

Abstract
Most nucleosides in solution typically exist in equilibrium between two major sugar pucker forms, N-type and S-type, but bridged nucleosides can be locked into one of these conformations depending on their specific structure. While many groups have researched these bridged nucleosides for the purpose of determining their binding affinity for antisense applications, we opted to look into the potential for biological activity within these conformationally-locked structures. A small library of 2′,4′- and 3′,4′-bridged nucleoside analogues was synthesized, including a novel 3′,4′-carbocyclic bridged system. The synthesized compounds were tested for antibacterial, antitumor, and antiviral activities, leading to the identification of nucleosides possessing such biological activities. To the best of our knowledge, these biologically active compounds represent the first example of 2′,4′-bridged nucleosides to demonstrate such properties. The most potent compound, nucleoside 33, exhibited significant antiviral activity against pseudoviruses SF162 (IC50 = 7.0 μM) and HxB2 (IC50 = 2.4 μM). These findings render bridged nucleosides as credible leads for drug discovery in the anti-HIV area of research.
1. Introduction
The first antiviral antisense oligodeoxynucleotides were discovered in 1978 by Zamecnik and Stephenson.1 Since then, interest in antisense technologies has been on the rise due to these molecules having the potential to treat diseases that small molecule drugs are incapable of targeting. Perhaps the most promising candidates for antisense drug therapy have been the 2′,4′-bridged nucleic acids (BNAs), as they have recently entered human clinical trials.2 Their success is due to their more rigid structures, which translate into higher affinities for their biological targets. Most sugars and nucleosides exist in solution in a fast equilibrium between two major sugar pucker forms, North (N-type) and South (S-type),3 but bridged nucleosides are locked intoone conformation: 2′,4′-bridged systems such as 1a exist in the C3′-endo (N-type) form 1b, while 3′,4′-systems such as 2a exist in the C2′-endo (S-type) conformation 2b (Figure 1). Therefore, 2′,4′-BNAs with the N-type conformation and 3′,4′-BNAs with the S-type conformation have a high binding affinity for complementary single-stranded RNA and DNA, respectively, giving the 2′,4′-BNAs a special advantage as promising antisense drug candidates.
Figure 1.
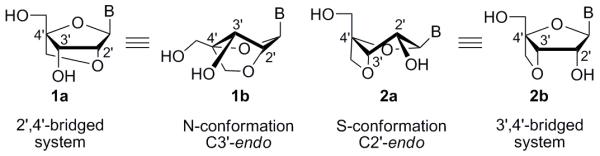
Locked conformations of 2′,4′-bridged system 1a/1b and 3′,4′-bridged system 2a/2b.
Due to this fact, several groups have directed their research efforts toward the synthesis of novel bridged nucleosides in an attempt to determine the best bridged nucleic acid for antisense drug therapy. Following the synthesis and evaluation of the 2′-O,4′-C-4 and 3′-O,4′-C-5 bridged nucleic acids, there have been reports on the synthesis of 2′-NO,4′-C systems,6 2′-N,4′-C-bridged systems,7 2′,4′-propylene-BNAs,8 carbocyclic-BNAs,9 and more.10 However, only a few groups have explored the biological activities of bridged nucleoside analogues themselves (see Figure 2 for selected examples). This appears to be a research area that warrants further attention, for what has been published does show potential for medicinal applications. Thus, and as shown in Figure 2, select epoxide- and cyclopropane-bridged systems have been shown to have significant antiviral activity (3–6),11 and act as adenosine A3 receptor inhibitors (7 and 8)12 and DNA polymerase modulating agents (9 and 10).13 Also reported were antiviral 3′,4′-oxetane bridged nucleoside analogue 11,14 cytotoxic 3′,4′-tetrahydrofuran bridged nucleoside derivative 12,15 and antiviral 2′,3′-hexahydroisobenzofuran bridged nucleosides 13.16 Additionally, a number of 2′,4′-bridged nucleoside analogues such as compounds 14 have demonstrated the ability to act as adenosine A3 receptor antagonists (see Figure 2),17 while other 2′,4′-bridged derivatives have shown no significant antiviral18 or anticancer19 activity.
Figure 2.

Previously synthesized bridged nucleoside analogues 3–14 and their biological activities. A3AR = adenosine A3 receptor. CC50 = Concentration at which compound is cytotoxic to half the amount of cells. MT-4 = metallothionein 4, a human T cell line. LA1 = lymphocytotropic strain of HIV-1.
Since there are a number of nucleoside analogue drugs currently in clinical use and in clinical trials, we felt it was important to further probe the biological activity of these bridged nucleoside systems while simultaneously developing new synthetic strategies for their continued development. Herein, we describe the synthesis of a library of 2′,4′- and 3′,4′-bridged nucleoside analogues which not only differ by the structure of the bridge, but also by the substitution on the adenine base (see compounds 15–51, Figure 3). Additionally, a new type of 3′,4′-bridged nucleoside was synthesized through a novel [2+2] cycloaddition pathway (see compounds 52 and 53, Figure 3). All compounds were tested for antibacterial, antitumor, and antiviral activities, with some compounds displaying significant biological activities.
Figure 3.

Synthesized compound library of bridged nucleoside analogues (15–53).
2. Results and discussion
2.1. Synthesis of 2′,4′-bridged nucleoside analogues
Beginning from commercially available diacetone D-glucose, oxidation, then reduction to invert the secondary alcohol, and subsequent benzylation afforded literature-known diacetonide 54 (Scheme 1).20 One-pot selective acetonide removal and oxidative cleavage with periodic acid furnished aldehyde 55 in 98% yield, from which differentially protected sugar 56 was prepared through a known sequence of steps.5c,21 Vorbrüggen coupling22 with 2,6-diaminopurine then installed the base onto diacetate 56; however, two different products resulted, depending on the specific conditions employed. The use of only two equivalents of N,O-bis(trimethylsilyl)acetamide (BSA) resulted in a quantitative yield of the thermodynamic product, N9-isomer 57. Increasing the amount of BSA to five equivalents caused the formation of compound 57 plus the kinetic product, unnatural N7-isomer 58, in a 1.6:1 ratio and 95% total yield. This appears to be due to the fact that excess BSA further silylates the 2,6-diaminopurine, resulting in a species that can attack the diacetate with either the N9 or N7 purine positions.23
Scheme 1.
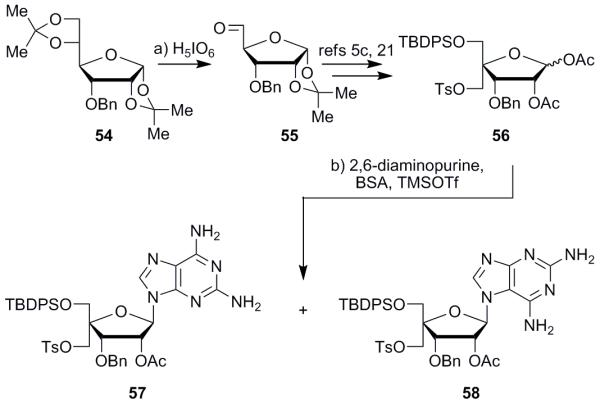
Synthesis of compounds 56 and 58. Reagents and conditions: a) H5IO6 (1.2 equiv), EtOAc; b) 2,6-diaminopurine (1.5 equiv), BSA (2.3 or 5.0 equiv), MeCN, 65 °C, 1.5 h; then TMSOTf (2.0 or 2.8 equiv), 65 °C, 3 h, 100% of 57 or 59% of 57 + 36% of 58. H5IO6 = periodic acid; EtOAc = ethyl acetate; MeCN = acetonitrile; Bn = benzyl; TBDPS = tert-butyldiphenylsilyl; Ts = para-toluenesulfonyl; Ac = acetyl; BSA = N,O-bis(trimethylsilyl)acetamide; TMSOTf = trimethylsilyl trifluoromethylsulfonate.
2′,4′-Bridged compound 59 was constructed through base-induced cyclization of N9-isomer 57 (aq. 2 M NaOH, 94% yield, Scheme 2). From this point, the synthesis diverged to produce a number of substituted amino analogues as shown in Scheme 2. Cyclization product 59 was peracylated with benzoyl chloride to afford tetrabenzoate 60 in 52% yield, along with a number of less substituted side products which were not isolated or characterized. Desilylation of compound 60 (HF·py) then led to the targeted nucleoside 15 in 50% yield. Compound 59 was also deprotected directly with HF·py to give diamino compound 16 in 85% yield. The use of boron trichloride to remove the benzyl ether from this intermediate furnished the expected β-nucleoside 17 in 85% yield, along with a 10% yield of the corresponding α-nucleoside (18), the mixture being a result of oxonium formation at the anomeric position. Additionally, the difference in the nucleophilicity of the amine groups in intermediate 59 was exploited to furnish, through reductive alkylation, mono-substituted derivatives 61–64 with ethyl, n-octyl, n-butyl, and isobutyl groups, respectively (see Scheme 2).24 The yields in this step were moderate (48, 29, 57, and 52%, respectively) due to incomplete reactions and decomposition of the alkyl aldehyde components. Desilylation (TBAF) of these products afforded compounds 19–21 and 23 in good yields. Further debenzylation of n-butylamine 21 and isobutylamine 23 with BCl3 furnished butylamine diol 22 and isobutylamine diol 24 in 64% and 71% yield, respectively. Due to the lack of biological activity of nucleosides 22 and 24, it was decided not to proceed with the deprotection of compounds 19 and 20.
Scheme 2.

Synthesis of compounds 15–24. Reagents and conditions: a) aq. 2 M NaOH, THF, 25 °C, 2 h, 94%; b) BzCl (4.0 equiv), py, 25 °C, 18 h, 52%; c) HF·py (5.0 equiv), THF, 25 °C, 12 h, 50% for 15, 85% for 16; d) BCl3 (2.0 equiv), CH2Cl2, 25 °C, 1 h, 85% of 17 + 10% of 18, 71% for 22, 64% for 24; e) alkyl aldehyde (8.0 equiv), NaBH3CN (6.0 equiv), MeOH, 25 °C, 48 h, 48% for 61, 29% for 62, 57% for 63, 52% for 64; f) TBAF (2.0 equiv), THF, 25 °C, 16 h, 79% for 19, 77% for 20, 77% for 21, 98% for 23. Bz = benzoyl; py = pyridine; THF = tetrahydrofuran; TBAF = tetra-n-butylammonium fluoride.
With N7-isomer 58 in hand, bridged nucleoside analogues with unnatural bases attached became synthetically accessible. Thus, in a similar fashion to the N9-isomer 57, N7-isomer 58 was cyclized with aqueous sodium hydroxide to produce 2′,4′-bridged tetrahydrofuran ring system 65 in 85% yield (Scheme 3). Reductive alkylation of the latter compound with isobutyraldehyde formed isobutylamine 66, which furnished targeted nucleoside 25 upon desilylation (TBAF) in 34% yield over the two steps. Alternatively, intermediate 65 could be desilylated directly with HF·py to generate analogue 26. Debenzylation (BCl3) then furnished nucleoside 27 in 46% yield over the two steps. Further explorations into these unnatural N7-isomeric analogues were not pursued due to instability problems with the nucleoside base during chemical transformations, which suggested that these compounds may also be too labile for biological applications.[Scheme 3]
Scheme 3.
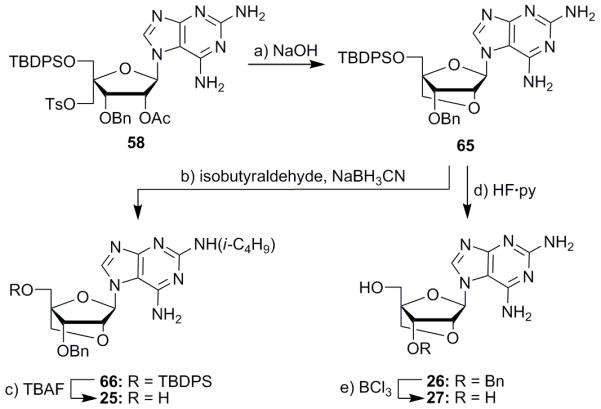
Synthesis of compounds 25–27. Reagents and conditions: a) aq. 2 M NaOH, THF, 25 °C, 2 h, 85%; b) isobutyraldehyde (8.0 equiv), NaBH3CN (6.0 equiv), MeOH, 25 °C, 48 h, 44%; c) TBAF (2.0 equiv), THF, 25 °C, 16 h, 77%; d) HF·py (5.0 equiv), THF, 25 °C, 12 h; e) BCl3 (2.0 equiv), CH2Cl2, 25 °C, 1 h, 46% over the two steps.
Bridged nucleoside analogues were also synthesized using nucleobases other than 2,6-diaminopurine (see Scheme 4). Vorbrüggen reaction of 2-chloroadenine with diacetate 56 produced nucleoside 67 as a single isomer in 85% yield. Deacetylation followed by cyclization to bridged intermediate 68 was effected with aq. NaOH; desilylation with HF·py then gave nucleoside 28 in 53% overall yield from compound 67. The use of methanesulfonic acid as a debenzylation agent was crucial to the successful generation of aminochloride diol 29 (46% yield),17 as the typical boron trichloride conditions proved to be too harsh for this analogue. Amino fluoride derivative 31 could also be prepared in a similar manner (see Scheme 4). Vorbrüggen coupling with diacetate 56 and 2-fluoroadenine led to nucleoside 69 (78% yield), which could then be deacetylated and cyclized with NaOH to produce intermediate 70 in 81% overall yield. Desilylation (HF·py), followed by debenzylation (BCl3) under the standard conditions, then furnished, sequentially, hydroxy benzyl ether 30 and diol 31 in 59% yield over the two steps. Intermediate 70 was converted into bridged analogue 32 through morpholine displacement of the fluoride moiety and desilylation (HF·py) in 76% yield over the two steps (see Scheme 4).
Scheme 4.

Synthesis of compounds 28–32. Reagents and conditions: a) 2-chloroadenine (1.5 equiv), BSA (2.3 equiv), MeCN, 65 °C, 1.5 h; then TMSOTf (2.0 equiv), 65 °C, 3 h, 85%; b) aq. 2 M NaOH, THF, 25 °C, 15 h, 87% for 68, 81% for 70; c) HF·py (5.0 equiv), THF, 25 °C, 12 h, 61% for 28, 85% for 30; d) MsOH (78 equiv), CH2Cl2, 0 °C, 1.5 h, 46%; e) 2-fluoroadenine (2.0 equiv), BSA (2.5 equiv), MeCN, 65 °C, 1.5 h; then TMSOTf (2.0 equiv), 65 °C, 3 h, 73%; f) BCl3 (2.0 equiv), CH2Cl2, 25 °C, 1 h, 69%; g) morpholine (2.0 equiv), DMSO, 95 °C, 24 h, 76% over the two steps. MsOH = methanesulfonic acid; DMSO = dimethyl sulfoxide.
2,6-Dichloropurine nucleoside 71 was synthesized in 64% yield through Vorbrüggen reaction of diacetate 56 and 2,6-dichloro-9H-purine as show in Scheme 5. This compound proved to be one of the most derivatizable intermediates in this study (see Scheme 5). The difference in the electrophilicity between the C2 and C6 carbons of nucleoside 71 was exploited to synthesize aminochloride 72, in 93% yield, via displacement of the C6 chlorine residue with benzylamine.25 Cyclization induced by NaOH then afforded, through the corresponding hydroxy tosylate, locked nucleoside 73 in 68% yield. Desilylation of the latter compound produced alcohol 33 (HF·py, 96% yield), and subsequent debenzylation of compound 33 (MsOH) furnished diol analogue 34 (61% yield). A similar sequence of steps could also be employed to synthesize nucleoside 36. Thus, compound 71 was reacted with N-methylbenzylamine to afford intermediate 74 lacking the acetoxy group in 82% yield (see Scheme 5). Cyclization of compound 74 through the action of NaOH then produced the bridged intermediate 75 (98% yield), followed by desilylation (HF·py) to form alcohol 35 (75% yield). Debenzylation (MsOH) of the latter compound then generated dihydroxy derivative 36 (75% yield). Displacement of both chlorine residues of intermediate 71 and concurrent ring closure were effected by exposure to NaOH in MeOH to afford locked nucleoside derivative 37 upon desilylation (HF·py), in 69% overall yield. Finally, debenzylation of the latter compound with BCl3 furnished dimethoxy nucleoside diol 38 (77% yield).
Scheme 5.

Synthesis of compounds 33–39. Reagents and conditions: a) 2,6-dichloro-9H-purine (2.0 equiv), BSA (2.5 equiv), MeCN, 95 °C, 1.5 h; then TMSOTf (2.0 equiv), 80 °C, 3 h, 64%; b) benzylamine (5.0 equiv) or N-methylbenzylamine (5.0 equiv), MeOH, 55 °C, 12 h, 93% for 72, 82% for 74; c) aq. 2 M NaOH, THF, 24 °C, 15 h, 68% for 73, 98% for 75; d) HF·py (5.0 equiv), THF, 25 °C, 12 h, 96% for 33, 75% for 35, 77% for 37, 31% over the three steps for 39; e) MsOH (78 equiv), CH2Cl2, 0 °C, 1.5 h, 61% for 34, 75% for 36; f) NaOH (15 equiv), MeOH:THF (1:1), 25 °C, 12 h, 90%; g) BCl3 (2.0 equiv), CH2Cl2, 25 °C, 1 h, 77%; h) tri-n-butyl(1-ethoxyvinyl)tin (2.0 equiv), Pd(PPh3)2Cl2 (0.10 equiv), DMF, 95 °C, 18 h. DMF = dimethylformamide.
Stille coupling reactions were employed to synthesize additional derivatives from dichloro compound 71 as shown in Schemes 5 and 6.26 Reaction of intermediate 71 with tri-n-butyl(1-ethoxyvinyl)tin and bis(triphenylphosphine)palladium(II) chloride followed immediately by base-induced deacetylation/cyclization and desilylation (HF·py) generated diketone nucleoside 39 in 31% yield over the three steps (Scheme 5). Intermediate 71 also underwent reaction with allyl(tri-n-butyl)tin to afford, upon subsequent cyclization with NaOH, compound 76 in 42% overall yield for the two steps (see Scheme 6). Formation of the conjugated olefin at the C6 position of the purine within compound 76 was presumed to have occurred as a result of palladium- and base-induced isomerization of the coupled allyl moiety. Desilylation of compound 76 (HF·py) furnished intermediate 40 (87% yield), which was converted to dipropyl derivative 41 (50% yield) by reduction and concomitant debenzylation with hydrogen and Pearlman’s catalyst. Stille reaction of dichloride 71 with 2-(tri-n-butylstannyl)furan, followed by ring closure (NaOH), afforded bridged product 77 (75% over the two steps) (Scheme 6). Sequential desilylation (HF·py) and debenzylation (BCl3) of compound 77 then produced, through the intermediacy of alcohol 42, difuran diol 43 in 90% yield over the two steps. Dichloride 71 was also reacted with 2-(tri-n-butylstannyl)thiophene to furnish the corresponding bis-thiophene derivative, which was converted to the bridged nucleoside 78 by reaction with NaOH in 71% overall yield (Scheme 6). Desilylation of the latter with HF·py gave intermediate 44 (99% yield), which was debenzylated (BCl3) to afford β-nucleoside 45 (40% yield), along with its α-anomer 46 (33% yield). Intermediate 44 was also converted to sulfamoyl derivative 47 in 35% yield by reaction with freshly prepared sulfamoyl chloride in the presence of pyridine.27
Scheme 6.

Synthesis of compounds 40–47. Reagents and conditions: a) allyl(tri-n-butyl)tin (3.0 equiv), Pd(PPh3)2Cl2 (0.10 equiv), DMF, 95 °C, 6 h; b) aq. 2 M NaOH, THF, 25 °C, 12 h, 42% over the two steps for 76, 76% for 77, 98% for 78; c) HF·py (5.0 equiv), THF, 25 °C, 12 h, 87% for 40, 100% for 42, 99% for 44; d) H2, Pd(OH)2 (10% w/w), EtOH, 50 °C, 12 h, 50%; e) 2-(tri-n-butylstannyl)furan (2.0 equiv), Pd(PPh3)2Cl2 (0.075 equiv), DMF, 95 °C, 7 h, 99%; f) BCl3 (2.0 equiv), CH2Cl2, 25 °C, 1 h, 90% for 43, 40% of 45 + 33% of 46; g) 2-(tri-n-butylstannyl)thiophene (2.0 equiv), Pd(PPh3)2Cl2 (0.12 equiv), DMF, 80 °C, 7 h, 72%; h) chlorosulfonyl isocyanate (4.3 equiv), formic acid (4.3 equiv), py (5.8 equiv), CH2Cl2, 25 °C, 24 h, 35%.
2.2. Synthesis of 3′,4′-bridged nucleoside analogues
The synthesis of the 3′-O,4′-C-bridged nucleoside analogues began with conversion of literature-known dimesylate 794e to dichloro nucleoside 80 through Vorbrüggen reaction with 2,6-dichloro-9H-purine (Scheme 7). The standard Vorbrüggen coupling procedure that had been employed in the synthesis of the other nucleosides mentioned above led to nucleoside 80 in low yield; however, an 84% yield of this nucleoside was achieved by the use of microwave irradiation instead of moderate heating.28 Debenzylation and concomitant deacetylation of compound 80 with boron trichloride led to a 90% yield of intermediate 81. Treatment of the latter compound with potassium carbonate in THF then caused smooth cyclization to afford compound 48 in 79% yield. At this point, all that remained to reach the targeted dichloro-locked nucleoside was manipulation of the mesyl group to form the 5′-hydroxyl functionality. However, attempts to accomplish this goal by reaction with NaOBz (followed by benzoate cleavage) resulted in additional displacement of the C6 chloride residue, producing dibenzoate 49 in 82% yield (see Scheme 7). The 5′-benzoate could not be cleaved without causing decomposition, thus an alternate route to these 3′,4′-bridged systems that did not involve chloride residues on the nucleobase was implemented as described below.
Scheme 7.

Synthesis of compounds 48–51. Reagents and conditions: a) 2,6-dichloro-9H-purine (2.0 equiv), BSA (3.5 equiv), MeCN, –40 °C, 5 min; then TMSOTf (1.5 equiv), μ-waves, 80 °C, 5 min, 84%; b) BCl3 (2.0 equiv), CH2Cl2, 25 °C, 1 h, 90% for 81, 82% for 83; c) K2CO3 (4.0 equiv), THF, 25 °C, 18 h, 79% for 48, 91% for 50; d) NaOBz (2.0 equiv), DMF, 90 °C, 4.5 h, 82%; e) 2-(tri-n-butylstannyl)furan (4.0 equiv), Pd(PPh3)2Cl2 (0.091 equiv), DMF, 95 °C, 3 h, 84%; f) aq. NaOMe, MeOH, 25 °C , 30 min, 69% for 51.
