

Abstract
Combinatorial library screens can identify a suitable ligand for a biological target of interest out of thousands or even millions of compounds, and can play a key role in the modern drug development process. While conventional high-throughput cell screens based on functional assays require expensive robotics, simple on-bead combinatorial assays for ligand binding to the target protein can be done far more cheaply. This article describes one such assay, developed using combinatorial peptoid libraries for targeting integral membrane receptors or other cell surface-exposed molecules. In addition to the reduced cost, a unique advantage of this assay is the direct identification of the most selective ligands for a cell surface receptor that is expressed in its natural environment.
INTRODUCTION
This article describes a general procedure for direct identification of highly selective synthetic ligands for cell surface receptors from large combinatorial libraries displayed on beads. In recent years, high throughput combinatorial screens have become one of the most important tools in the drug discovery process. Even though screening millions of compounds seems like a more efficient process to pick a potential ‘hit’ than working on one compound at a time, most of these approaches are associated with high cost and the necessity of specialized infrastructure. Also, the high rate of identifying poorly selective or promiscuous ‘hits’ and false positives makes post-validation a highly time- and resource-consuming process before finalizing a high affinity and selective lead molecule. Here we describe a cost effective and efficient method to screen millions of bead-bound peptoid compounds, directly identifying highly selective ligands for cell surface receptors.
The screen is based on the capability of the library compounds to recognize cells that express a target receptor (e.g., receptor ‘X’, Fig. 1) while ignoring otherwise identical cells that do not express this receptor (Udugamasooriya et al., 2008a). This is achieved by employing a cell type that does not express the receptor of interest, labeling these with a green dye, then introducing the receptor of interest into the same cells and labeling these with a red dye. These red and green stained cell groups can then be mixed in a 1:1 ratio and exposed to a one bead one compound library. If a bead is found with only red cells bound to it, this indicates that the peptoid on that bead is highly selective for receptor ‘X’ and does not bind to any other cell surface molecule (Fig. 1). If it binds to other cell surface molecules non-selectively, both red and green cells will be attracted to this bead, which is then discarded (Fig. 1). Identified ‘hit’ beads are manually picked and subjected to a strong cleaning process to get rid of cells and protein-based blocking agents. If automated Edman degradation is used for sequence identification, the cleaned ‘hit’ bead is used as is for on-bead sequencing. If mass spectroscopic sequencing (MS-MS) is applied, the compound is cleaved from the resin.
Figure 1.
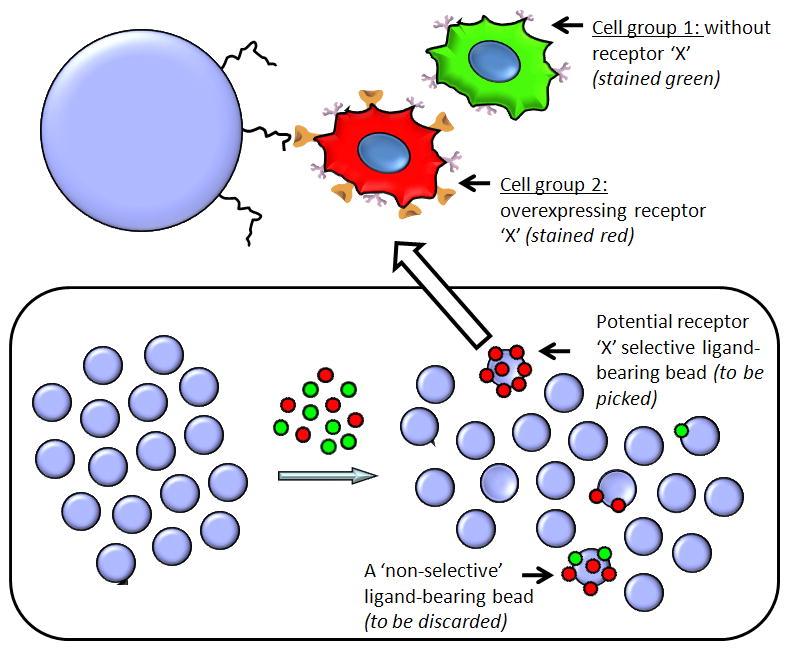
Schematic representation of the on-bead two-color (OBTC) assay for the identification of peptoid ligands for receptor ‘X’. Blue circles represent peptoid library beads. The single bead shown on top of the figure displays a red stained receptor ‘X’ overexpressing cell binding to a peptoid molecule on the bead through the specific interaction of the receptor ‘X’. Since this peptoid is highly specific and only binds to receptor ‘X’, it does not recognize any cell surface molecule on the parent cells (green stained) and hence no green cells attached to this bead. Schematic representation of the bottom panel depicts the actual equilibration of two stained cell types (red and green in 1:1 mixture) with OBOC library beads. A potential ‘hit’ (with only red cells) and a non specific peptoid carrying bead (with both cell types) is schematically shown in the bottom right.
