 35]. Notch interacts with Wnt/β-catenin signaling in synergic or antagonistic ways, depending on the context [24, 27]. The synergistic interactions generally involve ligand/CSL-dependent Notch signaling. For instance, Notch and β-catenin synergistically act to induce arterial endothelial cells and gene expression in an RBP-J dependent manner. [36]. The synergistic activity of Notch and Wnt/β-catenin signaling is also observed in early intestinal precursors and adenomas [37].
In contrast, ligand/CSL-independent Notch signaling is frequently associated with antagonism of Wnt/β-catenin signaling. In Drosophila, Wingless is required for the induction of Slouch (S59)+ muscle progenitors and its gain-of-function causes their expansion [18]. Notch-null mutation also leads to excessive numbers of muscle progenitors independent of ligand/CSL-mediated lateral inhibition, and deficiency of Notch -- but not of other canonical Notch pathway members -- restores the induction of Slouch+ progenitors in the absence of Wingless [18]. Similarly, decreased Notch or increased Wnt signaling promotes expansion of Evenskipped+ cardiac progenitors during development [38], although interaction of Notch and Wnt signaling was undetermined. This repressive role of Notch was also identified at the gene promoter level, where Notch inhibits Wingless activity on a mesodermal enhancer independent of CSL function in the peripheral nervous system as well as in epithelial cells [17, 24–26].
Interestingly, Notch antagonism of Wnt signaling that controls progenitor cell numbers appears to be conserved in mammalian stem and progenitor cells. In mouse embryos, Notch1 ablation in Islet1+ cardiac progenitor cells (CPCs) results in an expansion of CPCs with increased levels of active β-catenin protein and inhibits their cardiac differentiation [39]. Although the phenotype can be recapitulated in vivo or in vitro by stabilizing β-catenin or administering Wnt3a, but not by CSL deletion [5, 39, 40]. Conversely, the β-catenin-mediated expansion is rescued when NICD is co-expressed in CPCs [5], implying Notch negatively regulates β-catenin activity. In epithelial progenitor cells, Notch1 deletion causes epidermal hyperplasia, whereas increased levels of activated Notch1 leads to growth arrest and induction of early differentiation markers through a CSL-independent mechanism [41]. Likewise, Notch-mediated antagonism of Wnt/β-catenin signaling is also observed in embryonic stem cells (ESCs), neural stem cells and mesenchymal stem cells [5].
These findings may reveal an evolutionarily conserved role of non-canonical Notch signals in controlling stem/progenitor cell expansion mediated by canonical Wnt signaling and support the notion that Notch may function as a tumor suppressor [42]. For instance, Notch1 deletion in the epidermis causes epidermal and corneal hyperplasia, leading to skin carcinogenesis [43]. The hyperplasia is accompanied with increased Wnt/β-catenin signaling in epidermis, which can be reduced upon NICD overexpression [43]. In addition, although interaction with Wnt signaling hasn‘t been determined, Notch1-deficiency leads to high incidence and progression of pancreatic cancer, when the GTPase K-ras is activated [44]. Curiously Notch was proposed as an oncogene in a few other cancers [45]. However, Notch-mediated tumorigenesis requires activation of another oncoprotein, and therefore this discrepancy may represent a context-dependent nature of Notch signaling.
Together, these findings suggest that non-canonical Notch function may be closely associated with inhibition of canonical Wnt signaling during stem/progenitor cell development and oncogenesis. Nevertheless, the mechanisms by which Notch negatively regulates Wnt/β-catenin signaling through a ligand/CSL-independent pathway were not understood until recently.
Molecular Link between Non-Canonical Notch and Wnt Signals: Active β-Catenin
Cleaved NICD has long been thought to be the activated form of Notch, while uncleaved membrane-bound Notch is thought to be biologically inactive and constantly internalized for recycling or degradation through an endo-lysosomal pathway [46]. Interestingly, uncleaved full-length Notch1 in the plasma membrane—generated by inactivating the Notch-processing protease Furin, or site-specific mutagenesis of Furin target sequence in Notch—potently inhibits myogenesis of C2C12 myoblasts [8], which is also mediated by canonical Notch signaling [47]. However, unlike canonical Notch signaling, the uncleaved Notch mediates this event without affecting expression of the myogenic master transcription factor MyoD. These studies suggest that Notch may affect cell fates and differentiation in a non-canonical fashion. In 2005, it was demonstrated that a membrane-bound form of Notch physically interacts with β-catenin and modulates Wnt signaling by negatively regulating β-catenin activity in flies [30]. This study provided the first mechanistic clue in vivo for the antagonism of Wnt signaling by uncleaved Notch without involving Notch ligands and CSL.