Dichloride 80 was converted to difuran compound 82 via a Stille reaction with 2-(tri-n-butylstannyl)furan (84% yield), and the latter compound was deprotected (BCl3) to give diol 83 in 82% yield (see Scheme 7). Treatment of compound 83 with potassium carbonate in THF afforded intermediate 50 in 91% yield. This compound was successfully converted into diol 51 through the intermediacy of the corresponding 5′-benzoate (NaOBz; then NaOMe, 74% yield over the two steps). Other studies directed toward a number of different bridged nucleosides were thwarted, primarily due to difficulties in bringing about the desired oxetane formation. The ability of the nucleoside to undergo ring closure to the oxetane derivative was found to be strongly dependent on the actual substituents on the nucleobase. Additionally, after cyclization of the intermediate, the oxetane ring was found to be unstable to typical reaction conditions as well as general handling, causing concern about its stability during biological evaluation.
Faced with these difficulties, we opted to synthesize analogues with an alternate 3′,4′-bridged ring system which would be more stable than those containing the 3′,4′-oxetane bridge, and could be constructed through a flexible route to afford a variety of analogues. These analogues were synthesized via a [2+2] cycloaddition reaction as shown in Scheme 8. Esterification of known compound 8429 with trimethylsilyl diazomethane formed methyl ester 85 (88% yield). Elimination of the tosyl group (DBU) to form the α,β-unsaturated ester, followed by [2+2] cycloaddition of the resulting product with 1,2-cis-dichloroethylene under photoirradiation conditions generated compound 86 in 18% yield over the two steps as the major product, in addition to two other diastereomers (ca. 4:2:1) differing in the orientation of the chlorine residues. In all three diastereomers of compound 86, the cyclobutane ring occuppied the position opposite the acetonide moiety as expected on steric grounds. The trans relationship of the chlorine atoms within isomer 86, proven by NMR spectroscopy (ROESY), confirmed the radical nature of the [2+2] photocycloaddition. The stereochemistry of the two diastereomers of compound 86 was not determined. Following chromatographic separaion of comound 86, its acetonide moiety was cleaved and the resulting compound peracetylated in a one-pot reaction (AcOH, conc. H2SO4, Ac2O) to afford the corresponding bis-acetate. Subsequent Vorbrüggen reaction of the latter product with 2,6-diaminopurine and BSA provided bridged nucleoside 52 in 36% yield over the two steps. Deacetylation with potassium carbonate in methanol then provided the desired alcohol 53 in 83% yield.
Scheme 8.

Synthesis of compound 53. Reagents and conditions: a) TMSCHN2 (1.2 equiv), Et2O:MeOH (1:1), 0 °C, 30 min, 88%; b) DBU (1.2 equiv), benzene, 25 °C, 5 h; c) 1,2-cis-dichloroethylene (15.9 equiv), MeCN, hν, 25 °C, 3.5 d, 18% over the two steps; d) conc. H2SO4, Ac2O (12 equiv), AcOH, 25 °C, 16 h; e) 2,6-diaminopurine (1.5 equiv), BSA (2.3 equiv), MeCN, 65 °C, 1.5 h; then TMSOTf (2.0 equiv), 65 °C, 3 h, 36% over the two steps for 52; f) K2CO3 (0.30 equiv), MeOH, 25 °C, 30 min, 83% over one step or 38% over the three steps from compound 86; g) n-BuOH (1.2 equiv), DCC (1.2 equiv), DMAP (0.15 equiv), CH2Cl2, 25 °C, 24 h; then DBU (1.2 equiv), 25 °C, 24 h, 67%; h) 1,2-cis-dichloroethylene (16.9 equiv), MeCN, hν, 25 °C, 6.5 d, 33% based on 12% recovered starting material. TMSCHN2 = trimethylsilyl diazomethane; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; brsm = based on recovered starting material; Ac2O = acetic anhydride; AcOH = acetic acid; DCC = N,N′-dicyclohexylcarbodiimide; DMAP = 4-dimethylaminopyridine.
Although the targeted product 53 was accessed by this method, the yield of the key [2+2] cycloaddition reaction was disappointing. It was reasoned that the inefficiency of this process was, in part, due to the volatility of both the elimination product from compound 85 (step b) and the cycloaddition product 86. To circumvent the problem, a larger ester group was employed. Thus, substrate 84 was esterified with n-BuOH, DCC, and DMAP, and the resulting product was treated with DBU, leading to unsaturated n-butyl ester 87 in 67% yield (see Scheme 8). Light-induced [2+2] cycloaddition of compound 87 with 1,2-cis-dichloroethylene gave product 88 in an improved 33% yield (based on 12% recovered starting material), along with the other two diastereomers (27% combined yield). Intermediate 88 was then exposed to the same one-pot acetonide cleavage–bis-acetylation procedure and Vorbrüggen coupling to afford, upon potassium carbonate-induced deacetylation, desired product 53 in 38% overall yield for the three steps. This approach provided a more efficient and reliable access to these new 3′,4′-carbocyclic bridged nucleoside analogues.
2.3. Biological evaluation
The library of bridged nucleoside analogues 15–53 (Figure 3) were tested for antibacterial, antitumor, and antiviral properties. Of these thirty-nine compounds, six demonstrated significant biological activities (see Table 1 and Figure 4). Compound 36 was the only compound to demonstrate antibacterial activity, showing moderate potency against both E. coli (MIC = 16 μM) and S. aureus (MIC = 8 μM). It is interesting to note that several other compounds with very similar structures displayed no significant biological activity. The N-methylbenzylamine structural motif specifically appears to be essential, as compound 36 differs from compound 34 only by a methyl group, and compound 29 differs from nucleoside 36 by having no substituents on the C6 amino group. Neither of these two compounds (i.e. 34 and 29) displayed significant antibacterial activity. Additionally, benzylation of the 3′-hydroxyl group of compound 36 also results in complete loss of its biological activity since the resulting nucleoside (i.e. 35) is inactive against E. coli and S. aureus.
Table 1.
Antibacterial, anticancer, and antiviral activities of bridged nucleoside analogues.
| Compound | Antibacterial Activity MIC (μM)a |
Anticancer Activity IC50 (μM)b |
Antiviral Activity IC50 (μM)c |
|||
|---|---|---|---|---|---|---|
| E. coli | S. aureus | CCRF-CEM | Raji | SF162 | HxB2 | |
| 15 | NA | NA | 0.36 | 0.25 | NA | NA |
| 19 | NA | NA | NA | NA | 60.0 | NA |
| 21 | NA | NA | NA | NA | 68.0 | 28.0 |
| 33 | NA | NA | NA | NA | 7.0 | 2.4 |
| 36 | 16 | 8 | NA | NA | 27.9 | 24.9 |
| 40 | NA | NA | 7.6 | 5.8 | – d | 2.4e |
| cladribine30 | – | – | 0.0005 | 0.009 | – | – |
| AZT | – | – | – | – | 0.078 | 0.037 |
Minimum inhibitory concentration.
Concentration that causes 50% of cell growth inhibition.
Concentration that causes 50% neutralization of virus.
While an IC50 below 100 μM was calculated, this was determined to be from toxicity to the TMZ-bl cells, not neutralization of the virus.
Compound 40 is toxic to the TMZ-bl cells, however, below the toxicity limit it did display antiviral activity. If the activities due to toxicity at higher concentration of 40 are ignored, an IC50 of 2.4 μM can be determined. CCRF-CEM = Human T leukemic lymphoblasts derived from acute lymphoblastic leukemia. Raji = Human B lymphocytes derived from Burkitt’s lymphoma. SF162 and HxB2 = HIV-1 pseudoviruses. NA = not active at the highest concentration tested (64 μM for the antibacterial assay, 10 μM for the cytotoxicity assay, and 100 μM for the antiviral assay).
Figure 4.

Structures of biologically active, bridged nucleoside analogues 15, 19, 21, 33, 36, and 40 as compared to those of cladribine and azidothymidine (AZT).
Two bridged nucleosides were found to exhibit significant antitumor activity: compound 15 and compound 40 (see Table 1). Nucleoside 15 was found to possess the most potent activity of the two against CEM (IC50 = 0.36 μM) and Raji (IC50 = 0.25 μM) cancer cell lines. However, these activities are considerably lower than those of the anticancer nucleoside agent cladribine (CEM: IC50 = 0.5 nM; Raji: IC50 = 9 nM). Compound 40 (Figure 4) showed less potent activities than compound 15, with an IC50 of 7.6 μM against the T cell line CEM and an IC50 of 5.8 μM against the B cell line Raji. Important to note is the fact that the 2′,4′-bridged nucleoside analogue corresponding to the structure of cladribine, compound 29 (Figure 3), was also synthesized, but did not exhibit significant cytotoxicity below 10 μM. It appears that the act of locking the conformation of cladribine, or the addition of an extra CH2O moiety, changes the structure of the molecule enough to deplete its antitumor properties.
The most promising biological activities of the synthesized bridged nucleosides were discovered from screening the library against HIV-1 (see Table 1). Thus, compound 36 (Figure 4), which had also demonstrated antibacterial activity, showed moderate activity against both the SF162 (IC50 = 27.9 μM) and HxB2 (IC50 = 24.9 μM) pseudoviruses. Compound 19 (Figure 4), with an ethylamine functionality, also showed moderate activity, but only against SF162 (IC50 = 60.0 μM). Similarly, n-butylamine nucleoside 21 (Figure 4) displayed moderate activity against both pseudoviruses (SF162 IC50 = 68.0 μM; HxB2 IC50 = 28.0 μM). However, octyl analogue 20 (Figure 3) and isobutyl nucleoside 23 (Figure 3) did not exhibit any significant antiviral activities against the viruses. This suggests that an alkyl amine containing approximately two to four carbon atoms with no branching is an essential structural motif for antiviral properties.
The highest antiviral activities were demonstrated by compounds 33 and 40 (see Table 1). Nucleoside 40 (Figure 4), which had also displayed moderate antitumor activity, was found to be toxic to the TMZ-bl cells used in the antiviral assay at concentrations at or above 33 μM. However, compound 40 (Figure 4) did exhibit some actual antiviral activity against pseudovirus HxB2 below the concentration at which it was causing cell death. Therefore, an IC50 was approximated by ignoring the higher concentrations at which it was toxic; this approximation led to an IC50 against HxB2 of 2.4 μM for this analogue (40). Nucleoside 33 (Figure 4) exhibited the most potent antiviral activity without concomitant cellular toxicity (SF162: IC50 = 7.0 μM; HxB2: IC50 = 2.4 μM). Analogue 35 (Figure 3), which contains a N-methylbenzylamine moiety instead of a benzylamine substituent, did not display any antiviral activity. While compounds 33(Figure 4) and 40 (Figure 4) still remain approximately sixty times less potent than the antiviral nucleoside AZT (SF162: IC50 = 0.078 μM; HxB2: IC50 = 0.037 μM), these activities suggest considerable potential for these bridged nucleosides as possible lead compounds for further optimization.
3. Conclusion
In conclusion, we have synthesized a focused compound library of both 2′,4′- and 3′,4′-bridged nucleoside analogues with various modifications on the purine base. This study included the development of a [2+2] cycloaddition strategy to synthesize novel 3′,4′-carbocyclic bridged systems expediently and with the flexibility to target considerable molecular diversity. Some of the compounds synthesized were found to exhibit diverse but selective biological activities, with the best compounds displaying potent antiviral properties (e.g. compound 33, SF162 IC50 = 7.0 μM; HxB2 IC50 = 2.4 μM). While the mechanism of action of these nucleosides remains unknown, it has been shown that the biological activity is dependent on not only the bridged system on the sugar, but the specific modifications of the purine residue. These compounds are, to the best of our knowledge, the first examples of 2′,4′-bridged nucleosides exhibiting antibacterial, anticancer, or antiviral activity.
4. Experimental methods
4.1. Chemical synthesis
All reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Dry tetrahydrofuran (THF), toluene, benzene, diethyl ether (Et2O), N,N′-dimethylformamide (DMF), and methylene chloride (CH2Cl2) were obtained by passing commercially available pre-dried, oxygen-free formulations through activated alumina columns. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials, unless otherwise stated. Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm E. Merck silica gel plates (60F-254) using UV light as visualizing agent and an ethanolic solution of anisaldehyde and heat as developing agents. E. Merck silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography. Preparative thin-layer chromatography (PTLC) separations were carried out on 0.25 or 0.50 mm E. Merck silica gel plates (60F-254 for normal silica, RF-18 F-254 for C18-silica).
NMR spectra were recorded on Bruker AV-400, DRX-500, or DRX-600 instruments and calibrated using residual undeuterated solvent (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm; acetone-d6: δH = 2.05 ppm, δC = 29.84 ppm; CD3CN: δH = 1.94 ppm, δC = 1.32 ppm; CD3OD: δH = 3.31 ppm, δC = 49.00 ppm; D2O: δH = 4.79 ppm)31 as an internal reference. The following abbreviations were used to designate the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet, br = broad. Infrared (IR) spectra were recorded on a Perkin-Elmer 100 FT-IR spectrometer. High-resolution mass spectra (HRMS) were recorded on an Agilent ESI-TOF (time of flight) mass spectrometer using MALDI (matrix-assisted laser desorption ionization) or ESI (electrospray ionization). Optical rotations were recorded on a Perkin-Elmer Model 343 polarimeter at 589 nm, and are reported in units of 10−1 (deg cm2 g−1).
4.1.1. (2R,3S,5S)-4-(Benzyloxy)-5-((tert-butyldiphenylsilyloxy)methyl)-2-(2,6-diamino-9H-purin-9-yl)-5-(tosyloxymethyl)tetrahydrofuran-3-yl acetate (57)
Compound 56 (1.35 g, 1.81 mmol) and 2,6-diaminopurine (407 mg, 2.71 mmol) were suspended in MeCN (17.4 mL), and BSA (1.06 mL, 4.14 mmol) was added. The reaction was heated at 65 °C for 1.5 h, after which the reaction was cooled to 0 °C and TMSOTf (0.72 mL, 3.62 mmol) was added dropwise. The solution was then stirred at 65 °C for 3 h. The reaction mixture was quenched with cold sat. aq. NaHCO3 (10 mL) and extracted with CH2Cl2. The organics were washed with sat. aq. NaHCO3 (2 × 10 mL) and brine (2 × 10 mL), dried over MgSO4, filtered, concentrated, and purified by flash column chromatography (silica, EtOAc) to give compound 57 (1.51 g, 1.81 mmol, 100%). 57: white foam; Rf = 0.50 (silica, EtOAc); (MeCN, c = 1.57); FT-IR (film) νmax 3371, 2932, 1735, 1599, 1472, 1427, 1409, 1371, 1238, 1189, 1176, 1095, 1042, 975, 915, 813, 790, 741, 601, 665 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.70 (d, J = 8.3 Hz, 2 H), 7.64–7.59 (m, 4 H), 7.44 (s, 1 H), 7.44–7.31 (m 7 H), 7.28–7.25 (m, 4 H), 7.18 (d, J = 8.0 Hz, 2 H), 6.04 (t, J = 5.7 Hz, 1 H), 5.83 (d, J = 5.8 Hz, 1 H), 5.53 (s, 2 H), 4.78 (d, J = 5.5 Hz, 1 H), 4.58 (d, J = 11.4 Hz, 1 H), 4.52 (d, J = 11.4 Hz, 1 H), 4.42 (d, J = 10.5 Hz, 1 H), 4.35 (d, J = 10.6 Hz, 1 H), 4.27 (s, 2 H), 3.88 (d, J = 10.8 Hz, 1 H), 3.74 (d, J = 10.9 Hz, 1 H), 2.37 (s, 3 H), 2.03 (s, 3 H), 0.99 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 170.05, 159.58, 155.88, 151.61, 144.72, 137.39, 137.39, 137.19, 135.71, 135.65, 133.01, 132.85, 132.27, 130.08, 130.06, 129.74, 128.68, 128.31, 128.14, 128.09, 128.05, 127.99, 115.00, 86.21, 86.14, 78.26, 74.72, 73.57, 68.75, 63.86, 26.88, 21.71, 20.76, 19.22 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C45H49N6O8SSi+ 837.3096, found 837.3100.
4.1.2. (2R,3R,4S,5S)-4-(Benzyloxy)-5-((tert-butyldiphenylsilyloxy)methyl)-2-(2,6-diamino-7H-purin-7-yl)-5-(tosyloxymethyl)tetrahydrofuran-3-yl acetate (58)
Compound 56 (200 mg, 0.268 mmol) and 2,6-diaminopurine (61.8 mg, 0.412 mmol) were suspended in MeCN (2.6 mL), and BSA (0.34 mL, 1.33 mmol) was added. The reaction mixture was heated at 65 °C for 1 h, after which the reaction was cooled to 0 °C and TMSOTf (0.15 mL, 0.753 mmol) was added dropwise. The solution was then stirred at 65 °C for 3.5 h. The reaction was quenched with cold sat. aq. NaHCO3 (2 mL) and extracted with CH2Cl2. The organics were washed with sat. aq. NaHCO3 (2 × 2 mL) and brine (2 × 2 mL), dried over MgSO4, filtered, concentrated, and purified by flash column chromatography (silica, EtOAc) to give compound 58 (81.1 mg, 0.0964 mmol, 36%) along with isomer 57 (133 mg, 0.158 mmol, 59%). 58: white foam; Rf = 0.21 (silica, EtOAc); (CHCl3, c = 1.85); FT-IR (film) νmax 3342, 3191, 2932, 1735, 1668, 1625, 1576, 1470, 1428, 1359, 1234, 1189, 1176, 1105, 1045, 972, 812, 792, 741, 701, 665 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.67 (s, 1 H), 7.67–7.64 (m, 2 H), 7.53–7.51 (m, 2 H), 7.50– 7.48 (m, 2 H), 7.45–7.41 (m, 2 H), 7.35–7.31 (m, 7 H), 7.22 (d, J = 8.6 Hz, 2 H), 7.19 (dd, J = 3.8, 1.8 Hz, 2 H), 5.87 (d, J = 7.1 Hz, 1 H), 5.50 (s, 2 H), 5.45 (dd, J = 7.0, 5.8 Hz, 1 H), 4.92 (s, 2 H), 4.54 (d, J = 5.7 Hz, 1 H), 4.46 (dd, J = 11.1, 2.7 Hz, 2 H), 4.41 (d, J = 11.3 Hz, 1 H), 4.09 (d, J = 11.1 Hz, 1 H), 3.62 (s, 2 H), 2.39 (s, 3 H), 2.04 (s, 3 H), 0.99 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 159.95, 151.75, 145.11, 141.00, 136.26, 135.43, 135.25, 132.39, 131.73, 131.66, 130.13, 129.75, 128.60, 128.43, 127.93, 127.88, 127.69, 86.82, 85.84, 77.53, 75.21, 73.27, 67.64, 64.15, 26.72, 21.51, 20.30, 18.97 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C45H49N6O8SSi+ 837.3096, found 837.3096.
4.1.3. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-9H-purine-2,6-diamine (59)
Compound 57 (1.12 g, 1.34 mmol) was dissolved in THF (56 mL) at 0 °C, and 2 M NaOH (56 mL) was added. The reaction solution was warmed to rt and stirred for 2 h. The solution was extracted with CH2Cl2 and the organic layer was dried over MgSO4, filtered, and concentrated to give compound 59 (780 mg, 1.26 mmol, 94%). 59: white foam; Rf = 0.50 (silica, EtOAc); (CHCl3, c = 0.5); FT-IR (film) νmax 3330, 3187, 2931, 2857, 1591, 1471, 1427, 1408, 1363, 1279, 1198, 1111, 1038, 939, 791, 743, 701 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 7.75 (s, 1 H), 7.71–7.67 (m, 3 H), 7.45–7.37 (m, 4 H), 7.34 (t, J = 7.4 Hz, 3 H), 7.29 (dd, J = 5.4, 1.7 Hz, 2 H), 7.26–7.24 (m, 3 H), 5.92 (s, 1 H), 5.48 (s, 2 H), 4.75 (s, 2 H), 4.72 (s, 1 H), 4.64 (d, J = 11.6 Hz, 1 H), 4.56 (d, J = 11.6 Hz, 1 H), 4.28 (s, 1 H), 4.06 (d, J = 7.7 Hz, 1 H), 4.03 (d, J = 11.9 Hz, 1 H), 3.97 (d, J = 12.0 Hz, 1 H), 3.91 (d, J = 7.7 Hz, 1 H), 1.08 (s, 9 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 159.70, 155.67, 151.05, 137.20, 135.77, 135.68, 135.48, 132.74, 132.72, 130.08, 128.63, 128.17, 128.03, 127.98, 127.80, 114.85, 88.27, 86.44, 77.27, 77.03, 72.73, 72.49, 59.41, 26.91, 19.37 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C34H39N6O4Si+ 623.2796, found 623.2795.
4.1.4. N,N’-(9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-9H-purine-2,6-diyl)bis(N-benzoylbenzamide) (60)
Compound 59 (30 mg, 0.0483 mmol) was co-evaporated twice with anhydrous pyridine (2 × 0.6 mL) and then dissolved in anhydrous pyridine (0.6 mL). The solution was cooled to 0 °C, benzoyl chloride (0.023 mL, 0.193 mmol) was added, and the mixture was stirred at rt for 18 h. The mixture was diluted with EtOAc (4 mL) and washed with H2O (2 mL). The organics were dried over MgSO4, filtered, concentrated, and purified by preparative-plate chromatography (silica, hexanes:EtOAc 2:1) to give compound 60 (26 mg, 0.0251 mmol, 52%). 60: yellow oil; Rf = 0.49 (silica, hexanes:EtOAc 2:1); (CHCl3, c = 1.20); FT-IR (film) νmax 3068, 2932, 2858, 1704, 1599, 1576, 1491, 1449, 1428, 1365, 1244, 1177, 1112, 1048, 1027, 1001, 976, 932, 909, 864, 823, 794, 733, 701 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 8.13 (s, 1 H), 7.79 (dd, J = 8.4, 1.2 Hz, 4 H), 7.67–7.62 (m, 8 H), 7.51 (dd, J = 10.6, 4.3 Hz, 2 H), 7.43–7.30 (m, 19 H), 7.20 (dd, J = 6.8, 2.7 Hz, 2 H), 5.79 (s, 1 H), 4.47 (d, J = 11.3 Hz, 1 H), 4.38 (d, J = 11.3 Hz, 1 H), 4.16 (s, 1 H), 4.03 (d, J = 8.2 Hz, 2 H), 3.97 (d, J = 12.1 Hz, 1 H), 3.91 (d, J = 12.1 Hz, 1 H), 3.87 (d, J = 7.9 Hz, 1 H), 1.04 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 172.44, 172.30, 152.97, 152.58, 143.01, 137.31, 135.97, 135.86, 134.80, 134.44, 133.39, 133.01, 132.95, 132.90, 130.39, 130.36, 129.83, 129.58, 129.12, 129.04, 128.92, 128.50, 128.32, 128.26, 127.98, 126.17, 88.95, 87.19, 77.62, 77.00, 73.06, 72.68, 59.87, 27.23, 19.63 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C62 H55N6O8Si+ 1039.3845, found 1039.3826.