In the example shown in Fig. 2 and 3, vascular endothelial growth factor receptor-2 (VEGFR2) overexpressing porcine aortic endothelial (PAE/KDR) and VEGFR2 non-expressing PAE cells, stained with red and green quantum dots (cell cytosolic stain), respectively, were mixed with peptoid-displaying library beads (Udugamasooriya et al., 2008a). After washing away unbound cells, the beads were examined under a fluorescence microscope through a DAPI filter equipped with UV excitation and long pass emission. Under these conditions, the polystyrene-based beads fluoresce blue whereas the quantum dots emit red or green light, allowing simultaneous visualization of the beads and both cells lacking and containing VEGFR2. Since quantum dots have a large Stokes shift, when they are irradiated with UV light they emit far away from blue color and appear at the red and green region depending on the product selected. This allows the system to register all these colors (blue from beads, red and green from cells) through a single filter set (DAPI) in the fluorescence microscope (Fig. 1 and 2). Only five of the approximately 300,000 beads screened (approximately 0.0017% of the population) were observed to bind PAE/KDR (red-stained) cells alone (Fig. 2) indicating those bound through VEGFR2. In subsequent validation experiments, we found that all five of these peptoids bind to a single ‘hot spot’ on the VEGFR2 surface (Udugamasooriya et al., 2008c). We have dimerized one of those compounds to display strong anti-angiogeic properties both in vitro and in vivo mice models (Astle, 2008; Lynn et al., 2010; Roland et al., 2009; Udugamasooriya et al., 2008a; Udugamasooriya et al., 2008b) as well as attached Gd(III) based contrast agents to image VEGF receptors at the cellular level and in vivo mice models using MRI (De Leon-Rodriguez et al., 2010). As mentioned above, beads that bound both red and green cells were discarded (Fig. 3). We have further validated this approach in separate cell based systems to isolate highly specific ligands for T-cell receptors(Gocke et al., 2009), lung cancer cells and cancer stem cells (unpublished data).
Figure 2.
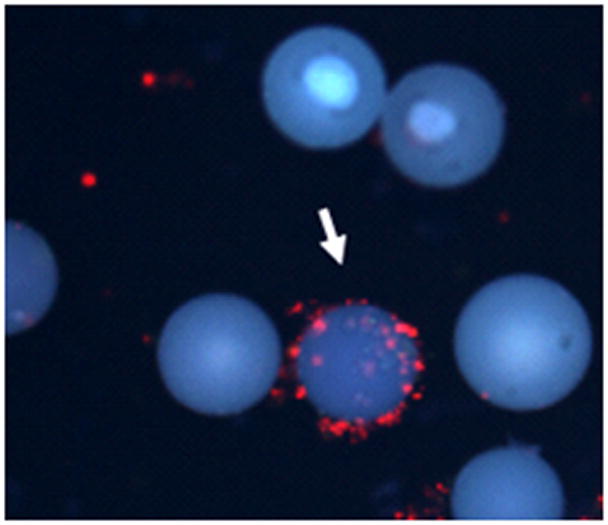
The fluorescence microscopic image of one of the selected ‘hit’ beads that binds only to PAE/KDR cells (10 × magnification; DAPI filter). The arrow indicates the red cells bound bead, while the majority of the surrounding beads do not have any cells bound, indicating the cleaner identification of the ‘hit’ bead (Reprinted with permission from UDUGAMASOORIYA, D.G., DINEEN, S.P., BREKKEN, R.A., KODADEK, T; A PEPTOID “ANTIBODY SURROGATE THAT ANTAGONIZES VEGF-VEGF RECEPTOR 2 INTERACTION, J. AMER. CHEM. SOC., 2008, 130, 5744–5752. Copyright 2008 American Chemical Society).
Figure 3.
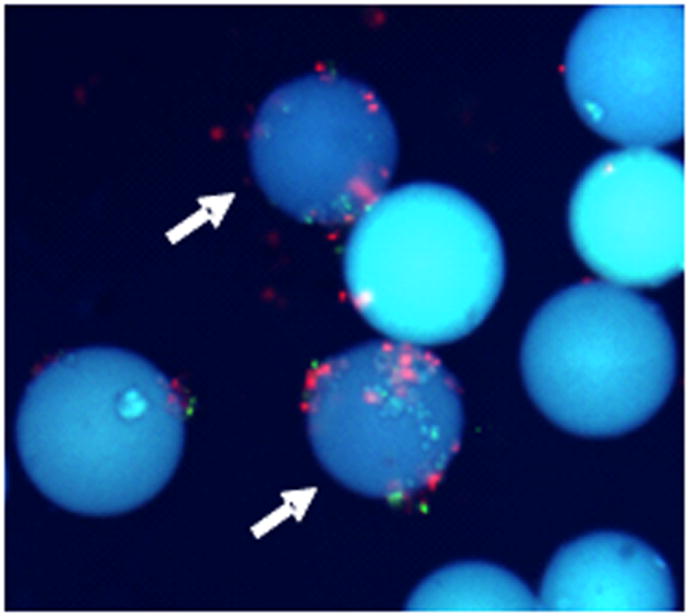
The fluorescence microscopic image of two non specific peptoid carrying beads (10 × magnification; DAPI filter). The arrows indicate both red and green cells bound beads and are discarded during the screen (Reprinted with permission from UDUGAMASOORIYA, D.G., DINEEN, S.P., BREKKEN, R.A., KODADEK, T; A PEPTOID “ANTIBODY SURROGATE THAT ANTAGONIZES VEGF-VEGF RECEPTOR 2 INTERACTION, J. AMER. CHEM. SOC., 2008, 130, 5744–5752. Copyright 2008 American Chemical Society).
As compared to many other combinatorial assay systems, this technology holds several key advantages. The use of a control cell line allows the elimination of non-selective binders at the very first step, leaving us to select only a handful of the most potent and promising candidates. This saves significant time and resources that might have been wasted on subsequent validation of non-selective ‘hits’ and false positives. As this assay does not require sophisticated instruments, other than a fluorescence microscope, it can easily be incorporated into any laboratory setting. In addition, since the receptors are exposed to the library compounds while they are expressed on live cells, these proteins are targeted in their natural environment. Since pre-requisites for this assay, such as the one bead one compound library development and exogenous receptor expression, are standard procedures in today’s research environment, we anticipate that our OBTC assay will be helpful for the rapid and cost effective discovery of novel ligands that can be developed into useful tool compounds and drug leads.