Recent in vivo and in vitro studies provided further insights into how Notch functions not only as a membrane-tethered transcription factor, but also post-translationally by lowering levels of the transcriptionally active form of β-catenin as a membrane-bound regulator [5, 15, 17]. This form of β-catenin is dephosphorylated at Ser37 and Thr41 and normally constitutes a small fraction of total β-catenin [48]. In mammalian stem and progenitor cells, Notch levels are inversely correlated with active β-catenin; increased levels of membrane Notch decrease active β-catenin levels and decreased levels of Notch increase active β-catenin levels. Notch regulation, however, does not appear to affect total β-catenin protein or transcript levels, but rather targets active β-catenin [30, 39]. In agreement with this, the physical association of Notch and β-catenin is mostly notable in cells with high levels of active β-catenin [5, 39]. The CSL binding domain of Notch, the RAM domain, was also required for the physical interaction and regulation of active β-catenin [5, 49], implying a dual role for canonical and non-canonical Notch function.
It was unexpected, however, that the membrane Notch regulation of active β-catenin occurred independent of GSK3β, a major component of the destruction complex, which acts through the ubiquitin-proteasome system [50, 51]. Genetic analyses revealed that membrane Notch was still able to oppose increased β-catenin activity resulting from GSK3β loss-of-function [30]. Similarly, membrane Notch could efficiently lower active β-catenin levels in stem cells treated with the GSK3β inhibitor, 6-bromoindirubin-3′-oxime (BIO) [5]. However, a newer study suggested that Axin and Apc -- other key components of the destruction complex -- participate in this regulation by modulating endocytosis and trafficking of membrane Notch [16]. In this process, Axin or Apc was necessary for normal trafficking of membrane Notch, which might contribute to the Notch-dependent lowering of active β-catenin levels in addition to the β-catenin destruction complex-mediated degradation [16]. It is unknown if Axin and APC are also involved in Notch endocytosis and trafficking in vertebrates.
Similar to the inhibitory role of uncleaved membrane Notch in differentiation of C2C12 myoblasts, increased levels of a membrane-tethered form of Notch in differentiating ESCs were shown to suppress induction of Brachyury+ mesodermal cells, dependent of Wnt/β-catenin signaling [52]. This may indicate that membrane-bound Notch modulates a Wnt/β-catenin-mediated cellular response in stem cells. Intriguingly, the phenotype was recapitulated when Notch endoproteolysis was blocked by treating cells with the γ-secretase inhibitor (GSI), DAPT, suggesting increased levels of endogenous membrane-bound Notch may negatively regulate active β-catenin levels [5, 53]. Indeed, DAPT treatment lowered active β-catenin levels and activity in various stem, progenitor and cancer cells [5]. Consistently, blocking α-secretase activity, required for ligand-mediated cleavage of NECD, showed a similar outcome [5]. This is surprising because γ-secretase inhibitors are widely used as a potent inhibitor of canonical Notch signaling, but paradoxically result in opposite biological effects: Wnt/β-catenin signaling is increased by Notch deficiency but decreased by DAPT. This may provide an explanation for some aspects of phenotypic differences between DAPT and other Notch loss-of-function mutations described earlier.
Multiple clinical studies demonstrated that a subset of non-steroidal anti-inflammatory drugs (NSAIDs) possess significant GSI activity, and their chronic use is associated with lowering the risk of developing various cancer cell types -- including human colorectal cancers -- whose tumorigenesis is initiated by an upregulation of active β-catenin [54–56]. Although their anti-cancer effects were generally attributed to anti-inflammatory function, NSAIDs with GSI activity also likely contribute to the beneficial effect [57, 58]. In fact, treating human colorectal cancer cells with ibuprofen, a common NSAID with GSI activity, lowered active β-catenin levels and activity in a Notch1-dependent manner [5], which agrees with the in vivo report that the number of intestinal adenomas is reduced by GSI treatment [59, 60]. Curiously, deleting CSL also results in reduction of intestinal adenomas [60], implying canonical Notch signaling may have oncogenic function in this context. If this holds true, although CSL can independently function as a transcriptional repressor [61] [62], GSI treatment may simultaneously inhibit the canonical Notch pathway and activate non-canonical Notch function, which might have better protective effects on tumorigenesis linked with high levels of Notch/CSL signaling and active β-catenin. Indeed, GSI treatment was shown to efficiently suppress expansion of intestinal adenoma cells caused by APC mutations in vivo and in vitro [60, 63].