4.1.5. N,N’-(9-((1S,3R,4R,7S)-7-(Benzyloxy)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-9H-purine-2,6-diyl)bis(N-benzoylbenzamide) (15)
Compound 60 (91 mg, 0.0880 mmol) was dissolved in THF (0.55 mL) and cooled to 0 °C. HF·pyridine (0.011 mL, 0.440 mmol) was added and the reaction solution stirred at rt for 12 h. The reaction solution was poured into cold aq. sat. NaHCO3 (1 mL) and stirred for 1 h. The resulting mixture was filtered through Celite and washed with CH2Cl2. The aqueous was extracted with CH2Cl2, and then the organics were dried over MgSO4, filtered, and concentrated. The residue was purified by preparative-plate chromatography (silica, EtOAc:hexanes 3:1) to give compound 15 (35.2 mg, 0.0440 mmol, 50%). 15: white foam; Rf = 0.46 (silica, EtOAc:hexanes 3:1); (CHCl3, c = 1.24); FT-IR (film) νmax 3506, 2947, 1702, 1598, 1577, 1491, 1449, 1407, 1365, 1244, 1177, 1143, 1052, 1002, 977, 932, 905, 864, 830, 795, 774, 732, 694 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 8.12 (s, 1 H), 7.81–7.75 (m, 4 H), 7.66–7.60 (m, 4 H), 7.51 (dd, J = 10.6, 4.3 Hz, 2 H), 7.43 (t, J = 7.5 Hz, 2 H), 7.34 (dt, J = 22.9, 7.7 Hz, 11 H), 7.25–7.22 (m, 2 H), 5.77 (s, 1 H), 4.49 (d, J = 11.5 Hz, 1 H), 4.42 (d, J = 11.4 Hz, 1 H), 4.20 (s, 1 H), 4.12 (s, 1 H), 4.06 (d, J = 8.0 Hz, 1 H), 3.90 (d, J = 12.6 Hz, 1 H), 3.84 (dd, J = 10.3, 7.0 Hz, 2 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 172.18, 172.01, 154.18, 152.75, 143.07, 137.10, 134.39, 134.02, 133.23, 132.75, 129.52, 129.30, 128.90, 128.77, 128.67, 128.29, 127.74, 125.61, 88.17, 86.92, 77.30, 76.95, 72.49, 72.47, 57.96 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C46H37N6O8+ 801.2667, found 801.2687.
4.1.6. ((1S,3R,4R,7S)-7-(Benzyloxy)-3-(2,6-diamino-9H-purin-9-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (16)
From compound 59, using the same procedure as for compound 15, compound 16 was obtained and purified by flash column chromatography to give nucleoside 16 (409 mg, 1.06 mmol, 85%). 16: white powder; Rf = 0.18 (C18 silica, 5% MeOH/DCM); (MeOH, c = 0.62); FT-IR (film) νmax 3332, 3195, 2927, 1595, 1455, 1407, 1280, 1201, 1036, 932, 908, 881, 809, 789, 740, 698 cm−1; 1H NMR (CD3OD, 500 MHz) δ = 7.86 (s, 1 H), 7.31–7.22 (m, 5 H), 5.85 (s, 1 H), 4.63 (s, 2 H), 4.58 (s, 1 H), 4.22 (s, 1 H), 4.05 (d, J = 7.8 Hz, 1 H), 3.93 (d, J = 1.8 Hz, 2 H), 3.87 (d, J = 7.8 Hz, 1 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 161.60, 157.34, 151.72, 138.80, 136.59, 129.39, 129.14, 128.99, 114.44, 89.49, 87.48, 78.44, 77.96, 73.49, 73.27, 58.15 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C18H21N6O4+ 385.1619, found 385.1614.
4.1.7. (1S,3R,4R,7S) and (1S,3S,4R,7S)-3-(2,6-Diamino-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (17 and 18)
Compound 16 (640 mg, 1.67 mmol) was partially dissolved in CH2Cl2 (3.3 mL) at 0 °C. Boron trichloride (3.33 mL, 1.0 M in DCM, 3.33 mmol) was slowly added dropwise and the solution was stirred at rt for 1 h. The reaction was quenched with MeOH (4 mL) and allowed to stir for 1.5 h, after which the reaction was evaporated and azeotroped with MeOH. Purification by flash column chromatography (C18 silica, 4% H2O/MeCN) was completed to give compound 17 (416 mg, 1.42 mmol, 85%) and epimer 18 (50 mg, 0.167 mmol, 10%). 17: characterization previously reported.32 18: white powder; Rf = 0.33 (C18 silica, 4% H2O/MeCN); (DMSO, c = 0.2); FT-IR (film) νmax 3318, 3142, 2922, 1632, 1594, 1481, 1460, 1410, 1386, 1286, 1226, 1178, 1140, 1119, 1035, 1009, 965, 941, 916, 872, 832, 808, 785 cm−1; 1H NMR (D2O, 600 MHz) δ = 7.94 (s, 1 H), 5.92 (s, 1 H), 4.63 (s, 1 H), 4.47 (s, 1 H), 4.07 (d, J = 8.5 Hz, 1 H), 4.01 (d, J = 10.2 Hz, 3 H) ppm; 13C NMR (D2O, 151 MHz) δ = 151.00, 137.32, 113.80, 89.21, 85.78, 80.29, 72.30, 71.07, 57.62 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C11H15N6O4+ 295.1149, found 295.1154.
4.1.8. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-N2-ethyl-9H-purine-2,6-diamine (61)
Compound 59 (70 mg, 0.112 mmol) and NaBH3CN (42 mg, 0.668 mmol) were suspended in MeOH (1.4 mL), and acetaldehyde (0.05 mL, 0.899 mmol) was added. The mixture was stirred at rt for 48 h. The solvent was evaporated and the residue was purified by preparative-plate chromatography (silica, hexanes:EtOAc:MeOH 1:1:0.1) to give compound 61 (35 mg, 0.0538 mmol, 48%) along with recovered starting material 59 (6.4 mg, 0.0101 mmol, 9%). 61: white foam; Rf = 0.42 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 1.03); FT-IR (film) νmax 3328, 3178, 3071, 2931, 2858, 1733, 1630, 1595, 1536, 1485, 1472, 1427, 1409, 1373, 1345, 1324, 1255, 1199, 1109, 1036, 938, 910, 885, 865, 823, 805, 789, 737, 701 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.73 (s, 1 H), 7.70 (t, J = 6.6 Hz, 4 H), 7.47–7.37 (m, 4 H), 7.34 (t, J = 7.5 Hz, 2 H), 7.28 (dd, J = 5.3, 1.9 Hz, 3 H), 7.23 (dd, J = 6.9, 2.5 Hz, 2 H), 5.95 (s, 1 H), 5.47 (d, J = 0.7 Hz, 2 H), 4.83 (t, J = 5.5 Hz, 1 H), 4.80 (s, 1 H), 4.63 (d, J = 11.5 Hz, 1 H), 4.54 (d, J = 11.5 Hz, 1 H), 4.27 (s, 1 H), 4.07 (d, J = 7.7 Hz, 1 H), 4.04 (d, J = 12.0 Hz, 1 H), 3.98 (d, J = 11.9 Hz, 1 H), 3.92 (d, J = 7.7 Hz, 1 H), 3.44–3.38 (m, 2 H), 1.21 (t, J = 7.2 Hz, 3 H), 1.08 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 159.66, 155.61, 151.27, 137.28, 135.87, 135.77, 134.98, 132.89, 132.84, 130.16, 128.68, 128.23, 128.12, 128.06, 127.88, 114.43, 88.28, 86.66, 77.46, 77.15, 72.87, 72.62, 59.62, 36.79, 27.01, 19.46, 15.31 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C36H43N6O4Si+ 651.3109, found 651.3103.
4.1.9. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-N2-octyl-9H-purine-2,6-diamine (62)
Compound 59 (70 mg, 0.112 mmol) and NaBH3CN (42 mg, 0.668 mmol) were suspended in MeOH (1.4 mL), and octyl aldehyde (0.14 mL, 0.899 mmol) was added. The mixture was stirred at rt for 48 h. The solvent was evaporated and the residue was purified by preparative-plate chromatography (silica, hexanes:EtOAc:MeOH 1:1:0.1) to give compound 62 (29 mg, 0.0325 mmol, 29%) along with recovered starting material 59 (21 mg, 0.0336 mmol, 30%). 62: yellow oil; Rf = 0.70 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 0.81); FT-IR (film) νmax 3325, 2927, 2855, 1635, 1599, 1537, 1470, 1427, 1408, 1362, 1324, 1263, 1198, 1112, 1036, 939, 911, 885, 862, 805, 789, 740, 701 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 7.75 (s, 1 H), 7.69 (dd, J = 8.0, 6.7 Hz, 4 H), 7.45–7.38 (m, 3 H), 7.35–7.27 (m, 6 H), 7.23 (dd, J = 6.8, 2.8 Hz, 2 H), 5.93 (s, 1 H), 5.69 (s, 2 H), 5.12 (s, 1 H), 4.77 (s, 1 H), 4.62 (d, J = 11.6 Hz, 1 H), 4.54 (d, J = 11.6 Hz, 1 H), 4.25 (s, 1 H), 4.06 (d, J = 7.7 Hz, 1 H), 4.03 (d, J = 12.0 Hz, 1 H), 3.97 (d, J = 12.0 Hz, 1 H), 3.91 (d, J = 7.7 Hz, 1 H), 3.37 (td, J = 7.0, 1.7 Hz, 2 H), 1.59–1.57 (m, 2 H), 1.29–1.25 (m, 10 H), 1.08 (s, 9 H), 0.88 (d, J = 5.1 Hz, 3 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 151.90, 138.37, 137.83, 136.50, 136.40, 133.46, 133.40, 130.82, 129.39, 129.32, 128.90, 128.76, 128.71, 128.65, 128.53, 88.98, 87.27, 73.47, 73.28, 63.98, 60.14, 42.66, 33.68, 32.69, 30.52, 30.27, 30.14, 27.89, 27.63, 23.53, 20.08, 14.98 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C42H55N6O4Si+ 735.4048, found 735.4053.
4.1.10. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-N2-butyl-9H-purine-2,6-diamine (63)
Compound 59 (145 mg, 0.234 mmol) and NaBH3CN (87 mg, 1.38 mmol) were suspended in MeOH (2.9 mL), and butyraldehyde (0.17 mL, 1.82 mmol) was added. The mixture was stirred at rt for 48 h. The solvent was evaporated and the residue was purified by flash column chromatography (silica, hexanes:EtOAc 1:1 then hexanes:EtOAc:MeOH 1:1:0.1) to give compound 63 (90 mg, 0.133 mmol, 57%) along with recovered starting material 59 (35 mg, 0.056 mmol, 24%). 63: yellow oil; Rf = 0.22 (silica, hexanes:EtOAc 1:1); (CHCl3, c = 0.20); FT-IR (film) νmax 3322, 2930, 2858, 1633, 1600, 1537, 1471, 1427, 1408, 1362, 1324, 1267, 1199, 1112, 1036, 938, 911, 823, 789, 740, 701 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 7.73–7.66 (m, 5 H), 7.46– 7.36 (m, 4 H), 7.33 (dd, J = 9.0, 5.9 Hz, 2 H), 7.27 (dd, J = 5.2, 1.9 Hz, 3 H), 7.25–7.21 (m, 2 H), 5.93 (s, 1 H), 5.32 (s, 2 H), 4.82–4.75 (m, 2 H), 4.62 (d, J = 11.5 Hz, 1 H), 4.53 (d, J = 11.5 Hz, 1 H), 4.26 (s, 1 H), 4.06 (d, J = 7.7 Hz, 1 H), 4.02 (d, J = 11.9 Hz, 1 H), 3.97 (d, J = 11.9 Hz, 1 H), 3.91 (d, J = 7.7 Hz, 1 H), 3.38 (d, J = 6.9 Hz, 1 H), 3.36 (d, J = 7.0 Hz, 1 H), 1.60–1.52 (m, 2 H), 1.40 (dq, J = 14.2, 7.2 Hz, 2 H), 1.07 (s, 9 H), 0.94 (t, J = 7.4 Hz, 3 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 159.78, 155.52, 151.19, 137.17, 135.78, 135.69, 134.83, 132.77, 132.73, 130.07, 128.59, 128.14, 128.03, 127.97, 127.80, 114.39, 88.16, 86.55, 77.31, 77.05, 72.77, 72.52, 59.49, 41.60, 32.05, 26.91, 20.29, 19.36, 14.07 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C38H46N6O4Si+ 679.3422, found 679.3417.
4.1.11. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-N2-isobutyl-9H-purine-2,6-diamine (64)
Compound 59 (140 mg, 0.225 mmol) and NaBH3CN (84 mg, 1.34 mmol) were suspended in MeOH (2.8 mL), and isobutyraldehyde (0.17 mL, 1.82 mmol) was added. The mixture was stirred at rt for 48 h. The solvent was evaporated and the residue was purified by flash column chromatography (silica, hexanes:EtOAc 1:1 then hexanes:EtOAc:MeOH 1:1:0.1) to give compound 64 (80 mg, 0.117 mmol, 52%) along with recovered starting material 59 (8 mg, 0.0135 mmol, 6%). 64: yellow oil; Rf = 0.38 (silica, 1:1 hexanes:EtOAc); (CHCl3, c = 2.0); FT-IR (film) νmax 3323, 2956, 1602, 1536, 1471, 1427, 1364, 1253, 1199, 1112, 1037, 939, 910, 789, 739, 702 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 7.73 (s, 1 H), 7.72–7.68 (m, 4 H), 7.45–7.37 (m, 5 H), 7.33 (t, J = 7.5 Hz, 2 H), 7.28 (dt, J = 4.6, 2.4 Hz, 2 H), 7.25–7.22 (m, 2 H), 5.94 (s, 1 H), 5.49 (s, 2 H), 4.96 (s, 1 H), 4.79 (s, 1 H), 4.62 (d, J = 11.5 Hz, 1 H), 4.54 (d, J = 11.5 Hz, 1 H), 4.25 (s, 1 H), 4.06 (d, J = 7.7 Hz, 1 H), 4.03 (d, J = 11.9 Hz, 1 H), 3.97 (d, J = 12.0 Hz, 1 H), 3.92 (d, J = 7.7 Hz, 1 H), 3.21 (t, J = 6.3 Hz, 2 H), 1.85 (m, 1 H), 1.08 (s, 9 H), 0.96 (d, J = 6.7 Hz, 6 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 155.45, 151.12, 137.14, 135.78, 135.69, 134.84, 132.76, 132.71, 130.07, 128.60, 128.16, 128.03, 127.98, 127.80, 114.22, 88.18, 86.54, 77.27, 77.03, 72.77, 72.55, 59.47, 49.45, 28.70, 26.91, 20.50, 19.36 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C38H46N6O4Si+ 679.3422, found 679.3424.
4.1.12. ((1S,3R,4R,7S)-3-(6-Amino-2-(ethylamino)-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (19)
Compound 61 (30 mg, 0.0461 mmol) was dissolved in THF (0.46 mL) at 0 °C, and tetra-n-butylammonium fluoride (0.092 mL, 0.0922 mmol, 1.0 M in THF) was slowly added dropwise. The reaction was allowed to warm to rt and stirred for 16 h. The solution was quenched with H2O (0.5 mL) and stirred for 10 min. The aqueous layer was extracted with 5% MeOH/DCM and the organics dried over MgSO4, filtered, and concentrated. Purification by preparative-plate chromatography (silica, hexanes:EtOAc:MeOH 1:1:0.1) was completed to give compound 19 (15 mg, 0.0364 mmol, 79%). 19: white foam; Rf = 0.21 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 0.50); FT-IR (film) νmax 3344, 2928, 1631, 1598, 1540, 1473, 1409, 1345, 1260, 1212, 1143, 1035, 935, 909, 789, 752, 698 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 7.55 (s, 1 H), 7.34–7.27 (m, 5 H), 5.89 (s, 1 H), 5.66 (s, 2 H), 4.93 (s, 1 H), 4.67 (d, J = 11.7 Hz, 2 H), 4.63 (d, J = 11.7 Hz, 1 H), 4.49 (s, 1 H), 4.12 (d, J = 7.7 Hz, 1 H), 4.04 (d, J = 12.6 Hz, 1 H), 3.96 (d, J = 12.6 Hz, 1 H), 3.91 (d, J = 7.7 Hz, 1 H), 3.43–3.36 (m, 2 H), 1.20 (t, J = 7.2 Hz, 3 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 159.37, 155.53, 151.17, 137.31, 135.11, 128.62, 128.20, 127.82, 114.02, 88.31, 86.99, 77.80, 72.58, 72.50, 57.76, 53.57, 36.70, 15.20 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C20H25N6O4+ 413.1932, found 413.1938.
4.1.13. ((1S,3R,4R,7S)-3-(6-Amino-2-(octylamino)-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (20)
From compound 62, using the same procedure as for compound 19, compound 20 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc:MeOH 1:1:0.1) was completed to give nucleoside 20 (15 mg, 0.0302 mmol, 77%). 20: white foam; Rf = 0.33 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 1.30); FT-IR (film) νmax 3335, 2924, 2854, 1634, 1598, 1539, 1467, 1408, 1366, 1326, 1264, 1206, 1143, 1034, 935, 909, 881, 809, 788, 738, 697 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.56 (s, 1 H), 7.32–7.27 (m, 5 H), 5.89 (s, 1 H), 5.54 (s, 2 H), 4.82 (t, J = 5.3 Hz, 1 H), 4.75 (s, 1 H), 4.66 (d, J = 11.6 Hz, 1 H), 4.62 (d, J = 11.7 Hz, 1 H), 4.45 (s, 1 H), 4.12 (d, J = 7.8 Hz, 1 H), 4.03 (d, J = 12.7 Hz, 1 H), 3.94 (d, J = 12.8 Hz, 1 H), 3.91 (d, J = 7.7 Hz, 1 H), 3.38–3.32 (m, 2 H), 1.59–1.52 (m, 2 H), 1.37–1.25 (m, 10 H), 0.87 (t, J = 6.9 Hz, 3 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 160.03, 155.94, 151.51, 137.62, 135.30, 128.89, 128.47, 128.10, 114.38, 100.01, 88.58, 87.28, 78.06, 72.87, 72.80, 58.08, 42.28, 32.25, 30.22, 29.83, 29.69, 27.47, 23.08, 14.52 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C26H37N6O4+ 497.2871, found 497.2872.
4.1.14. ((1S,3R,4R,7S)-3-(6-Amino-2-(butylamino)-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (21)
From compound 63, using the same procedure as for compound 19, compound 21 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc:MeOH 1:1:0.1) was completed to give nucleoside 21 (5 mg, 0.0114 mmol, 77%). 21: yellow oil; Rf = 0.30 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 0.45); FT-IR (film) νmax 3337, 2928, 2871, 1634, 1600, 1542, 1466, 1409, 1365, 1326, 1273, 1207, 1144, 1036, 935, 909, 882, 789, 736, 698 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.56 (s, 1 H), 7.34–7.27 (m, 5 H), 5.89 (s, 1 H), 5.82–5.66 (s, 1 H), 4.99 (dd, J = 3.5, 1.7 Hz, 1 H), 4.72 (s, 1 H), 4.66 (d, J = 11.7 Hz, 1 H), 4.64–4.60 (m, 1 H), 4.43 (s, 1 H), 4.15– 4.10 (m, 1 H), 4.03 (d, J = 12.8 Hz, 1 H), 3.95 (d, J = 12.7 Hz, 1 H), 3.91 (d, J = 7.8 Hz, 1 H), 3.36 (dt, J = 7.2, 4.7 Hz, 2 H), 2.08 (s, 1 H), 1.56 (dd, J = 8.5, 6.3 Hz, 2 H), 1.44–1.35 (m, 2 H), 0.94 (d, J = 7.3 Hz, 3 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 159.43, 155.53, 151.13, 137.25, 135.00, 128.63, 128.23, 127.85, 113.76, 88.26, 86.95, 77.73, 77.30, 72.61, 72.50, 57.84, 41.60, 31.96, 20.28, 14.05 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C22H29N6O4+ 441.2245, found 441.2249.
.1.15. (1S,3R,4R,7S)-3-(6-Amino-2-(butylamino)-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (22)
From compound 21, using the same procedure as for compound 64, compound 22 was obtained and purification by preparative-plate chromatography (C18 silica, 5% MeOH/DCM) was completed to give nucleoside 22 (18 mg, 0.0514 mmol, 71%). 22: yellow oil; Rf = 0.13 (C18 silica, 5% MeOH/DCM); (MeOH, c = 0.93); FT-IR (film) νmax 3258, 2957, 1683, 1624, 1512, 1463, 1416, 1367, 1327, 1222, 1130, 1031, 924, 902, 875, 833, 809, 762, 722, 675 cm−1; 1H NMR (CD3OD, 600 MHz) δ = 8.07 (s, 1 H), 5.86 (s, 1 H), 4.51 (s, 1 H), 4.32 (s, 1 H), 4.05 (d, J = 7.9 Hz, 1 H), 3.95 (s, 2 H), 3.87 (d, J = 8.0 Hz, 1 H), 3.45 (td, J = 6.9, 4.9 Hz, 2 H), 1.65 (dd, J = 10.0, 4.9 Hz, 2 H), 1.45 (dd, J = 15.0, 7.5 Hz, 2 H), 0.99 (t, J = 7.4 Hz, 3 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 152.90, 152.84, 139.61, 139.59, 112.88, 90.21, 87.51, 80.92, 72.85, 71.40, 58.08, 42.29, 32.09, 21.04, 14.12 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C15H23N6O4+ 351.1775, found 351.1781.