STRATEGIC PLANNING
There are two major pre-requisites that need to be fulfilled in the preparation phase of the assay. First is the development or use of an existing one bead one compound library. Any compound class, such as peptides, peptoids, nucleotides or even small organic molecules that can be synthesized in the one bead one compound format can be used here. The second important factor is the design of the cell pair. Since cells that are identical but differ only for the presence or absence of the target receptor are needed, this can simply be fulfilled by overexpressing the receptor of interest in cells that normally lack this molecule. The choice of the cell line depends on factors such as expression efficacy of the receptor of interest and their behavior, particularly regarding their “stickiness” (ability to form cell clumps) under the assay conditions. Common cell types such as HEK-293, CHO, Hela or other immortalized cancer cell types may be suitable candidates for the selection. If the interested receptor overexpressing cells are available (commercially or through collaborations), it is much easier to begin with those resources. If not, the receptor has to be overexpressed using transient or stable transfection methods. Here, the stably transfection method is recommended as it provides a steady amount of receptors on the cell surface. Nonetheless, it is important to confirm the receptor expression using standard methods, such as immunofluorescence, before the receptor-expressing cells are used in the assay.
Once the compound library and the cell pair are finalized, other factors need to be optimized to achieve the best results from the assay. The most important of these are cell and bead density, level of blocking for beads and cells, and equilibration time. These optimizations can be done by conducting small-scale studies in which each factor is individually varied. Typically, all these factors depend on the binding ability of the cells to compounds and beads. If cells are sticky and attach easily to beads, the cell density and incubation time should be reduced and the level of blocking should be increased. For example, adherent cell types such as endothelial cells need about 0.5 – 1.5 hours to complete the equilibrium with beads, while non-adherent cells such as T-cells need over night incubation at 37 °C with 5% CO2. Furthermore, when cancer cells are used, this incubation can even be reduced to 0.5 hours at room temperature. It is very important to avoid overly long incubations, which may result in the non-selective binding of control cell type (e.g. green stained cells) even to beads that display selective ligands for the target receptor.
For adherent cell types, 1X106 cells per 1 mL from each cell type is a good starting point for density of cells used in the incubation, but for non-adherent cells one may easily go up to ten times this amount (10X106 cells per 1 mL). Cells and beads are typically blocked using common blocking agents such as bovine serum albumin (BSA) or fetal bovine serum (FBS). The choice of blocking agent and concentration should be optimized for each system. BSA concentrations used for blocking usually range from 0.5% to 3%. If all these parameters are optimized, only about 10% of beads should show binding to cells (both red and green) at the end of the equilibration, ideally yielding only a handful of ‘hits’ that have only red cells bound.
Basic Protocol: Direct identification of highly selective peptoids for receptors of interest (e.g. VEGFR2) using the on-bead two-color (OBTC) assay
As previously mentioned, a suitable peptoid library and cell pair needs to be prepared prior to setting up the assay. For this purpose, an existing OBOC library or a newly customized library can be developed. For example, in the VEGFR2 targeting study, we developed a special library which has three lysine-like residues fixed at the C-terminus in every compound, as Lys is known to be important for natural ligand (VEGF) binding to VEGFR2. For this study, the cell pair of VEGFR2-overexpressing PAE/KDR cells and the parental PAE cells were commercially available.
For a single assay, a maximum amount of about 100,000 library beads (TentaGel beads: 140–170 μm diameter) can be used. The beads should be equilibrated in the final equilibration buffer at least 1 hour before introduction of the cells. In the meantime, the two cell groups are dissociated, counted and stained with red (Qtraker 655) and green (Qtraker 565) colored quantum dots. Confirmation of cell staining must be performed by observation under the fluorescence microscope before use of the cells in the assay. The two cell groups are then mixed at a 1:1 ratio, taking extra precaution to make sure there are no cell clumps formed. The 100,000 beads can be equally divided into 3 polypropylene tubes and a total of 2 mL cell mixture (red and green) containing media will be added to each tube. The optimized cell densities found in the initial small scale test studies should be applied here for each system. The beads and cell-containing tubes can be placed on a double deck oscillating shaker and equilibrated with gentle shaking for the desired/optimized time period that varies depending on the system. At the end of the equilibration, the beads will be washed and transferred to three 35 X 10mm tissue culture dishes for visualization under a fluorescent microscope. Only the individual beads containing fluorescently tagged red cells will be collected manually with a 20 μL pipet using medium size pipet tips as described below (steps 13 and 14). Selected beads are then washed and boiled in a 1% SDS solution to strip off cells and other debris to prepare them for automated Edman sequencing (Alluri et al., 2003; Lim et al., 2007; Udugamasooriya et al., 2008a), or mass spectroscopic sequencing (Paulick et al., 2006; Simpson et al., 2009).
Materials
100,000 (about 200mg) TentaGel (Rapp Polymere: TentaGel MB NH2 – cat # MB 160 002 – 140–170 μm diameter) one bead one compound library beads.
Two cell groups: e.g., PAE/KDR (VEGFR2 overexpressing) and parent (PAE) cell group (Sib Tech. Inc.)
GIBCO cell dissociation buffer when adherent cells are used. (Invitrogen cat. no. 13151-014)
Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen cat.no. 11965)
Dimethyl Formamide (DMF) (Sigma cat. no. 227056)
Bovine Serum Albumin - BSA (Sigma cat. no. A3803)
Fetal Bovine Serum - FBS (Invitrogen cat.no. 16000-044)
Qtraker 655 cell labeling Kit (Invitrogen, cat. no.Q25021MP)
Qtraker 565 cell labeling Kit (Invitrogen, cat. no.Q25031MP)
5ml polypropylene tubes (Falcon, cat. no. 35 2063)
70 μm cell strainer (BD falcon, cat. No. 352350).
35 X 10mm tissue culture dishes (Falcon, cat. no. 35 3001)
1% SDS solution
Double deck oscillating shaker
Light microscope
Fluorescence microscope equipped with a DAPI filter that has UV excitation and long pass emission (e.g., 11000V3 filter cube, Chroma Technology Corp.)
Prepare library beads
-
1
Take out 100, 000 library beads from the stock and wash twice with DMEM media. If dry beads are used, swell the beads overnight in DMF before washing them with media.