The degradation of active β-catenin protein by the destruction complex is well understood and involves phosphorylation of the N-terminus of β-catenin leading to proteasome-mediated degradation [50, 51]. Compromising the activity of the degradation complex did not prevent membrane Notch from suppressing active β-catenin protein levels and activity [5, 30], implying Notch shuttles active β-catenin to the proteosome in some other manner or may lead to lysosomal degradation. However, DAPT treatment effectively decreased active β-catenin in the presence of proteosome inhibitors, suggesting a proteosome-independent mechanism in which stem and cancer cells post-transcriptionally titrate the dosage of active β-catenin. A pulse-chase experiment supports the idea of lysosomal pathway-mediated degradation of active β-catenin; the authors show in the developing fly wing disc that membrane-bound Notch is actively endocytosed into the endosomal compartment in a ligand-independent fashion and that some of the internalized Notch molecules co-localize with β-catenin in endocytic vesicles [17]. The endocytosis and trafficking required the RAM-ANK domain, which was also important for the physical interaction of Notch and β-catenin in ESCs [5, 17]. However, it is unclear if high levels of active β-Catenin actively trigger the endocytosis and trafficking. A similar finding was reported in APC-mutated human colorectal cells, where a membrane-tethered form of Notch co-localizes with active β-catenin and the lysosomal protein Lamp1 [5]. Moreover, compromising lysosomal activity with bafilomycin A1, a specific inhibitor of vacuolar proton ATPases [64], abrogated the DAPT-induced reduction of active β-catenin levels in mouse ESCs [5]. Thus, convincing evidence exists that membrane-bound Notch controls the pools of active β-catenin by endo-lysosomal degradation (Figure 1).
Figure 1.

Post-Translational Regulation of β-Catenin Protein by Notch.
Notch can negatively regulate active β-catenin levels in a non-canonical fashion. In the presence of Wnts, membrane-bound Notch forms a complex with active β-catenin and degrades active β-catenin through an endo-lysosomal pathway. The degradation is independent of GSK3β-dependent destruction complex. Whether Notch is recycled back to the membrane is unclear. NICD can also regulate active β-catenin levels in a similar mechanism, but it is unknown whether endogenously processed NICD regulates active β-catenin protein. Protein interactions can be either direct or indirect.
While uncleaved Notch seems to modulate the levels and activity of active β-catenin through a lysosomal pathway, it is ambiguous if cleaved NICD also mediates the event through a similar mechanism. Several NICD overexpression studies suggest that NICD can also antagonize Wnt/β-catenin signaling by targeting active β-catenin and thereby affect cellular processes [15, 17]. In vertebrates, active β-catenin activity specifies dorsal cell fates during early embryogenesis, which is essential for establishing the dorso-ventral axis, and ventral overexpression of β-catenin causes dorsalization of ventral cells [65, 66]. It was reported that increased NICD levels ventralize frog embryos by opposing active β-catenin‘s dorsalizing activity [15]. As in the case of membrane-bound Notch, increased levels of NICD decrease levels of β-catenin in a manner that is insensitive to GSK3β activity [15], and this is also observed in other cell types including ESCs, CPCs and ST-2 stromal cells [5, 67]. GSK3β, on the other hand, was shown to protect NICD from proteasomal degradation [68]. NICD‘s nuclear localization appears to depend on β-catenin levels in the Xenopus blastula cells and cancer cells; overexpressed NICD localizes in the cytosol and nuclei but when co-expressed with β-catenin, NICD is not found in the nucleus but in cell-cell junctions [15], resembling the process mediated by membrane-bound Notch [17].
