

Abstract
We present an extension of the coarse-grained MARTINI model for proteins and apply this extension to amyloid- and elastin-like peptides. Atomistic simulations of tetrapeptides, octapeptides, and longer peptides in solution are used as a reference to parametrize a set of pseudodihedral potentials that describe the internal flexibility of MARTINI peptides. We assess the performance of the resulting model in reproducing various structural properties computed from atomistic trajectories of peptides in water. The addition of new dihedral angle potentials improves agreement with the contact maps computed from atomistic simulations significantly. We also address the question of which parameters derived from atomistic trajectories are transferable between different lengths of peptides. The modified coarse-grained model shows reasonable transferability of parameters for the amyloid- and elastin-like peptides. In addition, the improved coarse-grained model is also applied to investigate the self-assembly of β-sheet forming peptides on the microsecond time scale. The octapeptides SNNFGAIL and (GV)4 are used to examine peptide aggregation in different environments, in water, and at the water–octane interface. At the interface, peptide adsorption occurs rapidly, and peptides spontaneously aggregate in favor of stretched conformers resembling β-strands.
1. Introduction
Atomistic molecular dynamics (MD) simulations are useful computational methods in the study of biological systems. However, many phenomena, such as vesicle fusion, protein folding, and peptide aggregation occur at time scales much longer than those currently accessible using atomistic simulation and in some cases do not critically depend on an accurate atomistic representation.1 In such cases, coarse-grained (CG) models present an attractive alternative to atomistic simulations since they offer the possibility of investigating complex biological processes over relatively long periods of time and length scales at a reduced level of detail.
In CG models, groups of atoms are typically represented as one interaction site, which significantly decreases the total number of particles in the system. The reduced number of degrees of freedom and the use of smoother interaction potentials allow for longer time steps, which results in a significant increase in speed. A number of CG models are available for a variety of biomolecules, including lipids,2−4 proteins,5−8 DNA,9,10 and polymers.11,12 With reference to proteins, CG models have been developed at various levels of resolution differing in the number of degrees of freedom used to represent the protein backbone and side chains as well as the choice of potential energy functions that describe interactions in the system.13
Marrink and co-workers developed one CG model, coined the MARTINI force field,4,14 for the simulation of lipids and surfactants using a thermodynamics-based approach, i.e., its parameters were determined by reproducing free energies of partitioning between oil and aqueous phases. The MARTINI CG force field was later extended to proteins,7 fullerenes,15 and carbohydrates.16 The MARTINI model for peptides and proteins,7 in particular, was designed to provide a general model that is applicable to any class of protein and allows for discrimination between all amino acids through the use of more types of interaction sites than most other CG models. All amino acids in the MARTINI protein model are represented by at least one bead: The backbone of each amino acid residue is collectively described by one bead, and the side chains are represented by a variable number of beads depending on the dimensions of the side chain of each amino acid. Consistent with the philosophy of the original MARTINI force field, the MARTINI model for proteins was developed using the partitioning free energies of amino side chains between oil and aqueous phases to determine the appropriate nonbonded interaction parameters. The bonded parameters were derived systematically based on the distribution of bond lengths, angles, and dihedral angles calculated from the Protein Data Bank. Since the MARTINI force field was developed by calibrating a large number of chemical building blocks against thermodynamic data, it is applicable to a wide range of chemical systems without requiring further reparametrization. For specific applications, bonded parameters can be easily improved as required by targeting experimental data or atomistic models. In general, the parametrization of new chemical building blocks can be achieved relatively easily compared to other CG approaches because of its simple potential energy functions. Hence, the model can be extended in a straightforward manner to construct new molecular species while retaining its internal consistency and compatibility. In the past few years, the MARTINI force field has been successfully applied in numerous studies of peptide and peptide–lipid interactions,17,18 the assembly of micelles and bilayers around membrane proteins,19,20 and lipid phase separation.21
However, because little structural data were used during parametrization and the potential form is relatively simple, the current standard version of the MARTINI force field is limited in its ability to reproduce structural details of complex biomolecules and polymers.7,14 One approach to overcome this limitation is to use elastic network models on top of the CG parametrization in order to mimic structural and dynamical properties of a particular native or non-native structure. Periole and co-workers have introduced a modified MARTINI CG model, denoted as ELNEDIN,22 by mixing an elastic network model with the MARTINI force field. In this model, an elastic network was used to maintain the structure of a protein, and the MARTINI CG model was used to describe interactions in the system. The ELNEDIN protein model was found to be able to reproduce quantitatively both the structural and the dynamical properties of the proteins predicted by atomistic simulations. Recently, Singh and Tieleman have proposed an approach to optimizing parts of the MARTINI model, based on the partitioning free energies of amino acid side chains at the lipid–water interface for the Wimley–White hydrophobicity scale peptides.23 The authors established a well-defined experimental test system and simulation protocol for guiding future improvements of the MARTINI model.23 Side chain parameters have also been improved using potentials of mean force between two side chains in solution.66
Other approaches are being developed to further enhance the accuracy and applicability of CG models. Hybrid simulations24−26 in which CG and atomistic models are combined have been applied to study peptide–membrane interactions27 and proteins.28 In this approach, essential parts of molecular systems are represented at a detailed atomistic resolution, while the remaining parts are modeled at a reduced CG level.
Another important limitation in the current implementation of the MARTINI force field7 is that secondary structure transformations are not modeled. Changes in protein secondary structure cannot be modeled with the current MARTINI force field because backbone bonded parameters are dependent on predefined secondary structures. Secondary structure elements of protein are fixed at strand, helix, or coil structures throughout simulations through the use of angle and dihedral potential energy functions. However, to accurately describe numerous biological phenomena which involve the folding and unfolding of secondary structures, a representative sampling of the different secondary structure elements must be achieved during the dynamics. Currently, such processes lie outside the applicability of MARTINI forcefield. Our work is a first step toward overcoming this limitation by introducing internal flexibility on the peptide backbone during CG simulations.
In the present work, we test whether incorporating potentials of mean force derived from atomistic simulations in the backbone of a set of CG peptides improves the conformations generated by the CG simulations. We then use these potentials to simulate the dynamics of aggregation of amyloid- and elastin-like peptides over long time scales and investigate the transferability of the backbone potentials to longer peptides and peptides in different environments at a water–hydrophobic interface and as part of aggregates. Our test systems are peptides that have elastin- or amyloid-like properties.
Elastin is a polymeric structural protein that provides extensibility and elastic recoil to tissues, such as lungs, large arteries, and skin.29,30 Tropoelastin, the monomeric form of elastin, is composed of hydrophobic and cross-linking domains, which alternate along the sequence of the protein. Hydrophobic domains are rich in nonpolar amino acids, such as glycine, proline, and valine; they are thought to be responsible for elastin’s temperature-induced self-aggregation and elasticity.31,32 The sequences used in this paper are modeled after those of the hydrophobic domains of elastin.33 In contrast, amyloid fibrils represent a pathogenic form of the assembly of soluble proteins associated with tissue-degenerative diseases, such as Alzheimer’s, Parkinson’s, and the prion diseases.34,35 All these proteins differ in amino acid sequences and adopt different structures in their monomeric forms.36−39 Despite the dissimilarity of their monomeric precursors, amyloid fibrils have been observed to share a common insoluble cross-β-sheet structure with the β-strands perpendicular to the fiber axis based on data from X-ray scattering and solid-state NMR experiments.40,41
We have previously studied the physical properties of elastomeric and amyloid fibrils by atomistic molecular simulations of monomeric and aggregated states of peptides.33 An extensive set of such simulations33,42 is used to parametrize a backbone potential for the MARTINI model for elastin- and amyloid-like peptides in the present work. We then use these parameters to simulate additional peptides and peptides in different environments and compare the results to atomistic simulations.