TentaGel MB NH2 beads contain 500, 000 beads per gram and therefore a 200 mg portion should contain about 100, 000 beads. Dry beads can be measured with more accuracy and a spatula can be used for transfer from the original stock library. If the library beads are stored in a solvent such as DMSO, it is important to remove the solvent as completely as possible before weighing. This can be done after pipetting out beads with the solvent into a 1.5-ml eppendorf tube (use a large pipette tip with 1 mL pipette, so that beads have fewer tendencies to stick to the pipette tip). Then use a medium pipette tip with 100 μL pipette to remove the solvent above the settled beads. Finally, push the pipette tip to the bottom of the tube (in between settled beads), to carefully remove the remaining media without the loss of beads. -
2
Distribute the beads equally into three, 5 mL polypropylene tubes and equilibrate in 2 mL of 3% BSA containing DMEM media with 10% FBS for at least 1 hour.
It is easier to perform equal distribution of weighed beads using a beads suspension. For example, if a total of 3 mL media is added, pipette out 1 mL of the bead suspension to each tube using a large pipette tip (1 mL pipette).The amount of BSA and FBS used is highly variable. Ideally this would be the same cocktail that should be used in the final cell-bead equilibrium, but this is not always the case. If the library contains high amounts of positively charged residues such as lysine-like residues, it is advisable to use higher % of BSA (3% or more) to avoid a high % of non-specific cell attraction towards beads.
Dissociate cells from tissue culture plates (for adherent cells only)
-
3
Rinse the cells with PBS and add 2ml of GIBCO enzyme free cell dissociation buffer to each 35 X 10mm tissue culture plate containing cells and incubate at 37C° with 5% CO2.
It is extremely important to use only the enzyme free dissociation buffer instead of standard dissociation solutions like trypsin. This is mainly to avoid the cleavage of cell surface protein molecules by those enzymes, as we use these cells immediately for the assay which essentially needs an unmodified cell surface.Incubation time varies depending on cell type. For cell types such as PAE-KDR, PAE, HEK-293, BHK cells, 5 –10 min incubation is sufficient. But for highly adherent cancer cell types or normal cell types such as HBEC cells, this may be increased to even 15–20 min. -
4
Gently pipette and take the cells off from the plates and collect into 15 ml falcon tubes.
In order to collect almost all of the cells, wash the plates twice with respective medium and collect all media into the falcon tubes. -
5
Centrifuge the tubes at 480×g for 5 minutes. Aspirate the supernatant and resuspend pellet in 1 mL DMEM media.
-
6
Count the cells (Using a hemacytometer) and resuspend each cell line in DMEM medium with 10% FBS to a final density of 1X106 cells per mL. The subsequent cell staining procedure requires 1X106 cells in 1 mL portions and therefore the cell suspension can be divided into required amount of 1 mL portions.
At this point it is important to have a proper calculation of how many cells are needed for the final equilibrium as that determines the number of 1 mL portions needed for the cell staining. For example, for PAE/KDR and PAE cells the optimized cell density is 1X106 cells per 1 mL from each cell type (that is 2X106 cells per 1 mL since both cell types are in the final cocktail). A total of 2 mL media is needed for each tube meaning 6 mL is needed for all 3 tubes. Therefore, a total of 6 million cells are needed of each cell type. In all, a total of twelve 1 mL cell suspension portions (6 from each) needs to be prepared for the cell staining. -
7
Stain the two cell groups with red and green fluorescence quantum dots using the standard protocol provided by Invitrogen (see Support Protocol 1).
Equilibrate cells with beads
-
8
After the final washing step of the cell staining process, resuspend each of the PAE/KDR and PAE cell line in 3 mL of 1% BSA containing DMEM media with 5% FBS for a final density of 2× 106 cellcells/mL.
We have recently noticed that higher % of FBS and BSA can cause cell clumping, which is unsuitable for the assay. Therefore in such a situation, it would be advisable to use a lesser % of FBS and BSA. This can be optimized by starting with a no BSA and FBS condition and then increasing both of them in different combinations on a small scale. Our recent experience suggests that 0.5 – 1 % BSA could be more suitable at this stage while reducing FBS to 5%, for many of the common cell types such as HEK-293, CHO, Hela. -
9
Mix the labeled cells together in a 1:1 ratio to prepare a total of 6 mL of cell suspension with a final density of each cell line at 1X106 cells/ mL. The overall cell density (i.e. of both cell lines together) will be 2X106 cells / mL. Gently pipette the cell suspension to dissociate any clumps.
Dissociation of cell clumps is a very important step. This is because during the assay the most desirable situation is to have individual cells floating around library beads and if the receptor recognizes a peptoid on a single bead, it should be able to bind independently and not as a cell clump. If a cell clump is formed with a red and green cell mixture, this complicates the visualization step by identifying that bead as a non-specific bead with both cells bound. Therefore after trying to break cell clumps by pipetting the cell suspension, it is important to filter the cells through a 40 – 70 μm cell strainer to make sure no clumps are present. The cell strainer can be placed on the top of a 50 mL conical vial and the cell mixture can be pipetted out to a larger pipette tip using 1 mL pipette. The cell containing pipette tip should be pressed onto the cell strainer and the cells should be released through the strainer collecting into the conical vial. -
10
Remove the equilibration media from each tube containing ~ 33,000 library beads. Add 2 mL portions of the cell suspension mixture to each polypropylene tube.
Here also the pipette tip can be pushed to the bottom of the tube (in between settled beads), to completely remove the media without losing beads. Then the cell mixture containing media can be gently added to these bead-containing tubes. -
11
Close the caps of the polypropylene tubes and equilibrate tubes at 37 °C with gentle shaking (40–50 reversals per min.) on a double deck oscillating shaker kept in an incubator for about 1 hour.
