

Abstract
IκBNS has been identified as a member of the IκB family of NF-κB inhibitors, which undergoes induction upon T cell receptor (TCR) signaling. Mice carrying a targeted gene disruption of IκBNS demonstrate dysregulation of cytokines in T cells, macrophages and dendritic cells. IκBNS mediates both positive and negative gene regulation, depending on individual cell type and/or cytokine. Here we demonstrate an additional role for IκBNS in the B cell lineage. B cells from IκBNS knockout (KO) mice are impaired in proliferative responses to LPS and anti-CD40. IgM and IgG3 immunoglobulins are drastically reduced in the serum of IκBNS KO mice although IκBNS KO B cells exhibit a higher level of surface IgM than wild type (WT) mice. Switching to IgG3 is significantly reduced in IκBNS KO B cells. The in vitro induction of plasma cell development demonstrates that progression to antibody secreting cells is impaired in IκBNS KO B cells. In agreement with this finding, the number of antibody secreting cells in the spleens of IκBNS KO mice is reduced and production of antigen-specific immunoglobulins is lower in IκBNS KO mice after influenza infection as compared to WT mice. In addition, IκBNS KO mice lack B1 B cells and exhibit a reduction in marginal zone B cells. Thus, IκBNS significantly impacts the development and functions of B cells and plasma cells.
Introduction
NF-κB is a transcription factor that was first identified in B cells (1) but is now recognized as a master controller of multiple genes in virtually every cell type. In particular, NF-κB plays a key role in the overall regulation of the immune system and the inflammatory response. NF-κB consists of homodimers or heterodimers formed by five different NF-κB family members (2-5). Transcriptional control via NF-κB is extremely rapid due to the unique mechanism of regulation of NF-κB by inhibitory proteins termed IκB's. The first IκB protein identified and best characterized, IκBα, binds NF-κB proteins and positions them in the cytoplasm and away from the nucleus, thus preventing DNA binding and gene regulation (6, 7). Upon triggering of cell surface receptors that signal through NF-κB including the T cell receptor (TCR), B cell receptor (BCR), TNF Receptor (TNFR) 1/2 or many other input signals, the IκBα protein is phosphorylated by IκB kinase (IKK) and then ubiquitinated, leading to degradation of IκBα and release of the NF-κB heterodimer enabling nuclear translocation followed by gene regulation. In turn, IκBα is itself a target of NF-κB regulation such that degradation of IκBα releases NF-κB, which then acts to induce synthesis of new IκBα proteins that can begin another cycle of sequestering NF-κB and shutting down the transcriptional activity. This mechanism of NF-κB activation has been termed the “classical” activation pathway. A pathway activating relB, termed the “non-classical” pathway, involves the partial proteolysis of p100 to p52, which translocates to the nucleus with relB (reviewed in (8, 9). Thus the extremely rapid response to the over 150 stimuli that induce NF-κB activity is a result of the release from IκB inhibition of the pre-existing NF-κB proteins and NF-κB activation occurs without the need for transcription or translation allowing cells to respond very rapidly to cell surface signals.
Five forms of NF-κB proteins have been identified (2-5) but detailed analysis of their specific individual roles has been complicated due to the overlapping tissue distributions and redundant functions of the various homo- and heterodimeric NF-κB pairs. The p65, c-Rel and relB NF-κB proteins contain transactivation domains (TAD) capable of activating gene transcription, while p50 and p52 lack TADs. Thus homodimers of p50 and p52 are thought to inhibit gene transcription by blocking κB-binding sites. Strict regulation of NF-κB activation is necessary for proper immune cell function and avoidance of tumor formation (10) and abnormal levels of NF-κB subunits lead to a variety of cancers (reviewed in (11) including various B cell leukemias (reviewed in (12). Targeted gene disruption or transgenic overexpression of NF-κB genes has aided more precise delineation of the roles of each of these subunits. In particular, deletion of each of the NF-κB subunits affects the immune response in some way, underscoring the importance of NF-κB in cells of the immune system (reviewed in (13). The phenotypes of mice carrying deletions of NF-κB genes include effects on both T and B cells, but herein our concentration is on B lymphocytes. Deletion of rela is embryonic lethal and reconstitution of rag-1-/- or SCID mice with day E13 rela-/- fetal liver cells demonstrates that RelA is required for mitogen-induced lymphocyte proliferation and isotype switching. Various defects in B cell activation result from the deletion of rel and isotype switching is also affected, perhaps as a result of reduced transcription through the heavy chain locus. Isotype switching and Ig secretion are normal when assayed in vitro on relb-/- cells indicating that the humoral defect of impaired IgG responses to T cell-dependent antigens observed in relb-/- mice is most likely a secondary consequence due to the absence of specific dendritic cell populations. Thus while normal hemopoiesis does not require any of the other NF-κB proteins, the development of specific dendritic cell populations requires relB. In nfkb1-/- mice, immunoglobulin secretion, immunogloblulin switching and proliferation are impaired and quiescent nfkb1-/- B cells turnover more rapidly, suggesting that p50 is required for survival of non-activated B cells. Mice lacking p52/p100 proteins develop normally but display disruption of splenic and lymph node architecture. However, nfkb2-/- lymphocytes activated in vitro exhibit only mildly impaired proliferative responses coupled with normal antibody or cytokine production. Thus while the failure of nfkb2-/- mice to mount a normal T cell-dependent antibody response is associated with an inability to form germinal centers, intrinsic B-cell defects are excluded.
With regard to the NF-κB family of proteins no new family members have been identified for some time. In contrast, the family of IκB inhibitors has continued to expand with the identification of IκBNS (14) and IκBζ (15-17). Prior to this, seven NF-κB inhibitor proteins were known: IκBα, IκBβ, IκBε IκBγ Bcl-3, p100 and p105 (reviewed in 2). IκBNS and IκBζ show more sequence homology to each other and to Bcl-3 than to IκBα and, like Bcl-3, these proteins appear to be nuclear rather than cytoplasmic. IκBζ (18), Bcl-3 (2, 19, 20) and IκBNS (21-23) have been shown to regulate promoters of cytokines controlled by NF-κB and to be involved in inflammatory responses. Their mechanisms of action are different from that of IκBα since their expression is transcriptionally rather than post-translationally regulated and more restricted in cell expression patterns (14-19, 24, 25), unlike the more ubiquitous NF-κB and other IκB family members.
We previously created a knockout mouse line of IκBNS and demonstrated that T cells in IκBNS KO mice exhibit defective proliferation and cytokine production (23). For example, IκBNS KO T cells produce less IL-2, a key cytokine in survival and activation of T cells during the immune response (23). Additional cytokines including IFNγ are also affected by IκBNS (23). Others have shown independently that IκBNS suppresses the inflammatory cytokines IL-6 and IL-12 in macrophages and dendritic cells (21, 22). In contrast, IκBζ increases expression of IL-6 and IL-12 as well as other inflammatory cytokines (18). Both IκBNS and IκBζ bind the p50 NF-κB family member (14, 18). Thus, these newly identified members of the IκB family serve to focus the action of the ubiquitous NF-κB transcription factors. Their tissue specificity explains the mechanism by which various cell surface receptors act through the NF-κB pathway to induce different genes in different cell types.
Herein we describe the effects of the deletion of IκBNS on B cells. Our initial analysis of the IκBNS KO mice showed slightly lower total B cell numbers but the difference was not significant between WT and IκBNS KO mice (23). However, further detailed analysis has revealed a disturbance in individual B lineage subsets of IκBNS KO mice. Furthermore, we find that IκBNS KO B cells are deficient in proliferation, immunoglobulin production and plasma cell differentiation.
Materials and Methods
Mice
IκBNS KO mice on a C57BL/6 background were established and maintained as described (23). C57BL/6 (B6) and B6Ly5.1 mice were purchased from Taconic (Germantown, NY). Mice were maintained and bred under specific pathogen-free conditions in the animal facility of the Dana-Farber Cancer Institute under a protocol reviewed and approved by the Institutional Animal Care and Use Committee.
Antibodies and flow cytometric analysis
The following mAbs were used: FITC-anti-Ly5.1 (A20), FITC-anti-CD19 (1D3), FITC-anti-CD5 (53-7.3), FITC- or PE-Cy7-anti-IgM (R6-60.2), PE-anti-CD45.2 (104), PE-anit-CD21/35 (7G6), PE-anti-CD138 (Syndecan-1) (281-2), PE- or APC-Cy7-anti-B220 (RA3-6B2), biotin-anti-CD23 (B3B4), PE-Cy7- or APC-streptavidin (BD Biosciences). Single cell suspensions were prepared in a FACS buffer solution (1× PBS containing 2% FCS and 0.05 % NaN3). Cells were pretreated for 10 min at 4°C with anti-FcγRII/III (clone 2.4G2; 1 μg/ml) to reduce nonspecific staining. Staining for cell-surface antigen expression was performed at saturating antibody concentrations for 20 min at 4°C. Cells were washed once in FACS buffer solution and incubated with second-step reagent if necessary. A FACScan (BD Bioscience) and CELLQuest software were used for analysis of triple stained samples, and FACSAria and FlowJo software (Tree Star) were used for 5-color samples. Dead cells were excluded from the analysis by forward and side scatter gating. While C57BL/6 mice do not have the IgG2a isotype but rather, the IgG2c isotype, the anti-IgG2a antibody crossreacts with IgG2c, and therefore detects IgG2c in our mice.