4.1.16. ((1S,3R,4R,7S)-3-(6-Amino-2-(isobutylamino)-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (23)
From compound 64, using the same procedure as for compound 19, compound 23 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc:MeOH 1:1:0.1) was completed to give nucleoside 23(38 mg, 0.0863 mmol, 98%). 23: white foam; Rf = 0.22 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 1.83); FT-IR (film) νmax 3339, 2956, 1633, 1601, 1542, 1485, 1467, 1409, 1384, 1354, 1277, 1207, 1143, 1036, 935, 909, 863, 807, 789, 734, 698 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.61 (s, 1 H), 7.32–7.26 (m, 5 H), 5.88 (s, 1 H), 5.84 (s, 2 H), 5.10 (s, 1 H), 4.77 (s, 1 H), 4.65 (d, J = 11.6 Hz, 1 H), 4.61 (d, J = 11.6 Hz, 1 H), 4.38 (s, 1 H), 4.12 (d, J = 7.7 Hz, 1 H), 4.03 (d, J = 12.9 Hz, 1 H), 3.94 (d, J = 12.9 Hz, 1 H), 3.91 (d, J = 7.8 Hz, 1 H), 3.19 (dd, J = 9.2, 3.3 Hz, 2 H), 2.07 (s, 1 H), 1.84 (dp, J = 13.4, 6.8 Hz, 1 H), 0.94 (d, J = 6.7 Hz, 6 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 159.73, 155.58, 151.09, 137.31, 134.99, 128.60, 128.17, 127.79, 113.77, 99.72, 88.38, 86.82, 77.38, 72.59, 72.54, 57.63, 49.44, 28.63, 20.46 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C22H29N6O4+ 441.2245, found 441.2242.
4.1.17. (1S,3R,4R,7S)-3-(6-Amino-2-(isobutylamino)-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (24)
Compound 23 (19.8 mg, 0.0449 mmol) was dissolved in CH2Cl2 (0.45 mL) at 0 °C, and boron trichloride (0.09 mL, 1.0 M in DCM, 0.0899 mmol) was added slowly dropwise. The solution was allowed to stir at rt for 16 h. The reaction was quenched with MeOH (1 mL) and allowed to stir for 1.5 h, after which the reaction was evaporated and azeotroped with MeOH. Purification by flash column chromatography (C18 silica, 5% MeOH/DCM) was completed to give compound 24 (10.2 mg, 0.0287 mmol, 64%). 24: white powder; Rf = 0.29 (C18 silica, 5% MeOH/DCM); (MeOH, c = 0.17); FT-IR (film) νmax 3274, 2958, 1684, 1627, 1575, 1512, 1465, 1420, 1385, 1283, 1223, 1175, 1132, 1032, 928, 904, 808 cm−1; 1H NMR (CD3OD, 600 MHz) δ = 7.90 (s, 1 H), 5.85 (s, 1 H), 4.52 (s, 1 H), 4.36 (s, 1 H), 4.04 (d, J = 7.8 Hz, 1 H), 3.93 (s, 2 H), 3.88 (d, J = 7.8 Hz, 1 H), 3.19 (ddd, J = 26.4, 13.1, 6.9 Hz, 2 H), 1.94–1.88 (m, 1 H), 0.96 (d, J = 6.7 Hz, 6 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 152.05, 136.47, 114.05, 89.82, 87.30, 81.06, 72.88, 71.56, 58.35, 50.21, 29.63, 20.66 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C15H23N6O4+ 351.1775, found 351.1781.
4.1.18. 7-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-7H-purine-2,6-diamine (65)
From compound 58, using the same procedure as for compound 59, compound 65 (636 mg, 1.02 mmol, 85%) was obtained. 65: white foam; Rf = 0.21 (silica, EtOAc); (CHCl3, c = 1.15); FT-IR (film) νmax 3330, 3182, 2931, 2857, 1574, 1470, 1427, 1401, 1361, 1217, 1105, 1036, 923, 883, 856, 823, 792, 744, 699, 666 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.56 (dd, J = 10.4, 3.8 Hz, 4 H), 7.44–7.30 (m, 13 H), 5.75 (s, 1 H), 5.45 (s, 2 H), 4.81 (d, J = 0.9 Hz, 2 H), 4.72 (d, J = 12.0 Hz, 1 H), 4.57 (d, J = 12.0 Hz, 1 H), 4.54 (s, 1 H), 4.14 (d, J = 7.5 Hz, 2 H), 3.98 (t, J = 6.9 Hz, 3 H), 0.95 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 160.34, 151.92, 139.19, 136.80, 135.57, 135.52, 132.86, 132.54, 130.13, 130.09, 128.95, 128.76, 128.13, 127.96, 106.10, 89.93, 86.19, 78.07, 76.64, 73.10, 72.95, 59.59, 26.70, 19.34 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C34H39N6O4Si+ 623.2796, found 623.2778.
4.1.19. 7-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-N2-isobutyl-7H-purine-2,6-diamine (66)
From compound 65, using the same procedure as for compound 17, compound 66 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 1:1 then hexanes:EtOAc:MeOH 1:1:0.1) was completed to give nucleoside 66 (43 mg, 0.0634 mmol, 44%). 66: white foam; Rf = 0.22 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 2.13); FT-IR (film) νmax 3331, 2956, 1629, 1588, 1470, 1427, 1385, 1361, 1233, 1112, 1039, 910, 856, 823, 791, 736, 701 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.58 (td, J = 8.4, 1.3 Hz, 4 H), 7.45–7.31 (m, 12 H), 5.77 (s, 1 H), 5.68 (d, J = 0.4 Hz, 2 H), 4.72 (d, J = 11.9 Hz, 1 H), 4.63–5.59 (m, 2 H), 4.14 (dd, J = 7.0, 2.2 Hz, 2 H), 4.00 (d, J = 1.8 Hz, 2 H), 3.98 (d, J = 8.0 Hz, 1 H), 3.31–3.24 (m, 2 H), 2.04 (s, 1 H), 1.91 (dt, J = 13.4, 6.7 Hz, 1 H), 0.99–0.95 (m, 15 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 151.86, 139.01, 136.86, 135.60, 132.91, 132.64, 130.16, 130.14, 129.00, 128.81, 128.25, 128.01, 128.00, 105.38, 99.74, 90.08, 86.29, 78.03, 76.70, 73.17, 73.00, 59.61, 49.37, 28.58, 26.78, 20.42, 19.40 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C38H47N6O4Si+ 679.3422, found 679.3421.
4.1.20. ((1S,3R,4R,7S)-3-(6-Amino-2-(isobutylamino)-7H-purin-7-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (25)
From compound 66, using the same procedure as for compound 19, compound 25 was obtained and purification by preparative-plate chromatography (silica, 5% MeOH/DCM) was completed to give nucleoside 25 (20 mg, 0.0454 mmol, 77%). 25: white foam; Rf = 0.11 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 0.98); FT-IR (film) νmax 3351, 2956, 1630, 1579, 1530, 1488, 1467, 1411, 1362, 1229, 1147, 1095, 1039, 911, 856, 790, 733, 699 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.39 (ddd, J = 27.1, 17.2, 7.2 Hz, 5 H), 7.07 (s, 1 H), 5.63 (s, 1 H), 5.48 (s, 2 H), 4.97–4.90 (m, 1 H), 4.87 (d, J = 12.1 Hz, 1 H), 4.77 (d, J = 12.1 Hz, 1 H), 4.65 (d, J = 11.9 Hz, 1 H), 4.57 (s, 1 H), 4.15 (d, J = 7.9 Hz, 1 H), 4.05 (d, J = 13.3 Hz, 1 H), 3.97 (d, J = 13.3 Hz, 1 H), 3.91 (d, J = 7.9 Hz, 1 H), 3.21 (dt, J = 12.8, 6.2 Hz, 1 H), 3.14–3.07 (m, 1 H), 2.15 (s, 1 H), 1.82 (dt, J = 13.5, 6.7 Hz, 1 H), 0.93 (d, J = 6.7 Hz, 6 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 159.67, 151.56, 138.80, 137.58, 128.87, 128.54, 128.17, 105.58, 99.74, 90.76, 85.89, 77.91, 77.04, 73.08, 57.57, 49.37, 28.49, 20.45, 20.41 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C22H29N6O4+ 441.2245, found 441.2245.
4.1.21. ((1S,3R,4R,7S)-7-(Benzyloxy)-3-(2,6-diamino-7H-purin-7-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (26)
From compound 65, using the same procedure as for compound 15, compound 26 was obtained and purification for characterization was completed by preparative-plate chromatography (C18 silica, MeCN/H2O 10:1). 26: Rf = 0.12 (C18 silica, 5% MeOH/DCM); (MeOH, c = 1.11); FT-IR (film) νmax 3115, 1648, 1545, 1455, 1389, 1316, 1219, 1143, 1047, 937, 884, 858, 733, 696, 676 cm−1; 1H NMR (D2O, 600 MHz) δ = 7.83 (s, 1 H), 7.37 (s, 5 H), 6.03 (s, 1 H), 4.75 (s, 1 H), 4.70 (s, 1 H), 4.67 (d, J = 11.7 Hz, 1 H), 4.11 (s, 1 H), 4.09 (d, J = 8.5 Hz, 1 H), 4.04 (d, J = 8.5 Hz, 1 H), 4.01 (d, J = 13.4 Hz, 1 H), 3.96 (d, J = 13.4 Hz, 1 H) ppm; 13C NMR (D2O, 151 MHz) δ = 153.65, 153.35, 150.03, 140.93, 136.10, 128.77, 128.75, 104.92, 89.66, 85.90, 77.11, 76.27, 72.70, 72.16, 56.43 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C18H21N6O4+ 385.1619, found 385.1624.
4.1.22. (1S,3R,4R,7S)-3-(2,6-Diamino-7H-purin-7-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (27)
From compound 26, using the same procedure as for compound 17, compound 27 was obtained and purification by flash column chromatography (C 18 silica, 4% H2O/MeCN) was completed to give nucleoside 27 (100 mg, 0.469 mmol, 46% over two steps). 27: white semi-solid; Rf = 0.39 (C18 silica, 20% H2O/MeOH); (MeOH, c = 0.21); FT-IR (film) νmax 3129, 1648, 1466, 1392, 1224, 1172, 1091, 1039, 1012, 975, 929, 874, 832, 764 cm−1; 1H NMR (D2O, 400 MHz) δ = 8.16 (s, 1 H), 6.02 (s, 1 H), 4.97 (s, 1 H), 4.42 (s, 1 H), 4.07 (d, J = 10.6 Hz, 2 H), 3.97 (s, 2 H) ppm; 13C NMR (D2O, 126 MHz) δ = 154.29, 153.86, 150.48, 142.01, 141.78, 90.69, 86.27, 79.34, 72.06, 70.81, 57.03 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C11H15N6O4+ 295.1149, found 295.1157.
4.1.23. (2R,3R,4S,5S)-2-(6-Amino-2-chloro-9H-purin-9-yl)-4-(benzyloxy)-5-((tert-butyldiphenylsilyloxy)methyl)-5-(tosyloxymethyl)tetrahydrofuran-3-yl acetate (67)
Compound 56 (400 mg, 0.526 mmol) and 2-chloroadenine (136 mg, 0.803 mmol) were dissolved in MeCN (5.2 mL), and BSA (0.30 mL, 1.23 mmol) was added. The mixture was heated to 65 °C for 1.5 h. The reaction mixture was then cooled to 0 °C and TMSOTf (0.24 mL, 1.07 mmol) was added, after which the reaction was heated at 65 °C for 3 h. The reaction solution was quenched with cold sat. aq. NaHCO3 (2 mL) and extracted with CH2Cl2. The organics were washed with sat. aq. NaHCO3 (2 × 5 mL) and brine (2 × 5 mL), dried over MgSO4, filtered, concentrated, and purified by flash column chromatography (silica, hexanes:EtOAc 1:1) to give compound 67 (390 mg, 0.447 mmol, 85%). 67: white foam; Rf = 0.44 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 1.64); FT-IR (film) νmax 3320, 3174, 2931, 2858, 1745, 1643, 1593, 1497, 1461, 1428, 1360, 1308, 1227, 1189, 1175, 1105, 978, 936, 909, 813, 790, 731, 700, 666 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 7.75 (s, 1 H), 7.71–7.67 (m, 2 H), 7.58–7.52 (m, 4 H), 7.45– 7.27 (m, 11 H), 7.20 (d, J = 8.0 Hz, 2 H), 6.06 (s, 2 H), 5.94 (d, J = 4.9 Hz, 1 H), 5.71 (dd, J = 5.7, 5.0 Hz, 1 H), 4.71 (d, J = 5.8 Hz, 1 H), 4.54 (s, 1 H), 4.51 (s, 1 H), 4.36 (d, J = 10.7 Hz, 1 H), 4.28 (d, J = 10.7 Hz, 1 H), 3.76 (d, J = 11.0 Hz, 1 H), 3.73 (d, J = 11.1 Hz, 1 H), 2.38 (s, 3 H), 2.04 (s, 3 H), 1.00 (s, 9 H) ppm; 13C NMR (CDCl3, 101 MHz) δ = 169.99, 156.22, 154.35, 147.46, 144.85, 139.78, 137.03, 135.72, 135.58, 132.92, 132.63, 132.32, 130.09, 130.07, 129.83, 128.67, 128.38, 128.27, 128.13, 128.00, 127.90, 86.79, 86.54, 78.15, 74.86, 74.67, 68.68, 64.75, 27.01, 21.73, 20.74, 19.31 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C43H47ClN5O8SSi+ 856.2598, found 856.2587.
4.1.24. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-2-chloro-9H-purin-6-amine (68)
Compound 67 (29 mg, 0.0339 mmol) was dissolved in THF (3.42 mL) at 0 °C, and 2 M NaOH (0.28 mL) was added. The mixture was allowed to warm to rt with stirring over 15 h. The reaction solution was worked up by extraction with CH2Cl2, drying over MgSO4, filtration, and concentration. Purification by flash column chromatography was completed to produce compound 68 (19 mg, 0.0285 mmol, 87%). 68: white foam; Rf = 0.53 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 1.55); FT-IR (film) νmax 3314, 3170, 2931, 2858, 1645, 1592, 1571, 1498, 1456, 1427, 1345, 1310, 1245, 1202, 1112, 1038, 937, 909, 857, 823, 804, 735, 701 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 8.01 (s, 1 H), 7.69 (ddd, J = 8.0, 3.7, 1.4 Hz, 4 H), 7.48–7.32 (m, 7 H), 7.29–7.27 (m, 2 H), 7.26–7.22 (m, 2 H), 6.09 (s, 2 H), 6.02 (s, 1 H), 4.79 (s, 1 H), 4.66 (d, J = 11.6 Hz, 1 H), 4.57 (d, J = 11.6 Hz, 1 H), 4.24 (s, 1 H), 4.07 (d, J = 7.8 Hz, 1 H), 4.04 (d, J = 11.9 Hz, 1 H), 3.98 (d, J = 12.0 Hz, 1 H), 3.90 (d, J = 7.8 Hz, 1 H), 1.08 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 156.18, 154.34, 138.69, 137.03, 135.75, 135.64, 132.74, 132.66, 130.12, 128.59, 128.23, 128.06, 128.00, 127.88, 88.62, 86.72, 77.36, 76.88, 72.71, 72.56, 59.31, 26.92, 19.38 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C34H36ClN5O4Si+ 642.2298, found 642.2280.
4.1.25. ((1S,3R,4R,7S)-3-(6-Amino-2-chloro-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (28)
From compound 68, using the same procedure as for compound 15, compound 28 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc:MeOH 1:1:0.1) was completed to give nucleoside 28(28 mg, 0.0695 mmol, 61%). 28: white semi-solid; Rf = 0.18 (silica, hexanes:EtOAc:MeOH 1:1:0.1); (CHCl3, c = 0.56); FT-IR (film) νmax 3322, 3176, 2926, 2248, 1644, 1593, 1572, 1497, 1455, 1347, 1309, 1249, 1203, 1182, 1143, 1097, 1035, 987, 933, 907, 883, 821, 789, 729, 697, 681 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.84 (s, 1 H), 7.34–7.27 (m, 5 H), 6.49 (s, 2 H), 5.96 (s, 1 H), 4.78 (s, 1 H), 4.72 (d, J = 11.8 Hz, 1 H), 4.68 (d, J = 11.9 Hz, 1 H), 4.43 (s, 1 H), 4.13 (d, J = 7.9 Hz, 1 H), 4.04 (d, J = 12.2 Hz, 2 H), 3.96 (d, J = 7.8 Hz, 1 H), 3.91 (d, J = 7.8 Hz, 1 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 156.19, 154.45, 138.83, 137.25, 128.63, 128.26, 127.88, 99.71, 88.57, 87.08, 77.56, 77.27, 72.52, 57.69 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C18H19ClN5O4+ 404.1120, found 404.1120.
4.1.26. (1S,3R,4R,7S)-3-(6-Amino-2-chloro-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (29)
Compound 28 (5 mg, 0.0124 mmol) was dissolved in CH2Cl2 (0.13 mL) at 0 °C, and methanesulfonic acid (0.06 mL, 0.963 mmol) was added. The reaction was stirred at this temperature for 1.5 h. The reaction was neutralized with concentrated aq. NaOH (0.13 mL, 0.963 mmol) and the mixture concentrated. The residue was diluted with CH2Cl2 (2 mL) and MeOH (0.5 mL) then filtered through Celite to remove most salts. Purification by preparative-plate chromatography (C18 silica, EtOAc) was completed to give compound 29 (1.8 mg, 0.00570 mmol, 46%). 29: white powder; Rf = 0.61 (C18 silica, EtOAc); (MeOH, c = 0.15); FT-IR (film) νmax 3179, 2925, 1667, 1597, 1571, 1498, 1460, 1445, 1354, 1311, 1265, 1204, 1180, 1131, 1034, 1009, 934, 900, 877, 848, 821, 786, 749, 725, 678 cm−1; 1H NMR (CD3OD, 500 MHz) δ = 8.20 (s, 1 H), 5.92 (s, 1 H), 4.48 (s, 1 H), 4.30 (s, 1 H), 4.03 (d, J = 7.9 Hz, 1 H), 3.92 (s, 2 H), 3.86 (d, J = 7.9 Hz, 1 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 158.08, 155.48, 139.99, 90.18, 87.55, 81.01, 72.82, 71.36, 58.16 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C11H13ClN5O4+ 314.0651, found 314.0657.
4.1.27. (2R,3R,4S,5S)-2-(6-Amino-2-fluoro-9H-purin-9-yl)-4-(benzyloxy)-5-((tert-butyldiphenylsilyloxy)methyl)-5-(tosyloxymethyl)tetrahydrofuran-3-yl acetate (69)
Acetylated compound 56 (200 mg, 0.268 mmol) was dissolved in dry MeCN (2.0 mL) along with 2-fluoro-9H-purine-6-amine (81 mg, 0.530 mmol) and N,O-bis(trimethylsilyl)acetamide (0.17 mL, 0.670 mmol). The reaction mixture was heated to 80 °C for 1.5 h. The reaction mixture was cooled to 0 °C and TMSOTf (0.132 mL, 0.530 mmol) was slowly added. Heating was resumed for an additional 3 h at 80 °C. The reaction mixture was quenched with sat. aq. NaHCO3 (2 mL) and extracted with EtOAc. The combined organic extracts were washed with brine (2 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography (silica, hexanes:EtOAc 4:1) was completed to give compound 69 (176 mg, 0.210 mmol, 78%). 69: light yellow oil; Rf = 0.49 (silica, hexanes:EtOAc 2:3); (MeCN, c = 0.30); FT-IR (film) νmax 3459, 2931, 1652, 1496, 1438, 1410, 1387, 1255, 1224, 1094, 1062, 865, 659 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 7.74 (s, 1 H), 7.68 (d, J = 8.2 Hz, 2 H), 7.55 (dd, J = 11.5, 6.8 Hz, 4 H), 7.42–7.30 (m, 9 H), 7.26–7.23 (m, 2 H), 7.19 (d, J = 8.0 Hz, 2 H), 5.89 (d, J = 4.8 Hz, 1 H), 5.74–5.71 (m, 1 H), 4.78 (s, 2 H), 4.73 (d, J = 5.8 Hz, 1 H), 4.54 (d, J = 11.3 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.36 (d, J = 10.7 Hz, 1 H), 4.26 (d, J = 10.7 Hz, 1 H), 3.77 (d, J = 11.0 Hz, 1 H), 3.71 (d, J = 11.0 Hz, 1 H), 2.37 (s, 3 H), 2.02 (s, 3 H), 0.99 (s, 9 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 176.75, 169.98, 144.91, 139.65, 139.63, 136.96, 135.70, 135.54, 132.77, 132.57, 132.27, 130.09, 130.08, 129.83, 128.65, 128.35, 128.24, 128.20, 128.13, 128.10, 127.99, 127.90, 86.64, 86.48, 77.95, 74.78, 74.46, 68.64, 64.58, 26.95, 21.73, 20.74, 19.29 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C43H47N5O8SSi+ 840.2893, found 840.2892.
4.1.28. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-2-fluoro-9H-purin-6-amine (70)
From compound 69, using the same procedure as for compound 30, compound 70 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 4:1) was completed to give nucleoside 70 (6 mg, 0.00964 mmol, 81%). 70: colorless foam; Rf = 0.60 (silica, hexanes:EtOAc 2:3); (MeCN, c = 0.30); FT-IR (film) νmax 3490, 2293, 2253, 1443, 1375, 1039, 918, 749 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 7.95 (s, 1 H), 7.71–7.65 (m, 4 H), 7.49–7.43 (m, 2 H), 7.42–7.35 (m, 4 H), 7.32–7.25 (m, 5 H), 6.32 (s, 2 H), 5.92 (s, 1 H), 4.70 (s, 1 H), 4.65 (d, J = 11.8 Hz, 1 H), 4.59 (d, J = 11.8 Hz, 1 H), 4.38 (s, 1 H), 4.08–4.01 (m, 2 H), 3.98 (d, J = 7.9 Hz, 1 H), 3.87 (d, J = 7.9 Hz, 1 H), 1.02 (s, 9 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 160.56, 159.19, 158.48, 158.34, 139.07, 139.05, 138.59, 136.35, 136.29, 133.67, 133.60, 130.89, 129.23, 128.76, 128.68, 128.58, 88.97, 87.00, 78.11, 77.69, 73.09, 72.70, 60.22, 27.01, 19.66 ppm; HRMS (ESI-TOF) (m/z): [M+Na]+ calcd. for C34H36FN5O4SiNa+ 648.2413, found 648.2411.