During the incubation period, check for cells binding to the beads starting from around 30 min (continue to do so in 10–15 min intervals after that) to make sure not to over incubate, which could increase non-specific binding of cells to beads (this is a simple quick look at the equilibrating beads, still in the tubes). This can be done even using a light microscope and when about 10% of beads have cells bound, the incubation should be stopped.When highly adherent cancer cells such as lung cancer cells are used, it is important to check the beads as early as 15 min and continue to do so in 10–15 min intervals.The actual conditions for this incubation depends on the cell system applied. For general adherent cell types such as PAE-KDR, PAE, HEK-293, BHK can be equilibrated at 37 °C or at room temperature with gentle shaking. For non-adherent cell types such as T-cells this should be elongated to overnight at 37 °C with 5% CO2 and gentle shaking. -
12
After the incubation, wash the beads gently two times with DMEM medium (with 5–10% FBS -to reduce surface tension). Gently pipette the beads out from the polypropylene tubes and place them in 35X10mm tissue culture dishes for evaluation by fluorescence microscopy.
Use a 1 mL pipette with a larger pipette tip to avoid physical damage to the cells bound on beads when transferring these beads. The contents of one tube can be placed in one dish. The amount of media should be optimized such that beads can form only a monolayer on the plate and at the same time they are fully suspended. During this transfer process, extra care should be taken to avoid applying strong physical forces that could cause detachment of bound cells from beads.Also, it is important to make sure not to lose beads. Be sure to wash each of the bead caps, as well as collect every single bead adhering to the tube surface.
Picking up the ‘hits’
-
13
Place each tissue culture dish (one at a time) with washed beads in fresh media on a secondary container (preferably on the cover of a large cell culture plate) and screen under a fluorescence microscope equipped with DAPI filter (10 X magnification). Then, start screening from one corner of the dish, continuing up and down through imaginary parallel columns until the whole surface area has been observed.
This should be continued without missing any of the fields on the plate. In order to complete this whole process successfully, it is recommended to put a mark on the top side of the plate. This is important if the plate has to be removed from the secondary container during the ‘hit’ picking up process (described below), and when placing it back at the original position to continue the screen. -
14
When a ‘hit’ (only red cells bound to a bead) is found, this needs to be picked up manually. Before starting this process, it is important to take a proper picture of the ‘hit’ bead and surrounding beads. The ‘hit’ bead can be extracted in 2 ways:
If the bead is in an isolated area (no other beads found very close by), use a 20 μL pipette with a medium size pipette tip to collect it. Adjust the volume of the pipette to about 15–20 μL and bend the pipette tip about 1cm from its end. This allows placing of the pipette tip directly above the ‘hit’ bead while holding the pipette perpendicular to the bent portion (it is better to handle the pipette with both hands to increase the precision). Then pipette out the ‘hit’ bead in to the bent portion of the tip. Collect the bead into a 1.5 mL microcentrifuge tube containing about 30 μL of media.
If the ‘hit’ bead is surrounded by other beads, first try to push the other beads away from the ‘hit’ bead with the help of the pipette tip or a small needle (while looking under the microscope). If this is successful, use the method described in step 14.a to pick the ‘hit’ bead. If this is not successful (a few beads still around the ‘hit’ bead), collect all of those beads on that particular field where the ‘hit’ bead resides using a 100 uL pipette with a medium size pipette tip and place them on a glass slide. Now place this glass slide under the microscope and separate out the unwanted beads freely to isolate the ‘hit’ bead. Picking up only the ‘hit’ bead is one of the most challenging and important steps in this process (this is common for all on-bead combinatorial assay types, whether it is a cell or direct protein screen). Since these beads are very small, it is possible to pick up two or more beads at the same time. Therefore, to make sure whether only the ‘hit’ bead is collected and no other bead is present in the microcentrifuge tube, place the bottom of the tube under the microscope (bead will clearly be illuminated in blue color, with red colored cells attached). If there is two or more beads present, transfer the bead mixture to a glass slide as explained above and isolate only the ‘hit’ bead.
Preparing beads for sequencing
-
15
Once the ‘hit’ beads are collected, remove the attached cells and debris fully from the beads before subjecting it for sequencing. There are two methods for sequencing (a) Edman sequencing and (b) mass spectroscopic sequencing. There are differences in preparing the beads for each of these methods and are explained in support protocols 2 and 3 below.
Support protocol 1 Cell labeling procedure (from Invitrogen)
The specific procedure provided by the Invitrogen is used for quantum dot labeling of cells. At the end of the procedure cell permeable quantum dot products are end up inside cells simply providing the color.
Materials
Two cell groups: e.g., PAE/KDR (VEGFR2 overexpressing) and parent (PAE) cell group (Sib Tech. Inc.)
Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen cat.no. 11965)
Fetal Bovine Serum - FBS (Invitrogen cat.no. 16000-044)
Qtraker 655 cell labeling Kit (Invitrogen, cat. no.Q25021MP)
Qtraker 565 cell labeling Kit (Invitrogen, cat. no.Q25031MP)
Vortex
Cell incubator
-
Fluorescence microscope equipped with a DAPI filter that has UV excitation and long pass emission (e.g., 11000V3 filter cube, Chroma Technology Corp.)
-
For each color (red and green), premix 1 μL each of Qtracker reagent A and B (provided by Invitrogen) in a 1.5 mL microcentrifuge tube and incubate for 5 minutes at room temperature.
This is sufficient to prepare 10 nM labeling solution for 1X106 cells. The number of tubes to be prepared depends on the amount of cells used for the experiment and is explained in step 6 of the Basic Protocol. Add 0.2 mL of DMEM medium with 10% FBS to each tube and vortex for 30 seconds.
Add 1X106 PAE-KDR cells to each tube containing Qtracker 655 (red) and 1X106 PAE cells to Qtracker 565 (green) containing tubes and incubate at 37 °C with 5% CO2 for 45–60 minutes.