The bulk of these findings indicate that increased levels of NICD may inhibit active β-catenin levels in a similar mechanism as membrane-bound Notch does. Under endogenous conditions, however, NICD might mostly translocate to the nucleus after membrane cleavage, while overexpression of NICD results in aberrant localization of significant NICD in the cytosol. Thus, while NICD interacts with active β-catenin for lysosome-mediated degradation in the cytosol, this may not be its normal function under physiologic conditions.
Curiously, Notch was also shown to physically associate with the endocytic protein Numb and required Numb and its homologue Numblike to regulate active β-catenin activity in ES cells [5, 69]. These findings indicate Numb may be a key component of the Non-canonical Notch pathway. The potential role and mechanisms of Numb will be discussed in the next section.
Potential Role of Numb in Notch and β-Catenin Regulation
While Notch had been defined as a fundamental mediator of extrinsic factors for cell-fate specification, Numb was identified as the primary intrinsic factor that antagonized Notch in classic studies in Drosophila [69]. This interaction depends on the spatio-temporal distribution of Numb during cell division to one pole of the cell resulting in asymmetric cell division in which daughter cells retained distinct properties and different fates [70, 71].
Numb may inhibit canonical Notch activity by direct interaction or as a mediator recruiting other factors to prevent the nuclear translocation of Notch protein [69, 72]. One mechanism of inhibition requires Numb to bind the NICD of membrane-bound Notch with a third party to sequester Notch [73]. For instance, α-adaptin, a component of adaptor complex 2, is asymmetrically distributed with Numb, and these proteins interact to induce endocytosis of Notch at specific sites [73]. Numb-dependent regulation of Notch may also occur via endosome-independent pathways [74]. For example, Numb interacts with E3 ligases to promote ubiquitination of membrane-bound Notch, leading to its subsequent degradation [75].
Membrane-bound Notch is constantly internalized through endocytosis and then sorted for endosome-mediated recycling to the membrane or for lysosomal degradation by Numb [76]. Since Numb and its homologue, Numblike, appear to be necessary for degradation of active β-catenin by membrane Notch [5], it is reasonable to consider that the Notch-β-catenin complex is being trafficked into lysosomes for degradation. This is in agreement with the recent finding that Notch associates with active β-catenin, and together are endocytosed into the endosomal compartment [17]. It remains to be determined if Numb shares common molecular machinery for the regulation of Notch and active β-catenin, and if so, how Numb selectively affects the activity and levels of Notch and active β-catenin (Figure 2).
Figure 2.

Proposed Model for Numb Regulation of Notch and β-Catenin
Numb may bind directly to Notch independent of α-adaptin (a) or may bind via an α-adaptin-dependent mechanism (b) with subsequent targeting of the Numb-Notch complex for lysosomal degradation. In both cases it may be possible that activated β-catenin could also be targeted for lysosomal destruction either as an innocent bystander or through an active process with unknown partners. Downregulation of Notch may occur through Numb-mediated targeting via ubiquitination intermediaries, such as E3-ligase, for proteasome-mediated degradation (c).
Physiological Significance
During the last decade increasing evidence has suggested that a complex functional relationship exists between Notch and Wnt signaling, particularly during establishment of stem and progenitor cell fate determination and cancer formation. The recent findings of how membrane-bound Notch post-translationally regulates Wnt/β-catenin signaling provide novel insight of this complex relationship during fundamental biological and disease processes, such as proliferation, differentiation, lineage decisions, and tumorigenesis. Although increased levels of membrane Notch were shown to significantly affect key cellular events including proliferation and differentiation, the physiological role of membrane Notch remains to be elucidated. Further investigation is now required to understand the endogenous function of the membrane Notch/β-catenin pathway.
Concluding remarks
It has been puzzling that endogenous Notch protein is mostly detected at the cell membrane and/or cytoplasm but rarely seen in the nucleus. With accumulating evidence, it is now becoming apparent that Notch can function in non-nuclear environments, where it affects canonical Wnt signaling by titrating active β-catenin levels. Although active β-catenin has emerged as a conserved mediator of a ligand/CSL-independent Notch pathway across species, it is likely that Notch interacts with additional key players, such as Numb and Numblike, to control cellular processes outside the nucleus. Thus, it will be critical to identify these molecules and determine their roles in the non-canonical pathway. Nevertheless, the functional and molecular interactions of Notch and active β-catenin provide a potential explanation for many aspects of non-canonical Notch effects described, and make active β-catenin levels and activity useful readouts for non-canonical Notch activity.