In the following sections, we describe the model systems and the potential energy functions in the MARTINI force field, followed by the force field parametrization procedure for new dihedral angle potentials. We also describe the simulation conditions for the CG and atomistic simulations. Next, the performance of the model is assessed in terms of its ability to reproduce structural properties. We present results for a range of test cases of amyloid- and elastin-like peptides. Then, we discuss the performance of the model in terms of the transferability of parameters between different lengths of peptides. Subsequently, we present amyloid peptide aggregation at the water–octane interface to validate the extension of the CG MARTINI model and investigate the transferability of the newly derived parameters in different environments.
2. Computational Methods
2.1. The Systems
We used a set of octapeptides consisting of amyloid- and elastin-like sequences: SNNFGAIL,43,44 (GA)4, (GV)4, GVGVAGVG, GVGVGGVG, GVGVAGGV, GVGGVGGV (amyloid), GVGVPGVG, GVPGVPGV, GVAPGVGV, and GVGGVPGV (elastin).33 Our study on the elastin-like octapeptides was motivated by the investigations of Rousseau et al.45 on neutral peptides. In particular, the behavior of the peptide fragments, representing part of hydrophobic domains, is of interest in this work. For consistency, all of the octapeptides investigated in this paper were capped with an acetyl group at the N-terminus and an amide group at the C-terminus, and they are neutral. The sequences of the elastin-like peptides are formed by the pairwise combination of four fragments: PGV, GGV, GV, and GVA.33 In addition, tetrapeptide fragments of the octapeptides listed above as well as longer peptides ((GV)18, (PGV)12) are used to test the performance of our model in terms of accuracy and parameter transferability. The sequences of all model peptides are shown in Table 1.
Table 1. Sequence of Model Peptides.
| peptides | tetrapeptides |
|---|---|
| (GA)4 | GAGA, AGAG |
| (GV)4 | GVGV, VGVG |
| SNNFGAIL | SNNF, NNFG, NFGA, FGAI, GAIL |
| GVGVAGVG | GVGV, VGVA, GVAG, VAGV, AGVG |
| GVGVGGVG | GVGV, VGVG, GVGG, VGGV, GGVG |
| GVAGVAGV | GVAG, VAGV, AGVA |
| GVGVAGGV | GVGV, VGVA, GVAG, VAGG, AGGV |
| GVGGVGGV | GVGG, VGGV, GGVG |
| GVGVPGVG | GVGV, VGVP, GVPG, VPGV, PGVG |
| GVPGVPGV | GVPG, VPGV, PGVP |
| GVAPGVGV | GVAP, VAPG, APGV, PGVG, GVGV |
| GVGGVPGV | GVGG, VGGV, GGVP, GVPG, VPGV |
| (GV)18 | GVGV, VGVG |
| (PGV)12 | PGVP, GVPG, VPGV |
2.2. The Model
2.2.1. Interaction Sites
The basic parameters for the CG peptide model are taken from the MARTINI protein force field.7 In the MARTINI model, a single interaction site generally represents a group of four heavy atoms, with an exception for ring-like molecules. There are four main types of interaction sites in the model: polar (P), nonpolar (N), apolar (C), and charged (Q). Each type has a number of subtypes, which describe more accurately the chemical nature of the underlying atomic structure. The mapping of all protein amino acids is available in the original MARTINI protein force field paper.7 As an illustration, the mapping of SNNFGAIL is shown in Figure 1.
Figure 1.
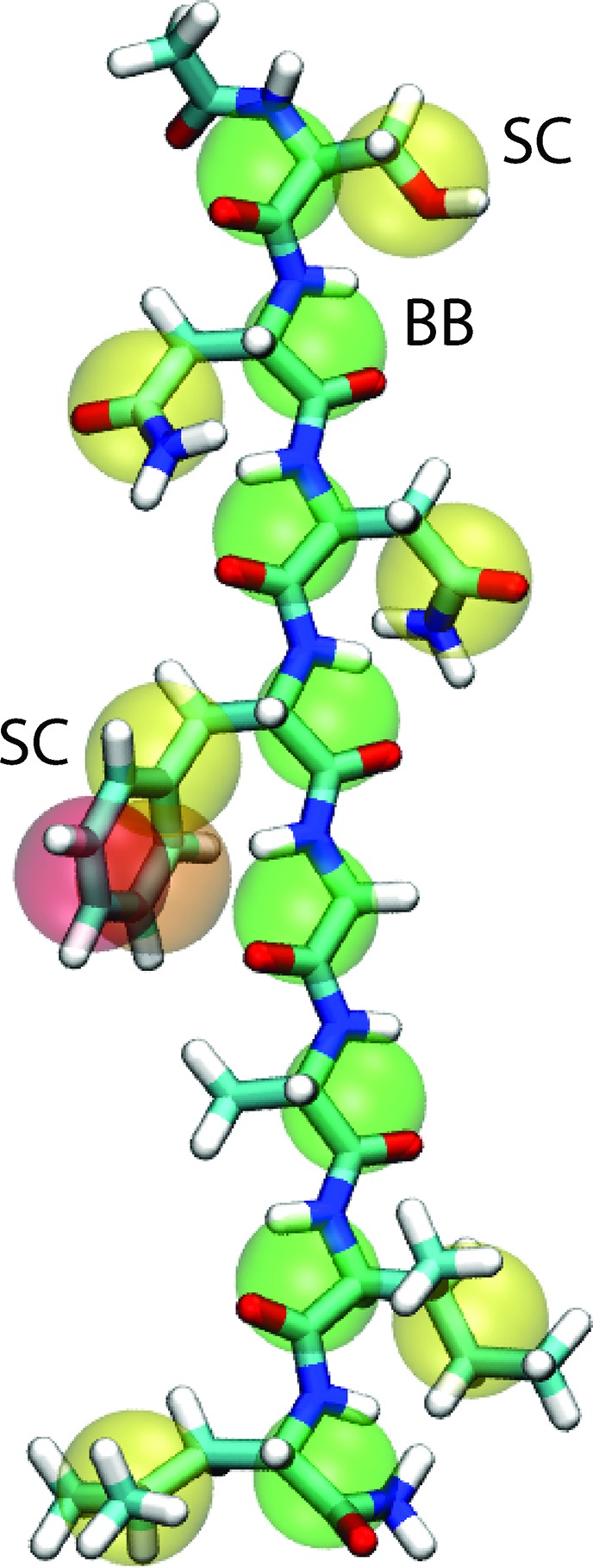
Mapping between the atomistic structure and the CG model for SNNFGAIL. Backbone beads are indicated by “BB” and side chain beads by “SC”. All pictures presented in this paper were generated with VMD.65
2.2.2. Bonded Interactions
Bonded interactions in the original MARTINI peptide force field7 are described by a set of potential energy functions: the bond potential Vb, the angle potential Va, and dihedral angle potential Vd. The bond stretching between two bonded sites i and j is represented by a harmonic potential Vb
| 1 |
with a force constant Kb about an equilibrium distance rb. A cosine-based angle potential Va is used to represent bond angle bending between bonded sites i, j, and k:
| 2 |
where Ka and θa are the harmonic force constant for bond angle bending and equilibrium angle, respectively. For dihedrals, the proper dihedral angle potential acting between bonded sites i, j, k, and l is given by
| 3 |
where Kd and ϕd are the torsional rotation force constant and the phase angle, respectively. The parameter n controls the periodicity. The procedure of force field parametrization for dihedral angle potentials is described in Section .