B cell proliferation assay
Splenic B cells from IκBNS KO or B6 WT mice were purified by negative selection using anti-CD43-conjugated micro beads (Miltenyi Biotec). The CD43 negative cells were suspended in PBS containing 0.5 % BSA, stained with 1 μM CFSE at 37°C for 10 min, washed three times in complete medium, and cultured in the presence of 0.1, 1 or 10 μg/ml LPS (Sigma), 0.1, 1 or 5 μg/ml anti-CD40 (BD Biosciences) or 10 μg/ml anti-IgM (μ chain) (Jackson ImmunoResearch Laboratories). The cells were harvested on different days as indicated, and CFSE intensity monitored by flow cytometry. For determination of B cell proliferation by [3H]-thymidine incorporation, assays were carried out as previously described (23) and 37 KBq per well of [3H]-thymidine was added for the last 18 h of culture, and the incorporated radioactivity measured by scintillation counter (PerkinElmer Micro Beta Jet).
Immunizations
To measure the immune response to T-independent and T-dependent antigens, 8-12 week old mice were inoculated with an intraperitoneal injection of 100 μg of TNP-Ficoll or 75 μg of TNP-KLH (Bioresearch Technologies) mixed with Imject Alum (Pierce) as an adjuvant. Serum was collected every 7 days after immunization and analyzed for antigen-specific antibodies.
For analysis of germinal center formation, 5 WT and 5 IκBNS KO mice received an intraperitoneal injection of 5 × 105 sheep red blood cells (Innovative Research). Mice were sacrificed 5 or 10 days later and spleens examined for PNA reactivity.
Immunohistochemical analysis
Spleens were removed, frozen in Tissue-Tec OCT compound (Sakura Finetek), and subjected to immunofluorescent staining. Cryosections (5 mm thick) were mounted onto slides, air-dried for 1 hr and fixed with ice-cold acetone for 10 min. Sections were stained with TRITC-anti-mouse IgM (SouthernBiotech) and FITC-MOMA-1 (AbD Serotec). The slides were mounted with low fluorescent glycerol (Invitrogen), and observed with a fluorescent microscope (Olympus).
Surface and intracellular immunoglobulin staining
Cells were stained for surface expressing immunoglobulin with biotinylated antibodies and streptavidin-APC. Purified anti-mouse Ig antibodies were then used to block surface immunoglobulin. The membrane-labeled cells were treated with fixation and permeabilization reagents (BD Pharmingen) and then stained with FITC-labeled anti-immunoglobulin diluted in permeabilization medium (0.05% saponin-2% FBS-PBS). The cells were washed and resuspended in PBS containing 2% fetal bovine serum, and immediately analyzed on a flow cytometer.
Influenza virus inoculation
Mice received an intraperitoneal injection of 104 MOI influenza A virus (PR8). Fifteen days after the inoculation, serum was collected to measure the virus-specific antibody levels, and mice were intranasally infected with 104 MOI of PR8. Survival was monitored for 20 days after the infection.
Serum Ig titration
Ig isotype levels of serum were measured by enzyme-linked immunosorbent assay (ELISA) with 2.5 μg/ml anti-IgGAM coated immunoplates and HRP-conjugated mouse Ig isotype-specific antibodies (Southern Biotech). For TNP-specific antibody, the plates were pre-coated with 10 μg/ml of TNP-BSA. The TMB substrate set (BD Pharmingen) was used, and absorbance at 450 nm measured. For detection of Influenza-specific antibody, plates coated overnight with 100 μl of 106 MOI/ml UV-inactivated Influenza virus in PBS were used.
Adoptive transfer of bone marrow cells
Bone marrow cells were prepared from the tibia and femur of 2-3 month-old WT or IκBNS KO mice. T and B cells were eliminated from total bone marrow cells by CD4, CD8- and B220- negative separation using magnetic beads. Bone marrow cells (2 ×106) were i.v. injected into irradiated (900 rad) Ly-congenic mice (B6Ly5.1). Six weeks after the injection, recipient mice were sacrificed, and spleen, peripheral blood and peritoneal cavity cells were FACS analyzed for donor-derived Ly5.2+ cells. For competitive reconstruction assays of B cell subsets, one million bone marrow cells prepared from B6 Ly5.1 and IκBNS KO mice were mixed and transferred together into 900 rad irradiated B6 Ly5.1 mice.
Reconstitution of B and T cells in Rag-2-deficient mice
B cells were purified from splenocytes of WT and IκBNS KO mice by positive selection of B220-expressing cells using anti-B220-conjugated MACS magnetic beads (Miltenyi Biotec). T cells were purified from lymph nodes of WT mice by depleting B cells with anti-B220-conjugated magnetic beads. FACS analysis with B220, CD4 and CD8 staining showed that B cell preparations contained > 98 % B220+ cells, while T cell preparations contained > 95 % (CD4+ and CD8+) cells. The purified B cells from WT and IκBNS KO mice were mixed with T cells from WT mice (1 × 107 each) and injected intravenously into Rag-2-/- mice. The reconstituted mice were challenged with 75 μg of alum-precipitated TNP-KLH given intraperitoneally 24 h after the transfer. Serum was collected every 7 days after immunization and the spleen was harvested on day 14 post-immunization.
ELISPOT assay for antibody-secreting cells
Nitrocellulose membranes were coated with anti-IgGAM antibody at 2.5 μg/ml in PBS for two hours and then blocked with 5% BSA in PBS for four hours both at room temperature. Single cell suspensions were prepared from spleens, red blood cells depleted and the cells incubated with the membranes in 10% FBS, 50 μM 2ME, RPMI1640 at 37°C for 18 hours. Membranes were washed in PBS and incubated with HRP-conjugated goat anti-mouse IgG or IgM for one hour at room temperature followed by detection using an ECL kit (GE Healthcare/Amersham). The results are expressed as the mean of Ig-secreting cells/106 spleen cells for three to five experiments.
Semi-quantitative RT-PCR analysis
Semi-quantitative RT-PCR analysis was performed to compare the relative mRNA levels of μM, μS, γ3 and γ1 in stimulated B cells from WT and IκBNS KO mice. Purified splenic B cells from B6 or IκBNS KO mice were incubated with LPS (5 μg/ml) alone or in combination with IL-4 (10 ng/ml) for three-five days and RNA isolated using TRIzol reagent (Invitrogen). One μg total RNA was used for cDNA synthesis (Roche). Semi-quantitative RT-PCR was performed using β-actin or HPRT as an internal control. Primers for μM and μS (26) and for germline and post-switch γ3 and γ1 (27) were as previously described.
Real-time PCR
RNA isolated using TRIzol from purified WT or IκBNS KO B cells was used for both semi-quantitative and quantitative real time PCR analysis. Real-time PCR was performed using the Applied Biosystems TaqMan Universal PCR Master Mix. TaqMan Gene Expression pre-made probes were used for Blimp-1, Xbp1, Irf4 and β-actin. Probes for IκBNS were custom ordered and sequences are available upon request. The expression level of β-actin was used to normalize the template input. When the relative gene expression levels in WT and IκBNS KO B cells were compared, the mean value of WT B cells was set at 1. Assays were performed in triplicate.
Statistical Analysis
The results are shown as the mean ± SD of values obtained from three to five separate experiments. For proliferation, results of a single representative experiment are provided. ELISA data were analyzed by the Student's t-test to asses the significance of the differences between IκBNS KO and WT groups.
Results
Altered B Cell Compartments in the Absence of IκBNS
We had previously noted a modest reduction in the number of B cells in the spleen and lymph nodes of the IκBNS KO mice (23). This was confirmed after backcrossing the IκBNS KO mice onto a C57BL/6 background for 11 generations. Analyses of cells isolated from the lymph node and spleen of C57BL/6 and IκBNS KO mice using an antibody against the B cell marker, B220, further demonstrates that the percentage of B cells is lower in the IκBNS KO spleen and LN compared to WT. This reduction was even more dramatic in the blood of these animals (Fig. 1A). Furthermore, within the IκBNS KO B220+ population, the distribution of IgM is skewed compared to WT. The geometric mean fluorescence intensity of IgM is 216 on IκBNS KO peripheral blood B220+ cells and 37 on WT B cells (Fig. 1A and S1). These observations led us to perform a more detailed analysis of IκBNS KO B cells as follows.
Figure 1.
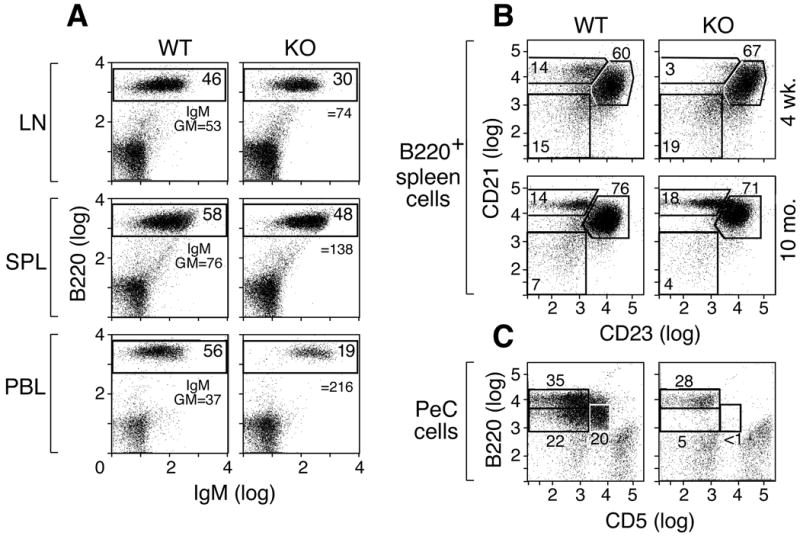
Altered B cell profiles in IκBNS KO mice. A) IκBNS KO mice exhibit a reduction in the percentage of B220+ B cells in the periphery. Cells prepared from the lymph nodes, spleens or blood of 9 month old C57BL/6 or IκBNS KO mice were examined for B220 and IgM surface expression by FACS. Numbers in boxes indicate the percentage of B220-positive cells, and the mean fluorescent intensity of IgM-FITC on B220+ cells is given for each sample. B) Abnormal generation of marginal zone B cells in IκBNS KO mice. B220+ splenic B cells from 4-week or 10-month old WT and IκBNS KO mice were examined for CD21 and CD23 by flow cytometry. Young IκBNS KO mice show a reduced percentage of CD21hiCD23lo marginal zone B cells. C) IκBNS KO mice lack B1 B cells in the peritoneal cavity. Peritoneal cavity cells isolated from WT or IκBNS KO mice were analyzed for B220+ CD5+ B cells. Gates show B2 (CD5-B220hi), B1a (CD5+B220int/lo) and B1b (CD5-B220lo) cells. B1a and B1b cell populations are absent from the IκBNS KO mouse peritoneal cavity. Three age-matched animals were examined in each set of experiments.
