

Abstract
Active exchangers dissipate the gradient of one substrate to accumulate nutrients, export xenobiotics and maintain cellular homeostasis. Mechanistic studies suggested that all exchangers share two fundamental properties: substrate binding is antagonistic and coupling is maintained by preventing shuttling of the empty transporter. The CLC Cl−: H+ exchangers control the homeostasis of cellular compartments in most living organisms but their transport mechanism remains unclear. We show that substrate binding to CLC-ec1 is synergistic rather than antagonistic: chloride binding induces protonation of a critical glutamate. The simultaneous binding of H+ and Cl− gives rise to a fully-loaded state incompatible with conventional mechanisms. Mutations in the Cl− transport pathway identically alter the stoichiometries of Cl−: H+ exchange and binding. We propose that the thermodynamics of synergistic substrate binding determine the stoichiometry of transport rather than the kinetics of conformational changes and ion binding.
Keywords: Isothermal titration calorimetry, channel, CLC, alternating access, chloride, proton
The coordinated generation and dissipation of ionic gradients by active transporters and ion channels allows cells to control and finely tune their internal ionic composition. Primary and secondary active transporters create the gradients at the expense of energy in the form of ATP hydrolysis for the former and dissipation of other electrochemical gradients for the latter. Depending on whether substrates travel together or in opposite directions, transporters are further classified as symporters or exchangers. While active transporters differ in energy sources, substrate specificities, oligomeric organization and structures, they all function according to the archetypal paradigm of alternating access 1-7. In this model substrate translocation is mediated by concerted conformational rearrangements that alternatively expose the ligand binding sites to either side of the membrane. Careful mechanistic investigations have shown that exchangers operate following ping-pong, or double displacement, kinetics 2,8-10 (Fig. 1a). In these models the transporters interact with one substrate at a time and the empty transporter does not alternate exposure of the binding sites 2,3,11. Thus, the exchange cycle is comprised of two half-cycles during which the transporter binds and translocates each substrate (Fig. 1a). Simultaneous binding schemes have rarely been proposed 12,13 and found little support since non-canonical behaviors could also originate from allosteric substrate binding sites on a sequential exchanger 10. The recent surge in structural information and functional data on a number of primary and secondary active exchangers 5,14-21 has greatly refined our understanding of transport mechanisms and has provided strong support to ping-pong exchange models. One ambiguous case is the recently identified family of CLC H+:Cl− exchangers 22-27 for which the applicability of a ping-pong alternating access scheme has been questioned 22,28,29. In these transporters substrate movement takes place along two partially congruent translocation pathways 28,30. This organization is completely different from that found in conventional exchangers, in which substrates take turns occupying a single pathway. It has been speculated that the existence of these two pathways might allow the CLC exchangers to function by simultaneously binding both substrates 28. The Cl− pathway is defined by three anionic binding sites which span the length of the membrane 14,31 (Fig. 1b) while only the extremities of the H+ pathway, Glu148 and Glu203, have been identified 22,28 (Fig. 1b). Crystallographic work 14,29,31 has shown that Glu148 can adopt three conformations: it can occupy Sex with ions bound to Sin and Scen (Fig. 1b), when protonated its side chain moves outside of the pathway and all three sites are occupied by Cl− ion (Fig. 1c) and it can adopt a conformation in which the Glu148 side chain occupies Scen while Cl− ions are found in Sin and Sex (Fig. 1d). Furthermore, in the structures the Cl− pathway is not completely occluded at the intracellular side 14,29, leading to the proposal that their exchange stoichiometry of 2 Cl− for 1 H+ arises from the kinetics of Cl− unbinding rather than from a conformational change 29. In contrast, some functional characteristics of the CLC exchangers could be described using classical schemes 32 suggesting that their transport mechanism is more conventional. Overall, the mechanism of substrate coupling and transport of these transporters remains unclear. Recent studies have shown that the strict coupling between Cl− and H+ is essential to allow these proteins to regulate endosomal and lysosomal homeostasis in vivo: mice where a CLC transporter is replaced with a mutant mediating H+-uncoupled Cl− fluxes display the same pathologies as those seen in the corresponding knock-out animals 33. Therefore, understanding the molecular basis for substrate coupling in the CLC exchangers is essential to elucidate their transport mechanism as well as their physiological role.
Figure 1.
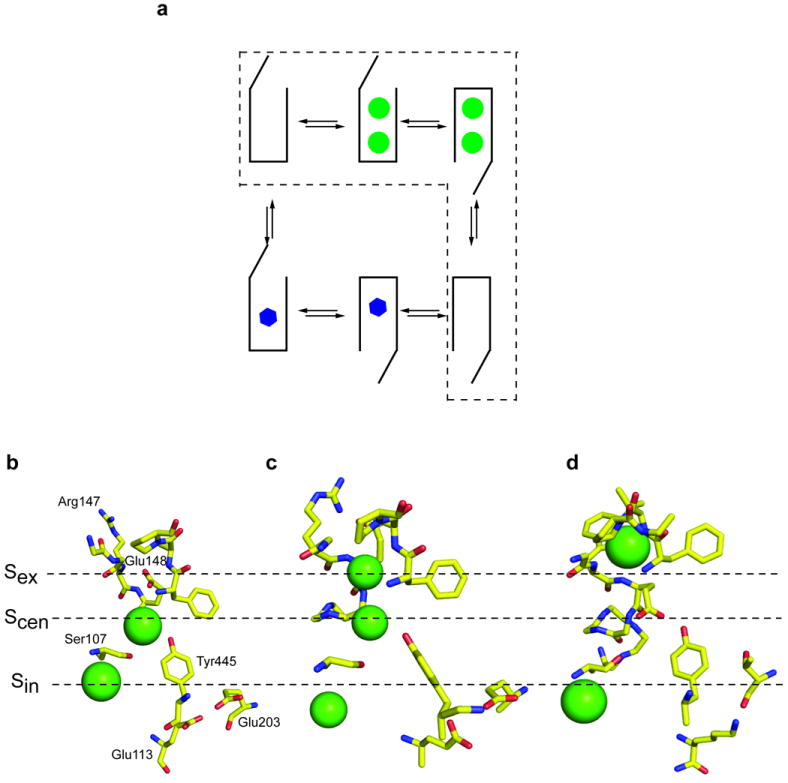
Alternating access transport (a) Schematic representation of a canonical alternating access scheme for a 2 Cl− (green circles): 1 H+ (blue hexagons) exchanger. The dashed line highlights the Cl− half-cycle. (b-d) Close up view of the ion binding region of WT CLC-ec1 (b), of the E148Q mutant of CLC-ec1 (c) and of WT cmCLC (d). The Cl− ions are shown as green spheres. Critical residues are labeled. For WT cmCLC the residues equivalent to those labeled in (b) are displayed: Arg147 is Trp209, Glu113 is Lys171 and Glu203 is Thr269. (PDB accession codes are WT CLC-ec1: 1OTS 31; E148Q CLC-ec1: 1OTU 31; WT cmCLC: 3ORG 29)
Here we investigate the coupling mechanism of the CLC exchangers using Isothermal Titration Calorimetry (ITC) and free energy calculations. We asked if binding of Cl− and H+ to CLC-ec1 is linked, and if so whether it is antagonistic, as demanded by canonical double displacement mechanisms, or not. We find that substrate binding to CLC-ec1 is synergistic (Cl− binding to the transport pathway facilitates protonation of a critical glutamate residue), sequential (Cl− binds first, followed by H+) and simultaneous (both substrates coexist bound to the protein).