2.2.3. Parametrization of the Dihedral Angle Potential
To develop the dihedral angle potential of peptides in CG representation, extensive conformational sampling was performed by atomistic simulations,33,42 and various structural properties calculated from these atomistic trajectories were used as reference for our CG simulations. The CG simulations of amyloid- and elastin-like peptides were performed in aqueous solution using the original MARTINI model. The main goal of the reparametrization of the CG dihedral angle potential is to reproduce the conformational ensemble of the atomistic system. The dihedral angle was defined by four consecutive backbone beads (BB), and the dihedral angle distributions were calculated for every quartet of residues possible for each peptide. Using the center of mass of the group of atoms corresponding to CG beads, the distributions of the dihedral angles were calculated for every peptide from atomistic trajectories. Then, the corresponding potentials of each dihedral angle were extracted from the probability distributions by using the Boltzmann inversion method,46 i.e,
| 4 |
where kB is Boltzmann’s constant, T is the temperature, and Pi is the normalized probability distribution of the ith dihedral angle. In the next step, potential energies Ui were fitted to a sum of cosine and sine terms given by the following function:
| 5 |
where ϕ is the dihedral angle, and Ci and Si values are the force constants. Figure 2 shows the probability distributions for all possible backbone dihedral angles of SNNFGAIL obtained from atomistic simulations (Figure 2A), and the corresponding potentials and fitting energy functions (Figure 2B). The corresponding potentials and fitting energy functions for all other octapeptides are in Figure SI3, Supporting Information. For convenience, this functional form was converted into the periodic dihedral potential function in eq 6 already implemented in the GROMACS molecular simulation package.47
| 6 |
where Ki is the force constant, ϕi is the phase angle, and the parameter ni controls the periodicity.
Figure 2.
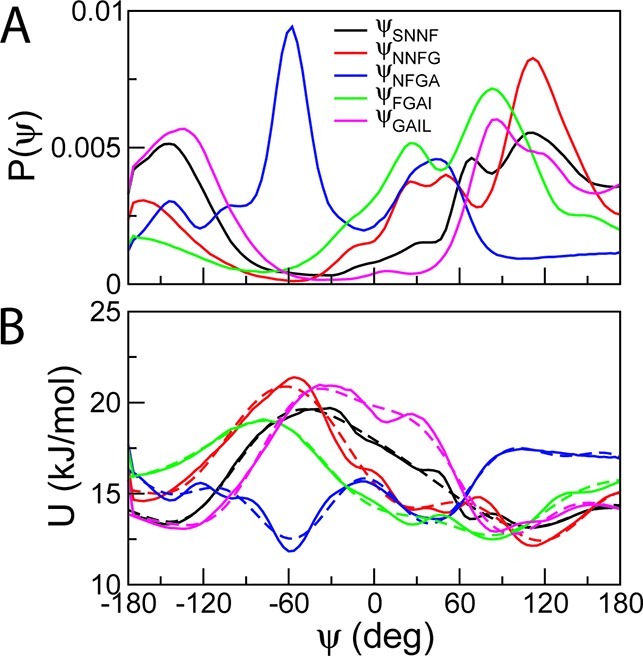
(A) Probability distributions for all possible BB–BB–BB–BB dihedral angles of SNNFGAIL obtained from atomistic simulations at 305 K. (B) The corresponding potentials of mean force (solid lines) and fitting energy functions (dotted lines).
CG simulations of amyloid- and elastin-like peptides were reperformed in solution using the modified model. Parameters for bond stretching and bending potentials were taken from the original MARTINI force field.7 The distributions of dihedral angles were computed from CG simulations using the original MARTINI and the modified force fields and compared to those extracted from atomistic simulations. The result is shown in Figure 3 for the peptide SNNFGAIL. The distributions obtained from the original MARTINI model are significantly different from the results of atomistic simulations, but the fitted potential accurately reproduces the distributions of dihedral angles from atomistic simulations.
Figure 3.

Probability distributions for backbone dihedral angles of SNNFGAIL obtained from atomistic (AT) and CG simulations at 305 K.
2.3. Simulation Details
2.3.1. Atomistic Simulations
All atomistic simulations except those of GVGVPGVG, (GA)4, and (GV)4 have not been published previously. The atomistic simulations of all octapeptides were carried out using simulated tempering distributed replica sampling (STDR).42,48 For each octapeptide, the simulation system consisted of the octapeptide in a 3 × 3 × 3 nm cubic box with explicit water molecules. The octapeptides were capped with an acetyl group at the N-terminus and an NH2 group at the C-terminus. The temperatures were spaced exponentially between 280 and 694 K, resulting in an acceptance ratio of approximately 35%. The same fully extended starting structure was used for all temperatures. Simulations were performed using the GROMACS MD simulation package, version 3.3.147,49 with the OPLS-AA/L force field50,51 for the solute and the TIP3P model for water.52 Periodic boundary conditions were applied. The switch function of GROMACS was used for Lennard-Jones interactions, which corresponds to the usual Lennard-Jones function until 1.3 nm, after which it is switched to reach zero at 1.4 nm. No scaling factors were used for nonbonded interactions. An initial energy minimization was performed in GROMACS using the steepest descent method. Covalent bonds involving hydrogen atoms were constrained with the SHAKE algorithm.53 Calculations of electrostatic forces utilized the particle mesh Ewald (PME) summation method54,55 with a Fourier spacing of 0.15 nm and a fourth-order interpolation. The real-space Coulombic cutoff was 1.49 nm. All MD simulations were performed in the canonical ensemble. Peptide and solvent were coupled to the same reference temperature bath with a time constant of 2 ps using the Nosé–Hoover method.56,57 An integration step size of 2 fs was used, and coordinates were stored every 1 ps. For the tetrapeptides, simulation conditions were identical to the octapeptides simulations. The only difference is that simulations of all tetrapeptides were performed in the canonical ensemble at a single temperature (305 K).
The atomistic simulations of the longer peptides, (GV)18 and (PGV)12, were also performed using the STDR algorithm. The details of these simulations are the same as our previously published work.58 In particular, an exponentially spaced list of 105 temperatures between 266 and 749 K was used. All simulations of the octapeptides involved between 120 and 170 ns of sampling at each temperature. The simulations of the two longer peptides had an average of 800 ns per temperature, for a total simulation time of 84 μs per peptide. Coordinates were saved every 1 ps for the octapeptide simulations and every 10 ps for the tetrapeptide simulations.