B cells consist of a number of different subpopulations including terminally differentiated plasma cells, germinal center B cells and marginal zone B cells. In the periphery, surface IgMhigh B cells contain immature or “transitional’ B cells and marginal zone (MZ) B cells. Therefore, we further analyzed the developmental status of B cells in the absence of IκBNS. As shown in Figure S2, the IκBNS KO has a lower percentage of sIgDhighsIgMlow cells, which contain the most mature B cells, but B cells from the IκBNS KO spleen have slightly more CD93 (AA4.1)-positive immature B cells (7.0 % in IκBNS WT, 8.2 % in IκBNS KO at 2-3 months of age (n=3)). Examination of marginal zone (MZ) B cells (B220+ CD21hi CD23lo) revealed a deficit of this population in the spleens of 4-week old IκBNS KO mice (Fig. 1B). However, as 10 month old IκBNS KO mice have MZ B cells, the development of this population seems delayed rather than blocked in the absence of IκBNS. Histochemical analysis of frozen sections from the spleens of WT and IκBNS KO mice was performed to further examine MZ B cells. In both WT and IκBNS KO mice, the MZ could be observed as layer outside of the B cell rich follicle separated by a layer of MOMA-1+ MZ metallophilic macrophages. The MZ area of IκBNS KO spleens was reduced compared to WT in young mice (Figure S3). In addition, IκBNS KO mice lack B1 B cells, a B220lo population found mainly in the peritoneal and pleural cavities (28) (Fig. 1C). Neither the B1a (CD5+) nor B1b (CD5-) cells are detected in the absence of IκBNS.
When bone marrow cells from IκBNS KO mice were adoptively transferred to irradiated Ly5.1+ congenic WT hosts, the donor-derived Ly5.2+ cells recapitulated the marginal zone B cell deficiency observed in the IκBNS KO mice (Fig. 2A) demonstrating that this developmental defect is inherent to the IκBNS KO cells. In addition, we noted reduced generation of germinal center (Fas+ PNA+) B cells when donor bone marrow cells were derived from IκBNS KO mice (Fig. 2A). These results are confirmed in competitive reconstitution assays. When donor cells consisted of Ly5.1+ WT plus Ly5.2+ IκBNS KO bone marrow cells, the resulting B220+ CD21hi CD23lo MZ cells and Fas+ PNA+ germinal center cells were Ly5.1+ and thus derived from WT but not IκBNS KO bone marrow (Figure 2B). Furthermore, analysis of peritoneal cavity cells after adoptive transfer of WT or IκBNS KO bone marrow shows that CD5+ and CD5- B220lo cells are not repopulated when the donor cells are from IκBNS KO mice (Fig. 2A).
Figure 2.
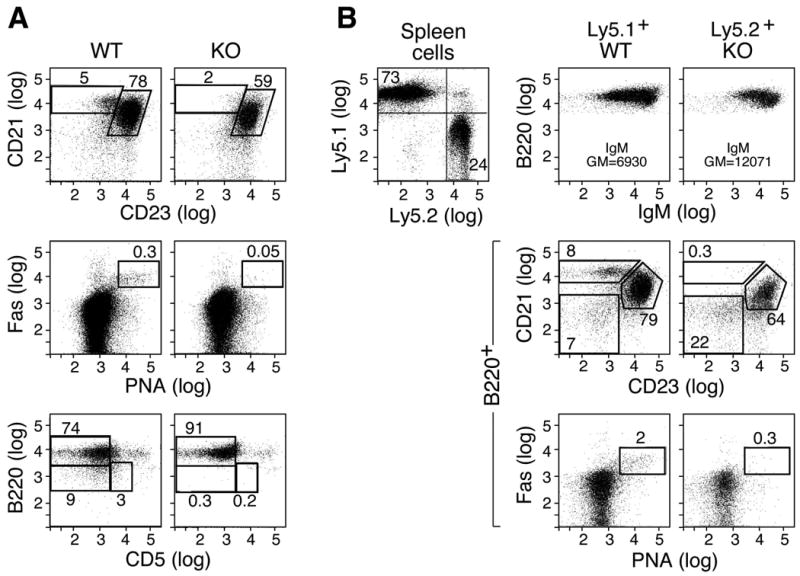
A developmental defect of marginal zone, germinal center and B1 B cells is an intrinsic property of IκBNS KO B cells. A) T- and B-depleted bone marrow cells (2 × 106) from WT or IκBNS KO mice (Ly5.2) were adoptively transferred to lethally irradiated (900 rads) B6Ly5.1 hosts and Ly5.2+ donor-derived cells examined at 6 weeks post-transfer. IκBNS KO-derived bone marrow produced reduced CD21hiCD23lo MZ and PNAhiFas+ GC B cells after gating on B220+ cells. In addition, KO-derived bone marrow failed to repopulate B1a (CD5+B220int/lo) and B1b (CD5-B220lo) cells. Two independent experiments were performed and similar results obtained. B) Competitive reconstitution of B cell populations in bone marrow chimera mice. T- and B-depleted bone marrow cells (2 × 106) from WT (Ly5.1) or IκBNS KO mice (Ly5.2) were adoptively transferred to lethally irradiated (900 rads) B6Ly5.1 hosts and donor-derived B220+ cells examined as described in A. In the upper panel, spleen cells from one bone marrow chimera show Ly5.1 WT- and Ly5.2 KO-derived populations, although fewer KO cells are present. WT Ly5.1+ bone marrow-derived cells repopulated both MZ (CD21hiCD23lo) and GC (PNA+Fas+) subsets whereas IκBNS KO Ly5.2+ bone marrow-derived cells did not. Two recipient animals of each type were analyzed.
IκBNS KO B Cells Are Altered in Proliferation
The expression of IκBNS is induced by TCR mediated signals in T cells (23) and TLR mediated signals in macrophages and dendritic cells (21, 22). We therefore examined the ability of various stimuli to induce IκBNS expression in B cells. Purified splenic B cells were exposed to LPS, anti-CD40 and anti-IgM. Using real-time PCR we demonstrated that IκBNS RNA expression is induced upon stimulation with each of these mitogens (Fig. 3A). Splenic B cells stimulated with LPS, anti-CD40 and anti-IgM increase expression of IκBNS within 0.5 to 6 hours and RNA expression peaks at about 2 hours. The increased RNA levels lead to increased IκBNS protein levels (Fig. 3B).
Figure 3.
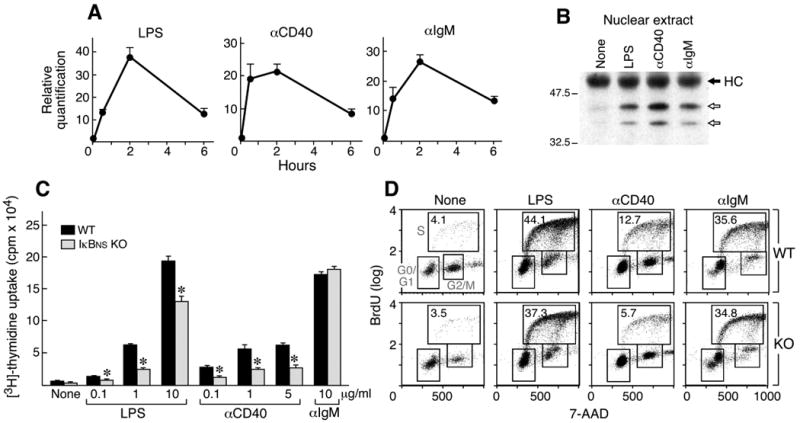
Induction of IκBNS and proliferation of IκBNS KO and WT B cells in response to LPS, anti-IgM and anti-CD40. A) RNA was isolated from WT B cells 0, 0.5, 2 and 6 hours after stimulation by the indicated mitogens and examined for IκBNS expression by real-time PCR. B) Splenic B cells (CD43-) were treated for 2 hours with the indicated reagent or left untreated (None). Nuclear lysates (50μg) were immunoprecipitated using anti-IκBNS antisera. After transfer to PVDF membranes, IκBNS was identified using an anti-IκBNS mAb for Western blotting. Arrows indicate two isoforms. HC indicates the heavy chain of the immunoprecipitating IgG. C) Splenic B cells (CD43-) were incubated for 48 hours in the presence of media only (None) or the indicated reagents (0.1, 1 or 10 μg/ml LPS, 0.1, 1 or 5 μg/ml anti-CD40, 10 μg/ml anti-IgM). Proliferation was measured by 3H-thymidine incorporation. *p<0.01 D) BrdU cell cycle analysis. Cells were pulsed with BrdU, then stained with anti-BrdU antibody and 7-AAD. Percentages of BrdU-positive S phase cells are indicated. Two independent experiments were performed and similar results obtained.