Results
We used ITC to determine whether binding of Cl− to CLC-ec1 is linked to the exchange of a H+. The total enthalpy measured during an ITC experiment, ΔHtot, is due to several protein-specific reactions (Cl− binding, conformational changes and protonation or deprotonation of the protein) plus the ionization of the buffer donating or accepting the protons exchanged by the protein 34-36. Therefore ΔHtot can be written as:
where ΔHprot denotes the sum of the enthalpies of the protein specific reactions, ΔHbuff is the enthalpy of ionization of the buffer and N is the number of protons exchanged between protein and buffer per Cl− binding event. ΔHbuff is a buffer specific quantity and can be directly measured in the absence of protein (Supplementary Table 1). Therefore, the slope of the linear relationship between ΔHtot and ΔHbuff is the stoichiometry of coupled Cl− and H+ binding. If binding of substrates to CLC-ec1 is not coupled then ΔHtot should not vary when measured in buffers with different ΔHbuff so that N = 0. On the other hand, a direct coupling between substrate binding would be revealed by a variable ΔHtot. A negative slope would indicate that Cl− binding induces unbinding of a proton from CLC-ec1, antagonistic binding, while a positive slope would indicate that Cl− binding promotes protonation of CLC-ec1, or synergistic binding.
Cl− binding induces protonation of CLC-ec1
We measured Cl− binding to wildtype CLC-ec1 in three buffers, Tris, Hepes and Phosphate, whose ΔHbuff span ~6.6 kCal Mol−1 (Supplementary Table 1) and fit the liberated heats with single site binding isotherms 37. Chloride binds with comparable affinities in all three buffers (Table 1) indicating that the buffer molecules do not interact with the binding sites. We found that ΔHtot increases monotonically with the ionization enthalpy of the buffers: the enthalpy in Phosphate, −8.3±0.2 kcal mol−1, is larger than that in Hepes, −7.5±0.2 kcal mol−1, and Tris, −5.1±0.3 kcal mol−1 (Fig. 2, Table 1). Thus, chloride binding to the transport pathway induces protonation of CLC-ec1 indicating that substrate binding is synergistic and, at equilibrium, H+ and Cl− simultaneously bind to the protein. The stoichiometry of this coupling is derived by fitting ΔHtot to Eq. 1 and is N = 0.5±0.1. Thus, for every ~2 chloride binding events 1 proton associates to CLC-ec1.
Table 1.
Thermodynamic parameters for Cl− binding to WT and mutant CLC-ec1 in different buffers. Kd and ΔH were obtained from a fit to a binding isotherm, while ΔG and TΔS were calculated from ΔG = RT lnKd and TΔS = ΔH−ΔG. We fixed the number of binding sites to n = 1 for WT, E203Q and Y445L. We let n vary freely for the E148A mutant since for this mutant we could achieve c values where n can be independently determined. In all cases ΔHtot represents the total enthalpy generated during the binding reaction rather than the enthalpy for each independent binding site. Fort WT, Glu203 and Y445L the two quantities coincide while for the E148A mutant ΔHtot = nΔH. The final column reports the number of independent experiments performed in each condition. Values are reported as the mean ± s.e.m. of the independent experiments.
| Buffer | n | ΔHtot (kcal mol−1) | TΔS (kcal mol−1) | ΔG (kcal mol−1) | Kd (μM) | # exp | |
|---|---|---|---|---|---|---|---|
| WT | Tris | 1 | −5.1 ± 0.3 | −0.9 ± 0.2 | −4.3 ± 0.1 | 780 ± 60 | 9 |
| Hepes | 1 | −7.5 ± 0.2 | −3.2 ± 0.2 | −4.3 ± 0.1 | 640 ± 40 | 8 | |
| Phos. | 1 | −8.3 ± 0.2 | −3.9 ± 0.2 | −4.4 ± 0.1 | 680 ± 80 | 7 | |
| E148A | Tris | 1.8 ± 0.1 | −12.0 ± 1.0 | −0.1 ± 0.5 | −6.6 ± 0.1 | 15 ± 1 | 5 |
| Hepes | 1.98±0.03 | −12.9 ± 0.5 | −0.1 ± 0.3 | −6.7 ± 0.1 | 13 ± 1 | 3 | |
| Phos. | 1.5 ± 0.1 | −13.5 ± 1.3 | −2.3 ± 0.5 | −6.6 ± 0.1 | 14 ± 2 | 5 | |
| E203Q | Tris | 1 | −2.1 ± 0.1 | −0.8 ± 0.1 | −2.9 ± 0.1 | 7300± 760 | 3 |
| Hepes | 1 | −3.7 ± 0.6 | −0.7 ± 0.6 | −3.0 ± 0.1 | 5800± 220 | 3 | |
| Phos. | 1 | −5.1 ± 0.3 | −2.1 ± 0.3 | −3.1 ± 0.1 | 6300 ±340 | 4 | |
| Y445L | Tris | 1 | −3.9 ± 0.2 | −0.75 ± 0.2 | −3.2 ± 0.1 | 4600± 400 | 4 |
| Hepes | 1 | −4.6 ± 0.3 | −1.3 ± 0.3 | −3.3 ± 0.1 | 4100± 270 | 4 | |
| Phos. | 1 | −4.7 ± 0.1 | −1.4 ± 0.1 | −3.4 ± 0.1 | 4100 ± 50 | 4 |
Figure 2.
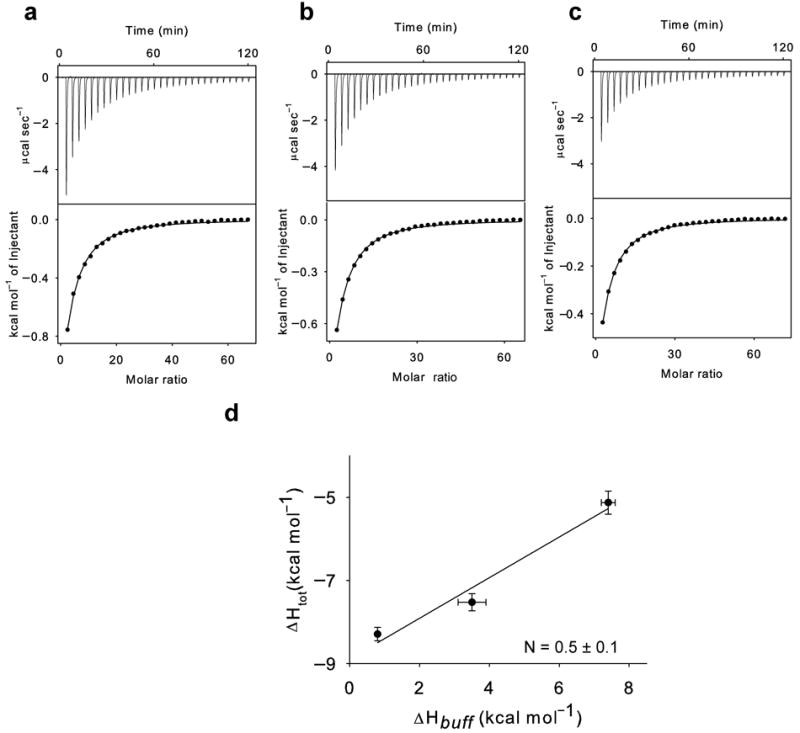
Synergistic binding of H+ and Cl− to WT CLC-ec1. (a-c): Top panels: heat liberated when a 25 mM KCl solution is injected into the experimental chamber containing protein and Phosphate (a), Hepes (b) and Tris (c). Each downward deflection corresponds to one injection. Bottom panels: the area underneath each deflection is integrated and represents the total heat exchanged (squares). Black lines are the best fits to a single site binding isotherm. The averaged thermodynamic parameters are reported in Table 1. d) Plot of the total enthalpy of Cl− binding, ΔHtot, as a function of the enthalpy of buffer ionization, ΔHbuff. Squares: experimental data. Solid line: fit to Eq. 1 with ΔHprot = −8.9±0.4 kcal mol−1 and N = 0.5±0.1. The errors for the experimental points are the s.e.m. while those on ΔHprot and N represent the uncertainty of the fit.