4.1.29. ((1S,3R,4R,7S)-3-(6-Amino-2-fluoro-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (30)
From compound 70, using the same procedure as for compound 15, compound 30 was obtained and purification by flash column chromatography (silica, EtOAc:hexanes 8:1) was completed to give nucleoside 30 (21 mg, 0.0543 mmol, 85%). 30: colorless oil; Rf = 0.25 (silica, 10% MeOH/DCM); (MeCN, c = 0.10); FT-IR (film) νmax 3311, 2949, 2837, 1646, 1408, 1113, 1014 cm−1; 1H NMR (CD3OD, 400 MHz) δ = 8.13 (s, 1 H), 7.29–7.24 (m, 5 H), 5.92 (s, 1 H), 4.97 (s, 1 H), 4.63 (s, 2 H), 4.22 (s, 1 H), 4.06 (d, J = 7.8 Hz, 1 H), 3.94 (d, J = 1.8 Hz, 2 H), 3.89 (d, J = 7.8 Hz, 1 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 161.19, 159.80, 159.10, 139.81, 138.73, 129.35, 129.16, 129.00, 89.70, 87.69, 78.38, 78.03, 73.52, 73.30, 58.08 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C18H19FN5O4+ 388.1416, found 388.1411.
4.1.30. (1S,3R,4R,7S)-3-(6-Amino-2-fluoro-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (31)
From compound 30, using the same procedure as for compound 12, compound 31 was obtained and purification by flash column chromatography (silica, 5% MeOH/DCM) was completed to give nucleoside 31 (5.3 mg, 0.0178 mmol, 69%). 31: white foam; Rf = 0.12 (silica, 10% MeOH/DCM); (MeCN, c = 0.10); FT-IR (film) νmax 3343, 2948, 2836, 2502, 2238, 2073, 1655, 1449, 1119, 1021, 977 cm−1; 1H NMR (CD3OD, 600 MHz) δ = 8.21 (s, 1 H), 5.92 (s, 1 H), 4.50 (s, 1 H), 4.34 (s, 1 H), 4.05 (d, J = 7.9 Hz, 1 H), 3.94 (s, 2 H), 3.88 (d, J = 7.9 Hz, 1 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 161.30, 159.91, 139.83, 90.14, 87.50, 80.97, 72.84, 71.42, 58.18 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C11H13FN5O4 298.0946, found 298.0945.
4.1.31. ((1S,3R,4R,7S)-3-(6-Amino-2-morpholino-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (32)
Compound 70 (18 mg, 0.0288 mmol) was dissolved in dry DMSO (0.30 mL) along with morpholine (0.0050 mL, 0.0570 mmol). The reaction mixture was heated to 95 °C for 24 h. The mixture was placed under vacuum to remove solvent, and the residue was dissolved in dry THF (0.2 mL). The reaction was cooled to 0 °C and treated with HF·pyridine (0.03 mL, 0.280 mmol). After 1 h at 0 °C, the reaction mixture was quenched with sat. aq. NaHCO3 (1 mL) and extracted with CH2Cl2. The combined organic extracts were washed brine (2 × 1 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, 5% MeOH/DCM) was completed to give compound 32 (10 mg, 0.0220 mmol, 76% over two steps). 32: light yellow oil; Rf = 0.54 (silica, 5% MeOH/DCM); (MeCN, c = 0.10); FT-IR (film) νmax 3399, 2962, 1605, 1474, 1408, 1293, 1056, 1017 cm−1; 1H NMR (CD3CN, 500 MHz) δ = 7.69 (s, 1H ), 7.34–7.25 (m, 5 H), 5.83 (s, 1 H), 5.73 (s, 2 H), 4.67 (s, 1 H), 4.61 (s, 2 H), 4.29 (s, 1 H), 3.99 (d, J = 7.9 Hz, 1 H), 3.88 (dd, J = 7.7, 3.2 Hz, 2 H), 3.84 (d, J = 7.9 Hz, 1 H), 3.67 (s, 8 H), 3.19–3.15 (m, 1 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 160.12, 156.51, 152.08, 138.84, 136.40, 129.26, 128.70, 128.60, 88.78, 86.85, 78.39, 78.12, 73.06, 72.73, 67.34, 58.23, 45.72 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C22H27N6O5+ 455.2037, found 455.2050.
4.1.32. (2R,3R,4S,5S)-4-(Benzyloxy)-5-((tert-butyldiphenylsilyloxy)methyl)-2-(2,6-dichloro-9H-purin-9-yl)-5-(tosyloxymethyl)tetrahydrofuran-3-yl acetate (71)
Peracetylated compound 56 (500 mg, 0.669 mmol) was dissolved in dry MeCN (5.0 mL) along with 2,6-dichloro-9H-purine (240 mg, 1.33 mmol) and BSA (0.41 mL 1.67 mmol). The reaction mixture was heated to 95 °C for 1.5 h. After cooling to 0 °C, TMSOTf (0.33 mL, 1.33 mmol) was slowly added and heating was resumed for 3 h at 80 °C. The reaction mixture was brought to rt, quenched with sat. aq. NaHCO3 (5 mL) and extracted with EtOAc. The combined organic extracts were washed with brine (2 × 5 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, hexanes:EtOAc:acetone 8:1:1) was completed to give compound 71 (374 mg, 0.428 mmol, 64%). 71: yellow foam; Rf = 0.51 (silica, 10% MeOH/ DCM); (CHCl3, c = 0.10). FT-IR (film) νmax 2932, 1750, 1595, 1557, 1428, 1360, 1227, 1189, 1177, 1112, 980, 883, 814, 744, 702, 667 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 8.09 (s, 1 H), 7.69 (d, J = 8.2 Hz, 2 H), 7.55–7.49 (m, 4 H), 7.40–7.32 (m, 8 H), 7.28 (d, J = 5.8 Hz, 2 H), 7.22–7.19 (m, 3 H), 6.02 (d, J = 5.2 Hz, 1 H), 5.67 (t, J = 6.0 Hz, 1 H), 4.60 (d, J = 5.9 Hz, 1 H), 4.55 (d, J = 11.4 Hz, 1 H), 4.48 (d, J = 11.4 Hz, 1 H), 4.36 (d, J = 10.7 Hz, 1 H), 4.26 (d, J = 10.7 Hz, 1 H), 3.76 (d, J = 10.8 Hz, 1 H), 3.71 (d, J = 10.8 Hz, 1 H), 2.38 (s, 3 H), 2.05 (s, 3 H), 0.99 (s, 9 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 176.98, 170.23, 153.42, 152.57, 152.49, 145.20, 144.80, 135.92, 135.77, 133.04, 132.60, 132.35, 131.71, 130.39, 130.08, 128.97, 128.77, 128.55, 128.32, 128.23, 128.09, 87.60, 87.28, 77.99, 77.58, 74.77, 68.68, 64.86, 27.22, 21.99, 20.92, 19.49 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C43H45Cl2N4O8SSi+ 875.2099, found 875.2097.
4.1.33. ((3S,4R,5R)-5-(6-(Benzylamino)-2-chloro-9H-purin-9-yl)-3-(benzyloxy)-2-((tert-butyldiphenylsilyloxy)methyl)-4-hydroxytetrahydrofuran-2-yl)methyl 4-methylbenzenesulfonate (72)
Compound 71 (104 mg, 0.119 mmol) was dissolved in MeOH (7.0 mL) and treated with benzylamine (0.065 mL, 0.594 mmol). The reaction mixture was heated at 55 °C for 12 h. The solvent was evaporated and purification of the residue by flash column chromatography (silica, hexanes:EtOAc 1:1) was completed to give compound 72 (100 mg, 0.111 mmol, 93%). 72: white foam; Rf = 0.48 (silica, hexanes:EtOAc 1:1); (CHCl3, c = 1.07); FT-IR (film) νmax 3325, 3069, 2931, 2858, 1619, 1581, 1533, 1496, 1472, 1454, 1428, 1354, 1310, 1189, 1176, 1103, 1029, 1019, 977, 910, 813, 788, 736, 701, 667 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.75–7.71 (m, 2 H), 7.54 (ddd, J = 17.0, 8.0, 1.4 Hz, 5 H), 7.42– 7.28 (m, 16 H), 7.23 (d, J = 7.9 Hz, 2 H), 6.36 (s, 1 H), 5.65 (d, J = 4.9 Hz, 1 H), 4.77 (s, 2 H), 4.70 (d, J = 11.4 Hz, 1 H), 4.64 (d, J = 11.4 Hz, 1 H), 4.59 (dd, J = 11.3, 6.0 Hz, 1 H), 4.48 (d, J= 5.9 Hz, 1 H), 4.38 (q, J = 10.5 Hz, 2 H), 3.77 (d, J = 10.8 Hz, 1 H), 3.71 (d, J = 6.7 Hz, 1 H), 3.66 (d, J = 10.8 Hz, 1 H), 2.39 (s, 3 H), 1.00 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 144.93, 139.28, 136.88, 135.68, 135.58, 132.86, 132.69, 132.31, 130.07, 129.88, 128.89, 128.84, 128.58, 128.44, 128.17, 128.14, 127.97, 127.89, 127.86, 89.73, 86.91, 79.47, 74.82, 74.55, 69.08, 64.92, 26.99, 21.75, 19.30 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C48H51ClN5O7SSi+ 904.2961, found 904.2961.
4.1.34. N-Benzyl-9-((1R,3R,4R,7S)-7-(benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-2-chloro-9H-purin-6-amine (73)
From compound 72, using the same procedure as for compound 68, compound 73 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 7:3) was completed to afford nucleoside 73 (59 mg, 0.0806 mmol, 68%). 73: white foam; Rf = 0.87 (silica, hexanes:EtOAc 1:1); (MeCN, c = 0.10); FT-IR (film) νmax 2930, 1619, 1577, 1455, 1428, 1346, 1311, 1208, 1112, 1046, 937, 858, 824, 741, 701 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.94 (s, 1 H), 7.68 (ddd, J = 8.0, 3.6, 1.4 Hz, 4 H), 7.46–7.26 (m, 15 H), 7.25–7.23 (m, 1 H), 6.32 (s, 1 H), 6.02 (s, 1 H), 4.81 (d, J = 9.3 Hz, 3 H), 4.66 (d, J = 11.6 Hz, 1 H), 4.57 (d, J = 11.6 Hz, 1 H), 4.23 (s, 1 H), 4.07 (d, J = 7.8 Hz, 1 H), 4.03 (d, J = 12.0 Hz, 1 H), 3.97 (d, J = 12.0 Hz, 1 H), 3.90 (d, J = 7.8 Hz, 1 H), 1.07 (s, 9 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 137.99, 137.10, 135.75, 135.65, 132.75, 132.68, 130.10, 128.91, 128.59, 128.25, 128.19, 128.04, 127.99, 127.92, 127.87, 99.72, 88.54, 86.67, 77.41, 76.91, 72.71, 72.53, 59.36, 26.90, 19.35 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C41H43ClN5O4Si+ 732.2767, found 732.2765.
4.1.35. ((1S,3R,4R,7S)-3-(6-(Benzylamino)-2-chloro-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (33)
From compound 73, using the same procedure as for compound 15, compound 33 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 7:3) was completed to give nucleoside 33 (39 mg, 0.0790 mmol, 96%). 33: light yellow oil; Rf = 0.17 (silica, hexanes:EtOAc 1:1); (MeCN, c = 0.50); FT-IR (film) νmax 3316, 2930, 1703, 1618, 1578, 1496, 1454, 1348, 1310, 1212, 1145, 1097, 1050, 931, 883, 857, 787, 739, 698, 679 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 7.73 (s, 1 H), 7.38–7.28 (m, 10 H), 6.56 (s, 1 H), 5.95 (s, 1 H), 4.80 (s, 2 H), 4.70 (d, J = 13.3 Hz, 2 H), 4.66 (d, J = 11.8 Hz, 1 H), 4.40 (s, 1 H), 4.11 (d, J = 7.8 Hz, 1 H), 3.98 (d, J = 12.5 Hz, 1 H), 3.88 (d, J = 7.7 Hz, 2 H), 3.38 (d, J = 0.5 Hz, 1 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 155.08, 154.97, 149.05, 138.05, 137.69, 137.21, 128.92, 128.63, 128.28, 128.22, 127.99, 127.95, 119.09, 88.47, 87.19, 77.83, 77.16, 72.59, 72.47, 57.84, 44.95 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C25H25ClN54O+ 495.1589, found 495.1591.
4.1.36. (1S,3R,4R,7S)-3-(6-(Benzylamino)-2-chloro-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (34)
From compound 33, using the same procedure as for compound 39, compound 34 was obtained and purification by preparative-plate chromatography (C18 silica, EtOAc) was completed to give nucleoside 34 (10 mg, 0.0248 mmol, 61%). 34: yellow oil; Rf = 0.71 (C18 silica, EtOAc); (MeOH, c = 0.86); FT-IR (film) νmax 3269, 2926, 1619, 1577, 1533, 1453, 1351, 1300, 1205, 1138, 1094, 1040, 930, 876, 847, 821, 786, 694, 678 cm−1; 1H NMR (CD3OD, 500 MHz) δ = 8.25 (s, 1 H), 7.46 (d, J = 7.3 Hz, 2 H), 7.39 (t, J = 7.5 Hz, 2 H), 7.32 (t, J = 7.2 Hz, 1 H), 6.02 (s, 1 H), 4.82 (s, 2 H), 4.57 (s, 1 H), 4.40 (s, 1 H), 4.12 (d, J = 7.9 Hz, 1 H), 4.02 (s, 2 H), 3.95 (d, J = 7.9 Hz, 1 H) ppm; 13C NMR (CD3OD, 126 MHz) δ = 149.14, 138.90, 138.55, 128.55, 127.84, 127.80, 127.34, 89.15, 86.53, 80.06, 71.83, 70.41, 57.19 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C18H19ClN5O4+ 404.1120, found 404.1122.
4.1.37. ((2S,3S,4R,5R)-5-(6-(Benzyl(methyl)amino)-2-chloro-9H-purin-9-yl)-3-(benzyloxy)-2-((tert-butyldiphenylsilyloxy)methyl)-4-hydroxytetrahydrofuran-2-yl)methyl 4-methylbenzenesulfonate (74)
Compound 71 (107 mg, 0.122 mmol) was dissolved in MeOH (2.14 mL), and N-benzylmethylamine (0.08 mL, 0.610 mmol) was added. The reaction was heated to 55 °C for 3 h. Evaporation and purification by flash column chromatography (silica, hexanes:EtOAc 3:1) were completed to give compound 74 (95 mg, 0.100 mmol, 82%). 74: white foam; Rf = 0.3 (silica, hexanes:EtOAc 2:1); (CHCl3, c = 0.70); FT-IR (film) νmax 2930, 2858, 1592, 1454, 1427, 1402, 1359, 1313, 1237, 1210, 1189, 1176, 1095, 974, 941, 908, 812, 730, 699, 666 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.73 (d, J = 8.3 Hz, 2 H), 7.60 (s, 1 H), 7.57–7.51 (m, 4 H), 7.43–7.28 (m, 15 H), 7.23 (d, J = 8.0 Hz, 3 H), 5.65 (d, J = 4.8 Hz, 1 H), 4.71 (d, J = 11.4 Hz, 1 H), 4.64 (d, J = 11.4 Hz, 1 H), 4.58 (dd, J = 11.5, 6.0 Hz, 1 H), 4.49 (d, J = 5.9 Hz, 1 H), 4.38 (d, J = 10.5 Hz, 1 H), 4.36 (d, J = 10.5 Hz, 1 H), 3.77 (d, J = 10.8 Hz, 1 H), 3.65 (dd, J = 8.7, 4.7 Hz, 3 H), 3.17 (s, 1 H), 2.39 (s, 3 H), 0.99 (s, 9 H) ppm; 13C NMR (CDCl3, 151 MHz) δ 144.94, 136.94, 135.71, 135.60, 132.83, 132.70, 132.34, 130.07, 129.89, 128.85, 128.80, 128.56, 128.46, 128.21, 127.99, 127.88, 127.70, 119.42, 89.85, 86.93, 79.58, 74.83, 74.68, 69.16, 64.93, 27.00, 21.79, 19.31 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C49H53ClN5O7SSi+ 918.3124, found 918.3110.
4.1.38. N-Benzyl-9-((1R,3R,4R,7S)-7-(benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-2-chloro-N-methyl-9H-purin-6-amine (75)
From 74, using the same procedure as for 68, compound 75 was obtained (73 mg, 0.0979 mmol, 98%). 75: white foam; Rf = 0.77 (silica, hexanes:EtOAc 2:1); (CHCl3, c = 1.18); FT-IR (film) νmax 3030, 2930, 2857, 1590, 1582, 1524, 1495, 1472, 1454, 1427, 1402, 1354, 1314, 1241, 1207, 1110, 1037, 939, 910, 885, 856, 824, 806, 784, 736, 701, 677 cm−1; 1H NMR (CD3CN, 500 MHz) δ = 7.92 (s, 1 H), 7.66 (dd, J = 9.3, 8.1 Hz, 4 H), 7.43 (dd, J = 10.6, 4.2 Hz, 2 H), 7.40– 7.24 (m, 14 H), 5.94 (s, 1 H), 4.73 (s, 1 H), 4.66 (d, J = 11.8 Hz, 1 H), 4.59 (d, J = 11.8 Hz, 1 H), 4.35 (s, 1 H), 4.06 (d, J = 12.1 Hz, 1 H), 4.03 (d, J = 12.1 Hz, 1 H), 3.98 (d, J = 8.0 Hz, 1 H), 3.88 (d, J = 7.9 Hz, 1 H), 3.64 (s, 1 H), 3.12 (s, 1 H), 1.00 (s, 9 H) ppm; 13C NMR (CD3CN, 126 MHz) δ = 138.66, 137.49, 136.43, 136.38, 133.81, 133.77, 130.98, 130.96, 129.61, 129.33, 128.86, 128.80, 128.69, 128.37, 120.10, 89.12, 87.12, 78.31, 77.80, 73.23, 72.87, 60.36, 27.16, 19.80 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C42H45ClN5O4Si+ 746.2929, found 746.2922.
4.1.39. ((1S,3R,4R,7S)-3-(6-(Benzyl(methyl)amino)-2-chloro-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (35)
From compound 75, using the same procedure as for compound 15, compound 35 was obtained and preparative-plate chromatography (silica, EtOAc) was completed to give nucleoside 35 (36 mg, 0.0710 mmol, 75%). 35: white foam; Rf = 0.67 (silica, EtOAc); (CHCl3, c = 1.13); FT-IR (film) νmax 3378, 2942, 1592, 1454, 1403, 1354, 1313, 1211, 1145, 1039, 938, 910, 856, 783, 751, 700, 677 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.75 (s, 1 H), 7.35–7.26 (m, 10 H), 5.99 (s, 1 H), 5.53 (s, 1 H), 5.00 (s, 1 H), 4.72–4.63 (m, 3 H), 4.42 (s, 1 H), 4.13 (d, J = 7.8 Hz, 1 H), 4.05–3.99 (m, 1 H), 3.98–3.93 (m, 1 H), 3.92 (t, J = 5.5 Hz, 1 H), 3.65 (s, 1 H), 3.21 (s, 1 H), 2.78 (s, 1 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 155.12, 154.13, 137.24, 136.52, 128.82, 128.62, 128.25, 127.97, 127.72, 119.39, 88.21, 87.12, 77.89, 77.36, 77.29, 72.58, 72.47, 58.12 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C26H27ClN5O4+ 508.1746, found 508.1750.
4.1.40. (1S,3R,4R,7S)-3-(6-(Benzyl(methyl)amino)-2-chloro-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (36)
From compound 35, using the same procedure as for compound 29, compound 36 was obtained and purification by preparative-plate chromatography (silica, EtOAc) was completed to give nucleoside 36 (36 mg, 0.0863 mmol, 75%). 36: white semi-solid; Rf = 0.34 (silica, EtOAc); (MeOH, c = 0.80); FT-IR (film) νmax 3354, 2928, 1592, 1453, 1403, 1356, 1313, 1215, 1131, 1038, 936, 905, 874, 845, 820, 783, 738, 701, 676 cm−1; 1H NMR (CD3OD, 600 MHz) δ = 8.15 (s, 1 H), 7.35–7.22 (m, 5 H), 5.96 (s, 1 H), 5.56 (s, 2 H), 4.49 (s, 1 H), 4.32 (s, 1 H), 4.05 (d, J = 7.9 Hz, 1 H), 3.94 (s, 2 H), 3.88 (d, J = 7.9 Hz, 1 H), 3.65 (s, 3 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 156.23, 155.04, 138.25, 129.68, 129.35, 129.17, 128.81, 128.55, 120.20, 90.10, 87.48, 81.03, 72.81, 71.37, 58.16 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C19H20ClN5O4+ 418.1277, found 418.1280.
4.1.41. ((1S,3R,4R,7S)-7-(Benzyloxy)-3-(2,6-dimethoxy-9H-purin-9-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (37)
Compound 71 (150 mg, 0.171 mmol) was dissolved in MeOH (2.0 mL) and THF (2.0 mL) and solid NaOH (100 mg, 2.50 mmol) was added. The reaction mixture was stirred at rt for 12 h. The reaction mixture was diluted with water and extracted with EtOAc. The combined organic extracts were washed with brine (2 × 4 mL), dried over MgSO4, filtered, and concentrated to give the cyclic compound (10 mg, 0.153 mmol, 90%), which was taken to the next step without further purification. The oil (10 mg, 0.153 mmol) was dissolved in dry THF (2.0 mL) along with HF·pyridine (0.20 mL, 2.00 mmol) at 0 °C. The reaction mixture was stirred at rt for 12 h. The reaction was quenched with sat. aq. NaHCO3 (1 mL) and extracted with CH2Cl2. The combined organic extracts were washed with brine (2 × 4 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, 7:3 hexanes:acetone) was completed to afford compound 37 (49 mg, 0.0118 mmol, 77% over two steps). 37: colorless semi-solid; Rf = 0.41 (silica, 10% MeOH/DCM); (CHCl3, c = 0.50); FT-IR (film) νmax 3300, 2951, 1594, 1480, 1397, 1363, 1255, 1144, 1040, 791, 742, 700 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 7.85 (d, J = 8.5 Hz, 1 H), 7.35–7.27 (m, 5 H), 6.01 (d, J = 3.2 Hz, 1 H), 4.68–4.57 (m, 3 H), 4.33 (s, 1 H), 4.19– 4.12 (m, 4 H), 4.06–3.97 (m, 5 H), 3.93 (dd, J = 7.8, 3.5 Hz, 1 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 162.42, 162.19, 152.48, 138.35, 137.27, 128.89, 128.57, 128.19, 118.07, 88.48, 87.02, 77.90, 72.93, 72.70, 58.15, 55.65, 54.76 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C20H23N5O4+ 415.1612, found 415.1629.