Given that we observed a defect in proliferation of the IκBNS KO T cells (23) and that known B cell mitogens induce IκBNS expression, we next analyzed the proliferation of IκBNS KO B cells. As seen in Fig. 3C, proliferation measured by incorporation of 3H-thymidine in response to LPS and anti-CD40 is reduced in IκBNS KO B cells while the response to anti-IgM is slightly higher. This experiment was performed five times and the response of the IκBNS KO B cells to IgM was consistently equivalent or higher. The alterations in proliferation in the IκBNS KO B cells were further confirmed using flow cytometery and CFSE labeling (data not shown). The lower proliferative response for LPS and anti-CD40 in IκBNS KO B cells results from reduced S phase progression in the absence of IκBNS (Fig. 3D). Signaling induced by LPS, anti-CD40 and anti-IgM involves the NF-κB pathway (reviewed in 4) consistent with the fact that disruption of IκBNS impacts the proliferative response in B cells. Since all three agents induce expression of IκBNS, the fact that the proliferation induced by anti-IgM is not reduced in the IκBNS KO seems a contradiction. However, as described above and shown in Fig. 1, IκBNS KO B cells exhibit an altered distribution of IgM compared to WT B cells. The increased number of IgM molecules on a cell may mask a decreased proliferative response to the crosslinking of IgM in the IκBNS KO B cells.
Defective Immunoglobulin Production in IκBNS KO Mice
As a primary function of B cells is to produce antibody, we determined the level of immunoglobulins in sera from IκBNS KO and WT mice (Fig. 4A). Levels of IgM and IgG3 are dramatically lower in the IκBNS KO mice and levels of IgG2c appear slightly reduced. IgG1 and IgG2b immunoglobulin levels are equal to WT, however. While the surface levels of IgM appear higher on the IκBNS KO B cells, less IgM is secreted in vivo. To examine the possibility that the regulation of membrane vs. secretory IgM production is defective in IκBNS KO B cells, we performed flow cytometric analysis of surface vs. intracellular IgM in B cells purified from the spleens of WT and IκBNS KO mice. While cytoplasmic IgM is virtually undetectable in stimulated and unstimulated IκBNS KO B cells, the surface IgM levels are much higher compared to WT (Figure S4) suggesting a defect in IgM secretion. Both the lack of B1 B cells and the delayed differentiation of marginal zone (MZ) B cells as described above, two major cellular sources of serum IgM (28, 29), as well as a decreased level of the secreted form of IgM mRNA (Fig. 4B) likely account for the low serum IgM in IκBNS KO mice.
Figure 4.
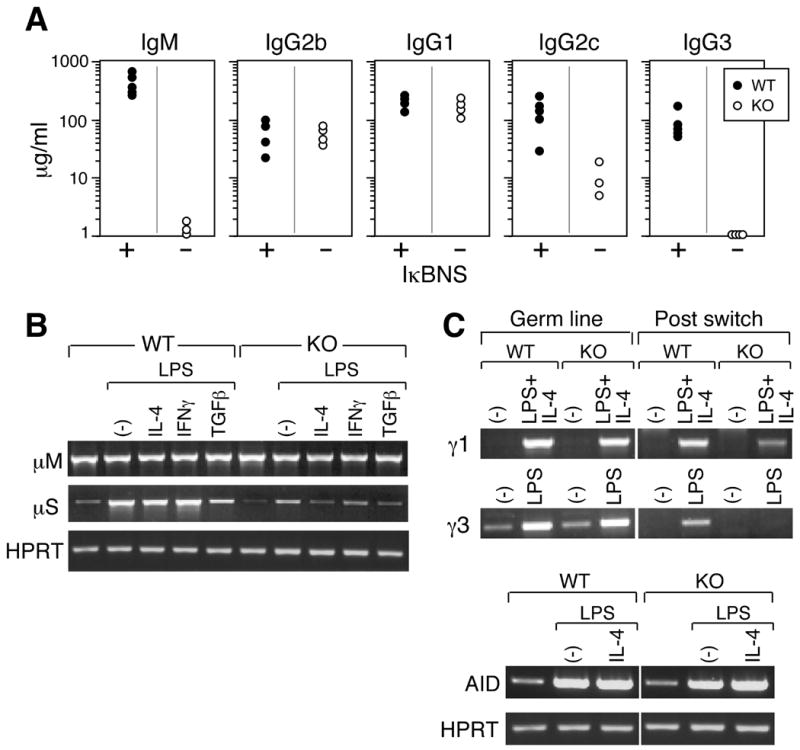
IκBNS KO mice manifest B cell defects in serum immunoglobulin levels and IgG3 switch recombination. A) Basal immunoglobulin isotype levels in the sera from naïve IκBNS KO (-) mice and age-matched WT/HET mice (+) were measured by ELISA. Immunoglobulin levels less than 0.1 μg/ml were plotted on the base line. The serum IgA levels of IκBNS KO mice were similar to that of C57BL/6 mice (data not shown). p<0.05 for IgG2c; p<0.01 for IgM and IgG3. All mice are on a C57BL/6 background: 1 mouse was WT, 4 mice were IκBNS heterozygous and 4 mice were IκBNS KO. B) Splenic B cells (CD43-) from WT and IκBNS KO mice were stimulated in vitro with LPS (5 μg/ml) alone or LPS plus cytokines (10 ng/ml) as indicated for 4 days. Semi-quantitative RT-PCR analysis of μM, μS and HPRT mRNA was performed. C) Germline and post-switch transcripts for IgG1 and IgG3 were assessed in cDNA prepared from cells cultured in the presence and absence of LPS alone or in combination with with IL-4, respectively. Lower panels: AID and HPRT expression before and after stimulation with LPS or LPS plus IL-4 were examined. AID expression confirms that switch recombination processes have initiated and HPRT expression confirms equivalent loading of RNA in each sample. For B and C, two independent experiments were performed and similar results were obtained.
The production of classes of immunoglobulins other than IgM requires switching at the DNA level, and we therefore investigated switching using an in vitro assay. Spleen B cells were exposed to LPS or LPS + IL-4 to induce production of IgG3 or IgG1, respectively (Fig. 4C). Measurement of both germline and post-switch transcription in B cells was carried out by PCR using primers designed to produce distinct DNA products arising from either the germline or recombined DNA configurations. Treatment of both WT and IκBNS KO B cells with LPS or LPS + IL-4 results in increased transcription of Activation-Induced Cytidine Deaminase (AID) to an equal extent relative to the HPRT control (Fig. 4C lower panel). AID is required for the process of immunoglobulin heavy chain class switch recombination (27) and the equal induction of this enzyme in both WT and IκBNS KO B cells implies that the process of class switching has been successfully initiated, at least with respect to this enzyme. Transcription from the germline configuration is approximately equal for the WT and IκBNS KO B cells and the level of transcription from the germline configuration is increased by the addition of LPS + IL-4 for IgG1 and by addition of LPS alone for IgG3 in both WT and IκBNS KO cells (Fig. 4C). The level of transcription from the post-switch configuration of IgG1 appears to be reduced in the IκBNS KO cells while transcription from the post-switch IgG3 configuration is undetectable. The defect in class switching was confirmed when anti-CD40 was used to stimulate IgG3 and IgG1 class switching in presence and absence of IL-4, respectively (Figure S5). Thus the absence of IκBNS dramatically impairs class switching to the IgG3 heavy chain gene as reflected in the lack of detectable IgG3 in the IkBNS KO mouse serum.
Given these deficiencies, are the IκBNS KO mice capable of mounting a humoral immune response? We performed ELISA assays to measure the TNP-specific IgM and IgG produced by IκBNS KO and WT mice before and after intraperitoneal immunization with TNP-Ficoll, a Type II T-independent antigen. At one and two weeks post-immunization, IκBNS KO mice produce no detectable TNP-specific IgM or IgG3 while such antibodies were detected in WT mice at both time points (Fig. 5A). Using TNP-KLH as a T-dependent antigen, we next measured the TNP-specific IgM and IgG1 produced by IκBNS KO and WT mice one and two weeks after immunization. As shown in Fig. 5B, the IκBNS KO mice produce little, if any, TNP-specific IgM. Production of antigen-specific IgG1 appears to be delayed by a week although the final levels of IgG1 are equivalent to those of WT mice. Since we previously showed that the absence of IκBNS also alters T cell functions, we performed adoptive transfer assays into B6.Rag-2-/- mice using purified B cells from WT or IκBNS KO spleen mixed with WT T cells. Upon immunization with TNP-KLH, the production of IgM by IκBNS KO B cells remains decreased showing that the decreased production of IgM is a B-cell intrinsic defect (Figure S6). However, in the presence of WT T cells, production of IgG1 from IκBNS KO B cells is equivalent to that of WT B cells indicating that the defective IκBNS T cells alter the production of IgG1 by the IκBNS KO B cells.
Figure 5.