Wash the cells twice with DMEM medium, by centrifugation at 1200 rpm for 5 min each time.
PAE-KDR cells labeled with Qtracker 655 should be visualized in red color and PAE cells labeled with Qtracker 565 should be visualized in green color via DAPI filter that has UV excitation and long pass emission in the fluorescence microscope.
-
Support Protocol 2. Preparation of ‘hit’ beads for Edman Sequencing
Isolated ‘hit’ beads should be free from all proteins and cell debris for the sequencing and therefore needs to be cleaned thoroughly. The Edman sequencing can be processed using the single ‘hit’ bead without cleaving off the compound.
Materials
‘Hit’ beads with bound red cells (output from Basic Protocol 1)
1% SDS solution
Milli-Q purified water
Dissecting microscope
-
Edman sequencing cartridge
Wash the individual beads on each of the microcentrifuge tube three times with aqueous 1% SDS solution (100 μL X 3 times).
Boil the beads with about 200 μL of 1% SDS solution for 45 minutes.
Wash the beads with Milli-Q purified water 100 μL X 3 times. Use a dissecting microscope to confirm that the beads are not lost during the washing.
Place each bead in an Edman sequencing cartridge using a 20 μL pipette and medium size tip and submit for sequencing.
Support Protocol 3. Preparation of ‘hit’ beads for mass spectroscopic sequencing
Isolated ‘hit’ beads should be free from all proteins and cell debris for the sequencing and therefore needs to be cleaned thoroughly. The compound needs to be cleaved off the bead for mass spectroscopic sequencing.
Materials
‘Hit’ beads with bound red cells (output from Basic Protocol 1)
GIBCO cell dissociation buffer
1% SDS solution
Acetonitrile: Water (1:1) V/V mixture
Milli-Q purified water
Cyanogen bromide (CNBr) 30mg/mL in 5:4:1 Acetonitrile : Acetic acid : Water mixture
-
Dissecting microscope
Wash individual beads on each of the microcentrifuge tubes three times with 1% SDS solution (100 μL X 3 times).
Boil the beads with about 200 μL of 1% SDS solution for 2 hours.
Wash the beads with Milli-Q purified water 100 μL X 3 times.
Add 200 μL of cell dissociation buffer and leave at RT for 30 minutes.
Wash the beads with Acetonitrile: Water (1:1) mixture 100 μL X 3 times. Use a dissection microscope to confirm that the beads are not lost during each of washing steps.
Cleave off the peptoid by treating with 20 μL of CNBr cleavage cocktail (described in the materials above) overnight.
Evaporate the solvent by SpeedVac system or blowing air over it and redissolve the peptoid in 10 μL of Acetonitrile: Water (1:1) mixture. Use this mixture for mass spectroscopy sequencing.
COMMENTARY
Background Information
This protocol avoids a common problem in targeting integral membrane receptors, which is the poor solubility and biochemical properties of many such proteins, often necessitating the use of detergents, micelles or other problematic reagents in screening experiments using recombinant receptors. In this case, the receptor is displayed in a relatively natural environment on the surface of the cell. Thus, this protocol is less likely to yield hits that do not bind the receptor under native conditions. Previously, Lam and co-workers (Peng et al., 2006) reported a screen of an encoded-bead-based (on-bead-cell growth) screening of peptidomimetic compounds against α4β1 integrin that employed integrin-expressing Jurkat cells and there are a few more examples of this kind (Aina et al., 2002). In these cases however, the isolation of a specific receptor ligand was dependent on the subsequent screen of the receptor-lacking cells using the ‘hits’ found from first screen, making the overall process longer and more time consuming. The use of different colored cells that do and do not express the target receptor removes this limitation and allows this approach to be applied to almost any cell surface target. More importantly, use of quantum dots for staining (Olivos et al., 2003)_ENREF_7_ENREF_7 solved one of the main bottlenecks on this type of a multicolor approach used on TentaGel beads which arises due to the inherent broad emission spectra of these beads. This is explained in detail under the critical parameters section below.
There are other different types of cell-based assays reported in the literature (Black et al., 2011; Cooper, 2004; Maynard et al., 2009; Minor, 2005, 2008; Osmond et al., 2010). Most of these high throughput screens monitor cellular events dependent on receptor function, such as activation of a downstream reporter gene that is triggered by a small molecule. The biggest drawback of these assays is that it requires spatial separation of the cells and molecules into different wells of a microtiter plate and a significant robotics infrastructure is needed to carry out screens on a large numbers of compounds, making it an immensely expensive process. Furthermore, there is always a possibility to isolate molecules that modulate the reporter event in some way other than by binding to the receptor through these screens. In contrast, our screen registers only selective ligands binding to the target receptor, requires no specialized equipment other than a fluorescence microscope, and can easily accommodate libraries containing hundreds of thousands of molecules.
When compared to the other reported combinatorial screens, the most important features unique to this assay are the ‘direct’ identification of a ‘fewer number’ of the ‘most selective’ ligands for the receptor of interest. This is accomplished by the presence of the parental cells that display all other common bio-molecules on the cell surface, and filtering out any ligands binding to these targets. This is an extremely important feature to avoid the identification of false-positives and molecules that bind non-selectively, which is the most common and difficult problem to overcome in high throughput screens.
In addition to the applications of targeting a known receptor, this assay can be applied to identify ligands in an ‘unbiased’ approach. This essentially bypasses the conventional drug development approach where knowledge of the target is a prerequisite. This has enormous potential to identify ligands for a particular cell surface without even knowing the targeted biomarker over other cells and we have successfully validated the OBTC assay in several of these applications recently (unpublished data). Also, the use of a ‘multi-color’ approach is feasible instead of two-color approach, highlighting many different applications of this assay in multi-cell systems in the future.