2.3.2. CG Simulations
All CG simulations described in this paper were performed using the GROMACS MD package, version 4.0.4.47 All conformations at 305 K from each replica in atomistic simulations were used for CG parametrization. The CG simulations of monomeric amyloid- and elastin-like peptides (tetrapeptides, octapeptides, and longer peptides) were performed in solution using the original MARTINI model7 as well as using the MARTINI model with the newly derived dihedral potentials (eq 6). The CG water model provided by the MARTINI force field was used.14 In this model, one bead represents four water molecules. The CG topologies for peptides were generated from the atomistic structures. The acetyl group at the N-terminus and an NH2 group at the C-terminus were not present in the CG simulations. No secondary structure was imposed on the peptides. Backbone bonded parameters are consistent with random coil, and the particle type P5 was used for the backbone in all peptides. The CG simulations were run for 1.5 μs. The simulations using the original MARTINI model were run with an integration time step of 20 fs. An integration time step of 7 fs was used for stability in simulations employing the modified MARTINI model. This relatively small time step compared to a more typical MARTINI time step of 20 fs is a limitation of the implementation of a standard dihedral potential over four BB, which, unlike normal atomistic force fields, have a significant probability of having three beads colinear, i.e., when three beads are colinear, any dihedral angle including these three beads is undefined. When the beads approach colinearity, small displacements lead to large changes in the dihedral term, which forces a smaller time step to be used. An alternative implementation that prevents this artifact is under development.
In the CG simulations, a cutoff of 1.2 nm (rcut) was used in the calculation of nonbonded interactions with a shifted function. The Lennard-Jones potential is shifted from 0.9 to 1.2 nm. The electrostatic potential is shifted from 0.0 to 1.2 nm. Both the energy and the force vanish at the cutoff distance. The temperature is kept constant using the Berendsen temperature coupling algorithm59 with a coupling time constant of 1 ps. Isotropic pressure coupling was applied using the Berendsen algorithm59 with a reference pressure of 1 bar. A coupling constant of 5.0 ps and a compressibility of 4.5 × 10–5 bar–1 were used. Bond lengths in aromatic amino acid side chains and the backbone side chain bonds for Val were constrained using the LINCS algorithm.47 All simulations were run at T = 305 K. The improved model was applied to the self-assembly of amyloid-like peptides as a simple model system for aggregation. The CG simulations of peptides SNNFGAIL and (GV)4 were performed at 305 K both in water and at the water–octane interface with a different number of peptides: 1, 8, and 64. The topology and parameters for water and octane are taken from the MARTINI force field data set.14 Each simulation was carried out for 1 μs.
3. Results and Discussion
CG simulations were performed using both the original MARTINI model and the model with the addition of new dihedral potentials. To validate our model, we compared the CG results with the results obtained from atomistic simulations.32,42 The backbone dihedral angle probability distributions were used to assess statistical convergence of atomistic and CG simulations (Figures SI1 and SI2, Supporting Information).
3.1. Structural Properties
The performance of our model was assessed by comparing the distributions of various structural properties with their counterparts from atomistic simulations. We calculated the size-dependent behavior of the models on two global structural properties of the peptides: the radius of gyration Rg and the end-to-end distance dee. Results are shown in Table 2. In general, values of Rg and dee computed with the new model show a good agreement with the atomistic results. The performance is also comparable to that of the original model. For longer peptides, we observed that (GV)18 is on average more expanded than (PGV)12, while in the atomistic simulations they have approximately the same average Rg. We have not found the exact reason for this discrepancy between atomistic and CG results, but it is not affected by the addition of the new dihedral parameters. Therefore, the addition of new dihedral potentials to the model does not significantly change either Rg or dee of the peptides, suggesting that the global structural properties are primarily determined by the nonbonded parameters in the force field.
Table 2. Radius of Gyration Rg and the End-to-End Distance dee for Various Peptidesa.
|
Rg (nm) |
dee (nm) |
|||||
|---|---|---|---|---|---|---|
| peptide | original CG | new CG | AT | original CG | new CG | AT |
| (GA)4 | 0.48 ± 0.08 | 0.47 ± 0.07 | 0.52 ± 0.09 | 1.16 ± 0.40 | 1.07 ± 0.39 | 0.90 ± 0.36 |
| (GV)4 | 0.58 ± 0.07 | 0.58 ± 0.07 | 0.55 ± 0.08 | 1.17 ± 0.38 | 1.15 ± 0.45 | 1.11 ± 0.40 |
| SNNFGAIL | 0.61 ± 0.05 | 0.60 ± 0.06 | 0.58 ± 0.07 | 1.29 ± 0.39 | 1.12 ± 0.36 | 1.03 ± 0.37 |
| GVGVAGVG | 0.56 ± 0.06 | 0.55 ± 0.06 | 0.56 ± 0.09 | 1.30 ± 0.37 | 1.23 ± 0.36 | 1.03 ± 0.41 |
| GVGVGGVG | 0.56 ± 0.06 | 0.54 ± 0.07 | 0.55 ± 0.09 | 1.33 ± 0.37 | 1.16 ± 0.37 | 0.97 ± 0.40 |
| GVGVAGGV | 0.58 ± 0.07 | 0.57 ± 0.07 | 0.57 ± 0.10 | 1.29 ± 0.38 | 1.24 ± 0.37 | 1.06 ± 0.41 |
| GVGGVGGV | 0.58 ± 0.07 | 0.55 ± 0.07 | 0.57 ± 0.10 | 1.38 ± 0.38 | 1.14 ± 0.38 | 1.04 ± 0.40 |
| GVGVPGVG | 0.55 ± 0.06 | 0.49 ± 0.05 | 0.59 ± 0.09 | 1.28 ± 0.39 | 1.34 ± 0.37 | 1.13 ± 0.42 |
| GVPGVPGV | 0.56 ± 0.07 | 0.56 ± 0.07 | 0.61 ± 0.09 | 1.21 ± 0.39 | 1.19 ± 0.38 | 1.17 ± 0.43 |
| GVAPGVGV | 0.57 ± 0.07 | 0.54 ± 0.07 | 0.57 ± 0.10 | 1.26 ± 0.39 | 1.08 ± 0.38 | 1.09 ± 0.40 |
| GVGGVPGV | 0.57 ± 0.07 | 0.56 ± 0.06 | 0.60 ± 0.10 | 1.25 ± 0.39 | 1.12 ± 0.36 | 1.13 ± 0.41 |
| (GV)18 | 1.18 ± 0.24 | 1.18 ± 0.23 | 0.82 ± 0.04 | 1.50 ± 0.52 | 1.46 ± 0.48 | 1.33 ± 0.47 |
| (PGV)12 | 0.87 ± 0.07 | 0.87 ± 0.07 | 0.88 ± 0.05 | 1.63 ± 0.63 | 1.52 ± 0.49 | 1.31 ± 0.56 |
The results obtained from atomistic (AT) and CG trajectories using the original and the new CG models are shown. Error bars represent standard deviations associated with average properties computed over all trajectories, for both AT and CG simulations.