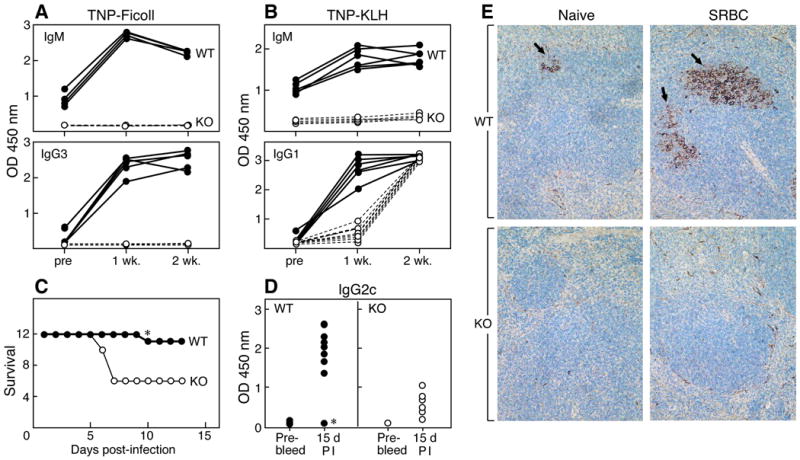
Defective production of antigen-specific antibodies in IκBNS KO mice. 8-12 wk-old WT and IκBNS KO mice were injected intraperitoneally with A) 100 μg of TNP (2,4,6 trinitro-phenyl)-Ficoll (T-independent antigen) or B) 75 μg of TNP-KLH (T-dependent antigen). Sera were collected at 7 and 14 days post-immunization. TNP-specific IgM and IgG3 or TNP-specific IgM and IgG1 in the sera were determined by ELISA. One representative data set from two independent experiments is shown. Similar results were obtained in both experiments. C) 12 age-matched (8-12 weeks old) male WT and IκBNS KO mice were immunized intraperitoneally with influenza virus PR8 at an MOI of 1 × 104. 15 days after immunization, mice were intranasally infected with 1 × 104 PR8 virus and survival followed. Day 1 on the survival graph is the day of infection. D) Virus-specific IgG2c antibodies were determined by ELISA in WT and IκBNS KO mice before immunization (Pre-bleed) and 15 days post-immunization (PI). Closed circles are WT samples and open circles are IκBNS KO samples. The asterisk in C and D denotes one wild type mouse that succumbed to viral infection and did not produce IgG2c in response to PR8. At 15 days post-immunization, p<0.01 for WT vs. KO. E) Spleen sections were prepared from WT and IκBNS KO mice 5 days after i.p. injection of sheep red blood cells (SRBC). Sections were stained with PNA and with hematoxylin and eosin. Arrows indicate PNA positive areas in active germinal centers in WT mice. IκBNS KO mice did not develop active germinal centers. Five WT and five KO mice were examined with similar results.
The humoral response to infection in the absence of IκBNS was assayed using a murine-adapted influenza A virus, PR8. WT and IκBNS KO mice were immunized i.p. and then infected intranasally approximately 2 weeks later with influenza virus. The survival curve in Fig. 5C shows that IκBNS KO mice are more severely affected by influenza A, with 50% succumbing to infection. The asterisk in Fig. 5C indicates the single death of one WT mouse after receiving a sublethal viral challenge. Antigen-specific antibody production in response to influenza immunization was then determined (Fig. 5D). While IκBNS KO mice are deficient in production of influenza-specific antibodies of the IgM, IgG1 and IgG2b classes (data not shown), the most significant deficiency is in IgG2c immunoglobulin as shown in Fig. 5D. This is noteworthy as this class of immunoglobulins affords a prominent protective response against influenza A in the murine system (30, 31).
To examine the B cell response histologically, we assayed germinal center formation using immunization with sheep red blood cells in WT and IκBNS KO mice (Fig. 5E). Five days after immunization, spleens were removed and sections prepared by staining with hematoxylin and eosin followed by PNA staining to visualize germinal centers where core-1 glycans are O-linked without sialic acid adducts and, hence, PNA-reactive. While germinal centers were clearly evident in the spleens of WT mice, no germinal center formation was detected in the spleens of the five IκBNS KO mice examined. Given the fact that the TNP-KLH IgG1 response appeared delayed, this experiment was repeated and germinal center formation examined at 10 days after immunization. Again, no germinal center formation was observed in the IκBNS KO mice while areas of PNA reactivity were clear in the WT mice (data not shown).
IκBNS KO B Cells Exhibit Reduced Plasma Cell Differentiation
One path of B cell differentiation culminates in the production of plasma cells, which are terminally differentiated, non-dividing antibody-secreting cells. Determination of the ex vivo number of antibody-secreting cells from the spleens of WT and IκBNS KO mice shows that in the IκBNS KO spleen there are fewer B cells secreting all of the types of the immunoglobulins assayed but the most dramatic differences are in IgM-, IgG3-, and IgG2b-producing splenic cells. Fig. 6 shows results of an individual ELISPOT assay and statistics compiled from 3-5 experiments in spleen (Panel B) and bone marrow (BM) (Panel C). Note that IgM- and IgG3-antibody-secreting cells (ASC) are reduced in both organs.
Figure 6.
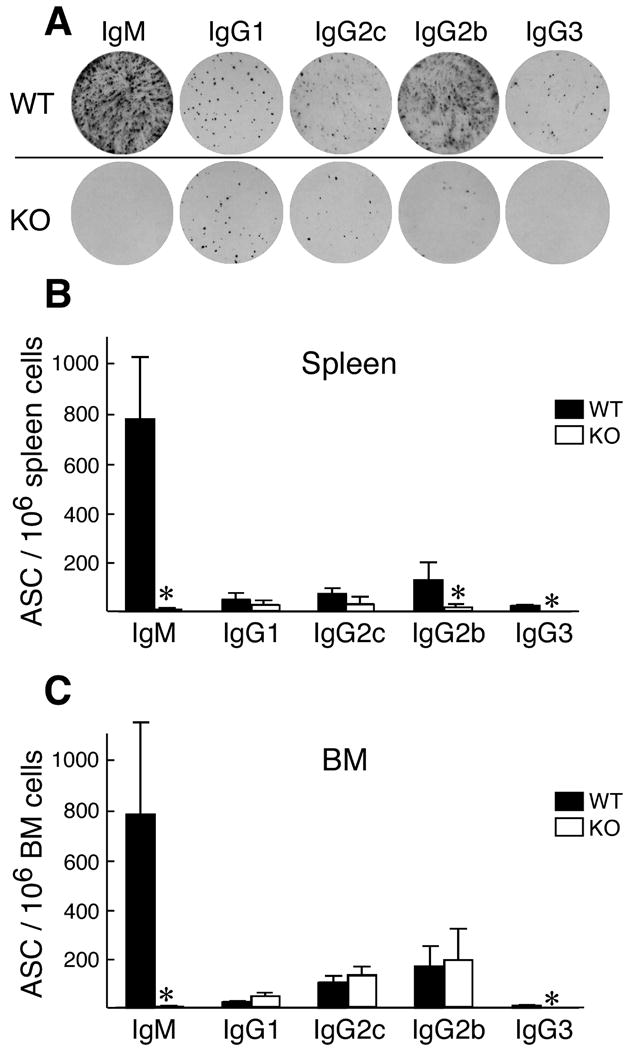
Splenic antibody-secreting cells are reduced in IκBNS KO mice. A) Immunoglobulin-secreting cells for WT and IκBNS KO mice were compared using an ELISPOT assay on total splenocytes after RBC lysis. The number of antibody secreting cells (ASC) per 106 spleen cells (B) and bone marrow cells (C) are shown graphically. Results are a compilation of three to five independent assays. Statistical analysis was performed using the Student's t-test.
The finding that there are fewer ASC in the spleens of IκBNS KO mice led us to examine the process of plasma cell differentiation in IκBNS KO B cells. B cells from spleens were cultured for five days in the presence of LPS to induce plasma cell differentiation. Proliferation was followed using CFSE staining while plasma cell differentiation was measured by CD138 (syndecan-1) expression. As can be seen in Fig. 7A, LPS-stimulated plasma cell differentiation is decreased in IκBNS KO B cells. Addition to the culture of IL-4, IFNγ or TGFβ in combination with LPS results in production of IgG1+, IgG2c+ or IgG2b+ cells, respectively, in the culture as determined by FACS of cell surface IgG (Supplemental Table I). Cells incubated with LPS alone express surface IgG3 (Supplemental Table I). The percentage of IgG3+ or IgG2c+ cells is lower in the IκBNS KO cultures than in the WT cultures, but the percentages of IgG1+ or IgG2b+ cells are approximately equivalent (Supplemental Table I). However, the percentage of CD138 positive cells is 4-6 fold lower in the IκBNS KO cultures under every culture condition (Fig. 7B) indicating that IκBNS KO B cells are defective in plasma cell differentiation.
Figure 7.
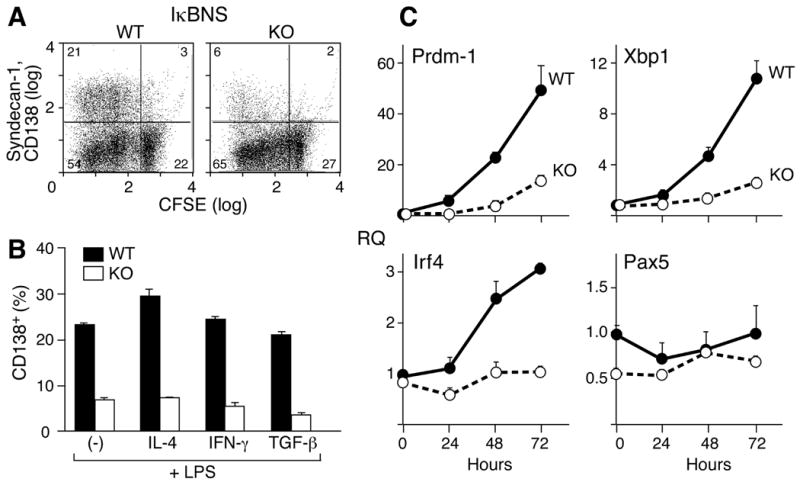
In vitro induction of CD138+ plasma cells from WT and IκBNS KO B cells. A) Splenic CD43- B cells from IκBNS KO and WT mice were CFSE labeled and then cultured for three days in the presence of 5 μg/ml LPS. Cells were examined for proliferation and CD138 expression by flow cytometry. B) Cells were cultured as described for A. On day 3, 10 ng/ml of IL-4, IFNγ or TGFβ were added for two additional days to stimulate isotype switching to IgG1, IgG2a or IgG2c, respectively. Cultures were then examined for CD138 and IgG expression. The graph indicates the percentage of CD138 positive cells under each condition in the WT (filled bar) and IκBNS KO (open bar) cultures. C) Enriched CD43- splenic B cells (1 × 105 cells/200 μl/well) were stimulated with LPS (5 μg/ml) for the indicated periods, and total RNA was isolated for quantitative real-time PCR analysis. All data were normalized to β-actin expression. Fold differences relative to day 0 WT levels are shown. Open circles represent KO samples and closed circles represent WT samples. Two independent experiments were performed in A, B, C and similar results were obtained.