The identity of the stoichiometries of transport and binding suggests that these linked binding events are part of the exchange cycle. An alternative explanation could be that the absorbed heat reflects protonation of one or more residues outside of the anion pathway with a serendipitous coincidence of the stoichiometries. To test whether this is the case, we introduced mutations that alter the stoichiometry of transport and asked how they affect the stoichiometry of binding. We mutated three residues, Glu148, Glu203 and Tyr445, which line the Cl− pathway (Glu148 and Tyr445) or the H+ pathway (Glu148 and Glu203) and are involved in H+ coupling 22,28,38. Mutating either Glutamate residue to Alanine or Glutamine completely abolishes H+ transport 22,28 while the Y445L mutation degrades the transport stoichiometry from 2 Cl− :1 H+ to ~6:1 38. Chloride binding to the purified E148A transporter is nearly iso-enthalpic (Fig. 3, Table 1): the total enthalpies are indistinguishable (p>0.4), ΔHtot(Phosphate) = −13.5±1.3 kcal mol−1~ΔHtot (Hepes) = −12.9 ± 0.5 kcal mol−1~ΔHtot (Tris) = −12.0±1.0 kcal mol−1. Therefore, the E148A mutation completely abolishes coupling between H+ and Cl− in both the transport cycle and in binding. The Y445L mutation also influences both processes to a similar extent: Cl− binding still promotes protonation but the stoichiometry is drastically reduced to N = 0.12±0.07 (Fig. 4, Table 1), a value within experimental error of the stoichiometry of transport of ~0.17 seen in this mutant 38. Thus, two mutations in the anion transport pathway have parallel effects on the ability of the protein to couple Cl− and H+ transport and binding. In contrast, the E203Q mutation leaves the stoichiometry of binding nearly unaltered, N = 0.43±0.05 (Fig. 5, Table 1), while it eliminates coupled exchange. Therefore, our data suggests that binding of Cl− induces protonation of a single residue within the transport pathway, Glu148, as mutating this residue is sufficient to abolish coupled binding and transport. In contrast, protonation of Glu203 is independent of Cl− binding, although it is an essential step in the transport cycle.
Figure 3.
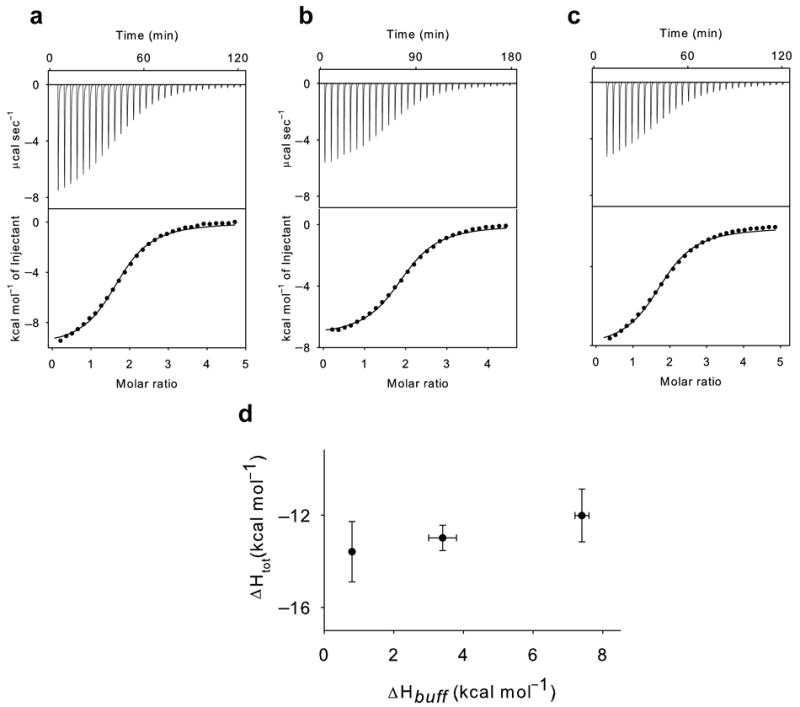
The E148A mutant uncouples Cl− and H+ binding. (a-c): Panels as in Figure 2 with the difference that a 5 mM KCl solution is injected into the experimental chamber. In these fits n was kept as a free parameter rather than fixed. The averaged thermodynamic parameters are reported in Table 1. (d) Plot of the total enthalpy of Cl− binding, ΔHtot, as a function of the enthalpy of buffer ionization, ΔHbuff. Squares: experimental data. The data points are indistinguishable, p>0.4, t-test. The errors for the experimental points are the s.e.m.
Figure 4.
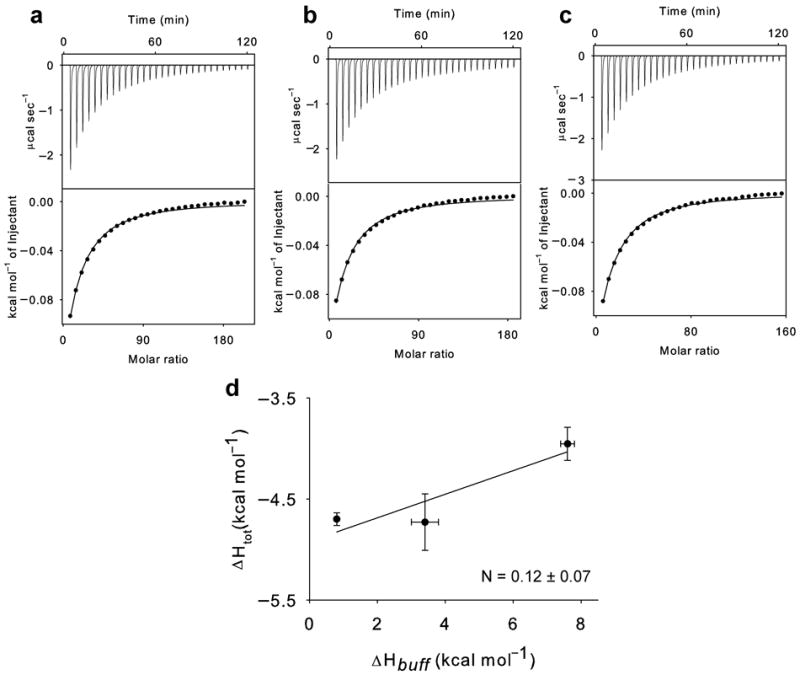
Cl−-coupled H+ binding to the Y445L mutant. (a-c): Panels as in Figure 2 with the difference that 75-100 mM KCl is titrated into the experimental chamber. The averaged thermodynamic parameters are reported in Table 1. (d) Plot of the total enthalpy of Cl− binding, ΔHtot, as a function of the enthalpy of buffer ionization, ΔHbuff. Squares: experimental data. Solid line: fit to Eq. 1 with ΔHprot = −4.9±0.3 kcal mol−1 and N = 0.12±0.07. The errors for the experimental points are the s.e.m. while those on ΔHprot and N represent the uncertainty of the fit.
Figure 5.
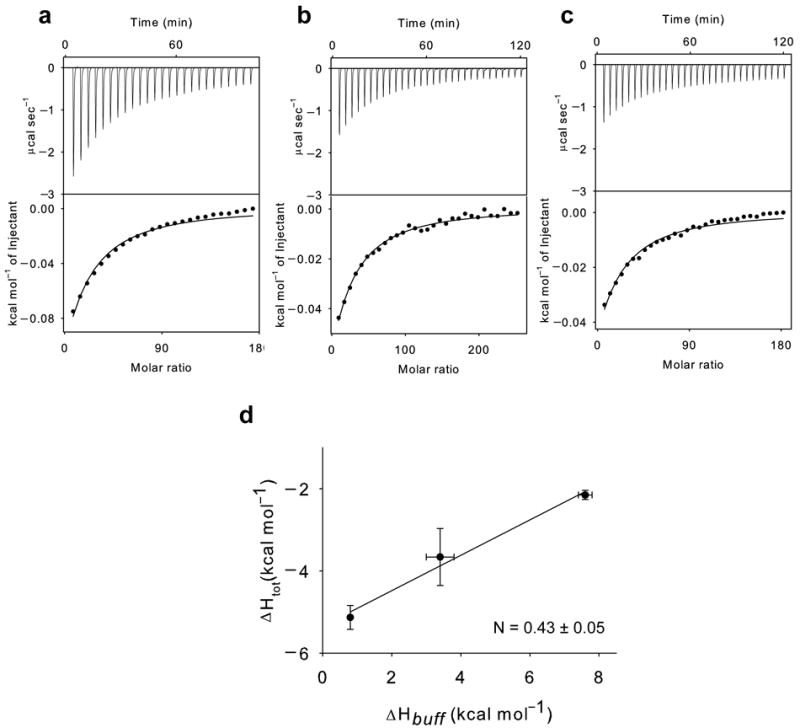
Cl−-coupled H+ binding to the E203Q mutant. (a-c): Panels as in Figure 2 with the difference that 75-100 mM KCl is titrated into the experimental chamber. The averaged thermodynamic parameters are reported in Table 1. (d) Plot of the total enthalpy of Cl− binding, ΔHtot, as a function of the enthalpy of buffer ionization, ΔHbuff. Squares: experimental data. Solid line: fit to Eq. 1 with ΔHprot = −5.4±0.3 kcal mol−1 and N = 0.43±0.05. The errors for the experimental points are the s.e.m. while those on ΔHprot and N represent the uncertainty of the fit.