In Figure 4, we show the contact maps of BB of four selected peptides, GVAPGVGV, GVPGVPGV, (GV)4, and SNNFGAIL obtained from CG and atomistic simulations. The contact maps display the probability of two residues forming a contact as a function of their residue numbers. Distance cutoffs of 0.6 and 0.65 nm for atomistic and CG trajectories, respectively, were employed to generate the contact maps based on probability distributions representing averages over all possible BB–BB distances for each sequence. These distances are the first minimum in radial distribution functions. BB distances were calculated from the center of mass of the appropriate group of atoms for the atomistic data. The contact maps show the fraction of total simulation time a contact is present. The color scheme for the contact maps and representative structures with contacts from the trajectory of GVAPGVGV is shown in Figure 4E. For all cases, the new model reproduced the most populated contact of atomistic results: the A3–V6 contact in peptide GVAPGVGV, the V2–V5 contact in peptide GVPGVPGV, the G3–V6 contact in peptide (GV)4, and the N3–A6 contact in peptide SNNFGAIL. Several other contacts with lower populations were also reproduced with the new model. Although the original CG model is able to identify the majority of contacts observed in atomistic simulations for all cases, its performance is inferior to the new model at reproducing the atomistic probabilities of contact formation on a quantitative level. Our results indicate that the accuracy of the model was significantly improved with the addition of new dihedral potentials. It also suggests that the addition of dihedral angle potentials on peptide backbones mainly affects the local structural properties of peptides.
Figure 4.

Backbone beads contact maps at 305 K. Each square in the matrix (i,j) corresponds to a contact between the BB of residues i and j: (A) GVAPGVGV, (B) GVPGVPGV, (C) (GV)4, and (D) SNNFGAIL. (E) Representative snapshots of GVAPGVGV showing the presence of significantly populated contacts. In the color scheme, each color represents a range of probabilities of contact formation. On each map, the atomistic map lies above the diagonal, and the corresponding CG map, obtained from the simulations using either original MARTINI model (a) or the new model with the addition of dihedral potentials (b), lies below the diagonal.
3.2. Transferability of Parameters
It is important to test the transferability of the results obtained from short peptides to longer ones since it is significantly more challenging to obtain complete conformational sampling for longer peptides compared to shorter peptides with atomistic models.42 We carried out the conformational analysis of two elastin-like peptides GVGVPGVG and (GVPGV)7, using atomistic simulations, and computed the probability distributions of the distance between Cα(i) and Cα(i + 3) of the five distinct four-residue fragments from the two peptides (Figure 5). Comparison of the distributions showed that they were all similar, suggesting that the properties of disordered peptides can be deduced from those of short fragments. Accordingly, the parametrization of the MARTINI force field may rely on the transferability of fragment properties obtained for short peptides.
Figure 5.

Conformational analysis of two elastin-like peptides GVGVPGVG and (GVPGV)7 from atomistic simulations in water at 296 K. (A) Probability distributions of the distance (r) between Cα(i) and Cα(i + 3) of the four-residue fragments from the two peptides. (B) Representative conformations of GVGVPGVG: (a) VPGV turn, (b) GVGV turn, (c) both VPGV and GVGV turns (“s” shape), and (d) neither turn (extended state).
We constructed several peptides of different lengths ((GV)4, GVPGVPGV, (GV)18, (PGV)12). Tetrapeptide fragments, i.e., each quartet of amino acids residues of the amyloid- and elastin-like octapeptides listed were prepared. Subsequently, we extended the periodic sequence to generate longer peptides ((GV)18, (PGV)12), which were used to test the transferability of parameters derived from simulations of short peptides to simulations of longer peptides.
Atomistic simulations were carried out for all tetrapeptides in solution, and the resulting trajectories were used to derive parameters of backbone dihedral angle potentials for the CG model (see Computational Methods Section). The dihedral angle potentials were then employed in CG simulations of octapeptides and longer peptides to model their torsions. The distributions of dihedral angles of possible quartets were calculated from atomistic and CG trajectories. Figures 6 and 8 show the average dihedral angle distributions of four-residue fragments of amyloid-like (GVGV and VGVG) and elastin-like (GVPG, VPGV, and PGVP) peptides, respectively. For example, the distributions of dihedral angles for the GVGV fragments of (GV)4, shown in Figure 6 (A), were computed by averaging the distributions for the three GVGV fragments present in the octapeptide.
Figure 6.

Average dihedral distributions of fragments GVGV (A) and VGVG (B) in peptides (GV)4 and (GV)18. Atomistic distributions obtained from tetrapeptides GVGV and VGVG are also shown. Error bars on distributions represent standard deviations in the distributions of all possible GVGV (A) and VGVG (B) fragments in (GV)4 and (GV)18.
Figure 8.

Average dihedral distributions of fragments GVPG (A), VPGV (B), and PGVP (C) in peptides GVPGVPGV and (PGV)12. Atomistic distributions obtained from tetrapeptides GVPG, VPGV, and PGVP are also shown. Error bars represent standard deviations computed from distributions of all possible fragments in GVPGVPGV and (PGV)12 for GVPG (A), VPGV (B), and PGVP (C).
The CG distribution of dihedral angles for the GVGV fragments of (GV)4, shown in Figure 6(A), is similar to that of (GV)18, both featuring a broad peak at 30°. The corresponding atomistic distributions, on the other hand, are characterized by two prominent peaks at −60°and 25°. Comparison of the atomistic and CG distributions of dihedral angles for the tetrapeptide shows that the maximum at −60° is also absent from the CG distribution, although it is visible in the atomistic distribution. It is likely that the difference between the atomistic and CG results for (GV)4 and (GV)18 is the result of a limitation of the CG parameters in modeling torsional changes about −60° during the simulations. For the VGVG fragment (Figure 6B), the distribution of dihedral angles for the tetrapeptide computed from atomistic trajectories displays a sharp peak at 70° in addition to a shoulder around 0° and a minimum around −90°. All features are well reproduced by the corresponding CG distribution, although slight differences are visible in the relative probabilities of the dihedral angles around the shoulder and the maximum. The overall shape and features of the distributions are retained by the longer peptides, both for atomistic and CG trajectories. The CG distributions of the VGVG fragment for (GV)4 and (GV)18 are strikingly similar, as previously noticed for the GVGV fragment in Figure 6A. The excellent agreement between the atomistic and CG distributions for (GV)4 and (GV)18 indicates that the CG parameters are highly transferable for the VGVG fragment. The performance is in contrast with results obtained for the GVGV fragment. To better understand this difference, we examined the quality of the fits described by eq 5 generated during the parametrization procedure of the tetrapeptides in Figure 7. It can be observed that the description of torsional motion afforded by the fitting function is more accurate for VGVG than for GVGV. The dihedral potential of the GVGV tetrapeptide is characterized by numerous low-lying barriers which require functions more complex than eq 5 or the use of numerical tables instead of fitting functions to accurately model torsional motions. Dihedral potential energies of the GVGV tetrapeptide computed from numerical tables, shown in eq 5, confirm the superior quality of such fits. These would result in better transferability of parameters for amyloid-like peptides. Taken together, our observations suggest that it is possible to derive CG parameters for longer peptides based on short amyloid-like peptides.
Figure 7.