4.1.42. (1S,3R,4R,7S)-3-(2,6-Dimethoxy-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (38)
From compound 37, using the same procedure as for compound 17, compound 38 was obtained and purification by flash column chromatography (silica, DCM:MeOH 99:1) was completed to give nucleoside 38 (18 mg, 0.0555 mmol, 77%). 38: colorless oil; Rf = 0.18 (silica, 10% MeOH/DCM); (MeCN, c = 0.20); FT-IR (film) νmax 3341, 2951, 1594, 1507, 1480, 1397, 1362, 1255, 1240, 1138, 1082, 1034, 1012, 959, 937, 905, 875, 829, 806, 791, 770, 742, 722, 677 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 7.96 (s, 1 H), 5.90 (s, 1 H), 4.48 (s, 1 H), 4.40 (s, 1 H), 4.08 (s, 3 H), 4.01–3.95 (m, 4 H), 3.84 (dt, J = 17.7, 6.1 Hz, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 162.32, 162.17, 153.15, 139.10, 89.39, 86.95, 80.46, 72.43, 71.60, 58.11, 55.56, 54.77 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C13H17N4O +6 325.1143, found 325.1151.
4.1.43. 1,1′-(9-((1S,3R,4R,7S)-7-(Benzyloxy)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-9H-purine-2,6-diyl)diethanone (39)
Compound 71 (65 mg, 0.0742 mmol) was dissolved in dry, degassed DMF (2.0 mL) along with tributyl(1-ethoxyvinyl)tin (0.050 mL, 0.148 mmol) and Pd(PPh3)2Cl2 (5 mg, 0.00713 mmol). The reaction mixture was stirred at 95 °C for 18 h. It was then allowed to cool down and directly passed through a silica gel plug, affording a mixture of inseparable compounds that was taken to the next step without further purification. The oil was dissolved in THF (3.3 mL) along with 2 M NaOH (0.30 mL) and stirred at rt for 12 h. The reaction mixture was diluted with H2O (5 mL) and extracted with EtOAc. The combined organic extracts were washed with brine (2 × 5 mL), dried over MgSO4, filtered, and concentrated to afford a yellow foam, which was dissolved in dry THF (0.50 mL) along with HF·pyridine (0.10 mL, 0.860 mmol) at 0 °C. The reaction mixture was stirred at rt for 6 h. It was then quenched with sat. aq. NaHCO3 (3 mL) and extracted with CH2Cl2. The combined organic extracts were washed with brine (2 × 3 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, hexanes:EtOAc:acetone 5:4:1) was completed to give compound 39 (10 mg, 0.0228 mmol, 31% over three steps). 39: colorless oil; Rf = 0.44 (silica, 10% MeOH/DCM); (MeOH, c = 0.10); FT-IR (film) νmax 3408, 2925, 1708, 1580, 1363, 1210, 1138, 1054, 1033, 700 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.66 (s, 1 H), 7.32–7.23 (m, 5 H), 6.16 (s, 1 H), 4.75 (s, 1 H), 4.60 (s, 2 H), 4.30 (s, 1 H), 4.07–4.02 (m, 1 H), 3.93 (m, 3 H), 2.86 (s, 3 H), 2.81 (s, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 199.20, 197.38, 177.55, 149.42, 148.99, 138.77, 129.23, 128.69, 128.62, 89.53, 87.59, 78.22, 78.09, 73.21, 72.78, 57.99, 28.45, 27.58 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C22H23N4O6+ 439.1612, found 439.1612.
4.1.44. 2-Allyl-9-((1R,3R,4R,7S)-7-(benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-6-((E)-prop-1-enyl)-9H-purine (76)
Compound 71 (150 mg, 0.171 mmol) was dissolved in dry, degassed DMF (0.50 mL) along with allyl(tri-n-butyl)tin (0.16 mL, 0.513 mmol) and Pd(PPh3)2Cl2 (12 mg, 0.0171 mmol). The reaction mixture was stirred at 95 °C for 6 h. It was allowed to cool down and directly passed through a silica gel plug, affording a mixture of inseparable compounds that were taken to the next step without further purification. The yellow oil residue was dissolved in THF (0.60 mL) along with 2 M NaOH (0.10 mL) and stirred at rt for 12 h. The reaction mixture was diluted with H2O (1 mL) and extracted with EtOAc. The combined organic extracts were washed with brine (2 × 2 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, 4:1 hexanes:EtOAc) was completed to give compound 76 (48 mg, 0.0714 mmol, 42% over two steps). 76: light yellow oil; Rf = 0.90 (silica, 5% MeOH/DCM); (MeOH, c = 0.20); FT-IR (film) ν 2931, 1579, 1428, 1365, 1211, 1112, 1038, 911, 823, 741, 702 cm−1; 1 max H NMR (CD3CN, 600 MHz) δ = 8.20 (s, 1 H), 7.68 (dd, J = 6.7, 4.9 Hz, 4 H), 7.66–7.60 (m, 1 H), 7.47– 7.42 (m, 2 H), 7.37 (dt, J = 12.8, 7.4 Hz, 4 H), 7.29–7.23 (m, 5 H), 6.90 (dd, J = 15.7, 1.7 Hz, 1 H), 6.22 (ddt, J = 17.0, 10.2, 6.8 Hz, 1 H), 6.04 (s, 1 H), 5.16 (dd, J = 17.2, 1.9 Hz, 1 H), 5.09 (dd, J = 10.1, 1.9 Hz, 1 H), 4.82 (s, 1 H), 4.65 (d, J = 11.8 Hz, 1 H), 4.59 (d, J = 11.8 Hz, 1 H), 4.44 (s, 1 H), 4.08 (d, J = 12.1 Hz, 1 H), 4.04 (d, J = 12.1 Hz, 1 H), 4.00 (d, J = 7.9 Hz, 1 H), 3.90 (d, J = 7.9 Hz, 1 H), 3.71 (dt, J = 6.8, 1.5 Hz, 2 H), 2.04 (dd, J = 6.9, 1.7 Hz, 3 H), 1.01 (s, 9 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 163.74, 154.46, 152.40, 142.59, 140.85, 138.66, 136.71, 136.40, 136.35, 133.74, 133.69, 130.94, 130.00, 129.27, 128.82, 128.81, 128.72, 128.57, 127.63, 116.70, 89.02, 87.15, 78.46, 77.79, 73.22, 72.80, 60.35, 44.52, 27.08, 19.73, 19.18 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C40H45N4O4Si+ 673.3204, found 673.3211.
4.1.45. ((1S,3R,4R,7S)-3-(2-Allyl-6-((E)-prop-1-enyl)-9H-purin-9-yl)-7-(benzyloxy)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (40)
From compound 76, using the same procedure as for compound 40, compound 40 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 1:1) was completed to give nucleoside 40 (28 mg, 0.0645 mmol, 87%). 40: colorless oil; Rf = 0.31 (silica, 5% MeOH/chloroform); (MeOH, c = 0.70); FT-IR (film) νmax 3347, 2941, 1653, 1578, 1492, 1454, 1368, 1288, 1213, 1142, 1096, 1054, 1033, 976, 909, 882, 860, 811, 738, 698 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.19 (s, 1 H), 7.68–7.58 (m, 1 H), 7.32–7.22 (m, 5 H), 6.92–6.85 (m, 1 H), 6.21 (ddt, J = 17.0, 10.2, 6.8 Hz, 1 H), 6.02 (s, 1 H), 5.16 (ddd, J = 17.2, 3.4, 1.6 Hz, 1 H), 5.10 (ddd, J = 10.1, 1.9, 1.4 Hz, 1 H), 4.71 (s, 1 H), 4.64–4.56 (m, 2 H), 4.34 (s, 1 H), 4.02 (d, J = 7.8 Hz, 1 H), 3.93– 3.87 (m, 3 H), 3.70 (dd, J = 6.8, 1.5 Hz, 2 H), 3.46 (s, 1 H), 2.04 (dd, J = 6.9, 1.7 Hz, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 163.64, 154.47, 152.52, 142.83, 140.88, 138.81, 136.70, 130.09, 129.24, 128.68, 128.59, 127.62, 116.76, 89.17, 87.41, 78.31, 78.29, 73.13, 72.73, 58.13, 44.47, 19.17 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C24H27N4O4+ 435.2027, found 435.2015.
4.1.46. (1S,3R,4R,7S)-3-(2,6-Dipropyl-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (41)
Protected compound 40 (10 mg, 0.0230 mmol) was dissolved in wet ethanol (1.2 mL) along with Pd(OH)2 (1 mg, 0.00712 mmol). The mixture was degassed and put under hydrogen atmosphere. The reaction mixture was heated to 50 °C for 12 h. The mixture was filtered and the solvent was concentrated under reduced pressure. Purification by flash column chromatography (silica, DCM:MeOH 95:5) was completed to give compound 41 (4 mg, 0.0115 mmol, 50%). 41: colorless oil; Rf = 0.19 (silica, hexanes:acetone 7:3); (MeCN, c = 0.10); FT-IR (film) νmax 3300, 2927, 1592, 1490, 1368, 1050, 810, 738, 698 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.20 (s, 1 H), 5.99 (s, 1 H), 4.47 (s, 1 H), 4.43 (s, 1 H), 4.00 (d, J = 7.9 Hz, 1 H), 3.88 (d, J = 4.6 Hz, 2 H), 3.84 (d, J = 8.0 Hz, 1 H), 3.06–3.03 (m, 2 H), 2.92–2.88 (m, 2 H), 1.88–1.82 (m, 4 H), 0.97 (t, J = 7.4 Hz, 6 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 165.96, 162.85, 151.35, 142.25, 131.84, 89.23, 86.70, 80.49, 72.49, 71.74, 58.27, 41.80, 35.63, 22.95, 22.44, 14.28, 13.95 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C17H25N4O4+ 349.187, found 349.1884.
4.1.47. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-2,6-di(furan-2-yl)-9H-purine (77)
Compound 71 (83 mg, 0.0948 mmol) was dissolved in dry, degassed DMF (2.0 mL) along with 2-(tributylstannyl)furan (0.060 mL, 0.189 mmol) and Pd(PPh3)2Cl2 (5 mg, 0.00712 mmol). The reaction mixture was stirred at 95 °C for 13 h. It was then allowed to cool to rt and directly passed through a silica gel plug, affording the compound as a yellow oil (88 mg, 0.0937 mmol, 99%), which was taken to the next step without further purification. The yellow oil (85 mg, 0.0905 mmol) was dissolved in THF (5.0 mL) along with 2 M NaOH (0.40 mL) and stirred at rt for 12 h. The reaction mixture was diluted with H2O (5 mL) and extracted with EtOAc. The combined organic extracts were washed with brine (2 × 5 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, hexanes:EtOAc 4:1) was completed to give compound 77(50 mg, 0.0690 mmol, 76%). 77: yellow foam; Rf = 0.94 (silica, hexanes:EtOAc 7:3); (MeOH, c = 1.00); FT-IR (film) ν 3602, 2937, 1584, 1053, 1033, 1012, 702 cm−1 max ; 1H NMR (CD3CN, 500 MHz) δ = 8.31 (s, 1 H), 7.92–7.86 (m, 2 H), 7.72 (dd, J = 1.7, 0.9 Hz, 1 H), 7.71–7.66 (m, 4 H), 7.46–7.42 (m, 2 H), 7.41–7.34 (m, 6 H), 7.27–7.23 (m, 4 H), 6.75 (dt, J = 2.9, 1.4 Hz, 1 H), 6.65 (dt, J = 2.9, 1.4 Hz, 1 H), 6.13 (s, 1 H), 4.91 (s, 1 H), 4.68 (d, J = 11.8 Hz, 1 H), 4.64 (d, J = 11.8 Hz, 1 H), 4.48 (s, 1 H), 4.09 (q, J = 12.1 Hz, 2 H), 4.03 (d, J = 7.9 Hz, 1 H), 3.94 (d, J = 7.9 Hz, 1 H), 1.03 (s, 9 H) ppm; 13C NMR (CD3CN, 126 MHz) δ = 153.68, 152.84, 152.61, 150.49, 147.13, 146.62, 145.89, 143.67, 138.74, 136.45, 136.40, 133.83, 133.76, 130.97, 129.30, 128.86, 128.74, 128.61, 128.46, 118.75, 113.72, 113.69, 113.19, 89.23, 87.34, 78.51, 77.90, 73.30, 72.88, 60.45, 27.14, 19.79 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C42H41N4O6Si+ 725.2790, found 725.2798.
4.1.48. ((1S,3R,4R,7S)-7-(Benzyloxy)-3-(2,6-di(furan-2-yl)-9H-purin-9-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (42)
From compound 77, using the same procedure as for compound 15, compound 42 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 7:3) was completed to give nucleoside 42 (27 mg, 0.0552 mmol, 100%). 42: light yellow foam; Rf = 0.38 (silica, hexanes:EtOAc 7:3); (MeOH, c = 0.50); FT-IR (film) νmax 3375, 2943, 1584, 1487, 1367, 1208, 1144, 1102, 1050, 1012, 938, 895, 885, 827, 801, 747, 698 cm−1; 1H NMR (1% D2O/CD3CN, 400 MHz) δ = 8.34 (s, 1 H), 7.90 (dd, J = 3.5, 0.8 Hz, 1 H), 7.87 (dd, J = 1.8, 0.8 Hz, 1 H), 7.73 (dd, J = 1.8, 0.9 Hz, 1 H), 7.35 (dd, J = 3.4, 0.9 Hz, 1 H), 7.29–7.22 (m, 5 H), 6.75 (dd, J = 3.5, 1.8 Hz, 1 H), 6.65 (dd, J = 3.4, 1.8 Hz, 1 H), 6.10 (s, 1 H), 4.80 (s, 1 H), 4.62 (s, 2 H), 4.34 (s, 1 H), 4.06–4.04 (m, 1 H), 3.93–3.91 (m, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 153.54, 152.67, 152.48, 150.32, 147.00, 146.44, 145.78, 143.71, 138.74, 129.17, 128.60, 128.52, 118.68, 113.59, 113.09, 89.21, 87.23, 78.26, 78.08, 73.13, 72.70, 58.05 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C26H23N4O6+ 487.1612, found 487.1623.
4.1.49. (1S,3R,4R,7S)-3-(2,6-Di(furan-2-yl)-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (43)
From compound 42, using the same procedure as for compound 12, compound 43 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 7:3) was completed to afford nucleoside 43 (11 mg, 0.0278 mmol, 90%). 43: off-white foam; Rf = 0.15 (silica, hexanes:EtOAc 7:3); (MeOH, c = 0.25); FT-IR (film) ν 3340, 2949, 2838, 1647, 1407, 1113, 1014, 550, 530 cm−1 max ; 1H NMR (CD3OD, 400 MHz) δ = 8.57 (s, 1 H), 7.91 (dd, J = 1.7, 0.7 Hz, 1 H), 7.83 (dd, J = 3.5, 0.7 Hz, 1 H), 7.75 (dd, J = 1.7, 0.8 Hz, 1 H), 7.40 (dd, J = 3.4, 0.8 Hz, 1 H), 6.76 (dd, J = 3.5, 1.7 Hz, 1 H), 6.65 (dd, J = 3.4, 1.8 Hz, 1 H), 6.18 (s, 1 H), 4.70 (s, 1 H), 4.45 (s, 1 H), 4.10 (d, J = 7.9 Hz, 1 H), 3.99 (s, 2 H), 3.94 (d, J = 7.9 Hz, 1 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 153.68, 153.57, 151.15, 147.59, 147.23, 146.10, 144.57, 118.24, 114.21, 113.68, 113.26, 90.27, 87.64, 81.06, 72.91, 71.58, 58.18 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C19H17N4O6+ 397.1143, found 397.1148.
4.1.50. 9-((1R,3R,4R,7S)-7-(Benzyloxy)-1-((tert-butyldiphenylsilyloxy)methyl)-2,5-dioxabicyclo[2.2.1]heptan-3-yl)-2,6-di(thiophen-2-yl)-9H-purine (78)
Compound 71 (40 mg, 0.0457 mmol) was dissolved in dry, degassed DMF (1.0 mL) along with 2-(tributylstannyl)thiophene (0.029 mL, 0.091 mmol) and Pd(PPh3)2Cl2 (4 mg, 0.00570 mmol). The reaction mixture was stirred at 80 °C for 7 h. It was then allowed to cool to rt and directly passed through a silica gel plug, affording the compound as yellow oil (32 mg, 0.0330 mmol, 72%), which was advanced to the next chemical transformation without further purification. The yellow oil (28 mg, 0.0289 mmol) was dissolved in THF (2.4 mL) along with 2 M NaOH (0.20 mL) and stirred at rt for 13.5 h. The reaction mixture was diluted with H2O (4 mL) and extracted with EtOAc. The combined organic extracts were washed with brine (2 × 5 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, hexanes:EtOAc 7:3) was completed to give compound 78 (21.3 mg, 0.0282 mmol, 98%). 78: white foam; Rf = 0.97 (silica, hexanes:EtOAc 7:3); (MeOH, c = 1.00); FT-IR (film) νmax 3477, 2979, 1709, 1422, 1360, 1221, 1066, 903 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.70–8.68 (m, 1 H), 8.34 (s, 1 H), 8.08–8.06 (m, 1 H), 7.80–7.78 (m, 1 H), 7.74–7.70 (m, 4 H), 7.61–7.59 (m, 1 H), 7.49–7.44 (m, 3 H), 7.41 (ddd, J = 21.6, 11.2, 3.8 Hz, 5 H), 7.35–7.33 (m, 1 H), 7.27–7.25 (m, 3 H), 7.23–7.20 (m, 1 H), 6.16 (s, 1 H), 4.97 (s, 1 H), 4.68 (d, J = 11.8 Hz, 1 H), 4.61 (d, J = 11.8 Hz, 1 H), 4.46 (s, 1 H), 4.15 (d, J = 12.0 Hz, 1 H), 4.11 (d, J = 12.1 Hz, 1 H), 4.06 (d, J = 7.8 Hz, 1 H), 3.97 (d, J = 7.8 Hz, 1 H), 1.06 (s, 9 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 177.49, 156.17, 152.74, 150.31, 144.42, 143.53, 140.94, 138.61, 136.42, 136.36, 133.79, 133.68, 132.33, 130.95, 130.58, 129.91, 129.43, 129.28, 129.25, 128.85, 128.83, 128.70, 128.58, 89.20, 87.40, 78.38, 77.87, 73.27, 72.85, 60.46, 27.12, 19.76 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C42H41N4O4S2Si 757.2333, found 757.2332.
4.1.51. ((1S,3R,4R,7S)-7-(Benzyloxy)-3-(2,6-di(thiophen-2-yl)-9H-purin-9-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methanol (44)
From compound 78, using the same procedure as for compound 15, compound 44 was obtained and purification by flash column chromatography (silica, hexanes:EtOAc 7:3) was completed to give nucleoside 44 (27 mg, 0.0521 mmol, 99%). 44: white foam; Rf = 0.43 (silica, hexanes:EtOAc 7:3); (MeOH, c = 0.50); FT-IR (film) νmax 3338, 2951, 1642, 1572, 1428, 1391, 1327, 1206, 1032, 1015, 820, 591, 541, 528 cm−1; 1H NMR (CD3CN, 400 MHz) δ = 8.68 (dd, J = 3.8, 1.2 Hz, 1 H), 8.34 (s, 1 H), 8.05 (dd, J = 3.7, 1.3 Hz, 1 H), 7.76 (dd, J = 5.0, 1.2 Hz, 1 H), 7.58 (dd, J = 5.0, 1.3 Hz, 1 H), 7.31 (dt, J = 6.1, 3.0 Hz, 1 H), 7.29–7.25 (m, 4 H), 7.25–7.21 (m, 1 H), 7.20 (dd, J = 5.0, 3.7 Hz, 1 H), 6.10 (s, 1 H), 4.85 (s, 1 H), 4.61 (m, 2 H), 4.34 (s, 1 H), 4.05 (d, J = 8.0 Hz, 1 H), 3.98–3.90 (m, 3 H), 3.27 (t, J = 6.0 Hz, 1 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 156.12, 152.74, 150.25, 144.42, 143.67, 140.93, 138.76, 133.64, 132.29, 130.55, 129.89, 129.40, 129.28, 129.22, 128.67, 128.59, 128.58, 89.28, 87.35, 78.32, 78.12, 73.19, 72.79, 58.15 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C26H22N4O4S2 519.1155, found 519.1151.
4.1.52. (1S,3R,4R,7S)-3-(2,6-Di(thiophen-2-yl)-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (45) and (1S,3S,4R,7S)-3-(2,6-Di(thiophen-2-yl)-9H-purin-9-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-ol (46)
From compound 44, using the same procedure as for compound 17, compounds 45 and 46 were obtained. Purification by flash column chromatography (silica, hexanes:EtOAc 7:3) was completed to give compound 45 (4.0 mg, 0.00934 mmol, 40%) and compound 46 (3.3 mg, 0.00770 mol, 33%). 45: white foam; Rf = 0.14 (silica, hexanes:EtOAc 7:3); (MeOH, c = 0.20); FT-IR (film) νmax 3361, 1572, 1535, 1443, 1426, 1376, 1222, 1038, 907, 823, 799, 711 cm−1; 1H NMR (CD3OD, 500 MHz) δ = 8.61 (d, J = 3.7 Hz, 1 H), 8.50 (s, 1 H), 8.04 (d, J = 3.5 Hz, 1 H), 7.75 (d, J = 5.0 Hz, 1 H), 7.56 (d, J = 4.9 Hz, 1 H), 7.32–7.25 (m, 1 H), 7.18–7.13 (m, 1 H), 6.13 (s, 1 H), 4.73 (s, 2 H), 4.48 (s, 1 H), 4.12 (d, J = 7.8 Hz, 1 H), 3.99 (s, 2 H), 3.96 (d, J = 7.8 Hz, 1 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 156.93, 152.85, 150.98, 144.73, 144.09, 141.36, 133.66, 132.29, 130.54, 129.61, 129.03, 128.54, 90.19, 87.63, 81.10, 72.97, 71.76, 58.30 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C19H17N4S2O4 429.0686, found 429.0691. 46: colorless oil; Rf = 0.54 (silica, hexanes:EtOAc 7:3); (MeOH, c = 0.40); FT-IR (film) νmax 3330, 2982, 1574, 1444, 1426, 1378, 1212, 1057, 1033, 800, 723 cm−1; 1H NMR (CD3OD, 400 MHz) δ = 8.68 (s, 1 H), 8.64 (dd, J = 3.7, 1.2 Hz, 1 H), 8.08 (dd, J = 3.7, 1.2 Hz, 1 H), 7.79 (dd, J = 5.0, 1.1 Hz, 1 H), 7.60 (dd, J = 5.0, 1.2 Hz, 1 H), 7.30 (dd, J = 5.0, 3.8 Hz, 1 H), 7.19 (ddd, J = 5.3, 3.7, 1.6 Hz, 1 H), 6.82 (d, J = 7.4 Hz, 1 H), 4.76 (dd, J = 7.4, 3.5 Hz, 1 H), 4.29 (d, J = 9.6 Hz, 1 H), 4.12 (d, J = 9.5 Hz, 1 H), 4.04 (d, J = 9.5 Hz, 1 H), 3.90–3.86 (m, 2 H) ppm; 13C NMR (CD3OD, 151 MHz) δ = 157.51, 153.16, 151.41, 144.91, 144.41, 141.01, 133.95, 132.67, 130.94, 129.96, 129.77, 129.13, 111.74, 89.41, 88.39, 77.81, 76.31, 67.80, 66.35 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C19H17N4O4S2 429.0686, found 429.0688.