Cl−-dependence of the free energy of protonation of Glu148
How does Cl− binding control protonation of Glu148? We used all-atom molecular dynamics simulations to calculate the protonation free energy of Glu148 and Glu203 considering different Cl− occupancy states of the transport pathway (see Supplementary Table 2 for a complete list of the calculations). When no ions are present in the pathway Glu148 relaxes to a position close to Scen (Supplementary Fig. 1a) where its protonation is extremely unfavorable, with a very high free energy barrier ΔG(0 Cl−) = 17.3±4.5 kcal mol−1. We then analyzed the effect that occupancy of a single ion binding site, either Sex or Scen, has on the free energy barrier to protonate Glu148. When a Cl− ion occupies Scen we see that Glu148 stably resides in Sex (Supplementary Fig. 1b) and that it protonation free energy is reduced by ~16 kcal mol−1 to ΔGSex(1 Cl−) = 1.1±0.7 kcal mol−1 (Fig. 6a). In contrast, occupancy of Sex by a Cl− ion has a much smaller effect on the energy required to protonate Glu148 positioned in Scen (Fig. 6a, Supplementary Fig. 1c) so that this process remains unfavorable as the associated free energy difference ΔGScen(1 Cl−) = 9.2±1.4 kcal mol−1. Finally, when we place 2 Cl− ions in the transport pathway, at Scen and Sex, protonation of the Glu148 side chain, located outside the pathway (Fig. 6a, Supplementary Fig. 1d), becomes strongly favored ΔGOut(2 Cl−) = −9.4±3.2 kcal mol−1.
Figure 6.
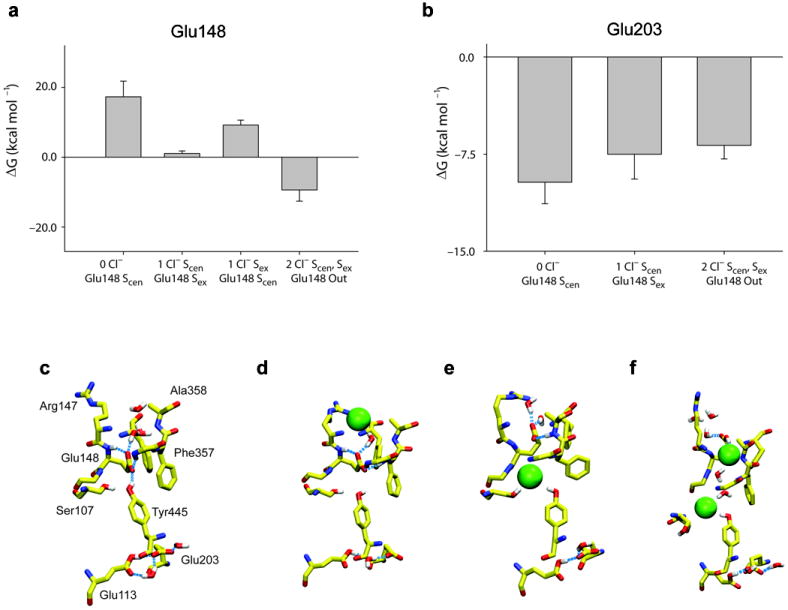
Modulation of the free energy of protonation of Glu148 and Glu203 by Cl− occupancy of the pathway. (a-b) Protonation free energy of residues Glu148 (a), and Glu203 (b) for different Cl− occupancy states of the transport pathway. The free energy values are the average of the ΔG of protonation and deprotonation calculated with protocols A and B (see Supplementary Table 2 for a complete list of the calculations). The meaning of the error bars is discussed in Supplementary Table 2. For Glu148 we considered Glu113 to be protonated and for Glu203 we considered no other residues protonated. (c-f) Representative conformation of the ion binding region in presence of 0 Cl− ions(c), 1 Cl− bound to Sex and Glu148 in Scen (d), 1 Cl− bound to Scen and Glu148 in Sex (e) or 2 Cl− (f) after at least 2 ns of MD simulations. The Glu148 side chain is protonated in the latter case. Only some key hydrogen atoms are shown.
In contrast to these results, our calculations suggest that the ΔG of protonation of Glu203 is independent from the Cl− occupancy of the transport pathway (Fig. 6b). It is worth emphasizing that several assumptions, such as the protonation state of individual residues of CLC-ec1, affect the absolute values of the calculated ΔGs. For example, the ΔG of protonation of Glu203 is exquisitely sensitive to the protonation state of Glu113 (Supplementary Table 2, see also Materials and Methods for a discussion of this). Therefore, the absolute values of the reported free energies of protonation in the different configurations might be difficult to interpret and their translation into pKa values is inappropriate. However, the effects of Cl− binding on the free energy of protonation for Glu148 and Glu203 is unaffected by these choices: protonation of Glu148 is unfavorable with 0 Cl− bound and favored by double occupancy of the pathway regardless of whether Glu113, Glu203 or both are protonated or not (Supplementary Table 2). Similarly, the work needed to protonate Glu203 is independent on the number of Cl− ions bound for all protonation states of E113, E202 and Glu148 (Supplementary Table 2).
Our simulations suggest that two factors play a key role in regulating the work required to protonate Glu148: the hydrogen bond network formed by Glu148 with the protein and the electrostatic interactions between Glu148 and the Cl− ions. When no Cl− ions are bound to the pathway the side chain of Glu148 relaxes to a position close to the central binding site (Supplementary Fig. 1a), similar to the conformation observed in the cmCLC structure 29 (Fig. 6c), where it is stabilized by a network of hydrogen bonds with proton donor groups from different residues (the backbone of Glu148, Gly149, Ile356, Phe357 and the hydroxyl group of Tyr445). This conformation limits the access of water molecules to the carboxylate group of Glu148 further stabilizing the unprotonated state of Glu148. This coordination network is only slightly affected by the presence of a Cl− ion bound to Sex, so that protonation of Glu148 in Scen remains an energetically unfavorable process (Fig. 6d). An ion in the central site stabilizes the Glu148 side chain in the external site where it still interacts with protein residues (Arg147, Glu148, Ala358) but becomes more accessible to water molecules so that its carboxylate group becomes relatively well solvated (Fig. 6e). The increased solvation together with the electrostatic influence of the ion bound to the central site drastically lower the free energy barrier for protonation by ~17 kcal mol−1. When the pathway is occupied by two ions and Glu148 side chain is extended towards the extracellular solution and interacts with the side chain of R147 and water molecules (Fig. 6f). The proximity of two negatively charged Cl− ions, together with the increased accessibility of water molecules to Glu148, greatly favor protonation as reflected by the drastically lowered ΔG (Fig. 6a; Supplementary Table 2). Once protonated, the side chain of Glu148 can either remain solvated or form a hydrogen bond with the Cl− ion in the external binding site (Fig. 6f).
pH dependence of Cl− uptake in WT and coupling-impaired mutants of CLC-ec1
These results suggest that Cl− binding modulates the protonation of Glu148 by directly perturbing its pKa. This would predict that there should be a correlation between degraded coupling of binding and altered pKa of transport. We tested this hypothesis by measuring the pH dependence of transport mediated by WT or mutant CLC-ec1. We used the 36Cl uptake assay rather than the Cl− efflux assay 39 because at pH>5.5 protein-free liposomes display substantial leak to Cl− once Valinomycin and FCCP are added to initiate efflux (Supplementary Figure 2). The rate of 36Cl uptake is a direct measure of the rate of the half-cycle of an exchanger (Fig. 2a) rather than of the full turnover. The rate of uptake mediated by the WT protein is strongly enhanced at acidic pH’s 40,41 (Fig. 7a) with an apparent pKa of 6.2±0.1 (Fig. 7). Uptake mediated by the E148A mutant is pH-independent 41 (Fig. 7b), while the other two mutants tested, E203Q and Y445L, maintain robust pH dependencies 28 (Fig. 7c, d). Consistent with our hypothesis, the apparent pKa of uptake mediated by the E203Q mutant is very close to that of the WT, 6.2±0.5, while that of the Y445L mutant is severely shifted to 4.7±0.1 (Fig. 7e, f). The strong correlation between disruption of the equilibrium coupling stoichiometry and the apparent pKa of the half cycle of CLC-ec1 is consistent with our hypothesis that Cl− binding modulates the protonation probability of Glu148 by shifting its pKa.