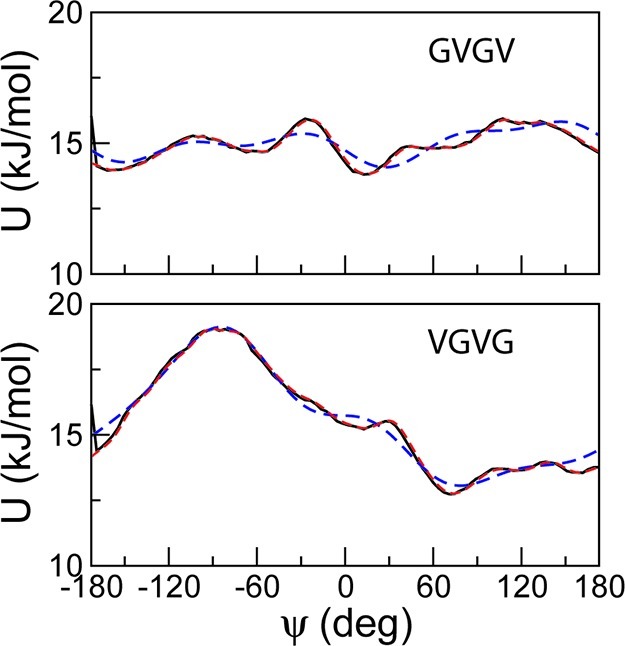
The potentials of mean force (black solid lines) and the fitting energy functions (blue dotted lines) of the GVGV and VGVG tetrapeptides. The fits with the use of numerical tables are also shown (red dotted lines). The potentials of mean force were extracted from the probability distributions of the dihedral angles calculated from atomistic trajectories.
For the three elastin-like peptide fragments, all atomistic distributions of tetrapeptides, octapeptide GVPGVPGV and (PGV)12 peak at the same values of dihedral angles with similar probabilities, namely at −110° for the GVPG fragment (Figure 8A), at −140°, −50°, and 30° for the VPGV fragment (Figure 8B), and at −170°, 70°, and 150° (Figure 8C) for the PGVP fragment. The appearance of a small additional peak around 60° in the atomistic distribution of dihedral angles of the GVPG fragment for (PGV)12 can be attributed to isomerization to cis-proline at high temperatures. Atomistic simulations of (PGV)12 were allowed to reach higher temperatures than simulations of the tetrapeptides and octapeptides. The highest temperature possible in the random walk was 749 K for the (PGV)12 simulation. In general, CG simulations reproduce all features of atomistic distributions for all the fragments in elastin-like peptides, showing that the effect of different lengths is comparable on atomistic and CG distributions. Therefore, our results show that the new CG model is reasonably transferable for elastin-like peptides from short to longer peptides.
3.3. Amyloid Peptide Aggregation
The study of protein aggregation is a biologically important and complex problem. Atomistic simulations of protein aggregation are currently limited to simulations of a small number of peptides on the nanosecond to microsecond time scale. Thus, simulations of the self-assembly and structural properties of large-scale peptide aggregates would benefit significantly from using CG parameters.60,61
Recently, Pomès and co-workers reported atomistic simulations of the self-aggregation of simple β-sheet forming peptides at a water–hydrophobic interface mimicking the core of lipid membranes.62 They investigated the influence of the water–hydrophobic interface on the aggregation of peptides (GV)4 and (GA)4 and observed an enhancing effect of the water–nonpolar interface on β-sheet formation compared to purely aqueous environments, in accordance with experimental evidence. They also proposed a general mechanism for β-sheet formation by hydrophobic interfaces during protein folding and amyloid self-organization based on their analysis of the physical and molecular aspects of the catalysis of amyloid formation. In this generic mechanism, the primary effect of the interface is to displace the conformational equilibrium of the peptides toward extended, β-strand-prone conformations.
To validate the extension of the CG MARTINI model and test the transferability of the parameters, we attempt to reproduce the extended structure of aggregated peptides at the water–hydrophobic interface using parameters derived in the aqueous phase. In particular, we examine whether our new model adequately reproduces the displacement in the conformational equilibrium of peptides during aggregation at the interface without additional adjustment of parameters in comparison with atomistic results. In addition, the compatibility of our model to reproduce the peptide conformational equilibrium is investigated in different environments, i.e., in water and at the water–octane interface.
CG models require certain elements to represent the secondary peptide structure in peptide aggregation. For instance, without elements such as backbone hydrogen bonding and chirality, the assembly of peptides cannot be modeled into a sheet.60 In order to describe chiral fibrillar peptide aggregation, the peptide model developed by Shea and co-workers retains most of the backbone degrees of freedom and introduces molecular chirality as an additional dihedral degree of freedom.60,63 Another CG model which has been successfully employed in the study of peptide aggregation is the optimal potential for efficient peptide-structure prediction force field.64 This model uses a single bead representation of side chains while keeping a detailed representation of all backbone atoms (N, H, Cα, C, and O) with potential energy functions, taking into account nonbonded interactions and hydrogen bonding interactions.61 In its current form, MARTINI does not have specific interactions that promote β-sheet or other secondary structure elements beyond the standard nonbonded and bonded interactions. Our modified backbone dihedral potential is one step toward including such interactions, but by itself likely remains insufficient to accurately simulate the formation of fibrils and other structures from individual peptides.
CG simulations of single and multiple peptides (8 and 64) of SNNFGAIL and (GV)4 were performed both in water and in the presence of a hydrophobic octane phase. Peptides were initially placed in water and peptide adsorption to the interface occurred rapidly. At the interface, the peptides aggregated into large clusters within tens of nanoseconds. Figure 9 shows the evolution of the size of the largest peptide cluster with respect to time for 64 SNNFGAIL. A peptide was defined as belonging to a cluster if the distance between the center of mass of two peptides is less than 1.2 nm. At initial time (t = 0), the system consists of 64 SNNFGAIL monomers. The peptides diffuse rapidly and aggregate into small clusters between 10 and 100 ns. Bigger clusters were formed after 100 ns, although their sizes were not consistent throughout the simulations. It has been reported that peptide monomers in water and at the interface adopted many different conformations, and they interconverted over the course of the simulations.62 To analyze the conformation of the peptides within the aggregates, we calculated the distribution of end-to-end distances, dee, for aqueous and adsorbed peptide monomers. Figure 10A presents the results obtained from simulations of 64 SNNFGAIL and (GV)4 monomers using both the original MARTINI model and the new model. Three conformation types62 were defined based on dee: short (S) for dee < 0.65 nm, intermediate (I) for 0.65 < dee > 1.2 nm, and extended (E) for dee > 1.2 nm. Representative snapshots of the peptide SNNFGAIL monomers are shown in Figure 10A. Comparison of the distributions of dee obtained from the original MARTINI model and the new model reveals that both CG models favor extended structures. With the new model, however, the distribution of peptides is skewed even further toward extended conformations resembling β-hairpins and β-strands at the water–octane interface during peptide aggregation (Figure 10A). We also show the distributions of dee for aqueous and adsorbed peptide monomers at different peptide concentrations with the newly developed CG model in Figure 10B. The plot shows that extended conformations are more populated at the interface than in water. The Pomès group reported a similar observation in the end-to-end distributions of amyloidogenic peptides, (GV)4 and (GA)4, calculated from atomistic trajectories.62 In particular, peptides at the interface displayed a preference for extended conformations compared to aqueous peptides, which indicates a shift in the conformational equilibrium of interfacial peptides toward stretched conformers upon adsorption. In addition, the population of peptides with short and intermediate end-to-end distances decreases as the concentration and time spent at the interface increase. The concurrent growth in the population of extended conformations suggests that short and intermediate conformations are gradually replaced by extended ones. Based on these observations and similar reports from the analysis of atomistic trajectories of the amyloidogenic (GV)4 and (GA)4 peptides, we conclude that the inclusion of torsional backbone flexibility in the new model provides an improved description of peptide conformational preferences during aggregation at the interface. Reproduction of the peptide conformational preferences at the interface with no parameter modification suggests that the backbone potentials to peptides in different environments are transferable.