4.1.53. ((1R,3R,4R,7S)-7-(Benzyloxy)-3-(2,6-di(thiophen-2-yl)-9H-purin-9-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl)methyl sulfamate (47)
Formic acid (0.055 mL, 1.45 mmol) was added to chlorosulfonyl isocyanate (0.13 mL, 1.45 mmol) at 0 °C with stirring. The resulting white solid was dissolved in CH2Cl2 (1.4 mL) and the solution warmed to rt and stirred for 14 h. The solution was cooled to 0 °C and a portion of this reagent (0.04 mL) was added dropwise to a solution of compound 44 (5 mg, 0.00964 mmol) in CH2Cl2 (0.08 mL) at 0 °C. Pyridine (0.005 mL, 0.0556) was added dropwise and the reaction mixture was warmed to rt and stirred 24 h. The solvent was evaporated and purification by preparative-plate chromatography (silica, hexanes:EtOAc 1:1) was completed to give compound 47 (2 mg, 0.00337 mmol, 35%). 47: white semi-solid; Rf = 0.37 (silica, hexanes:EtOAc 1:1); (acetone, c = 0.15); FT-IR (film) νmax 2925, 1572, 1535, 1487, 1443, 1426, 1376, 1245, 1183, 1148, 1094, 1039, 994, 911, 855, 818, 800, 782, 724 cm−1; 1H NMR (acetone-d6, 600 MHz) δ = 8.73 (dt, J = 3.7, 1.1 Hz, 1 H), 8.49 (d, J = 1.1 Hz, 1 H), 8.07 (dt, J = 3.6, 1.2 Hz, 1 H), 7.86 (dt, J = 5.0, 1.2 Hz, 1 H), 7.66 (dt, J = 5.0, 1.2 Hz, 1 H), 7.36–7.30 (m, 3 H), 7.25 (ddd, J = 7.1, 4.3, 1.1 Hz, 2 H), 7.21 (ddd, J = 6.1, 3.9, 1.2 Hz, 2 H), 7.01 (s, 2 H), 6.25 (s, 1 H), 5.09 (s, 1 H), 4.73 (ddd, J = 28.3, 14.7, 6.4 Hz, 3 H), 4.65 (d, J = 11.3 Hz, 2 H), 4.22 (d, J = 7.8 Hz, 1 H), 4.05 (d, J = 7.8 Hz, 1 H) ppm; 13C NMR (acetone-d6, 151 MHz) δ = 156.11, 152.60, 150.22, 144.37, 143.70, 140.96, 138.60, 133.70, 132.25, 130.47, 129.67, 129.33, 129.09, 129.02, 128.58, 128.54, 128.46, 87.55, 86.32, 78.78, 78.03, 73.06, 72.74, 65.93 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C26H24N5O6S3+ 598.0883, found 598.0885.
4.1.54. (2R,3R,4S)-4-(Benzyloxy)-2-(2,6-dichloro-9H-purin-9-yl)-5,5-bis((methylsulfonyloxy)methyl)tetrahydrofuran-3-yl acetate (80)
Literature-known compound 79 (290 mg, 0.569 mmol) was dissolved in dry MeCN (1.5 mL) along with 2, 6-dichloropurine (214 mg, 1.13 mmol) and BSA (0.51 mL, 1.99 mmol). After 5 min, the reaction mixture was cooled to –40 °C followed by slow addition of TMSOTf (0.21 mL, 0.850 mmol), and then heated to 80 °C in the microwave (CEM Discover, 300W). The reaction mixture was brought to rt, quenched with sat. aq. NaHCO3 (2 mL), and extracted with EtOAc. The combined organic extracts were washed with brine (3 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, hexanes:EtOAc 4:1) was completed to give compound 80 (30 mg, 0.478 mmol, 84%). 80: yellow foam; Rf = 0.56 (silica, 5% MeOH/DCM); (CHCl3, c = 0.10); FT-IR (film) νmax 3019, 1749, 1595, 1560, 1496, 1357, 1226, 1175, 1030, 963, 883, 826, 755, 702 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 8.18 (s, 1 H), 7.43– 7.32 (m, 5 H), 6.17 (d, J = 3.6 Hz, 1 H), 5.87 (dd, J = 5.9, 3.7 Hz, 1 H), 4.97 (d, J = 6.0 Hz, 1 H), 4.71–4.59 (m, 3 H), 4.42 (d, J = 10.9 Hz, 1 H), 4.35 (d, J = 11.7 Hz, 1 H), 4.30 (d, J = 10.9 Hz, 1 H), 3.04 (s, 3 H), 2.93 (s, 3 H), 2.14 (s, 3 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 207.26, 169.83, 153.77, 152.49, 151.89, 145.32, 136.39, 128.94, 128.82, 88.40, 84.96, 77.26, 75.08, 73.66, 67.48, 67.38, 38.00, 37.71, 31.10 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C22H25Cl2N4O10S2+ 639.0389, found 639.0388.
4.1.55. ((3S,4R,5R)-5-(2,6-Dichloro-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2,2-diyl)bis(methylene) dimethanesulfonate (81)
From compound 80, using the same procedure as for compound 17, compound 81 was obtained and purification by flash column chromatography (silica, hexanes:acetone 7:3) was completed to give nucleoside 81 (270 mg, 0.534 mmol, 90%). 81: colorless semi-solid; Rf = 0.40 (silica, 5% MeOH/DCM); (MeCN, c = 0.10); FT-IR (film) νmax 3368, 1726, 1660, 1596, 1561 1355, 1252, 1174, 1031, 999, 965, 884, 830 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.47 (d, J = 2.2 Hz, 1 H), 6.08 (dd, J = 6.3, 2.1 Hz, 1 H), 4.97– 4.93 (m, 1 H), 4.57–4.54 (m, 2 H), 4.45 (ddd, J = 15.6, 13.0, 6.5 Hz, 3 H), 4.21–4.09 (m, 2 H), 3.09 (t, J = 2.6 Hz, 6 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 153.97, 152.71, 151.45, 146.37, 132.10, 99.58, 89.05, 85.08, 73.50, 72.25, 69.17, 37.53, 29.24 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C13H17Cl2N4O9S2+ 506.9814, found 506.9810.
4.1.56. ((1R,3R,4R,5S)-3-(2,6-Dichloro-9H-purin-9-yl)-4-hydroxy-2,6-dioxabicyclo[3.2.0]heptan-1-yl)methyl methanesulfonate (48)
Compound 81 (200 mg, 0.364 mmol) was dissolved in wet THF (15 mL) along with K2CO3 (200 mg, 1.45 mmol). The reaction mixture was stirred at rt for 18 h. The reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with CH2Cl2. The combined organic extracts were washed with brine (2 × 10 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, hexanes:acetone 1:1) was completed to afford compound 48 (118 mg, 0.288 mmol, 79%). 48: colorless semi-solid; Rf = 0.48 (silica, 5% MeOH/DCM); (MeCN, c = 0.30); FT-IR (film) νmax 3615, 2293, 2253, 1443, 1375, 1039, 918, 750 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.51 (s, 1 H), 6.43 (d, J = 7.3 Hz, 1 H), 5.21 (dd, J = 16.9, 4.6 Hz, 1 H), 4.94–4.86 (m, 1 H), 4.67 (ddd, J = 21.2, 12.0, 6.5 Hz, 2 H), 4.54 (tt, J = 7.2, 3.5 Hz, 2 H), 3.75 (dd, J = 7.7, 1.4 Hz, 1 H), 3.07 (s, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 154.49, 153.31, 151.76, 146.28, 132.77, 89.19, 85.10, 84.51, 78.45, 75.12, 69.06, 37.78 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C12H13Cl2N4O6S+ 410.9933, found 410.9937.
4.1.57. ((1R,3R,4R,5S)-3-(6-(benzoyloxy)-2-chloro-9H-purin-9-yl)-4-hydroxy-2,6-dioxabicyclo[3.2.0]heptan-1-yl)methyl benzoate (49)
Compound 48 (100 mg, 0.244 mmol) was dissolved in dry DMF (0.50 mL) along with sodium benzoate (70 mg, 0.486 mmol). The reaction mixture was heated to 90 °C for 4.5 h. Direct purification of the mixture by flash column chromatography (silica, 7:3 hexanes:EtOAc) was completed to give compound 49 (105 mg, 0.201 mmol, 82%). 49: white foam; Rf = 0.45 (silica, 5% MeOH/DCM); (MeCN, c = 0.30); FT-IR (film) νmax 3618, 2293, 2253, 1443, 1375, 1038, 918, 749 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.08–7.99 (m, 5 H), 7.68–7.62 (m, 2 H), 7.52 (dt, J = 19.1, 7.7 Hz, 4 H), 6.74 (d, J = 6.8 Hz, 1 H), 5.68 (dt, J = 9.9, 4.6 Hz, 2 H), 5.01 (d, J = 8.3 Hz, 1 H), 4.78 (d, J = 8.3 Hz, 1 H), 4.73 (d, J = 12.2 Hz, 1 H), 4.69 (d, J = 12.2 Hz, 1 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 166.75, 166.03, 157.89, 149.13, 145.52, 139.43, 134.70, 134.42, 130.53, 130.51, 130.39, 129.75, 129.69, 129.64, 124.93, 87.16, 85.69, 84.48, 79.14, 77.35, 64.33 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C25H20ClN4O7+ 523.1015, found 523.1015.
4.1.58. (2R,3R,4S)-4-(benzyloxy)-2-(2,6-di(furan-2-yl)-9H-purin-9-yl)-5,5-bis((methylsulfonyloxy)methyl)tetrahydrofuran-3-yl acetate (82)
Compound 80 (50 mg, 0.0781 mmol) was dissolved in dry, degassed DMF (0.20 mL) along with 2-(tributylstannyl)furan (0.098 mL, 0.310 mmol) and Pd(PPh3)2Cl2 (5 mg, 0.0071 mmol). The reaction mixture was stirred at 95 °C for 3 h, quenched with a solution of potassium fluoride (4 mL), and extracted with EtOAc. The combined organic extracts were washed with brine (2 × 2 mL), dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, EtOAc:hexanes 4:1) was completed to give compound 82 (46 mg, 0.0655 mol, 84%). 82: yellow foam; Rf = 0.58 (silica, 10% MeOH/DCM); (MeOH, c = 0.20); FT-IR (film) νmax 1748, 1585, 1488, 1357, 1226, 1176, 966, 825, 753 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.31 (s, 1 H), 7.92 (d, J = 3.5 Hz, 1 H), 7.88 (d, J = 1.7 Hz, 1 H), 7.74 (d, J = 0.8 Hz, 1 H), 7.45–7.32 (m, 6 H), 6.76 (dd, J = 3.5, 1.7 Hz, 1 H), 6.65 (dd, J = 3.4, 1.7 Hz, 1 H), 6.34 (d, J = 3.4 Hz, 1 H), 6.16 (dd, J = 5.9, 3.4 Hz, 1 H), 5.28 (d, J = 5.9 Hz, 1 H), 4.75 (s, 2 H), 4.63 (d, J = 11.3 Hz, 1 H), 4.55 (d, J = 10.6 Hz, 1 H), 4.49 (d, J = 10.7 Hz, 1 H), 4.44 (d, J = 11.3 Hz, 1 H), 3.07 (s, 3 H), 2.91 (s, 3 H), 2.08 (s, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 170.51, 153.48, 152.78, 152.67, 150.19, 147.25, 146.83, 146.06, 145.76, 137.85, 129.28, 129.16, 129.09, 118.98, 113.74, 113.22, 88.77, 85.17, 79.59, 75.66, 74.33, 69.20, 69.14, 37.77, 37.34, 20.71 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C30H31N4O12S2+ 703.1374, found 703.1374.
4.1.59. ((3S,4R,5R)-5-(2,6-di(furan-2-yl)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2,2-diyl)bis(methylene) dimethanesulfonate (83)
From compound 82, using the same procedure as for compound 17, compound 83 was obtained and purification by flash column chromatography (silica, DCM:MeOH 95:5) was completed to give nucleoside 83 (20 mg, 0.0351 mmol, 82%). 83: white foam; Rf = 0.36 (silica, 10% MeOH/DCM); (MeOH, c = 0.10); FT-IR (film) νmax 3300, 2922, 1586, 1489, 1354, 1174, 1054, 1033, 1001, 832, 754 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.37 (s, 1 H), 7.94 (dd, J = 11.6, 3.5 Hz, 1 H), 7.88 (m, 1 H), 7.75 (dd, J = 1.7, 0.8 Hz, 1 H), 7.58 (s, 1 H), 7.38 (s, 1 H), 6.76 (ddd, J = 3.5, 1.7, 0.7 Hz, 1 H), 6.66 (ddd, J = 3.4, 1.7, 0.7 Hz, 1 H), 6.15 (d, J = 6.4 Hz, 1 H), 5.21 (d, J = 11.6 Hz, 1 H), 4.68 (s, 1 H), 4.60 (dd, J = 24.0, 10.8 Hz, 2 H), 4.50 (t, J = 11.1 Hz, 2 H), 4.12 (d, J = 12.5 Hz, 1 H), 3.10 (s, 3 H), 3.03 (s, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 153.56, 153.28, 152.86, 150.39, 147.21, 146.95, 146.11, 145.69, 128.69, 118.98, 113.80, 113.76, 113.22, 90.16, 89.38, 85.09, 73.69, 72.78, 69.34, 69.23, 37.69, 37.64 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C21H23N4O11S2+ 571.0805, found 571.0801.
4.1.60. ((1R,3R,4R,5S)-3-(2,6-Di(furan-2-yl)-9H-purin-9-yl)-4-hydroxy-2,6-dioxabicyclo[3.2.0]heptan-1-yl)methyl methanesulfonate (50)
From compound 83, using the same procedure as for compound 64, compound 50 was obtained and purification by flash column chromatography (silica, EtOAc:hexanes 8:1) was completed to give nucleoside 50 (7 mg, 0.0148 mmol, 91%). 50: slightly yellow foam; Rf = 0.31 (silica, 10% MeOH/DCM); (MeOH, c = 0.10); FT-IR (film) νmax 3300, 2922, 1586, 1354, 1175, 1054, 1033, 1001, 832 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 8.45 (s, 1 H), 7.94 (d, J = 3.5 Hz, 1 H), 7.89 (s, 1 H), 7.75 (s, 1 H), 7.40 (d, J = 3.3 Hz, 1 H), 6.77 (dd, J = 3.4, 1.7 Hz, 1 H), 6.66 (dd, J = 3.4, 1.7 Hz, 1 H), 6.53 (d, J = 7.2 Hz, 1 H), 5.30 (d, J = 4.6 Hz, 1 H), 4.94 (d, J = 8.4 Hz, 1 H), 4.84 (td, J = 7.1, 4.6 Hz, 1 H), 4.67 (d, J = 8.4 Hz, 1 H), 4.59 (q, J = 11.5 Hz, 1 H), 3.92 (d, J = 7.1 Hz, 1 H), 3.62–3.54 (m, 1 H), 3.10–2.94 (m, 3 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 153.72, 153.59, 153.05, 150.42, 147.21, 146.94, 146.06, 145.10, 126.37, 118.98, 113.84, 113.76, 113.24, 88.96, 85.60, 84.54, 78.60, 75.33, 69.40, 37.73 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C20H19N4O8S+ 475.0918, found 475.0927.
4.1.61. (1S,3R,4R,5S)-3-(2,6-Di(furan-2-yl)-9H-purin-9-yl)-1-(hydroxymethyl)-2,6-dioxabicyclo[3.2.0]heptan-4-ol (51)
Compound 50 (15 mg, 0.0316 mmol) was dissolved in dry DMF (0.20 mL) along with sodium benzoate (9.1 mg, 0.0630 mmol). The reaction mixture was heated to 90 °C for 4.5 h. The resulting white mixture was passed through a silica plug to give a white solid compound (15.3 mg, 0.0306 mmol, 97%). The benzoyl derivative (5 mg, 0.00999 mmol) was dissolved in MeOH (0.40 mL), and 0.1 M NaOMe (0.20 mL) was added. The mixture was stirred at rt for 30 min and quenched with a solution of 0.1 M HCl (1 mL). The reaction mixture was extracted with 10% MeOH:DCM, dried over MgSO4, filtered, and concentrated. Purification by flash column chromatography (silica, DCM:MeOH 9:1) was completed to give compound 51 (2.8 mg, 0.00707 mmol, 71%). 51: white semi-solid; Rf = 0.15 (silica, hexanes:acetone:MeOH 5:4:1); (MeOH, c = 0.10); FT-IR (film) νmax 3300, 2926, 1680, 1354, 1175, 1054, 1033, 1001, 830 cm−1; 1H NMR (CD3CN, 400 MHz) δ = 8.45 (s, 1 H), 7.97 (dd, J = 3.5, 0.8 Hz, 1 H), 7.91 (dd, J = 1.8, 0.8 Hz, 1 H), 7.76 (dd, J = 1.8, 0.9 Hz, 1 H), 7.42 (dd, J = 3.4, 0.9 Hz, 1 H), 6.79 (dd, J = 3.5, 1.8 Hz, 1 H), 6.68 (dd, J = 3.4, 1.8 Hz, 1 H), 6.47 (d, J = 7.3 Hz, 1 H), 5.25 (d, J = 4.5 Hz, 1 H), 4.89 (d, J = 8.1 Hz, 1 H), 4.80 (dd, J = 7.3, 4.5 Hz, 1 H), 4.60 (d, J = 7.6 Hz, 1 H), 3.90 (d, J = 12.3 Hz, 1 H), 3.81 (d, J = 12.3 Hz, 1 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 153.44, 153.41, 152.90, 150.37, 147.26, 147.00, 145.98, 145.68, 129.43, 119.10, 113.82, 113.78, 113.26, 90.33, 88.09, 86.33, 78.78, 75.55, 62.57 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd. for C19H17N4O6+ 397.1143, found 397.1145.
4.1.62. (3aR,5S,6R,6aR)-Methyl 2,2-dimethyl-6-(tosyloxy)tetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylate (85)
Literature-known compound 84 (770 mg, 2.15 mmol) was dissolved in dry Et2O (10 mL) and MeOH (10 mL) at 0 °C, and trimethylsilyldiazomethane (1.29 mL, 2.58 mmol, 2.0 M in hexanes) was slowly added. The reaction solution was stirred at this temperature for 30 min, then the solvent was evaporated. Purification by flash column chromatography (silica, hexanes:EtOAc 3:1) was completed to give compound 85 (700 mg, 1.88 mmol, 88%). 85: white foam; Rf = 0.67 (silica, hexanes:EtOAc 1:1); (CHCl3, c = 0.82); FT-IR (film) νmax 2988, 1769, 1741, 1597, 1495, 1440, 1372, 1294, 1215, 1190, 1176, 1163, 1094, 1063, 1034, 964, 901, 866, 846, 816, 772, 740, 704, 665 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 7.76 (d, J = 8.3 Hz, 2 H), 7.36 (d, J = 8.0 Hz, 2 H), 6.07 (d, J = 3.5 Hz, 1 H), 5.13 (d, J = 3.3 Hz, 1 H), 4.83 (d, J = 3.4 Hz, 1 H), 4.77 (d, J = 3.5 Hz, 1 H), 3.58 (s, 3 H), 2.46 (s, 3 H), 1.48 (s, 3 H), 1.31 (s, 3 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 166.73, 145.95, 133.22, 130.35, 128.42, 113.57, 105.78, 83.11, 82.45, 78.46, 52.81, 27.16, 26.72, 22.13 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C16H21O8S+ 373.0952, found 373.0957.
4.1.63. (1R,3S,4S,5S,6S,7S)-Methyl 6,7-dichloro-3,4-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-2-oxabicyclo[3.2.0]heptane-1-carboxylate (86)
Compound 85 (254 mg, 0.682 mmol) was dissolved in benzene (6.8 mL), and DBU (0.12 mL, 0.818 mmol) was added. The reaction solution was stirred at rt for 5 h. The mixture was worked up by evaporation to half volume, then elution through a silica gel plug with EtOAc. This residue was dissolved in MeCN (8 mL) under argon in a Quartz test tube tightly capped with a septum. 1,2-Cis-dichloroethylene (0.51 mL, 6.82 mmol) was added and the reaction degassed with argon for 20 min. The reaction solution was exposed to UV light (Hanovia 400W high-pressure Hg lamp) for 36 h. Additional 1,2-cis-dichloroethylene (0.3 mL, 4.0 mmol) was added and the solution degassed for 20 min. The reaction solution was exposed to UV light for an additional 48 h, then worked up by evaporation. Purification by flash column chromatography (hexanes:EtOAc 5:1) was completed to give compound 86 (36 mg, 0.123 mmol, 18% over two steps) along with a mix of two other diastereomers (27 mg, 0.0887 mmol, 13% over two steps). 86: yellow oil; Rf = 0.50 (silica, hexanes:EtOAc 4:1); (CHCl3, c = 2.70); FT-IR (film) νmax 2992, 1738, 1439, 1375, 1337, 1211, 1161, 1124, 1053, 990, 934, 899, 783, 708 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 6.08 (d, J = 3.8 Hz, 1 H), 5.08 (d, J = 3.8 Hz, 1 H), 4.50 (dd, J = 9.5, 6.4 Hz, 1 H), 4.17 (d, J = 6.6 Hz, 1 H), 3.87 (d, J = 0.7 Hz, 3 H), 3.79 (d, J = 9.7 Hz, 1 H), 1.41 (d, J = 15.5 Hz, 6 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 167.03, 114.98, 109.11, 88.90, 81.99, 65.55, 54.11, 53.04, 50.48, 27.93, 27.34 ppm; HRMS (ESI-TOF) (m/z): [M+Na]+ calcd for C11H14Cl2O5Na+ 319.0110, found 319.0103.