The differentiation of plasma cells involves a well-studied transcriptional program (reviewed in 32, 33) including Prdm-1 (Blimp-1), Xbp-1 and IRF-4. Given the role of IκBNS in gene regulation, we utilized real time PCR to examine the levels of expression of these transcription factors during LPS-stimulated culture of B cells from WT and IκBNS KO mice. Splenic B cells from WT and IκBNS KO mice were stimulated with LPS for 24, 48 and 72 hours. RNAs prepared at these time points were examined for levels of Blimp-1, Xbp1 and Irf4. As can be seen in Fig. 7C, levels of Prdm-1/Blimp-1 and Xbp-1 increase over 72 hours in the WT B cells but in the absence of IκBNS the increase in levels of these RNAs is a greatly diminished. By 72 hours, the levels of Prdm-1/Blimp-1, Xbp1 and Irf4 are four to five fold higher in WT than in IκBNS KO B cells. While IκBNS KO B cells show an increase, albeit much reduced, in Prdm-1/Blimp-1 and Xbp1 levels over time, there is no change in Irf4.
These results suggest that IκBNS plays a role in the induction of the transcriptional program leading to a terminally differentiated plasma cell. In the absence of IκBNS, the requisite increases in the plasma cell transcriptome, namely Prdm-1/Blimp-1, Xbp1 and Irf4 do not occur thereby resulting in decreased numbers of antibody-secreting cells (Fig. 7).
Discussion
Our findings that deletion of IκBNS, a member of the IκB family of NF-κB inhibitors, affects the development and function of B cells is consistent with the important role of NF-κB in this cell lineage. As stated above, NF-κB was first identified in B cells (1) and plays a major role in cells involved in inflammation and the immune response. B cells are primarily divided into B1 cells, follicular B cells and marginal zone (MZ) B cells (reviewed in 34). The origins of each cell type as well as the transitions that may occur between these cell types has long been an area of intense analysis. Follicular and MZ B cells are the most abundant mature B cells with most peripheral B cells being of the follicular type. Follicular B cells are mobile, circulate within the periphery and are found in the follicles of the splenic white pulp. They participate in both T cell-dependent and T cell-independent immune responses (35). MZ B cells, in contrast, remain at the marginal sinus of the spleen (29, 36) although they migrate to the follicles and back transporting immune complexes containing antigen to the follicles (37, 38). MZ cells participate in T cell-independent responses and respond rapidly to blood-borne pathogens, a property shared with B1 B cells (29, 39). B1 B cells comprise most of the B cells in the peritoneal and pleural cavities in mice (40, 41) and produce IgM and IgG3 serum antibodies (29, 42). The B1 subset is divided into B1a and B1b cells based on the levels of CD5 on these two cell populations. B1a cells, which express CD5, comprise the earliest B cells arising in the neonate to become a self-replenishing population (43, 44) that is the main source of ‘natural’ IgM antibodies (44). B1b cells arise from the neonate but are derived from adult bone marrow as well (45, 46). In addition to these developmental differences, B1a and B1b cells differ in function with B1a contributing to innate immunity and B1b to adaptive immunity (47).
As is true for T cells (23), the absence of IκBNS affects B cell proliferation in response to multiple stimuli including LPS, anti-CD40 and anti-IgM (Fig. 3). The diminution in LPS-triggered activation in the IκBNS KO is most obvious in 3H-TdR incorporation while that of anti-CD40 is more pronounced in the BrdU experiments. Consistent with this result, each of these stimuli induces IκBNS at both the protein and mRNA level (Fig. 3). LPS also induces IκBNS in gut and peritoneal macrophages (21, 22) presumably acting through the Toll-like receptor 4 (TLR-4) (48) in a MyD88-dependent manner (22). However, IκBNS KO B cells sustain a high level of surface IgM expression compared to WT B cells and, thus, even if the signal via each IgM molecule is weaker than in WT B cells, the abundance of IgM on the surface may compensate for the lack of IκBNS. These opposing effects may account for preservation of the anti-IgM signaling in IκBNS KO B cells. Activation via CD40 induces NF-κB and the reduced proliferation in the absence of IκBNS is consistent with this. Furthermore, the reduced response to anti-CD40 correlates with the reduction (or delay) in the response of IκBNS KO B cells to T-dependent antigens as shown in Fig. 5.
Production of immunoglobulin is also altered in IκBNS KO mice. Measurement of steady state serum levels (Fig. 4) show that IgM and IgG3 are drastically reduced, IgG2c is somewhat reduced and IgG1 and IgG2b are approximately equal compared to levels found in WT mice. As B1 B cells are the source of serum levels of IgM and IgG3 (reviewed in 49), the absence of these immunoglobulins can be partially explained by the lack of B1 B cells in these mice. In addition, B cells from the IκBNS KO mice produce less secreted IgM as evident by the lack of serum IgM and as assayed by PCR for the secreted versus membrane form of IgM mRNA (Fig. 4B). Given that the reduction in the response appears more profound than the reduction in MZ B cell numbers, perhaps this implies functional defects in residual cells.
Class switch recombination (CSR) to IgG3 at the DNA level is not detected in IκBNS KO B cells (Fig. 4) using an in vitro system that mimics CSR (reviewed in 50). In these experiments, LPS is used with or without additional cytokines to direct CSR to particular heavy chain isotypes. However, a low level of IgG3 protein expression is observed (Fig. 7). Presumably, the LPS/cytokine milieu directs germline transcription to distinct isotypes (reviewed in 51) resulting in modulation of CSR. LPS in combination with IL-4 induces CSR to IgG1 in both WT and IκBNS KO B cells and induction of AID is equivalent in both cell types indicating that at least this component of the CSR machinery is operational in IκBNS KO B cells. Clearly a specific effect of deletion of IκBNS is an alteration in IgG3 CSR. This does not appear to be due to a reduction in transcription through the IgG3 locus as germline transcription before and after LPS addition appears to be equivalent for both WT and IκBNS KO mice (Fig. 4). There is precedence for transcription factors regulating CSR. For example, T-bet acts either by controlling germline transcripts or by mediating IgH locus access to other transcription factors and recombination-inducing machinery (52) and affects T-independent IgG2a production (53). Ikaros was shown to repress transcription of IgG2a and IgG2b thus increasing transcription from and recombination to other heavy chain genes (54). Other proteins affecting CSR specificity include TGF-β, which induces Smad and Runx that promote CSR to IgG2b and IgA, and IL-4, which induces the transcriptional activator Stat6 whose activity promotes CSR to IgG1 and IgE (reviewed in 55). Perhaps more pertinent to our finding of the role of IκBNS in CSR, NF-κB proteins and, in particular, NF-κB p50 have been shown to contribute to isotype specificity in CSR (56-60). In addition to proteins that contribute to the isotype specificity of CSR, the switch region sequences that lie upstream of each CH gene also function to mediate antibody class switching (61 and references therein) and the roles of the various proteins and switch regions are an area of active investigation. The finding that IκBNS contributes to isotype switching extends further the list of involved proteins.
Along with the observed serum immunoglobulin and class switch phenotypes in the IκBNS KO mice, we found a reduction in ASC assayed ex vivo from the spleens of IκBNS KO mice (Fig. 6). The pattern of reduction of ASC corroborates the observed differences in serum immunoglobulin values in the IκBNS KO mice. Furthermore, the IκBNS KO mice failed to produce any antigen-specific IgM or IgG3 antibodies in response to TNP-Ficoll. Given that T-independent responses are predominantly mediated by MZ B cells (29, 39, 62), the lack of response to TNP-Ficoll correlates with the reduction in MZ B cells in the IκBNS KO mouse. Furthermore, the lack of antigen-specific IgM may result in part from the reduction in MZ B cells but also from the inability of IκBNS KO B cells to produce secreted IgM (Fig. 4). This is observed as well in the response to TNP-KLH, a T-dependent antigen, as there is a delayed production of antigen-specific IgG1 but no production of IgM (Fig. 5) although this may also be affected by intrinsic alterations in the IκBNS KO T cells (Fig. S6). When wild type T cells were mixed with either WT or IκBNS KO B cells and these mixtures used to reconstitute RAG-2-/- KO mice, the IgG1 response is delayed and the response in IκBNS KO mice reaches levels similar to that in WT mice, albeit the kinetics of the response are a bit slower in the reconstitution experiment (Fig. S6). The phenotype of the reduced secretion of IgM is quite clearly represented in both Fig. 5 and Fig. S6, however, and is thus an intrinsic B cell defect. In addition, flow cytometric analysis of spleen cells in the recipient mice in Fig. S6 showed 12.5±1.1% CD138+B220low plasma cells in recipients of WT B and T cells and 2.8±0.4% CD138+B220low plasma cells in recipients of IκBNS KO B and T cells (data not shown).
IκBNS KO B cells also exhibit a reduced response to influenza virus. WT mice immunized by intraperitoneal injection of influenza successfully withstood influenza infection and produced influenza-specific IgG2c antibodies, the major isotype found in mice surviving influenza infections (30, 31). IκBNS KO mice, in contrast, produce less influenza-specific IgG2c and are more susceptible to infection after immunization. The lack of formation of germinal centers (GC) in IκBNS KO mice upon immunization is additional evidence of a functional defect in IκBNS KO B cells and may explain the reduction in the antigen-specific immune response as the GC is where B cell affinity maturation and class switching occur resulting in antigen-specific antibody secreting cells (reviewed in 63). Impairment of GC formation is demonstrated by the lack of peanut agglutinin (PNA)-staining after immunization with a T-dependent antigen, sheep red blood cells (64). The presence of a low level of antigen-specific IgG2c antibodies can be explained by the fact that some antibody responses can be generated in the absence of GC formation (65). It is worth noting here that mice deficient for Bcl-3, an IκB family member to which IκBNS has a strong homology, also fail to develop a germinal center response and fail to produce influenza-specific antibodies although, in contrast to the IκBNS KO mice, they do produce influenza-specific IgM antibodies (66). In addition, the serum levels of all immunoglobulin types were similar between WT and Bcl-3 KO mice.