GSIs (such as DAPT) and NICD have been widely used to mimic canonical Notch loss-of-function and gain-of-function mutations, respectively, but it is important to acknowledge the fact that both can act as potent inhibitors of active β-catenin. While this observation could provide the foundation for novel therapeutic targets, caution regarding their effects on Wnt/β-catenin signaling is warranted when they are used for experimental or therapeutic purposes.
At present, the biology of membrane Notch has been minimally explored in the field of stem, progenitor and cancer cells. Notch and Wnt/β-catenin signaling are directly involved in, and essential to, nearly all known stem/progenitor cell self-renewal and differentiation processes, and in oncogenesis. As such, future investigation of the biological function and mechanism of the membrane Notch/β-catenin pathway will greatly expand our fundamental knowledge of stem, progenitor and cancer cell biology, and could eventually be leveraged for regenerative and therapeutic approaches.
Figure I.

Canonical vs. Non-Canonical Notch Signaling
Figure II.
Proposed Model for Numb Regulation of Notch and β-Catenin
Box 3. Outstanding Questions.
What proteins are associated with the membrane Notch–β-catenin complex and what are their roles?
What is the role of Numb in Notch regulation of active β-catenin?
What is the biological role of the membrane Notch/β-catenin pathway in stem/progenitor cell maintenance and lineage-specific differentiation?
Can the membrane Notch/β-catenin pathway be targeted for cancer therapeutics?
Is there a β-catenin-independent function and mechanism of membrane Notch?
Acknowledgments
We thank P. Cheng, D. Srivastava, and Kwon lab members for helpful discussions. This work was supported by grants from NHLBI/NIH and AHA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wharton KA, et al. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell. 1985;43:567–581. doi: 10.1016/0092-8674(85)90229-6. [DOI] [PubMed] [Google Scholar]
- 2.Morgan T. The theory of the gene. American Naturalist. 1917;51:513–544. [Google Scholar]
- 3.Yochem J, et al. The Caenorhabditis elegans lin-12 gene encodes a transmembrane protein with overall similarity to Drosophila Notch. Nature. 1988;335:547–550. doi: 10.1038/335547a0. [DOI] [PubMed] [Google Scholar]
- 4.Poulson DF. Effect of Notch deficiencies. Drosophila Information Services. 1939;12:64–65. [Google Scholar]
- 5.Kwon C, et al. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat Cell Biol. 2011;13:1244–1251. doi: 10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berechid BE, et al. Identification and characterization of presenilin-independent Notch signaling. J Biol Chem. 2002;277:8154–8165. doi: 10.1074/jbc.M108238200. [DOI] [PubMed] [Google Scholar]
- 7.Demehri S, et al. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol. 2008;6:e123. doi: 10.1371/journal.pbio.0060123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bush G, et al. Ligand-induced signaling in the absence of furin processing of Notch1. Dev Biol. 2001;229:494–502. doi: 10.1006/dbio.2000.9992. [DOI] [PubMed] [Google Scholar]
- 9.Nofziger D, et al. Notch signaling imposes two distinct blocks in the differentiation of C2C12 myoblasts. Development. 1999;126:1689–1702. doi: 10.1242/dev.126.8.1689. [DOI] [PubMed] [Google Scholar]
- 10.Shawber C, et al. Notch signaling inhibits muscle cell differentiation through a CBF1-independent pathway. Development. 1996;122:3765–3773. doi: 10.1242/dev.122.12.3765. [DOI] [PubMed] [Google Scholar]
- 11.Ordentlich P, et al. Notch inhibition of E47 supports the existence of a novel signaling pathway. Mol Cell Biol. 1998;18:2230–2239. doi: 10.1128/mcb.18.4.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodkinson PS, et al. Mammalian NOTCH-1 activates beta1 integrins via the small GTPase R-Ras. J Biol Chem. 2007;282:28991–29001. doi: 10.1074/jbc.M703601200. [DOI] [PubMed] [Google Scholar]
- 13.Endo Y, et al. Bimodal functions of Notch-mediated signaling are involved in neural crest formation during avian ectoderm development. Development. 2002;129:863–873. doi: 10.1242/dev.129.4.863. [DOI] [PubMed] [Google Scholar]
- 14.Endo Y, et al. Deltex/Dtx mediates NOTCH signaling in regulation of Bmp4 expression in cranial neural crest formation during avian development. Dev Growth Differ. 2003;45:241–248. doi: 10.1046/j.1524-4725.2003.693.x. [DOI] [PubMed] [Google Scholar]