Figure 9.
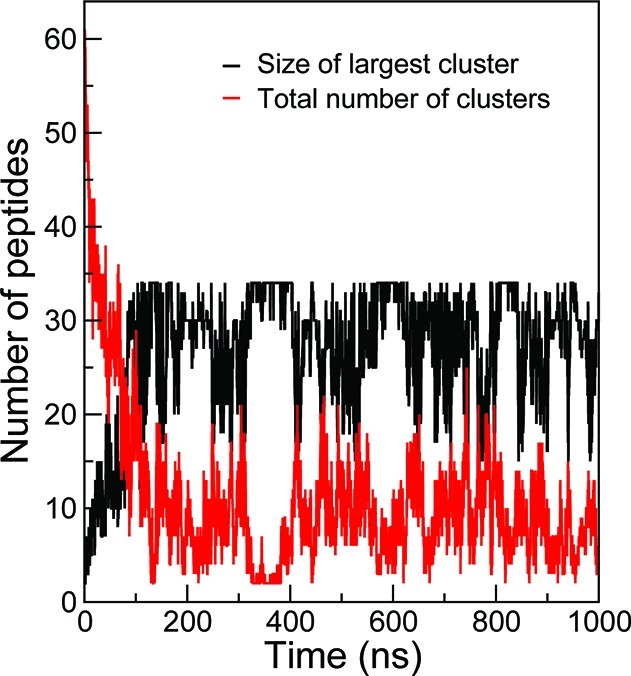
Plot of the size of the largest cluster and the total number of clusters as function of time for 64 SNNFGAIL.
Figure 10.

Distribution of end-to-end distance of SNNFGAIL and (GV)4 peptides. (A) Distribution of dee of 64 SNNFGAIL and (GV)4 at water–octance interface using the original and new CG models. Representative snapshots of SNNFGAIL in short and extended conformations are shown. Vertical lines at dee = 0.66 and 1.2 nm highlight the boundaries separating short, intermediate, and extended conformations. (B) Distribution of dee of SNNFGAIL at different concentration in water and at water–octane interface computed with the new model. Distribution of dee of 64 (GV)4 is also shown in water and at water–octane interface computed with the new model.
4. Conclusions
CG models enable the study of phenomena at longer time and length scales than more detailed atomistic models and their application to peptide aggregation allows for a direct examination of the process in silico. In the present work, we extended the CG MARTINI model to describe the backbone flexibility of proteins by introducing in the energy function a term that accounts for the dihedral angle potentials on the peptide backbone. Backbone flexibility is often required during simulations since biological processes, such as protein folding and aggregation, typically involve significant transitions between different types of secondary structure. The new model was applied to amyloid- and elastin-like peptides, and its performance was assessed based on its ability to reproduce various structural properties calculated from atomistic trajectories of peptides in water. Although little effect was observed on global structural properties, such as Rg and dee, when the new and original forms of the model were compared, peptide contact maps showed a significant improvement when torsional flexibility was allowed in the new model.
The transferability of the dihedral parameters was tested by employing the parameters derived from the atomistic trajectories of shorter fragments in simulations of longer peptides. Such a feature is highly desirable, since it offers the possibility of modeling longer flexible peptides with the new CG MARTINI model. Simulations of such systems using atomistic methods tend to be generally inefficient because they require extensive conformational sampling. Average distributions of dihedral angles were computed and compared for peptides of different lengths. Our results showed that the parametrization of longer elastin-like peptide sequences is feasible from shorter segments. For amyloid-like peptides, the transferability was observed to be very sensitive to the quality of the parameters. Overall, transferable parameters can be derived to model long peptides with the new CG MARTINI model. In particular, the approach used and presented in the present work can be directly applied to probe events which necessitate a flexible description of proteins over long time scales.
Finally, the improved model was subsequently employed to characterize the self-aggregation properties of peptides in water and at the water–octane interface. We investigated whether the simulations using the new model were able to reproduce the behavior observed in atomistic simulations. The new CG model was observed to successfully reproduce the shift in conformational equilibrium toward extended structures during aggregation at the interface without further parameter adjustment.
Angle and dihedral potential energy functions are used to introduce flexibility into the dynamics of peptide secondary structures in the current version of MARTINI model. It is noted that our work is a first step to describe more accurately the backbone flexibility of peptides. Further improvements in MARTINI are still needed to adequately incorporate changes in the secondary structure of proteins.
Acknowledgments
Computations were performed on the WestGrid and the GPC supercomputer at the SciNet HPC Consortium. SciNet is funded by the Canada Foundation for Innovation under the auspices of Compute Canada, the Government of Ontario, Ontario Research Fund–Research Excellence, and the University of Toronto. We also thank the Shared Hierarchical Academic Research Computing Network (SHARCNET) for a dedicated allocation of CPU resources and data storage. The Natural Sciences and Engineering Research Council (NSERC), the Canadian Institutes of Health Research (CIHR), PrioNet, and APRI are gratefully acknowledged for support. S.R. is funded by a Canada Graduate Scholarship from the NSERC and the Research Training Center at the Hospital for Sick Children. R.P. is a CRCP chairholder, and D.P.T. is an AHFMR scientist. M.S. thanks Bilkiss B. Issack for critical reading of the manuscript.