The GC response is dependent on T cell help provided to B cells through CD40 (reviewed in 63). We have shown that proliferation of IκBNS KO B cells through CD40 is reduced and the dampening of CD40 signaling in the absence of IκBNS may affect the ability of IκBNS KO B cells to participate in a GC response. Furthermore, B cells express Toll-like receptors (TLR), which participate in the innate and adaptive immune response (reviewed in 67). IκBNS plays a role in the downstream signaling of those TLRs that work through MyD88-dependent mechanisms (22). Thus the absence of IκBNS could affect both innate and adaptive immunity by altering TLR activation. In particular B1 and MZ B cells respond to TLR ligands in T-independent responses resulting in production of low affinity IgM (reviewed in 67).
Young IκBNS KO mice exhibit a reduced MZ B cell population but this population reaches WT levels after the mice are over 6 months old (Fig. 1 and data not shown). MZ B cell development is blocked in mice carrying a targeted gene deletion of the NF-κB p50 protein (56, 68, 69). We and others have shown that IκBNS interacts with p50 (14, 21) suggesting that this complex and genes regulated by it play a role in MZ B cell development. In addition to NF-κB p50, alterations in the expression of NF-κB p65 (RelA), c-Rel (68) and RelB (70) also affect MZ B cell development.
The absence of IκBNS also affects the development of plasma cells. Plasma cells are a final differentiation state of B cells and are basically an immunoglobulin-secreting factory. These cells emerge from a GC reaction and express antigen-specific high affinity immunoglobulin having undergone somatic hypermutation within the GC. Plasma cells are the source of persistent antigen-specific antibody titers (71) and, thus, the lack of development of plasma cells may contribute to the fact that the IκBNS KO mice have reduced anti-influenza specific antibodies. The differentiation of plasma cells has a defined transcriptional program including Prdm-1/Blimp-1, Xbp1 and Irf4 (reviewed in 32, 33). Our analysis of these factors in WT and IκBNS KO B cells during in vitro differentiation shows that in the absence of IκBNS, these transcription factors are decreased and in vitro plasma cell differentiation is impaired (Fig. 7). The frequency of antibody-secreting cells (ASC) found in the spleen of the IκBNS KO mice is also reduced (Fig. 6). The development and survival of plasma cells in special niches in the bone marrow is only partially understood, and the reduction in expression of Prdm-1/Blimp-1, Xbp1 and Irf4 in the absence of IκBNS suggests that IκBNS may control a point upstream of the plasma cell transcriptional program.
B1 B cells are missing from IκBNS KO mice. The origin of the B1 B cell lineage is controversial (42, 72) and recent findings suggest on the one hand that signal strength drives B cell fate (73) or that a specific progenitor exists for B1 B cells (46). Altered expression of molecules that affect signal strength through the BCR affect the B1 cell population in mice. For example, construction of transgenic mice using the LMP2A transgene in place of Igh demonstrated that mice with a high copy number of the transgene, which mimics strong BCR signaling, have higher numbers of B1 cells while mice carrying fewer LMP2A transgenes, consistent with a lower intensity BCR signal, have lower numbers of B1 B cells (73). Similarly, more B1 cells are also found in mice that lack negative regulators of BCR signaling (42) or express higher levels of BCR (72, 74). This suggests that IκBNS may play a role in strengthening the BCR signal and that in the absence of IκBNS a reduced BCR signal results in the lack of B1 B cells. A similar role for IκBNS was suggested in T cells (23). IκBNS was cloned as a gene that was induced in thymocytes after N15TCRtg RAG-2KO mice were injected with VSV8, a negatively selecting peptide for this TCR (14, 75). Positively selecting peptides did not induce expression of IκBNS (75). As negative selection is associated with a strong TCR signal and positive selection with a weaker TCR signal, the expression of IκBNS appeared to correlate with a strong TCR signal. Furthermore, production of IL-2 by T cells is thought to require a strong TCR signal and in the absence of IκBNS, IL-2 production is reduced (23), again associating IκBNS with receptor signal strength.
The phenotype of p50 KO mice has some commonalities with the phenotype of IκBNS KO mice, which is to be expected as IκBNS has been demonstrated to interact with p50 (14, 21). Specifically (as reviewed 13), p50 KO mice display normal development but are abnormal in immune function. As in the IκBNS KO, p50 KO B cells proliferate poorly to LPS and CD40 and display abnormal heavy chain switching due to lack of transcription through that region. In fact, p50 and p65 were found to bind the IgG3 switch region and p50 KO mice are specifically impaired in switching to IgG3 (59, 60, 76) and have a reduced MZ B cell population (68).
In summary, characterization of B cells in our IκBNS KO mouse uncovers a pleotropic phenotype, which is consistent with other factors that affect NF-κB activity. B cell functions including immunoglobulin production, plasma cell development and isotype switching are impaired and particular B cell populations such as B1 cells and MZ B cells are reduced in comparison to WT mice. Although not shown, the development of pro-B and pre-B cells in young adult bone marrow was not affected by IκBNS deletion arguing that the function of this regulator is targeted to the mantle zone and follicular B cell compartments, Ig switching processes and plasma cell development. The phenotype overlaps that of p50 KO mice correlating with the fact that IκBNS and p50 interact. That the IκBNS KO phenotype also overlaps in part that of the Bcl-3 KO is noteworthy since IκBNS and Bcl-3 are homologous members of the IκB family of NF-κB inhibitors. These results reveal a significant role for IκBNS in development of the B cell arm of the immune system and the humoral response in addition to the already defined role of IκBNS in T cells, macrophages and dendritic cells.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Klaus Rajewsky for helpful discussions and suggestions during the course of this work and Drs. Garnet Kelsoe, Emmanuel Derudder and Klaus Rajewsky for constructive criticism of the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants AI19807 (to E.L.R.) and AI51779 (to L.K.C.) and by KAKENHI (22790457), the Uehara Memorial Foundation and the Hayashi Memorial Foundation for Female Natural Scientists (to M.T.). F. S. is a postdoctoral fellow of the Global COE program supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan.
S.K. is a consultant for Medical and Biological Laboratories, Co. Ltd. The authors otherwise have no financial conflicts of interest.
References
- 1.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 3.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 4.Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 5.Sun SC, Ley SC. New insights into NF-κB regulation and function. Trends Immunol. 2008;29:469–478. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baeuerle P, Baltimore D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-κB transcription factor. Cell. 1988;53:211–217. doi: 10.1016/0092-8674(88)90382-0. [DOI] [PubMed] [Google Scholar]
- 7.Baeuerle P, Baltimore D. IκB: a specific inhibitor of the NF-κB transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 8.Pomerantz JL, Baltimore D. Two pathways to NF-κB. Mol Cell. 2002:693–701. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 9.Beinke S, Ley SC. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shisodia S, Aggarwal BB. Nuclear factor-kappaB: a friend or foe in cancer? Biochem Pharmacol. 2004;68:1071–1080. doi: 10.1016/j.bcp.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 11.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progresson. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 12.Goetz CA, Baldwin AS. NF-κB pathways in the immune system: control of the germinal center reaction. Immunol Res. 2008;41:233–247. doi: 10.1007/s12026-008-8033-1. [DOI] [PubMed] [Google Scholar]
- 13.Gerondakis S, Grossmann M, Nakamura Y, Pohl T, Grumont R. Genetic approaches in mice to understand Rel/NF-kappaB and IkappaB function: transgenics and knockouts. Oncogene. 1999;18:6888–6895. doi: 10.1038/sj.onc.1203236. [DOI] [PubMed] [Google Scholar]
- 14.Fiorini E, Schmitz I, Marissen WE, Osborn SL, Touma M, Sasada T, Reche PA, Tibaldi EV, Hussey RE, Kruisbeek AM, Reinherz EL, Clayton LK. Peptide-induced negative selection of thymocytes activates transcription of an NF-κB inhibitor. Mol Cell. 2002;9:637–648. doi: 10.1016/s1097-2765(02)00469-0. [DOI] [PubMed] [Google Scholar]
- 15.Kitamura H, Kanehira K, Okita K, Morimatsu M, Saito M. MAIL, a novel nuclear IκB protein that potentiates LPS-induced IL-6 production. FEBS Letters. 2000;485:53–56. doi: 10.1016/s0014-5793(00)02185-2. [DOI] [PubMed] [Google Scholar]
- 16.Haruta H, Kato A, Todokoro K. Isolation of a novel interleukin-1-inducible nuclear protein bearing ankyrin-repeat motifs. J Biol Chem. 2001;276:12485–12488. doi: 10.1074/jbc.C100075200. [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki S, Muta T, Takeshige K. A novel IκB protein, IκB-ζ, induced by proinflammatory stimuli, negatively regulates nuclear factor-κB in the nuclei. J Biol Chem. 2001;276:27657–27662. doi: 10.1074/jbc.M103426200. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, Saitoh T, Yamaoka S, Yamamoto N, Yamamoto S, Muta T, Takeda K, Akira S. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature. 2004;430:218–222. doi: 10.1038/nature02738. [DOI] [PubMed] [Google Scholar]
- 19.Kuwata H, Watanabe N, Miyoshik H, Yamamoto M, Kaisho T, Takeda K, Akira S. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-α production in macrophages. Blood. 2003;102:4123–4129. doi: 10.1182/blood-2003-04-1228. [DOI] [PubMed] [Google Scholar]
- 20.Wessells J, Baer M, Young HA, Claudio E, Brown K, Siebenlist U, Johnson PF. Bcl-3 and NF-κB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem. 2004;279:49995–50003. doi: 10.1074/jbc.M404246200. [DOI] [PubMed] [Google Scholar]