- 15.Acosta H, et al. Notch destabilises maternal beta-catenin and restricts dorsal-anterior development in Xenopus. Development. 2011;138:2567–2579. doi: 10.1242/dev.061143. [DOI] [PubMed] [Google Scholar]
- 16.Munoz-Descalzo S, et al. Modulation of the ligand-independent traffic of Notch by Axin and Apc contributes to the activation of Armadillo in Drosophila. Development. 2011;138:1501–1506. doi: 10.1242/dev.061309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanders PG, et al. Ligand-independent traffic of Notch buffers activated Armadillo in Drosophila. PLoS Biol. 2009;7:e1000169. doi: 10.1371/journal.pbio.1000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennan K, et al. Repression by Notch is required before Wingless signalling during muscle progenitor cell development in Drosophila. Curr Biol. 1999;9:707–710. doi: 10.1016/s0960-9822(99)80313-3. [DOI] [PubMed] [Google Scholar]
- 19.Rusconi JC, Corbin V. Evidence for a novel Notch pathway required for muscle precursor selection in Drosophila. Mech Dev. 1998;79:39–50. doi: 10.1016/s0925-4773(98)00170-1. [DOI] [PubMed] [Google Scholar]
- 20.Rusconi JC, Corbin V. A widespread and early requirement for a novel Notch function during Drosophila embryogenesis. Dev Biol. 1999;215:388–398. doi: 10.1006/dbio.1999.9483. [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee T, et al. Interaction between Notch and Hif-alpha in development and survival of Drosophila blood cells. Science. 2011;332:1210–1213. doi: 10.1126/science.1199643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becam I, et al. A role of receptor Notch in ligand cis-inhibition in Drosophila. Curr Biol. 2010;20:554–560. doi: 10.1016/j.cub.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 23.Zecchini V, et al. An activity of Notch regulates JNK signalling and affects dorsal closure in Drosophila. Curr Biol. 1999;9:460–469. doi: 10.1016/s0960-9822(99)80211-5. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence N, et al. Notch signaling targets the Wingless responsiveness of a Ubx visceral mesoderm enhancer in Drosophila. Curr Biol. 2001;11:375–385. doi: 10.1016/s0960-9822(01)00120-8. [DOI] [PubMed] [Google Scholar]
- 25.Ramain P, et al. Novel Notch alleles reveal a Deltex-dependent pathway repressing neural fate. Curr Biol. 2001;11:1729–1738. doi: 10.1016/s0960-9822(01)00562-0. [DOI] [PubMed] [Google Scholar]
- 26.Brennan K, et al. The abruptex mutations of notch disrupt the establishment of proneural clusters in Drosophila. Dev Biol. 1999;216:230–242. doi: 10.1006/dbio.1999.9501. [DOI] [PubMed] [Google Scholar]
- 27.Brennan K, et al. Wingless modulates the effects of dominant negative notch molecules in the developing wing of Drosophila. Dev Biol. 1999;216:210–229. doi: 10.1006/dbio.1999.9502. [DOI] [PubMed] [Google Scholar]
- 28.Shin HM, et al. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006;25:129–138. doi: 10.1038/sj.emboj.7600902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hing HK, et al. Modulation of wingless signaling by Notch in Drosophila. Mech Dev. 1994;47:261–268. doi: 10.1016/0925-4773(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 30.Hayward P, et al. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development. 2005;132:1819–1830. doi: 10.1242/dev.01724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson-Rawls J, et al. Activated notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol Cell Biol. 1999;19:2853–2862. doi: 10.1128/mcb.19.4.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross DA, Kadesch T. The notch intracellular domain can function as a coactivator for LEF-1. Mol Cell Biol. 2001;21:7537–7544. doi: 10.1128/MCB.21.22.7537-7544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artavanis-Tsakonas S, et al. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 34.Hayward P, et al. Wnt/Notch signalling and information processing during development. Development. 2008;135:411–424. doi: 10.1242/dev.000505. [DOI] [PubMed] [Google Scholar]
- 35.Couso JP, Martinez Arias A. Notch is required for wingless signaling in the epidermis of Drosophila. Cell. 1994;79:259–272. doi: 10.1016/0092-8674(94)90195-3. [DOI] [PubMed] [Google Scholar]