Critical parameters
Choice of the quantum dots as fluorescence probes
The TentaGel resin beads employed here have a very high level of intrinsic fluorescence, which is depicted by the characteristic broad emission spectra covering from ~ 500 nm to 700 nm (Alluri et al., 2003). This “background fluorescence” prohibits the use of any organic dyes, such as fluorescein, tetramethylrhodamine and Texas Red, making it impractical for screening experiments. In other words, if any of these organic dyes are used, the beads will also appear in the same color when it is screened on a particular filter set, affecting the proper identification of ‘hits’. The use of quantum dots provided a unique answer for this problem (Lim et al., 2007; Olivos et al., 2003; Udugamasooriya et al., 2008a). Since quantum dots have a large Stokes shift, when they are irradiated with UV light they emit far away from blue color and appear at the red-green region depending on the product selected. This allows the system to register all these colors (blue from beads, red and green from cells) through a single filter set in the fluorescence microscope (Fig. 1 and 2).
The other main advantage of the specific quantum dot products used in this OBTC assay is that they penetrate into the cells to provide the stain, thus avoiding any modification on the cell surface. This allows the exposure of the receptor of interest without any modifications while it is expressed in the natural environment, to the bead library.
Use of the special DAPI filter
Since we want to register all 3 of these colors through a single filter, we avoided using conventional filter sets made for standard organic dyes, which mostly have narrow excitation and emission wavelengths specific for a particular area (color). Here we use a simple and inexpensive DAPI filter (11000V3 filter cube – Chroma Technology Corp.) that has UV excitation and long pass emission. This allows the simultaneous visualization of all 3 colors.
Use of the dissociation buffer
Since the assay tries to register ligands binding to a cell surface receptor, it is very important to expose those cell surface protein molecules without any modifications to the library compounds. The standard cell dissociation methods apply trypsin, which can cleave these receptors from the cell surface. Therefore it is important to use an enzyme-free dissociation buffer that can dissociate cells without significant damage to the receptors on the cell surface.
Optimization of cell and bead density
The proper amount of beads and cell density is very important to obtain optimum results. If both cell density and bead density are too high, this might lead to an increase in non-specific binding. If both of these parameters are too low, the assay may not register any cell binding to beads. In these circumstances, the equilibration time might be extended, but most of the time this also can lead to cell deterioration and clumping, both of which will have adverse effects on the assay. In general terms, a density of 1 × 106 cells per 1 mL will be a good starting point for adherent cells; for non-adherent cells this could be about 10 × 106 cells per 1 mL. When a new system is used, it is best to start at this density and optimize accordingly. Having 33,000 beads in each vial is an optimized level for 140–170 μm diameter beads, for which indicates about 16,500 beads per 1 mL.
Troubleshooting
Cell preparation and avoid cell clumping
One of the main concerns associated with the assay is the possibility of cell clump formation. However, if the right conditions are used, this can be eliminated. First of all, the best media conditions (FBS and other supplements) that can keep the cells growing at appropriate rates should be used. If cell clumps appear when the two cell types are mixed, this might be removed by pipetting the cell mixture in-and-out. Once the clumps are reduced, a final filtration could remove the remaining clumps. But a difficult situation could arise if cell clumps start appearing during the bead equilibration. Most of the time, this arises when BSA is added as a blocking agent. Therefore, reducing BSA % may help to remove clumps in many cell types. If a persistent cell clumping occurs during the screen, this problem might need to be handled case-by-case. Reducing cell density, bead density and shaking speeds may help reducing clump formation. Since some cell types always tend to form clumps, and it is advisable to avoid these cell types for OBTC assay from the start.
Combinatorial library and sequencing related issues
Discussing a complete troubleshooting on one bead one compound combinatorial library synthesis is not appropriate herein as the focus is on the OBTC cell assay. But one of the initial critical steps is to have a properly validated one bead one compound library (this is done by either Edman or mass spectroscopic sequencing of random samples of the library beads). If the final sequencing of the ‘hits’ are not clean or not even successful, this can be due to the poor quality of the library. Therefore, validation of the library prior to use is a very important step. One other reason for poor sequencing results (in particular with mass spectroscopy analysis) may be the improper cleaning process of the ‘hit’. Following the exact cleaning process or even increasing the boiling time and cell dissociation buffer treatment steps may help to eliminate this problem. While peptoids are stable in these harsh conditions, it may be advisable to pay careful attention to peptides and other sensitive libraries when following or modifying these cleaning procedures.
This assay gives optimum results when the library beads are 140–170 μM in diameter. When smaller beads are used, this may not support proper displaying of ‘hits’ with fully bound red cells as shown in Fig. 2. Here, the reduced surface area may allow binding of a smaller number of cells to the bead. Along the same line, when the bead size gets smaller and smaller, obtaining reliable sequencing information from a single bead also becomes more problematic. Therefore, it is advisable to use 140–170 μM diameter macrobeads as the most suitable one bead one compound library format for this OBTC assay.
Anticipated results
A typical OBTC cell screen with 100,000 beads would ideally produce only about 1–5 ‘hits. Sometimes this 100,000 bead screen may not even produce any reliable ‘hits,’ indicating another round of 100,000 bead screen should be continued using a different batch from the library (provided the theoretical diversity of the library is greater than 200,000) or another new library. Usually about 10% of the beads will show up with both cell types bound as non specifics, while the majority (~90%) of the beads should not have any cells bound. If the library is of good quality and developed on 140–170 μm diameter beads, almost all of the ‘hits’ would be able to sequenced and have their structures identified.