Figure 7.
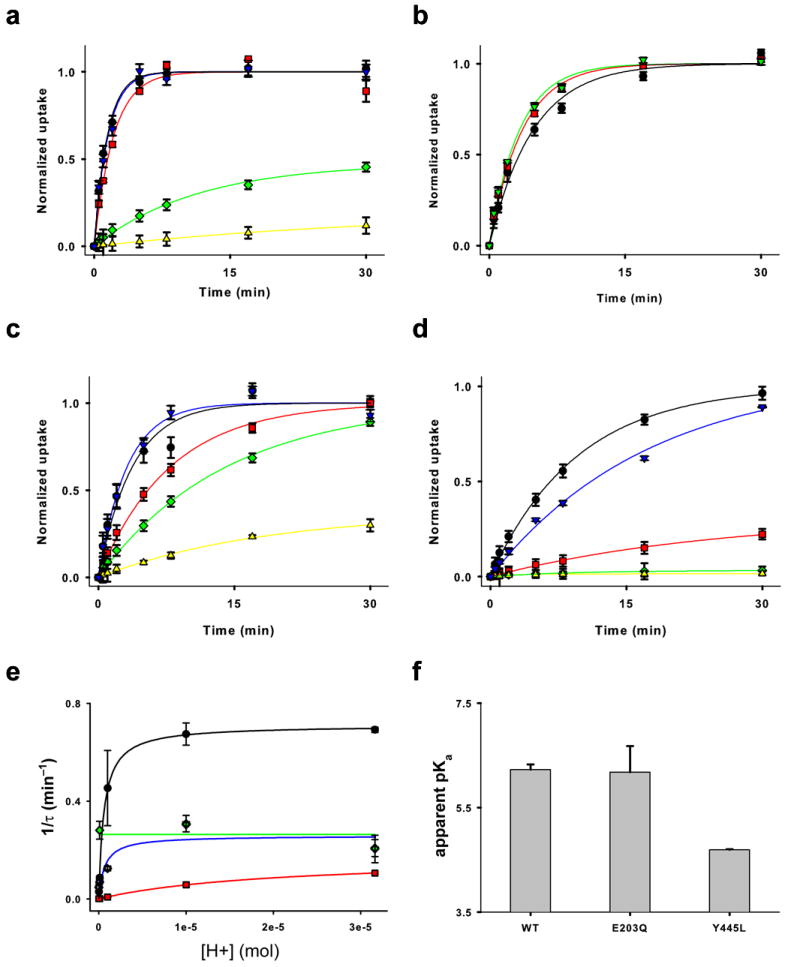
pH dependence of the Cl− half cycle of CLC-ec1. (a-d) Time course of 36Cl− uptake into proteoliposomes reconstituted with WT (a), E148A (b), E203Q (c) or Y445L (d) CLC-ec1 as a function of pH. Black: pH 4.5; Blue: pH 5; Red: pH 6; Green: pH 7 and Yellow: pH 8. Symbols and error bars represent the average of at least 3 independent time courses and the s.e.m. respectively. Solid lines are the fits to a single exponential or to a first order polynomial as discussed in the Materials and Methods section. (e) The rate of uptake (K = 1/τ) is plotted as a function of H+ concentration. Black: WT CLC-ec1; Green: E148A; Blue: E203Q and Red: Y445L. Solid lines represent fits to a single site binding curve of the form: K = A/(1+Kd/[H+]). (f) apparent pKa of the rate of 36Cl uptake for WT CLC-ec1 and its two mutants E203Q and Y445L. The errors represent the uncertainty of the fits in panel e.
pH dependence of Cl− binding to CLC-ec1
Our results suggest that Cl− binding and protonation of the Glu148 side chain are energetically linked. However, the high ΔG for protonation of Glu148 in the absence of Cl− (Fig. 6a) suggests that, at physiological concentrations, protons might not be able to drive Cl− binding. To test this hypothesis we measured Cl− binding in a wide pH range, 4.5-7.5 and found that a 3 orders of magnitude increase in H+ concentration is unable to promote Cl− binding (Supplementary Figure 3). This result is consistent with the hypothesis that a high free energy barrier prevents protonation of the empty pathway so that Cl− binding is not driven by H+. Thus, substrate association to CLC-ec1 is strictly sequential, at least at physiological pHs: Cl− binding is followed by protonation of Glu148.
Discussion
We used a combination of Isothermal Titration Calorimetry measurements and free energy calculations to investigate the mechanism of coupled substrate binding and transport in CLC-ec1, a H+:Cl− exchanger. We found that binding of the two substrates to the wildtype protein is energetically linked so that occupancy of the Cl− transport pathway determines the protonation state of the extracellular gating glutamate, Glu148. In contrast, the protonation state of the intracellular proton acceptor, Glu203, is unaffected by Cl− binding to the pathway. Our data shows that with no ions bound, protonation of Glu148 is extremely disfavored, while binding of 1 or 2 Cl− favors and stabilizes protonation. Our ITC measurements show that the linked binding of Cl− and H+ is stoichiometric, for every ~2 Cl− binding events there is 1 protonation event of Glu148. Several lines of evidence suggest that these coupled binding events are part of the transport cycle. First, the stoichiometries of transport and of linked binding coincide: for every 2 chloride ions that bind to the protein and are transported 1 proton binds and is exchanged (Fig. 2, Supplementary Figure 4). Mutating residues that directly line the Cl− pathway, Glu148 and Tyr445, have identical effects on both the stoichiometries of transport and coupled binding (Fig. 3, 4, Supplementary Figure 4). In contrast, mutating Glu203 which only lines the H+ pathway and is ~10 Å from the Cl− pathway (Fig. 1b), disables H+ transport but leaves coupled binding of Cl− and H+ unaltered (Fig. 5, Supplementary Figure 4). Second, the enhancement of the rate of 36Cl− uptake at low pH (Fig. 7a) implies that during the Cl− half cycle (Fig. 1a) a H+ also binds to CLC-ec1. Furthermore, there is a clear correlation between the effect of mutations on the binding stoichiometry (Fig. 2-4, Supplementary Figure 4) and shifts of the apparent pKa of Glu148 measured using the 36Cl− uptake assay (Fig. 7e,f). Third, the free energy barrier to protonate Glu148 is exquisitely sensitive to the number of Cl− bound to the pathway: double occupancy is required to stabilize a proton bound to this residue (Fig. 6a). In contrast, the work required to protonate Glu203 (Fig. 6b) and Glu113 (Supplementary Table 2) is insensitive to Cl− occupancy of the pathway. Taken together our data suggests that in CLC-ec1 coupled transport requires the formation of a state where both substrates coexist simultaneously bound to the transport pathway. This fully loaded state is incompatible with a canonical double-displacement exchange mechanism, as this paradigm requires antagonistic substrate binding. In contrast, our data shows that transport mediated by CLC-ec1 entails the sequential binding of at least one Cl− ion followed by protonation of Glu148.