- 36.Yamamizu K, et al. Convergence of Notch and beta-catenin signaling induces arterial fate in vascular progenitors. J Cell Biol. 2010;189:325–338. doi: 10.1083/jcb.200904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fre S, et al. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci U S A. 2009;106:6309–6314. doi: 10.1073/pnas.0900427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park M, et al. Mesodermal cell fate decisions in Drosophila are under the control of the lineage genes numb, Notch, and sanpodo. Mech Dev. 1998;75:117–126. doi: 10.1016/s0925-4773(98)00098-7. [DOI] [PubMed] [Google Scholar]
- 39.Kwon C, et al. A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol. 2009;11:951–957. doi: 10.1038/ncb1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwon C, et al. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc Natl Acad Sci U S A. 2007;104:10894–10899. doi: 10.1073/pnas.0704044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rangarajan A, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20:3427–3436. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dotto GP. Notch tumor suppressor function. Oncogene. 2008;27:5115–5123. doi: 10.1038/onc.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicolas M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 44.Hanlon L, et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010;70:4280–4286. doi: 10.1158/0008-5472.CAN-09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3:756–767. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- 46.Fortini ME, Bilder D. Endocytic regulation of Notch signaling. Curr Opin Genet Dev. 2009;19:323–328. doi: 10.1016/j.gde.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuroda K, et al. Delta-induced Notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis. J Biol Chem. 1999;274:7238–7244. doi: 10.1074/jbc.274.11.7238. [DOI] [PubMed] [Google Scholar]
- 48.van Noort M, et al. Wnt signaling and phosphorylation status of beta-catenin: importance of the correct antibody tools. Blood. 2007;110:2778–2779. doi: 10.1182/blood-2007-05-092445. [DOI] [PubMed] [Google Scholar]
- 49.Tamura K, et al. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 50.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 51.Winston JT, et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999;13:270–283. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindsley RC, et al. Canonical Wnt signaling is required for development of embryonic stem cell-derived mesoderm. Development. 2006;133:3787–3796. doi: 10.1242/dev.02551. [DOI] [PubMed] [Google Scholar]
- 53.Sastre M, et al. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2:835–841. doi: 10.1093/embo-reports/kve180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rostom A, et al. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med. 2007;146:376–389. doi: 10.7326/0003-4819-146-5-200703060-00010. [DOI] [PubMed] [Google Scholar]
- 55.Eriksen JL, et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan CM, et al. Celecoxib induces dose dependent growth inhibition in nasopharyngeal carcinoma cell lines independent of cyclooxygenase-2 expression. Biomed Pharmacother. 2005;59(Suppl 2):S268–271. doi: 10.1016/s0753-3322(05)80043-5. [DOI] [PubMed] [Google Scholar]
- 57.Bottone FG, Jr, et al. Gene modulation by the cyclooxygenase inhibitor, sulindac sulfide, in human colorectal carcinoma cells: possible link to apoptosis. J Biol Chem. 2003;278:25790–25801. doi: 10.1074/jbc.M301002200. [DOI] [PubMed] [Google Scholar]
- 58.Shiff SJ, et al. Sulindac sulfide, an aspirin-like compound, inhibits proliferation, causes cell cycle quiescence, and induces apoptosis in HT-29 colon adenocarcinoma cells. J Clin Invest. 1995;96:491–503. doi: 10.1172/JCI118060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci. 2007;64:2746–2762. doi: 10.1007/s00018-007-7164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 61.Dou S, et al. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol Cell Biol. 1994;14:3310–3319. doi: 10.1128/mcb.14.5.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waltzer L, et al. RBP-J kappa repression activity is mediated by a co-repressor and antagonized by the Epstein-Barr virus transcription factor EBNA2. Nucleic Acids Res. 1995;23:4939–4945. doi: 10.1093/nar/23.24.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwon C, et al. Notch post-translationally regulates β-catenin protein in stem and progenitor cells. Nature cell biology. 2011 doi: 10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tapper H, Sundler R. Bafilomycin A1 inhibits lysosomal, phagosomal, and plasma membrane H(+)-ATPase and induces lysosomal enzyme secretion in macrophages. J Cell Physiol. 1995;163:137–144. doi: 10.1002/jcp.1041630116. [DOI] [PubMed] [Google Scholar]