Time considerations
Once all necessary supplies are ready, this assay can be completed in a single day up to the level of the ‘hit’ identification and cleaning of those beads (excluding the sequencing) if adherent cells are used (for non-adherent cells, this will be 2 days in total). Apart from typical time frames needed for cell dissociation, counting, washing, staining and equilibration, the most time consuming portions are the microscopic screening and ‘hit’ picking up processes. For a typical 100,000 bead screen, this may vary from 2–4 hours. It needs to be emphasized that searching for a ‘hit’ is not difficult, since it is a matter of simply moving the plate up and down under the microscope. Since the majority of beads are without cells, when a cell-bound bead appears in a particular field, it can readily be identified (please refer to Fig. 2 and 3). But once a proper ‘hit’ with only red cells bound bead is found, it may takes 5 – 20 minutes to carefully pick it up. Once the ‘hits’ are found and cleaned on the day that the assay has been conducted, the sequencing can be completed within the next day or two, depending on the availability and the routine practices of Edman sequencing and mass spectroscopic facilities.
When the preparations for the assay are considered, the library development and receptor expressions could take weeks to months depending on the system. Also for a new system, it may take from few days to a few weeks to determine optimized conditions before proceeding to the 100,000 bead screen.
Acknowledgments
Financial support from the UT-Southwestern Medical Center and National Institutes of Health (RR02584, CA115531, and CA126608) is gratefully acknowledged.
Literature Cited
- Aina OH, Sroka TC, Chen ML, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66:184–199. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. Isolation of protein ligands from large peptoid libraries. J Am Chem Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- Astle JM, Udugamasooriya DG, Smallshaw JE, Kodadek T. A VEGFR2 antagonist and other peptoids evade immune recognition. Int J Pept Res Ther. 2008;14:223–227. [Google Scholar]
- Black CB, Duensing TD, Trinkle LS, Dunlay RT. Cell-based screening using high-throughput flow cytometry. Assay Drug Dev Technol. 2011;9:13–20. doi: 10.1089/adt.2010.0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MA. Advances in membrane receptor screening and analysis. J Mol Recognit. 2004;17:286–315. doi: 10.1002/jmr.675. [DOI] [PubMed] [Google Scholar]
- De Leon-Rodriguez LM, Lubag A, Udugamasooriya DG, Proneth B, Brekken RA, Sun X, Kodadek T, Dean Sherry A. MRI detection of VEGFR2 in vivo using a low molecular weight peptoid-(Gd)8-dendron for targeting. J Am Chem Soc. 2010;132:12829–12831. doi: 10.1021/ja105563a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocke AR, Udugamasooriya DG, Archer CT, Lee J, Kodadek T. Isolation of antagonists of antigen-specific autoimmune T cell proliferation. Chem Biol. 2009;16:1133–1139. doi: 10.1016/j.chembiol.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HS, Archer CT, Kodadek T. Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J Am Chem Soc. 2007;129:7750–7751. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn KD, Udugamasooriya DG, Roland CL, Castrillon DH, Kodadek TJ, Brekken RA. GU81, a VEGFR2 antagonist peptoid, enhances the anti-tumor activity of doxorubicin in the murine MMTV-PyMT transgenic model of breast cancer. BMC Cancer. 2010;10:397. doi: 10.1186/1471-2407-10-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard JA, Lindquist NC, Sutherland JN, Lesuffleur A, Warrington AE, Rodriguez M, Oh SH. Surface plasmon resonance for high-throughput ligand screening of membrane-bound proteins. Biotechnol J. 2009;4:1542–1558. doi: 10.1002/biot.200900195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor LK. Assays for membrane tyrosine kinase receptors: methods for high-throughput screening and utility for diagnostics. Expert Rev Mol Diagn. 2005;5:561–571. doi: 10.1586/14737159.5.4.561. [DOI] [PubMed] [Google Scholar]
- Minor LK. Label-free cell-based functional assays. Comb Chem High Throughput Screen. 2008;11:573–580. doi: 10.2174/138620708785204072. [DOI] [PubMed] [Google Scholar]
- Olivos HJ, Bachhawat-Sikder K, Kodadek T. Quantum dots as a visual aid for screening bead-bound combinatorial libraries. Chembiochem. 2003;4:1242–1245. doi: 10.1002/cbic.200300712. [DOI] [PubMed] [Google Scholar]
- Osmond RI, Crouch MF, Dupriez VJ. An emerging role for kinase screening in GPCR drug discovery. Curr Opin Mol Ther. 2010;12:305–315. [PubMed] [Google Scholar]
- Paulick MG, Hart KM, Brinner KM, Tjandra M, Charych DH, Zuckermann RN. Cleavable hydrophilic linker for one-bead-one-compound sequencing of oligomer libraries by tandem mass spectrometry. J Comb Chem. 2006;8:417–426. doi: 10.1021/cc0501460. [DOI] [PubMed] [Google Scholar]
- Peng L, Liu R, Marik J, Wang X, Takada Y, Lam KS. Combinatorial chemistry identifies high-affinity peptidomimetics against alpha4beta1 integrin for in vivo tumor imaging. Nature Chem Biol. 2006;2:381–389. doi: 10.1038/nchembio798. [DOI] [PubMed] [Google Scholar]
- Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS One. 2009;4:e7669. doi: 10.1371/journal.pone.0007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson LS, Burdine L, Dutta AK, Feranchak AP, Kodadek T. Selective toxin sequestrants for the treatment of bacterial infections. J Am Chem Soc. 2009;131:5760–5762. doi: 10.1021/ja900852k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. A peptoid “antibody surrogate” that antagonizes VEGF receptor 2 activity. J Am Chem Soc. 2008a;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- Udugamasooriya DG, Dunham G, Ritchie C, Brekken RA, Kodadek T. The pharmacophore of a peptoid VEGF receptor 2 antagonist includes both side chain and main chain residues. Bioorg Med Chem Lett. 2008b;18:5892–5894. doi: 10.1016/j.bmcl.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugamasooriya DG, Ritchie C, Brekken RA, Kodadek T. A peptoid antagonist of VEGF receptor 2 recognizes a ‘hotspot’ in the extracellular domain distinct from the hormone-binding site. Bioorg Med Chem. 2008c;16:6338–6343. doi: 10.1016/j.bmc.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]