- 21.Hirotani T, Lee PY, Kuwata H, Yamamoto M, Matsumoto M, Kawase L, Akira S, Takeda K. The nuclear IκB protein IκBNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. J Immunol. 2005;174:3650–3657. doi: 10.4049/jimmunol.174.6.3650. [DOI] [PubMed] [Google Scholar]
- 22.Kuwata H, Matsumoto M, Atarashi K, Morishita H, Hirotani T, Koga R, Takeda K. IκBNS inhibits induction of a subset of Toll-like receptor-dependent genes and limits inflammation. Immunity. 2006;24:41–51. doi: 10.1016/j.immuni.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Touma M, Antonini V, Kumar M, Osborn SL, Bobenchik AM, Keskin DB, Connolly JE, Grusby MJ, Reinherz EL, Clayton LK. Functional role for IκBNS in T cell cytokine regulation as revealed by targeted gene disruption. J Immunol. 2007;179:1681–1692. doi: 10.4049/jimmunol.179.3.1681. [DOI] [PubMed] [Google Scholar]
- 24.Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- 25.Bhatia K, Huppi K, McKeithan T, Siwarski D, Mushinski JF, Magrath I. Mouse bcl-3: cDNA structure, mapping and stage-dependent expression in B lymphocytes. Oncogene. 1991;6:1569–1573. [PubMed] [Google Scholar]
- 26.Ren H, Schmalstieg A, Yuan D, Gaynor RB. I-kappaB kinase beta is critical for B cell proliferation and antibody response. J Immunol. 2002;168:577–587. doi: 10.4049/jimmunol.168.2.577. [DOI] [PubMed] [Google Scholar]
- 27.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require Activation-Induced Cytidine Deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 28.Montecino-Rodriguez E, Dorshkind K. New perspectives in B-1 B cell development and function. Trends Immunol. 2006;27:428–433. doi: 10.1016/j.it.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol. 2002;2:323–335. doi: 10.1038/nri799. [DOI] [PubMed] [Google Scholar]
- 30.Coutelier JP, van der Logt JT, Heessen FW, Warnier G, Van Snick J. IgG2a restriction of murine antibodies elicited by viral infections. J Exp Med. 1987;165:64–69. doi: 10.1084/jem.165.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coutelier JP, van der Logt JT, Heessen FW, Vink A, Van Snick J. Virally induced modulation of murine IgG anitbody subclasses. J Exp Med. 1988;168:2373–2378. doi: 10.1084/jem.168.6.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 33.Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Annu Rev Immunol. 2008;26:133–169. doi: 10.1146/annurev.immunol.26.021607.090241. [DOI] [PubMed] [Google Scholar]
- 34.Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20:149–157. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 36.MacLennan ECM, Gray D, Kumararatne DS, Bazin H. The lymphocytes of splenic marginal zones: a distinct B-cell lineage. Immunol Today. 1982;3:305–397. doi: 10.1016/0167-5699(82)90032-9. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson AR, Youd ME, Corley RB. Marginal zone B cells transport and deposit IgM-containing immune complexes onto follicular dendritic cells. Int Immunol. 2004;16:1411–1422. doi: 10.1093/intimm/dxh142. [DOI] [PubMed] [Google Scholar]
- 38.Cinamon G, Zachariah MA, Lam OM, Foss FWJ, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pillai S, Carriappa A, Moran ST. Marginal zone B cells. Ann Rev Immunol. 2005;23:161–196. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- 40.Hayakawa K, Hardy RR, Herzenberg LA. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161:1554–1568. doi: 10.1084/jem.161.6.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lalor PA, Stall AM, Adams S, Herzenberg LA. Permanent alteration of the murine Ly-1 B repertoire due to selective depletion of Ly-1 B cells in neonatal animals. Eur J Immunol. 1989;19:501–506. doi: 10.1002/eji.1830190314. [DOI] [PubMed] [Google Scholar]
- 42.Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Ann Rev Immunol. 2002;20:252–300. doi: 10.1146/annurev.immunol.20.100301.064833. [DOI] [PubMed] [Google Scholar]
- 43.Kantor AB, Herzenberg LA. Origin of murine B cell lineages. Annu Rev Immunol. 1993;11:501–538. doi: 10.1146/annurev.iy.11.040193.002441. [DOI] [PubMed] [Google Scholar]
- 44.Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin Immunopathol. 2005;26:347–362. doi: 10.1007/s00281-004-0182-2. [DOI] [PubMed] [Google Scholar]
- 45.Hardy RR, Hayakawa K. A developmetal switch in B lymphopoiesis. Proc Natl Acad Sci USA. 1991;88:11550–11554. doi: 10.1073/pnas.88.24.11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montecino-Rodriguez M, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nat Immunol. 2006;7:293–301. doi: 10.1038/ni1301. [DOI] [PubMed] [Google Scholar]
- 47.Haas KM, Poe JC, Steeber DA, Tedder RF. B-1a and B-1b cells exhibit distinct developmental roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 48.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Toll-like receptor 5 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 49.Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol. 2005;23:367–386. doi: 10.1146/annurev.immunol.23.021704.115723. [DOI] [PubMed] [Google Scholar]
- 50.Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, Manis J, Alt FW. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 51.Manis J, Tian M, Alt FW. Mechanism and control of class-switch recombination. Trends Immunol. 2002;23:31–39. doi: 10.1016/s1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 52.Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci USA. 2002;99:5545–5550. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerth AJ, Lin L, Peng SL. T-bet regulates T-independent IgG2a class switching. Int Immunol. 2003;115:937–944. doi: 10.1093/intimm/dxg093. [DOI] [PubMed] [Google Scholar]
- 54.Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med. 2009;206:1073–1087. doi: 10.1084/jem.20082311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:22–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snapper CM, Zelazowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. B cells from p50/NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 57.Zelazowski P, Carrasco D, Rosas FR, Moorman MA, Bravo R, Snapper CM. B cells genetically deficient in the c-Rel transactivation domain have selective defects in germline CH transcription and Ig class switching. J Immunol. 1997;159:3133–3139. [PubMed] [Google Scholar]
- 58.Wuerffel RA, Ma L, Kenter AL. NF-kappa B p50-dependent in vivo footprings at IgS gamma 3 Da are correlated with mu->gamma 3 switch recombination. J Immunol. 2000;166:4552–4559. doi: 10.4049/jimmunol.166.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kenter AL, Wuerffel R, Dominguez C, Shanmugam A, Zhang H. Mapping of a functional recombination motif that defines isotype specificity for μ->γ3 switch recombination implicates NF-κB p50 as the isotype-specific switching factor. J Exp Med. 2004;199:617–627. doi: 10.1084/jem.20031935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang L, Wuerffel R, Kenter AL. NF-kappa B binds to the immunoglobulin S gamma 3 region in vivo during class switch recombination. Eur J Immunol. 2006;36:3315–3323. doi: 10.1002/eji.200636294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zarrin AA, Goff PH, Senger K, Alt FW. Sγ3 switch sequences function in place of endogenouse Sγ1 to mediate antibody class switching. J Exp Med. 2008;205:1567–1572. doi: 10.1084/jem.20080451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 63.Allen CDC, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacLennan ICM. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 65.Matsumoto M, Lo SF, Carruthers CJ, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MH, Chaplin DD. Affinity maturation without germinal centres in lymphotoxin-α-deficient mice. Nature. 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 66.Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, Love P, Sher A, Siebenlist U. Critical roles for the Bcl-3 oncoprotein in T cell–mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- 67.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 68.Cariappa A, Liou HC, Horwitz BH, Pillai S. Nuclear Factor κb is required for the development of marginal zone B lymphocytes. J Exp Med. 2000;192:1175–1182. doi: 10.1084/jem.192.8.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferguson AR, Corley RB. Accumulation of marginal zone B cells and accelerated loss of follicular dendritic cells in NF-kappaB p50-deficient mice. BMC Immunol. 2005;6:8. doi: 10.1186/1471-2172-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weih DS, Yilmaz ZB, Weih F. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J Immunol. 2001;167:1909–1919. doi: 10.4049/jimmunol.167.4.1909. [DOI] [PubMed] [Google Scholar]
- 71.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KGC, Dörner T, Hiepe F. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 72.Lam KP, Rajewsky K. B cell antigen receptor specificity and surface density together determine B-1 versus B-2 development. J Exp Med. 1999;190:471–478. doi: 10.1084/jem.190.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, Carroll MC, Rajewsky K. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5:317–327. doi: 10.1038/ni1036. [DOI] [PubMed] [Google Scholar]
- 74.Watanabe N, Nisitani S, Ikuta K, Suzuki M, Chiba T, Honjo T. Expression levels of B cell surface immunoglobulin regulate efficiency of allelic exclusion and size of autoreactive B-1 cell compartment. J Exp Med. 1999;190:461–469. doi: 10.1084/jem.190.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Clayton LK, Ghendler Y, Mizoguchi E, Patch RJ, Ocain TD, Orth K, Bhan AK, Dixit VM, Reinherz EL. T-cell receptor ligation by peptide/MHC induces activation of a caspase in immature thymocytes: the molecular basis of negative selection. EMBO J. 1997;16:2282–2293. doi: 10.1093/emboj/16.9.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horwitz BH, Zelazowski P, Shen Y, Wolcott KM, S ML, Baltimore D, Snapper CM. The p65 subunit of NF-κB is redundant with p50 during B cell proliferative responses, and is required for germline CH transcription and class switching to IgG3. J Immunol. 1999;162:1941–1946. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.