Supporting Information Available
Statistical convergence of atomistic and CG simulations; the potentials of mean force and fitting energy functions for all possible backbone dihedral angles of octapeptides; and the list of dihedral parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Ash W. L.; Zlomislic M. R.; Oloo E. O.; Tieleman D. P. Biochim. Biophys. Acta 2004, 1666, 158–189. [DOI] [PubMed] [Google Scholar]
- Smit B.; Hilbers P. A. J.; Esselink K.; Rupert L. A. M.; van Os N. M.; Schlijper A. G. Nature 1990, 348, 624–625. [Google Scholar]
- Saiz L.; Klein M. L. Acc. Chem. Res. 2002, 35, 482–489. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; de Vries A. H.; Mark A. E. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar]
- Tozzini V.; Rocchia W.; McCammon J. A. J. Chem. Theory Comput. 2006, 2, 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basdevant N.; Borgis D.; Ha-Duong T. J. Phys. Chem. B 2007, 111, 9390–9399. [DOI] [PubMed] [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S. J. J. Chem. Theory Comput. 2008, 4, 819–834. [DOI] [PubMed] [Google Scholar]
- DeVane R.; Shinoda W.; Moore P. B.; Klein M. L. J. Chem. Theory Comput. 2009, 5, 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper H. L.; Voth G. A. J. Chem. Phys. 2005, 122, 124906–124911. [DOI] [PubMed] [Google Scholar]
- Knotts T. A.; Rathore N.; Schwartz D. C.; de Pablo J. J. J. Chem. Phys. 2007, 126, 084901–084912. [DOI] [PubMed] [Google Scholar]
- Lee H.; de Vries A. H.; Marrink S. J.; Pastor R. W. J. Phys. Chem. B 2009, 113, 13186–13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi G.; Monticelli L.; Puisto S. R.; Vattulainen I.; Ala-Nissila T. Soft Matter 2011, 7, 698–708. [Google Scholar]
- Tozzini V. Curr. Opin. Struct. Biol. 2005, 15, 144–150. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. J. Phys. Chem. B 2007, 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Wong-Ekkabut J.; Baoukina S.; Triampo W.; Tang I.-M.; Tieleman D. P.; Monticelli L. Nat. Nanotechnol. 2008, 3, 363–368. [DOI] [PubMed] [Google Scholar]
- López C. A.; Rzepiela A. J.; de Vries A. H.; Dijkhuizen L.; Hünenberger P. H.; Marrink S. J. J. Chem. Theory Comput. 2009, 5, 3195–3210. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Sansom M. S. Curr. Opin. Struct. Biol. 2008, 18, 425–431. [DOI] [PubMed] [Google Scholar]
- Sansom M. S.; Scott K.; Bond P. Biochem. Soc. Trans. 2008, 36, 27–32. [DOI] [PubMed] [Google Scholar]
- Bond P.; Sansom M. S. J. Am. Chem. Soc. 2006, 128, 2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond P.; Holyoake J.; Ivetac A.; Khalid S.; Sansom M. S. J. Struct. Biol. 2007, 157, 593–605. [DOI] [PubMed] [Google Scholar]
- Risselada H.; Marrink S. J.; Mark A. E. Chem. Phys. Lipids 2005, 135, 223–244. [DOI] [PubMed] [Google Scholar]
- Periole X.; Cavalli M.; Marrink S. J.; Ceruso M. A. J. Chem. Theory Comput. 2009, 5, 2531–2543. [DOI] [PubMed] [Google Scholar]
- Singh G.; Tieleman D. P. J. Chem. Theory Comput. 2011, 7, 2316–2324. [DOI] [PubMed] [Google Scholar]
- de Jong D. H.; Periole X.; Marrink S. J. J. Chem. Theory Comput. 2012, 8, 1003–1014. [DOI] [PubMed] [Google Scholar]
- Christen M.; van Gunsteren W. F. J. Chem. Phys. 2006, 124, 154106. [DOI] [PubMed] [Google Scholar]
- Ayton G. S.; Noid W. G.; Voth G. A. Curr. Opin. Struct. Biol. 2007, 17, 192–198. [DOI] [PubMed] [Google Scholar]
- Rzepiela A. J.; Louhivuori M.; Peter C.; Marrink S. J. Phys. Chem. Chem. Phys. 2011, 13, 10437–10448. [DOI] [PubMed] [Google Scholar]
- Shi Q.; Izvekov S.; Voth G. A. J. Phys. Chem. B 2006, 110, 15045–15048. [DOI] [PubMed] [Google Scholar]
- Neri M.; Anselmi C.; Cascella M.; Maritan A.; Carloni P. Phys. Rev. Lett. 2005, 95, 218102. [DOI] [PubMed] [Google Scholar]
- Vrhovski B.; Weiss A. S. Eur. J. Biochem. 1998, 258, 1–18. [DOI] [PubMed] [Google Scholar]
- Muiznieks L. D.; Weiss A. S.; Keeley F. W. Biochem. Cell Biol. 2010, 88, 239–250. [DOI] [PubMed] [Google Scholar]
- Cox B.; Starcher B.; Urry D. J. Biol. Chem. 1974, 283, 255–264. [PubMed] [Google Scholar]
- Miao M.; Bellingham C.; Stahl R.; Sitarz E.; Lane C.; Ke J. Phys. Chem. B 2003, 278, 48553–48562. [DOI] [PubMed] [Google Scholar]
- Rauscher S.; Baud S.; Miao M.; Keeley F.; Pomès R. Structure 2006, 14, 1667–1676. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Nature 2003, 426, 884–890. [DOI] [PubMed] [Google Scholar]
- Stefani M.; Dobson C. M. J. Mol. Med. 2003, 81, 678–699. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. Trends Cell Biol. 1998, 8, 447–453. [DOI] [PubMed] [Google Scholar]
- Hou L.; Shao H.; Zhang Y.; Li H.; Menon N. K.; Neuhaus E. B.; Brewer J. M.; Byeon I.-J. L.; Ray D. G.; Vitek M. P.; Iwashita T.; Makula R. A.; Przybyla A. B.; Zagorski M. G. J. Am. Chem. Soc. 2004, 126, 1992–2005. [DOI] [PubMed] [Google Scholar]
- Legname G.; Baskakov I. V.; Nguyen H.-O. B.; Riesner D.; Cohen F. E.; DeArmond S. J.; Prusiner S. B. Science 2004, 305, 673–676. [DOI] [PubMed] [Google Scholar]
- Zahn R.; Liu A.; Luhrs T.; Riek R.; von Schroetter C.; Lopez Garcia F.; Billeter M.; Calzolai L.; Wider G.; Wuthrich K. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R.; Sawaya M. R.; Balbirnie M.; Madsen A. O.; Riekel C.; Grothe R.; Eisenberg D. Nature 2005, 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R.; Eisenberg D. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [DOI] [PubMed] [Google Scholar]
- Rauscher S.; Neale C.; Pomès R. J. Chem. Theory Comput. 2009, 5, 2640–2662. [DOI] [PubMed] [Google Scholar]
- Gedulin B. R.; Jodka C. M.; Herrmann K.; Young A. A. Regul. Pept. 2006, 137, 121–127. [DOI] [PubMed] [Google Scholar]
- Madine J.; Jack E.; Stockley P. G.; Radford S. E.; Serpell L. C.; Middleton D. A. J. Am. Chem. Soc. 2008, 130, 14990–15001. [DOI] [PubMed] [Google Scholar]
- Rousseau R.; Schreiner E.; Kohlmeyer A.; Marx D. Biophys. J. 2004, 86, 1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp W.; Kremer K.; Batoulis J.; Burger T.; Hahn O. Acta Polym. 1998, 49, 61–74. [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. J. Chem. Theory Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Rodinger T.; Howell P. L.; Pomès R. J. Chem. Theory Comput. 2006, 2, 725–731. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. J. Comput. Chem. 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. L. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Darden T.; York D.; Pedersen L. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Nosé S. Mol. Phys. 1984, 52, 255–268. [Google Scholar]
- Hoover W. G. Phys. Rev. A 1985, 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- Rauscher S.; Pomès R. J. Phys.: Conf. Ser. 2010, 256, 012011. [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M; van Gunsteren W. F.; DiNola A.; Haak J. R. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- Bellesia G.; Shea J.-E. J. Chem. Phys. 2007, 126, 245104. [DOI] [PubMed] [Google Scholar]
- Nasica-Labouze J.; Meli M.; Derreumaux P.; Colombo G.; Mousseau N. PLoS Comput. Biol. 2011, 7, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic A.; Baud S.; Rauscher S.; Pomès R. Proteins: Struct., Funct., Bioinf. 2011, 79, 1–22. [DOI] [PubMed] [Google Scholar]
- Bellesia G.; Shea J.-E. J. Chem. Phys. 2009, 130, 145103. [DOI] [PubMed] [Google Scholar]
- Tufféry M. J.; Mousseau N. Proteins 2007, 69, 394–408. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.