- 65.Heasman J, et al. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell. 1994;79:791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 66.Sokol SY. Wnt signaling and dorso-ventral axis specification in vertebrates. Curr Opin Genet Dev. 1999;9:405–410. doi: 10.1016/S0959-437X(99)80061-6. [DOI] [PubMed] [Google Scholar]
- 67.Deregowski V, et al. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J Biol Chem. 2006;281:6203–6210. doi: 10.1074/jbc.M508370200. [DOI] [PubMed] [Google Scholar]
- 68.Foltz DR, et al. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr Biol. 2002;12:1006–1011. doi: 10.1016/s0960-9822(02)00888-6. [DOI] [PubMed] [Google Scholar]
- 69.Guo M, et al. Control of daughter cell fates during asymmetric division: interaction of Numb and Notch. Neuron. 1996;17:27–41. doi: 10.1016/s0896-6273(00)80278-0. [DOI] [PubMed] [Google Scholar]
- 70.Uemura T, et al. numb, a gene required in determination of cell fate during sensory organ formation in Drosophila embryos. Cell. 1989;58:349–360. doi: 10.1016/0092-8674(89)90849-0. [DOI] [PubMed] [Google Scholar]
- 71.Knoblich JA, et al. Asymmetric segregation of Numb and Prospero during cell division. Nature. 1995;377:624–627. doi: 10.1038/377624a0. [DOI] [PubMed] [Google Scholar]
- 72.Spana EP, Doe CQ. Numb antagonizes Notch signaling to specify sibling neuron cell fates. Neuron. 1996;17:21–26. doi: 10.1016/s0896-6273(00)80277-9. [DOI] [PubMed] [Google Scholar]
- 73.Berdnik D, et al. The endocytic protein alpha-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev Cell. 2002;3:221–231. doi: 10.1016/s1534-5807(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 74.Tang H, et al. Numb proteins specify asymmetric cell fates via an endocytosis- and proteasome-independent pathway. Mol Cell Biol. 2005;25:2899–2909. doi: 10.1128/MCB.25.8.2899-2909.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGill MA, McGlade CJ. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem. 2003;278:23196–23203. doi: 10.1074/jbc.M302827200. [DOI] [PubMed] [Google Scholar]
- 76.McGill MA, et al. Numb regulates post-endocytic trafficking and degradation of Notch1. J Biol Chem. 2009;284:26427–26438. doi: 10.1074/jbc.M109.014845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rebay I, et al. Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell. 1993;74:319–329. doi: 10.1016/0092-8674(93)90423-n. [DOI] [PubMed] [Google Scholar]
- 78.Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- 79.Schroeter EH, et al. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 80.De Strooper B, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 81.Lu FM, Lux SE. Constitutively active human Notch1 binds to the transcription factor CBF1 and stimulates transcription through a promoter containing a CBF1-responsive element. Proc Natl Acad Sci U S A. 1996;93:5663–5667. doi: 10.1073/pnas.93.11.5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fortini ME, Artavanis-Tsakonas S. The suppressor of hairless protein participates in notch receptor signaling. Cell. 1994;79:273–282. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- 83.Petcherski AG, Kimble J. LAG-3 is a putative transcriptional activator in the C. elegans Notch pathway. Nature. 2000;405:364–368. doi: 10.1038/35012645. [DOI] [PubMed] [Google Scholar]
- 84.Brou C, et al. Inhibition of the DNA-binding activity of Drosophila suppressor of hairless and of its human homolog, KBF2/RBP-J kappa, by direct protein-protein interaction with Drosophila hairless. Genes Dev. 1994;8:2491–2503. doi: 10.1101/gad.8.20.2491. [DOI] [PubMed] [Google Scholar]
- 85.Bejsovec A. Wnt pathway activation: new relations and locations. Cell. 2005;120:11–14. doi: 10.1016/j.cell.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 86.Tolwinski NS, Wieschaus E. Rethinking WNT signaling. Trends Genet. 2004;20:177–181. doi: 10.1016/j.tig.2004.02.003. [DOI] [PubMed] [Google Scholar]