On the interpretation of the stoichiometry of linked binding
The most straightforward interpretation of the 2 Cl−: 1 H+ stoichiometry of linked binding would be that our ITC experiments report on binding to a state with 2 Cl− and 1 H+ bound (Fig. 1c). However, in our previous work 37 we described the calorimetric data for WT CLC-ec1 with a single site binding isotherm while this interpretation requires binding of two Cl− ions. Our earlier conclusion 37 was based on two considerations: first, the structures available at the time showed that a deprotonated Glu148 side chain occupies Sex (Fig. 1b) and that upon protonation it moves out of the pathway 31 (Fig. 1c) and, second, mutations that selectively disrupt binding to the central site 38,42 also weaken the binding affinity measured with ITC and equilibrium dialysis 37. Two new observations, however, suggest that other configurations could contribute to the heat absorbed during our ITC experiments. The recently published structure of cmCLC 29 revealed a state in which the gating glutamate (Glu210 in cmCLC) occupies Scen and a Cl− ion is bound to Sex (Fig. 1d). The similar affinity of Cl− for the central and external sites 37,43 further complicates the distinction between Sex and Scen. Thus the binding data could be due to Cl− binding to CLC-ec1 in a combination of these two states with a single Cl− ion bound (Fig. 1b and d). Second, as we show here, Cl− binding promotes protonation of Glu148. When this happens, binding of the Glu148 side chain to the pathway is destabilized, favoring a conformation similar to that of the E148Q mutant 31 (Fig. 1c). Therefore, we propose that at least part of the heat detected in our ITC experiments could be due to a state with 2 Cl− and 1 H+ bound. The main objection to this hypothesis is that Cl− binds to the E148A/Q mutants with a Kd of ~15 μM rather than the Kd of ~700 μM seen for the WT protein 37 (Table 1). However, these mutants are imperfect mimics of the protonated state of the gating glutamate since they cannot give up the proton and become charged. In these mutants the glutamate side chain is either absent (E148A) or does not bind to the pathway (E148Q) even in absence of Cl− 43. In contrast, in the WT protein the Glu148 side chain competes with the Cl− ions for both Scen and Sex. This competition is likely to lower the affinity of Cl− for those sites. In other words, in the WT protein the glutamate side chain acts as a tethered anion that competes with Cl− and lowers its affinity. Thus, we propose that the high Cl− affinity of the E148A/Q mutants arises from the lack of competition with the Glu148 side chain. This hypothesis offers a structural interpretation to the, otherwise puzzling, observation that these mutants have such a high affinity for Cl−, even though their structures are nearly superimposable to that of the WT protein 31.
The 2 Cl−: 1 H+ stoichiometry of linked binding could arise through three scenarios: (1) Cl− binding to both single ion occupancy states (Fig. 1b, d) increases the protonation probability of Glu148 by ~50%; (2) Cl− binding to one of the single ion occupancy states favors protonation of Glu148 while binding to the other state does not; (3) our ITC experiments report on binding to a state with 2 Cl− and 1 H+ bound (Fig. 1c). Our data suggest that protonation of Glu148 is unfavorable with no ions in the transport pathway, that protonation of the gating glutamate is favored by binding of a Cl− ion to Scen and is greatly stabilized by a second Cl− in Sex. In contrast, protonation of the Glu148 side chain bound to Scen remains unfavorable regardless of whether Sex is occupied by a Cl− or not. Thus, we can rule out the first mechanism, as the two single occupancy states have a very different impact on the protonation probability of Glu148. In contrast, our data suggest that both the second and third scenarios contribute to the measured 2:1 stoichiometry of linked binding. Finally, these results highlight the critical role of Cl− binding to Scen in the coupling mechanism in good agreement with previous experimental observations 38,42.
Relationship between coupled substrate binding and exchange
Our results have several implications for the transport mechanism of CLC-ec1, and most likely for all CLC transporters. One of the fundamental characteristics of the CLC exchangers is that they maintain a constant exchange stoichiometry of 2 Cl− for 1 H+ even though these proteins operate in a wide range of pHs, from 2 to 7, and at rates that range from 10 s−1 to nearly 105 s−1 30,39. This characteristic is essential to allow these transporters to fulfill their physiological function: losing H+ coupling leads to disease-like phenotypes in mouse models 33. Recently it has been proposed that coupling in the CLCs is kinetic 29 rather than arising from the coordinated conformational changes of two gates, like in other transporters. In this model the authors postulated that a high energy barrier slows down Cl− movement between the central and internal binding sites so that on average 2 Cl− ions transit per protonation or deprotonation event of Glu148. This argument, however, predicts that acidic pHs should degrade coupling, which is not observed in CLC-ec1 for pHs as low as 3 22,41 and in CLC-5 down to pH 5 23. Furthermore, in CLC-ec1 and CLC-5 32 the apparent pKa of the external glutamate, Glu148 in CLC-ec1, is shifted by nearly two pH units to ~6.2-6.5 (Fig. 7) so that its average protonated lifetime is ~10−4 s, assuming that the proton association rate protons is ~1010 M−1 s−1 29,44. Thus, in order to prevent substantial slippage, the maximal transport rate of these proteins should be less than 102 s−1 32, which is 10-1000 times slower than their measured transport rates 30,39. Our data offers an alternative, or complementary, mechanism to this kinetic model: that the 2:1 transport stoichiometry arises from the energetic requirement that 2 Cl− ions bind to the transport pathway in order to lower the free energy barrier for protonation of Glu148. This coupling mechanism allows the stoichiometry of transport to remain constant in all CLC transporters, provided that they share some basic characteristics, such as a glutamate at position Glu148, two conserved Cl− binding sites and a network of hydrogen bonds that shields Glu148 from extra- and intra-cellular protons. Therefore, we propose that the stoichiometry of transport is hardwired into all CLC transporters by two conserved features: the energetic interactions between substrates bound to the protein and by the structure of the CLC transport pathway. Our proposed mechanism also predicts and rationalizes the observation that a reduction of anion binding affinity, either through mutagenesis 38 or through substrate substitution 42,45, leads to a degraded stoichiometry of binding and of transport. We speculate that the extremely degraded Cl−: H+ coupling revealed by the irreversible gating of the CLC-0 channel 46,47 could result, at least in part, from the weakened Cl− binding affinity of these proteins 48. In other words, channels need to bind substrate weakly to sustain high rates of conduction but this weak binding impairs the energetic coupling of Cl− to protonation of the external glutamate.
We propose that the high free energy barrier which, in the absence of bound Cl−, prevents protons from reaching the carboxyl side chain of Glu148 is a key mechanistic feature that allows CLC-ec1 to maintain coupling, and presumably to preserve its proper function, even at very low pH 22,41. The absence of such a barrier would allow unhindered H+ access to Glu148 leading to nearly permanent protonation of this residue in extreme acidic conditions, such as those found in the stomach 40. This would lead to a constitutively open Cl− pathway, which would result in uncoupled Cl− transport. In contrast, the tight regulation of the protonation probability of Glu148 by the Cl− occupancy of the translocation pathway prevents this potentially lethal occurrence: protonation of Glu148 is permitted and stabilized by occupancy of the pathway by 2 Cl− ions and is destabilized by Cl− unbinding. Thus, during transport, binding of two Cl− ions will induce protonation of Glu148 giving rise to a fully loaded state of the protein. Unbinding of Cl− will destabilize the H+ bound to Glu148, causing its deprotonation, thus promoting closure of the transport pathway and preventing uncoupling. It is possible that in some CLC transporters the free energy barrier which prevents protonation of the external glutamate when the pathway is unoccupied by anions is lower than the one we found in CLC-ec1. This would lead to the uncoupling when exposed to an acidic environment, such as that recently documented for CLC-3 49.
In conclusion, our results suggest that in the CLC transporters stoichiometric coupling of the substrates arises from the direct energetic interactions between the ions bound to the protein rather than from kinetic steps as previously proposed. Thus, in the CLC exchangers the stoichiometry of transport appears to be uniquely determined by the thermodynamics of synergistic substrate binding.