4.1.64. (1R,3R,4R,5R,6S,7S)-Methyl 4-acetoxy-6,7-dichloro-3-(2,6-diamino-9H-purin-9-yl)-2-oxabicyclo[3.2.0]heptane-1-carboxylate (52)
Compound 86 (20 mg, 0.0675 mmol) was dissolved in acetic acid (0.72 mL) and acetic anhydride (0.08 mL, 0.810 mmol). Two drops of sulfuric acid were added and the reaction solution stirred at rt for 16 h. The reaction solution was poured into ice water (4 mL) and stirred for 30 min. Sat. aq. NH4Cl (4 mL) was added and the aqueous phase extracted with 5% MeOH/DCM. The combined organics were dried over MgSO4, filtered, and concentrated to give a residue. This crude material was exposed to the same procedure as for compound 57, and compound 52 was obtained as a crude oil. Purification by preparative-plate chromatography (C18 silica, 5% MeOH/DCM) was completed to give the title compound 52 (10.5 mg, 0.0243 mmol, 36% over two steps). 52: yellow oil; Rf = 0.71 (C18 silica, 5% MeOH/DCM); (MeCN, c = 0.90); FT-IR (film) νmax 3335, 3193, 2958, 1747, 1686, 1644, 1528, 1420, 1333, 1282, 1208, 1180, 1159, 1110, 1044, 1031, 884, 794, 760, 711 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 7.84 (s, 1 H), 7.40 (s, 2 H), 6.58–6.55 (m, 1 H), 6.25 (s, 2 H), 6.09 (d, J = 7.1 Hz, 1 H), 5.45 (d, J = 8.1 Hz, 1 H), 4.72 (t, J = 8.4 Hz, 1 H), 3.95–3.91 (m, 1 H), 3.82 (s, 3 H) ppm; 13C NMR (CD3CN, 600 MHz) δ = 170.47, 167.75, 155.10, 152.99, 152.74, 92.08, 87.27, 74.26, 66.18, 54.79, 53.42, 51.22, 20.73 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C15H16Cl2N6O5+ 431.0632, found 431.0630.
4.1.65. (3aR,6aR)-Butyl 2,2-dimethyl-3a,6a-dihydrofuro[2,3-d][1,3]dioxole-5-carboxylate (87)
Literature-known compound 84 (470 mg, 1.31 mmol) and anhydrous n-butanol (0.14 mL, 1.57 mmol) were dissolved in CH2Cl2 (11.8 mL), and DCC (325 mg, 1.57 mmol) and DMAP (24 mg, 0.196 mmol) were added. The reaction solution was stirred at rt for 24 h. DBU (0.22 mL, 1.57 mmol) was added and the solution stirred at rt for 24 h. The reaction solution was evaporated and purification by flash column chromatography (silica, hexanes:EtOAc 5:1) was completed to give compound 87 (200 mg, 0.878 mmol, 67%). 87: white semi-solid; Rf = 0.70 (silica, hexanes:EtOAc 2:1); (MeOH, c = 0.26); FT-IR (film) νmax 2993, 2957, 2935, 2873, 1722, 1639, 1627, 1575, 1458, 1397, 1381, 1371, 1332, 1320, 1305, 1256, 1248, 1234, 1213, 1156, 1127, 1082, 1039, 1009, 970, 897, 886, 852, 835, 805, 757, 668 cm−1; 1H NMR (CDCl3, 500 MHz) δ = 6.17 (d, J = 5.4 Hz, 1 H), 6.06 (d, J = 2.5 Hz, 1 H), 5.36 (dd, J = 5.4, 2.5 Hz, 1 H), 4.23 (t, J = 6.7 Hz, 2 H), 1.68 (dd, J = 9.9, 5.2 Hz, 2 H), 1.39 (dt, J = 14.8, 7.5 Hz, 2 H), 0.94 (t, J = 7.4 Hz, 3 H) ppm; 13C NMR (CDCl3, 126 MHz) δ = 159.88, 150.48, 113.22, 110.31, 106.84, 82.93, 65.74, 30.60, 28.10, 27.84, 19.18, 13.79 ppm; HRMS (ESI-TOF) (m/z): [M+Na]+ calcd for C12H18O5Na+ 265.1046, found 265.1034.
4.1.66. (1R,3S,4S,5S,6S,7S)-Methyl 6,7-dichloro-3,4-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-2-oxabicyclo[3.2.0]heptane-1-carboxylate (88)
Compound 87 (200 mg, 0.826 mmol) was dissolved in MeCN (16 mL) under argon in a Quartz test tube tightly capped with a septum. 1,2-Cis-dichloroethylene (0.86 mL, 8.86 mmol) was added and the reaction solution degassed with argon for 30 min. The mixture was exposed to UV light (Hanovia 400W high-pressure Hg lamp) for 108 h. Additional 1,2-cis-dichloroethylene (0.50 mL, 5.14 mmol) was added and the solution degassed for 30 min. The reaction mixture was exposed to UV light for an additional 48 h and then worked up by evaporation. Purification by flash column chromatography (silica, hexanes:EtOAc 20:1) was completed to give compound 76 (59 mg, 0.174 mmol, 21%) along with two other diastereomers (76 mg, 0.224 mmol, 27%) and recovered starting material 87 (23 mg, 0.0949 mmol, 12%). 88: yellow oil; Rf = 0.75 (silica, hexanes:EtOAc 4:1); (CHCl3, c = 1.00); FT-IR (film) νmax 2961, 2937, 2875, 1757, 1731, 1459, 1384, 1374, 1325, 1270, 1244, 1190, 1160, 1122, 1052, 987, 933, 899, 852, 782, 746, 707 cm−1; 1H NMR (CDCl3, 600 MHz) δ = 6.08 (d, J = 3.8 Hz, 1 H), 5.08 (d, J = 3.8 Hz, 1 H), 4.49 (dd, J = 9.7, 6.8 Hz, 1 H), 4.31–4.24 (m, 2 H), 4.16 (dd, J = 6.8, 1.2 Hz, 1 H), 3.77 (d, J = 9.7 Hz, 1 H), 1.69 (dq, J = 13.6, 6.7 Hz, 2 H), 1.43–1.38 (m, 8 H), 0.93 (t, J = 7.4 Hz, 3 H) ppm; 13C NMR (CDCl3, 151 MHz) δ = 166.61, 114.90, 109.03, 88.68, 81.95, 66.21, 65.58, 54.09, 50.56, 30.62, 27.90, 27.37, 19.14, 13.77 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C14H21Cl2O5+ 339.0760, found 339.0768.
4.1.67. (1R,3R,4R,5R,6S,7S)-Methyl 6,7-dichloro-3-(2,6-diamino-9H-purin-9-yl)-4-hydroxy-2-oxabicyclo[3.2.0]heptane-1-carboxylate (53)
Method 1: Compound 88 (4.3 mg, 0.00986 mmol) was dissolved in MeOH (0.47 mL), and K2CO3 (0.40 mg, 0.00296 mmol) was added. The reaction was stirred at rt for 30 min. The reaction mixture was quenched with one drop AcOH and purification by preparative-plate chromatography (C18 silica, 5% MeOH/DCM) was completed to give compound 53 (3.2 mg, 0.00818 mmol, 83%). Method 2: From compound 88, using the same procedures as for compound 52, a crude residue was obtained. This residue was then dissolved in MeOH (0.3 mL), and K2CO3 (0.26 mg, 0.00190 mmol) was added. The reaction mixture was stirred at rt for 1 h. The mixture was quenched with one drop AcOH and purification by preparative-plate chromatography (C18 silica, 5% MeOH/DCM) was completed to give compound 53 (2.2 mg, 0.00564 mmol, 38% over three steps). 53: white powder; Rf = 0.10 (silica, 5% MeOH/DCM); (MeOH, c = 0.26); FT-IR (film) νmax 3333, 2925, 1742, 1688, 1643, 1528, 1414, 1331, 1281, 1208, 1157, 1100, 1031, 959, 888, 791, 707 cm−1; 1H NMR (CD3CN, 600 MHz) δ = 7.93 (s, 1 H), 7.30 (s, 2 H), 6.32 (s, 2 H), 5.89 (d, J = 7.8 Hz, 1 H), 5.43 (t, J = 7.3 Hz, 1 H), 5.20 (dd, J = 8.0, 1.0 Hz, 1 H), 4.72–4.68 (m, 1 H), 4.21 (s, 1 H), 3.81 (s, 3 H), 3.66 (ddd, J = 6.8, 5.0, 1.1 Hz, 1 H) ppm; 13C NMR (CD3CN, 151 MHz) δ = 168.07, 154.25, 153.11, 152.15, 141.45, 125.82, 93.25, 86.46, 74.31, 66.88, 55.22, 53.30, 52.41 ppm; HRMS (ESI-TOF) (m/z): [M+H]+ calcd for C13H15Cl2N6O4+ 389.0526, found 389.0534.
4.2 Biological assays
4.2.1 Bacterial minimum inhibitory concentration (MIC) assays
Minimum inhibitory concentrations (MIC) were determined by CLSI micro-broth dilution guidelines.33 Briefly, compounds were serially diluted in 96-well plates containing 100 μL of cation-adjusted mueller hinton (MHB) broth per well. E. coli G1655 and S. aureus 8325 were inoculated into MHB from a fresh overnight plate scrape on MHA plates to a final concentration of 1 × 107 colony forming units ml−1, and 5 μL of this suspension was added to each well. The plates were incubated at 37 °C and the MIC determined after 24 h of growth. MICs were defined as the lowest concentration of the compound that yielded no observable growth, and reported values were the median value of at least three independent replicates with an error of no greater than two-fold.
4.2.2 Cytotoxicity assays
Raji and CEM cells were incubated for 72 h in 96-well plates with the test compounds. Cell proliferation by reduction of the yellow dye MTT [i.e., 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] to a blue formazan product was used to test viability. The amount of formazan dye formed is a direct indication of the number of metabolically active cells in the culture. The optical density of the blue formazan product was measured at 570 nm with an Infinite M200 (Tecan, Switzerland) and analyzed using one-way ANOVA using GraphPad Prism version 5.0b for OSX (GraphPad Software, San Diego, CA).
4.2.3 Pseudovirus neutralization assays
Pseudoviruses are generated in 293T cells and neutralization with single-round infectious pseudovirus is performed using TZM-bl cells as targets for infection as described previously.34 Briefly, pseudovirus is titrated on TZM-bl cells and a predetermined amount of virus that produces ~1 × 106 RLUs is incubated for 1 h at 37 °C with serially diluted samples and controls. TZM-bl cells are resuspended in media, washed, counted and plated at 1 × 105 cells per well over the incubated solution of virus and samples for an additional 48 h. The degree of virus neutralization by each sample is achieved by measuring luciferase activity. The wells are aspirated and washed once with PBS, and 60 μL of luciferase cell culture lysis reagent (Promega, Madison, WI) is added. The lysate is mixed by pipetting, and 50 μL is transferred to a round-bottom plate (Corning), and the plate is centrifuged at 1800 × g for 10 min at 4 °C. Then 20 μL is transferred to an opaque assay plate (Corning, Corning, NY), and the luciferase activity is measured on a luminometer (EG&G Berthold LB 96V; Perkin Elmer, Gaithersburg, MD) by using luciferase assay reagent (Promega, Madison, WI).
Supplementary Material
Acknowledgements
We thank Dr. D. H. Huang and Dr. L. Pasternack for NMR spectroscopic assistance and Dr. G. Siuzdak for mass spectrometric assistance. Financial support for this work was provided by the National Institutes of Health (U.S.A.) and the Skaggs Institute for research, along with fellowships from Novartis (to S.P.E.) and UCSD/SDSU IRACDA (to F.R.).
Footnotes
Supplementary data Supplementary data associated with this article can be found, in the online version, at doi: 10.016/j.bmc.2011,xxxxx.
References and notes
- 1.Zamecnik PC, Stephenson ML. Proc. Natl. Acad. Sci. USA. 1978;75:280. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2 (a).Petersen M, Wengel J. Trends Biotechnol. 2003;21:74. doi: 10.1016/S0167-7799(02)00038-0. Kurreck J. Eur. J. Biochem. 2003;270:1628. doi: 10.1046/j.1432-1033.2003.03555.x. Kaur H, Babu BR, Maiti S. Chem. Rev. 2007;107:4672. doi: 10.1021/cr050266u. Rahman SMA, Imanishi T, Obika S. Chem. Lett. 2009;38:512. D’Alonzo D, Guaragna A, Palumbo G. Chem. Biodiversity. 2011;8:373. doi: 10.1002/cbdv.201000303.
- 3 (a).Altona C, Sunderalingam MJ. Am. Chem. Soc. 1972;94:8205. doi: 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]; (b) Altona C, Sunderalingam MJ. Am. Chem. Soc. 1973;95:2333. doi: 10.1021/ja00788a038. [DOI] [PubMed] [Google Scholar]
- 4 (a).Koshkin AA, Singk SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, Olsen CE, Wengel J. Tetrahedron. 1998;54:3607. [Google Scholar]; (b) Koshkin AA, Rajwanshi VK, Wengel J. Tetrahedron Lett. 1998;39:4381. [Google Scholar]; (c) Rajwanshi VK, Håkansson AE, Sørensen MD, Pitsch S, Singh SK, Kumar R, Nielsen P, Wengel J. Angew. Chem. Int. Ed. 2000;39:1656. doi: 10.1002/(sici)1521-3773(20000502)39:9<1656::aid-anie1656>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]; (d) Obika S, Uneda T, Sugimoto T, Nanbu D, Minami T, Doi T, Imanishi T. Bioorg. Med. Chem. 2001;9:1001. doi: 10.1016/s0968-0896(00)00325-4. [DOI] [PubMed] [Google Scholar]; (e) Koshkin AA, Fensholdt J, Pfundheller HM, Lomholt C. J. Org. Chem. 2001;66:8504. doi: 10.1021/jo010732p. [DOI] [PubMed] [Google Scholar]
- 5 (a).Obika S, Morio K.-i., Nanbu D, Imanishi T. Chem. Commun. 1997:1643. [Google Scholar]; (b) Obika S, Morio K.-i., Hari Y, Imanishi T. Chem. Commun. 1999:2423. doi: 10.1016/s0960-894x(99)00028-1. [DOI] [PubMed] [Google Scholar]; (c) Obika S, Morio K.-i., Nanbu D, Hari Y, Itoh H, Imanishi T. Tetrahedron. 2002;58:3039. [Google Scholar]
- 6.Rahman SMA, Seki S, Obika S, Yoshikawa H, Miyashita K, Imanishi T. J. Am. Chem. Soc. 2008;130:4886. doi: 10.1021/ja710342q. [DOI] [PubMed] [Google Scholar]
- 7 (a).Umemoto T, Wengel J, Madsen AS. Org. Biomol. Chem. 2009;7:1793. doi: 10.1039/b901028a. [DOI] [PubMed] [Google Scholar]; (b) Astakhova IV, Lindegaard D, Korshun VA, Wengel J. Chem. Commun. 2010;46:8362. doi: 10.1039/c0cc03026k. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Xu J, Karimiahmadabadi M, Zhou C, Chattopadhyaya J. J. Org. Chem. 2010;75:7112. doi: 10.1021/jo101207d. [DOI] [PubMed] [Google Scholar]
- 9.Seth PP, Allerson CR, Berdeja A, Siwkowski A, Pallan PS, Gaus H, Prakash TP, Watt AT, Egli M, Swayze EE. J. Am. Chem. Soc. 2010;132:14942. doi: 10.1021/ja105875e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Review: Mathé C, Périgaud C. Eur. J. Org. Chem. 2008:1489.
- 11 (a).Webb TR, Mitsuya H, Broder S. J. Med. Chem. 1988;31:1475. doi: 10.1021/jm00402a038. [DOI] [PubMed] [Google Scholar]; (b) Okabe M, Sun R-C. Tetrahedron Lett. 1989;30:2203. [Google Scholar]; (c) Beard AR, Butler PI, Mann J, Partlett NK. Carbohydr. Res. 1990;205:87. doi: 10.1016/0008-6215(90)80130-u. [DOI] [PubMed] [Google Scholar]; (d) Rodriguez JB, Marquez VE, Nicklaus MC, Mitsuya H, Barchi JJ., Jr. J. Med. Chem. 1994;37:3389. doi: 10.1021/jm00046a024. [DOI] [PubMed] [Google Scholar]; (e) Park A-Y, Moon HR, Kim KR, Chun MW, Jeong LS. Org. Biomol. Chem. 2006;4:4065. doi: 10.1039/b612537a. [DOI] [PubMed] [Google Scholar]; (f) Moon HR, Park A-Y, Kim KR, Chun MW, Jeong LS. Nucleosides, Nucleotides Nucleic Acids. 2007;26:1653. doi: 10.1080/15257770701615599. [DOI] [PubMed] [Google Scholar]
- 12 (a).Tchilibon S, Joshi BV, Kim S-K, Duong HT, Gao Z-G, Jacobson KA. J. Med. Chem. 2005;48:1745. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jacobson KA, Joshi BV, Tchilibon S. International Patent WO 031505 A1. 2006 [Google Scholar]; (c) Tosh DK, Chinn M, Ivanov AA, Klutz AM, Gao Z-G, Jacobson KA. J. Med. Chem. 2009;52:7580. doi: 10.1021/jm900426g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eoff RL, McGrath CE, Maddukuri L, Salamanca-Pinzón SG, Marquez VE, Marnett LJ, Guengerich FP, Egli M. Angew. Chem. Int. Ed. 2010;49:1. doi: 10.1002/anie.201003168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O-Yang C, Kurz W, Eugui EM, McRoberts MJ, Verheyden JPH, Kurz LJ, Walker KAM. Tetrahedron Lett. 1992;33:41. [Google Scholar]
- 15.Kim MJ, Kim HO, Kim H-D, Kim JH, Jeong LS, Chun MW. Bioorg. Med. Chem. Lett. 2003;13:3499. doi: 10.1016/s0960-894x(03)00730-3. [DOI] [PubMed] [Google Scholar]
- 16.Díaz-Rodríguez A, Sanghvi YS, Fernández S, Schinazi RF, Theodorakis EA, Ferrero M, Gotor V. Org. Biomol. Chem. 2009;7:1415. doi: 10.1039/b818707j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17 (a).Ravn J, Qvortrup K, Rosenbohm C, Koch T. Bioorg. Med. Chem. 2007;15:5440. doi: 10.1016/j.bmc.2007.05.056. [DOI] [PubMed] [Google Scholar]; (b) Ravn J, Rosenbohm C, Qvortrup K, Koch T. International Patent WO 006369 A1. 2008 [Google Scholar]
- 18 (a).Olsen AG, Nielsen C, Wengel J. J. Chem. Soc., Perkin Trans. 1. 2001:900. [Google Scholar]; (b) Bryld T, Sørensen MH, Nielsen P, Koch T, Nielsen C, Wengel J. J. Chem. Soc., Perkin Trans. 1. 2002:1655. [Google Scholar]; (c) Nair V, Jeon G-S. ARKIVOC. 2004:133. [Google Scholar]
- 19 (a).Chun MW, Kim MJ, Kim HO, Kim H-D, Kim JH, Moon HR, Jeong LS. Nucleosides, Nucleotides Nucleic Acids. 2003;22:915. doi: 10.1081/NCN-120022685. [DOI] [PubMed] [Google Scholar]; (b) Hrdlicka PJ, Jepsen JS, Wengel J. Nucleosides, Nucleotides Nucleic Acids. 2005;24:397. doi: 10.1081/ncn-200059821. [DOI] [PubMed] [Google Scholar]
- 20 (a).Fleet GWJ, Ramsden NG, Dwek RA, Rademacher TW, Fellows LE, Nash RJ, St. Green DC, Winchester B. Chem. Commun. 1988:483. [Google Scholar]; (b) Yoshikawa M, Okaichi Y, Cha BC, Kitagawa I. Tetrahedron. 1990;46:7459. [Google Scholar]
- 21.Youssefyeh RD, Verheyden JPH, Moffatt JG. J. Org. Chem. 1979;44:1301. [Google Scholar]
- 22.Niedballa U, Vorbrüggen H. Angew. Chem. Int. Ed. Engl. 1970;9:461. doi: 10.1002/anie.197004612. [DOI] [PubMed] [Google Scholar]
- 23.Vorbrüggen H, Höfle G. Chem. Ber. 1981;114:1256. [Google Scholar]
- 24.Hirota K, Kazaoka K, Sajiki H. Bioorg. Med. Chem. 2003;11:2715. doi: 10.1016/s0968-0896(03)00234-7. [DOI] [PubMed] [Google Scholar]
- 25.Geng B, Breault G, Comita-Prevoir J, Petrichko R, Eyerman C, Lundqvist T, Doig P, Gorseth E, Noonan B. Bioorg. Med. Chem. Lett. 2008;18:4368. doi: 10.1016/j.bmcl.2008.06.068. [DOI] [PubMed] [Google Scholar]
- 26.Langli G, Gundersen L-L, Rise F. Tetrahedron. 1996;52:5625. [Google Scholar]
- 27.Thornton AR, Martin VI, Blakey SB. J. Am. Chem. Soc. 2009;131:2434. doi: 10.1021/ja809078d. [DOI] [PubMed] [Google Scholar]
- 28.Bookser BC, Raffaele NB. J. Org. Chem. 2007;72:173. doi: 10.1021/jo061885l. [DOI] [PubMed] [Google Scholar]
- 29 (a).Timoshchuk VA. Carbohyd. Res. 1985;145:131. [Google Scholar]; (b) Mezzina E, Savoia D, Tagliavini E, Trombini C, Umani-Ronchi A. J. Chem. Soc., Perkin Trans. 1. 1989:845. [Google Scholar]
- 30.Wilson PK, Szabados E, Mulligan SP, Christopherson RI. Int. J. Biochem. Cell Biol. 1998;30:833. doi: 10.1016/s1357-2725(98)00024-7. [DOI] [PubMed] [Google Scholar]
- 31.Gottlieb HE, Kotlyar V, Nudelman A. J. Org. Chem. 1997;62:7512. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 32 (a).Koshkin AA. J. Org. Chem. 2004;69:3711. doi: 10.1021/jo0303923. [DOI] [PubMed] [Google Scholar]; (b) Ramsing NB, Nielsen AT, Koshkin EA, Tolstrup N, Pfundheller HM, Lomholdt C. 0147924 A1 US Patent. 2006; (c) Pasternack A, Kierzek E, Pasternack K, Turner DH, Kierzek R. Nucleic Acids Res. 2007;35:4055. doi: 10.1093/nar/gkm421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. CLSI, Wayne; PA, USA: 2006. [Google Scholar]
- 34.Zwick MB, Wang M, Poignard P, Stiegler G, Katinger H, Burton DR, Parren PW. J. Virol. 2001;75:12198. doi: 10.1128/JVI.75.24.12198-12208.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.