Methods
Protein purification
Expression and purification of CLC-ec1 is performed according to the published protocols28,38,41. The protein was run on a Superdex 200 column (GE Healthcare) pre-equilibrated in 100 mM Na-K-Tartrate, 20 mM 2-Amino-2-hydroxymethyl-propane-1,3-diol (Tris) or 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes) or Phosphate–H2SO4, 5 mM n-Decyl-β-D-Maltoside (DM), pH 7.5 (buffer RT/H/P). Salts and buffers were purchased from Sigma while DM was purchased from Anatrace.
Binding measurements with Isothermal Titration Calorimetry
ITC measurements were performed using either a VP-ITC or an ITC200 Titration Calorimeter from Microcal, Inc. or a nano ITC from TA instruments. The thermodynamic parameters of binding measured with the three machines were comparable in all tested cases. Protein was concentrated to 80-180 μM and added to the experimental chamber. The injection syringe was filled with the appropriate buffer RT/H/P to which 5-100 mM of KCl was added based on the desired final molar ratio of injectant to CLC-ec1. Each experiment consisted of 20-35 injections of 1-10 μl of the ligand solution at 3-4 min intervals into the experimental chamber that is kept under constant stirring at 350 rpm and at 25.0±0.1 °C. All solutions were filtered and degassed prior to use. The protein was extensively dialyzed between runs and re-used.
Fitting ITC data
Fits were performed using the same procedure as described 37. Briefly, the dilution heats of the protein and of the salt were was determined independently and subtracted from each experiment. The data was fit to the Wiseman isotherm using the Origin ITC analysis package or the NanoAnalyze program from TA instruments. The different shapes of the ITC curves are due to the different affinities of Cl− for WT and mutant CLC-ec1 variants 37. The general shape of an ITC curve is determined by the value of the product of the association constant and the receptor concentration, called c-value 50. When c>5 then the curve takes a characteristic sigmoid shape, when c<5 then the curve becomes progressively more hyperbolic. For WT CLC-ec1 and the E203Q and Y445L mutants c<1, so the curves are hyperbolic. In contrast, the E148A mutant has a higher affinity for Cl−, so that c~9 and the curve is sigmoidal. The enthalpy of ionization of the various buffers, ΔHbuff, was determined experimentally (Supplementary Table 1) following established protocols 51.
Cl− uptake measurements were carried out as previously described 41,52. Individual uptake time courses were fitted to a saturating single exponential function of the form C(t) = aM × (1−exp(−t/τ). Where C(t) are the fractional counts at time t, aM is the maximum fraction of radioactivity trapped, t is time and τ is the time constant of uptake. Individual time courses were normalized to aM to yield the normalized uptake and averaged among different experiments. In some cases the time course of uptake was very slow and a fit with an exponential was inappropriate. In these cases we fit the data to C(t) = aM × t/τ and aM was kept fixed to the averaged value measured at pHs where saturation could be reached since the trapped volume does not depend on the pH of reconstitution 41.
Mutagenesis
All mutagenesis is carried out with the Quickchange method (Stratagene) and the mutated genes fully sequenced.
Free energy calculations
The molecular systems used for the calculations were assembled using the CHARMM-GUI web service 53. All systems are comprised by the ClC-ec1 dimer (PDB code 1OTS 31) embedded in a 242 DMPC bilayer and solvated by a ~150 mM KCl solution. The systems were equilibrated according to the different protonation and ion occupancy states listed in Supplementary Table 2. In the 2-ion state (Fig. 6f) both the central (Scen) and external (Sext) binding sites are occupied by Cl−, while in the 1-ion states (Fig. 6d, e) Scen and Sex are alternatively occupied by an ion. No restraint was applied to the glutamate residues during equilibration. The side chain of Glu148 spontaneously moves from Sext to Scen when no Cl− is present in the pore (Fig. 6c). A N-acetyl-Glu-(N’)-methyl-amide dipeptide was placed in solution, away from the membrane, and serves as a proton donor or acceptor. The reported free energy differences thus correspond to the work done to transfer a proton between this solvated di-peptide and a glutamic acid residue in the protein. Two different protocols were used for the staged free-energy perturbation calculations; protocol A: Δλ of 0.1, 11 windows, 20 ps per window, including 2 ps of equilibration; protocol B: Δλ of 0.05, 21 windows, 50 ps per window, including 10 ps of equilibration. Simulations were performed using the PERT module of the CHARMM software 54 with the PARAM27 force field 55. All simulation parameters were as described elsewhere for other membrane systems 56. The free energy difference between the end states was obtained by combining sampling from forward and backward simulations using the WHAM module 57 of CHARMM, which is in all point equivalent to the more recently popularized multistate Bennett acceptance ratio (MBAR) approach 58. According to the BAR analysis, the forward and backward perturbations are not required to give the same results for the final calculated free energy difference to be reliable. They are complementary in providing information on different portion of the configuration space 59. The forward and backward results are nevertheless reported in Supplementary Table 2 since they provide further information on the stability of the end states.
Unless otherwise stated we used for the protonatable residues their natural protonation state at pH 7, which is a value close to that used in the ITC experiments. This assumption is consistent with previous work 60 which showed that only Glu148, Glu113 and Glu203 appear to have severely shifted pKa values. It is worth noting that protonation of Glu203 when Glu113 is already protonated sometime leads to diverging results (in blue). In general, when Glu113 is protonated, the two residues form a hydrogen bond, which impedes the protonation of Glu203. However, the two residues can occasionally be linked by one or more water molecules. In such case, protonation of Glu203 can be favorable. These alternative conformations are possibly relevant to the proton permeation mechanism and will require further investigations.
Data analysis and figure preparation
Flux data was analyzed using the Ana (http://www.ge.ibf.cnr.it/~pusch.ibf/programs-mik.htm) and Sigmaplot (Systat Software, Inc.) softwares. Molecular models were prepared using Pymol (Open-Source PyMOL 0.99rc6)
Supplementary Material
Acknowledgments
The authors wish to thank Chris Miller (Brandeis University) for the generous gift of 36Cl, David Posson, Daniel Basilio, Mattia Malvezzi, Crina Nimigean and Harel Weinstein for helpful discussions and comments on the manuscript. This work was supported by grant 1R01GM085232 from the National Institutes of Health (NIH) to A.A and by a grant from the Swiss National Science Foundation to S. B. (SNF-Professorship #118928). Y.X. is supported by the China Scholarship Council. Part of the simulations was performed using the facilities of the Swiss National Supercomputing Centre (CSCS).
Footnotes
Author Contributions
A.P. performed the ITC measurements; A.A. and A.P. performed the flux experiments and analyzed data; Y.X., N.J. and S.B. performed and analyzed the simulations; A.A. designed research and wrote the paper; all authors contributed to the editing of the manuscript.
References
- 1.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 2.Jennings ML. Structure and function of the red blood cell anion transport protein. Annu Rev Biophys Biophys Chem. 1989;18:397–430. doi: 10.1146/annurev.bb.18.060189.002145. [DOI] [PubMed] [Google Scholar]
- 3.Arkin IT, et al. Mechanism of Na+/H+ antiporting. Science. 2007;317:799–803. doi: 10.1126/science.1142824. [DOI] [PubMed] [Google Scholar]
- 4.Khare D, Oldham ML, Orelle C, Davidson AL, Chen J. Alternating access in maltose transporter mediated by rigid-body rotations. Mol Cell. 2009;33:528–536. doi: 10.1016/j.molcel.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimamura T, et al. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010;328:470–473. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaback HR, Smirnova I, Kasho V, Nie Y, Zhou Y. The alternating access transport mechanism in LacY. J Membr Biol. 2011;239:85–93. doi: 10.1007/s00232-010-9327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radestock S, Forrest LR. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J Mol Biol. 2011;407:698–715. doi: 10.1016/j.jmb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Milanick MA. Ferret red cells: Na/Ca exchange and Na-K-Cl cotransport. Comp Biochem Physiol Comp Physiol. 1992;102:619–624. doi: 10.1016/0300-9629(92)90714-2. [DOI] [PubMed] [Google Scholar]
- 9.Sekler I, Lo RS, Kopito RR. A conserved glutamate is responsible for ion selectivity and pH dependence of the mammalian anion exchangers AE1 and AE2. J Biol Chem. 1995;270:28751–28758. doi: 10.1074/jbc.270.48.28751. [DOI] [PubMed] [Google Scholar]
- 10.Beaugè L, DiPolo R. The squid axon Na+/Ca2+ exchanger shows ping pong kinetics only when the Cai-regulatory site is saturated. Cell Physiol Biochem. 2009;23:37–42. doi: 10.1159/000204078. [DOI] [PubMed] [Google Scholar]
- 11.Adam Y, Tayer N, Rotem D, Schreiber G, Schuldiner S. The fast release of sticky protons: Kinetics of substrate binding and proton release in a multidrug transporter. Proc Natl Acad Sci U S A. 2007;104:17989–17994. doi: 10.1073/pnas.0704425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Restrepo D, Cronise BL, Snyder RB, Knauf PA. A novel method to differentiate between ping-pong and simultaneous exchange kinetics and its application to the anion exchanger of the HL60 cell. J Gen Physiol. 1992;100:825–846. doi: 10.1085/jgp.100.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Contreras-Jurado C, Sanchez-Morito N, Ruiz-Contreras A, Gonzalez-Martinez MT, Soler-Diaz A. Evidence for simultaneous 1Na+:1Mg2+ and ping pong 2Na+:1Mg2+ exchangers in rat thymocyte. Front Biosci. 2005;10:1639–1706. doi: 10.2741/1653. [DOI] [PubMed] [Google Scholar]
- 14.Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 15.Abramson J, et al. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 16.Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 17.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 18.Morth JP, et al. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- 19.Olesen C, et al. The structural basis of calcium transport by the calcium pump. Nature. 2007;450:1036–1042. doi: 10.1038/nature06418. [DOI] [PubMed] [Google Scholar]
- 20.Fang Y, et al. Structure of a prokaryotic virtual proton pump at 3.2 A resolution. Nature. 2009;460:1040–1043. doi: 10.1038/nature08201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Structure and mechanism of a Na+-independent amino acid transporter. Science. 2009;325:1010–1014. doi: 10.1126/science.1176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Accardi A, Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- 23.Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- 24.Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- 25.De Angeli A, et al. The nitrate/proton antiporter AtCLCa mediates nitrate accumulation in plant vacuoles. Nature. 2006;442:939–942. doi: 10.1038/nature05013. [DOI] [PubMed] [Google Scholar]
- 26.Graves A, Curran P, Smith C, Mindell J. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:5. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- 27.Matsuda J, et al. Overexpression of CLC-3 in HEK293T cells yields novel currents that are pH dependent. Am J Physiol Cell Physiol. 2008;294:251–262. doi: 10.1152/ajpcell.00338.2007. [DOI] [PubMed] [Google Scholar]
- 28.Accardi A, et al. Separate ion pathways in a Cl-/H+ exchanger. J Gen Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a Eukaryotic CLC Transporter Defines an Intermediate State in the Transport Cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zdebik AA, et al. Determinants of anion-proton coupling in mammalian endosomal CLC proteins. J Biol Chem. 2008;283:4219–4227. doi: 10.1074/jbc.M708368200. [DOI] [PubMed] [Google Scholar]
- 31.Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 32.Accardi A, Picollo A. CLC channels and transporters: proteins with borderline personalities. Biochim Biophys Acta. 2010;1798:1457–1464. doi: 10.1016/j.bbamem.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novarino G, Weinert S, Rickheit G, Jentsch TJ. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science. 2010;328:1398–1401. doi: 10.1126/science.1188070. [DOI] [PubMed] [Google Scholar]
- 34.Baker BM, Murphy KP. Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys J. 1996;71:2049–2055. doi: 10.1016/S0006-3495(96)79403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petrosian SA, Makhatadze GI. Contribution of proton linkage to the thermodynamic stability of the major cold-shock protein of Escherichia coli CspA. Protein Sci. 2000;9:387–394. doi: 10.1110/ps.9.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velazquez-Campoy A, et al. Thermodynamic dissection of the binding energetics of KNI-272, a potent HIV-1 protease inhibitor. Protein Sci. 2000;9:1801–1809. doi: 10.1110/ps.9.9.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picollo A, Malvezzi M, Houtman J, Accardi A. Basis of substrate binding and conservation of selectivity in the CLC family of channels and transporters. Nat Struct Mol Biol. 2009;16:1294–1301. doi: 10.1038/nsmb.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Accardi A, Lobet S, Williams C, Miller C, Dutzler R. Synergism between halide binding and proton transport in a CLC-type exchanger. J Mol Biol. 2006;362:691–699. doi: 10.1016/j.jmb.2006.07.081. [DOI] [PubMed] [Google Scholar]
- 39.Walden M, et al. Uncoupling and turnover in a Cl-/H+ exchange transporter. J Gen Physiol. 2007;129:317–329. doi: 10.1085/jgp.200709756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer R, Iverson TM, Accardi A, Miller C. A biological role for prokaryotic ClC chloride channels. Nature. 2002;419:715–718. doi: 10.1038/nature01000. [DOI] [PubMed] [Google Scholar]
- 41.Accardi A, Kolmakova-Partensky L, Williams C, Miller C. Ionic currents mediated by a prokaryotic homologue of CLC Cl- channels. J Gen Physiol. 2004;123:109–119. doi: 10.1085/jgp.200308935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguitragool W, Miller C. Uncoupling of a CLC Cl-/H+ exchange transporter by polyatomic anions. J Mol Biol. 2006;362:682–690. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Lobet S, Dutzler R. Ion-binding properties of the ClC chloride selectivity filter. Embo J. 2006;25:24–33. doi: 10.1038/sj.emboj.7600909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jenks WP. Catalysis in Chemistry and Enzymology. McGraw Hill Text; 1969. [Google Scholar]
- 45.Zifarelli G, Pusch M. Conversion of the 2 Cl(-)/1 H(+) antiporter ClC-5 in a NO(3)(-)/H(+) antiporter by a single point mutation. EMBO J. 2009;10:1111–1116. doi: 10.1038/emboj.2008.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richard EA, Miller C. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 1990;247:1208–1210. doi: 10.1126/science.2156338. [DOI] [PubMed] [Google Scholar]
- 47.Lísal J, Maduke M. The ClC-0 chloride channel is a ‘broken’ Cl-/H+ antiporter. Nat Struct Mol Biol. 2008;15:805–810. doi: 10.1038/nsmb.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen MF, Chen TY. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J Gen Physiol. 2003;122:133–145. doi: 10.1085/jgp.200308844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuda J, Filali M, Collins M, Volk K, Lamb F. The ClC-3 Cl-/H+ antiporter becomes uncoupled at low extracellular pH. J Biol Chem. 2009;285:2569–2579. doi: 10.1074/jbc.M109.018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ladbury JE, Doyle ML. Biocalorimetry 2 Applications of calorimetry in the biological sciences. Wiley; 2004. [Google Scholar]
- 51.Fukada H, Takahashi K. Enthalpy and heat capacity changes for the proton dissociation of various buffer components in 0.1 M potassium chloride. Proteins. 1998;33:159–166. [PubMed] [Google Scholar]
- 52.Maduke M, Pheasant DJ, Miller C. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J Gen Physiol. 1999;114:713–722. doi: 10.1085/jgp.114.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29 doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 54.Brooks BR, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacKerell ADJ, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 56.Boiteux C, Bernèche S. Absence of ion-binding affinity in the putatively inactivated low-[K+] structure of the KcsA potassium channel. Structure. 2011;19:70–79. doi: 10.1016/j.str.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 57.Souaille M, Roux B. Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Coputer Physics Communications. 2001;135:40–57. [Google Scholar]
- 58.Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129:123105. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pohorille A, Jarzynski C, Chipot C. Good Practices in Free-Energy Calculations. J Phys Chem B. 2010;114:10235–10253. doi: 10.1021/jp102971x. [DOI] [PubMed] [Google Scholar]
- 60.Faraldo-Gomez JD, Roux B. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J Mol Biol. 2004;339:981–1000. doi: 10.1016/j.jmb.2004.04.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.