

Abstract
Sulfurs in action
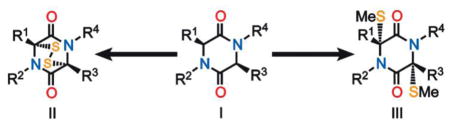
A practical and simple method for the introduction of sulfur atoms into 2,5-diketopiperazines (I) under mild conditions has been developed leading to more or less complex epidithiodiketopiperazines (II) and bis-methylthiodiketopiperazines (III).
Keywords: bis-methylthiodiketopiperazine, epidithiodiketopiperazine, natural product synthesis, sulfenylation
Sulfenylated 2,5-diketopiperazine structural motifs are found abundantly in nature as domains of a wide range of natural products.[1] Among them, the bis-methylthiodiketopiperazines [e.g. epicoccin G (1), Figure 1][2] and epidithiodiketopiperazines [e.g. aranotin (2), Figure 1][3] are the most common and important. These natural products are often endowed with important biological properties such as cytotoxic, antibacterial, antiviral, antiallergy and antimalarial activities.[4] Full biological investigations of several of these promising compounds are lacking, primarily due to their natural scarcity and difficulties associated with their chemical synthesis. The latter problems stem from the sensitivity of their sulfur moieties and the deficiencies of methods for their installation within the growing diketopiperazine scaffolds.[5] Herein we report a simple and practical method for the sulfenylation of 2,5-diketopiperazines to afford either epidithiodiketopiperazines or bis-methylthiodiketopiperazines through the use of elemental sulfur and sodium hexamethyldisilazide (NaHMDS).
Figure 1.

Molecular structures of epicoccin G (1) and aranotin (2).
Previous sulfenylation methods of 2,5-diketopiperazines involved installation of sulfur, either directly (i.e. NaNH2, S8, liq. NH3)[6] or indirectly through their 3,6-dibromodiketopiperazine,[7] 3,6-dimethoxydiketopiperazine[8] and 3,6-dihydroxydiketopiperazine[9] derivatives. These methods require either harsh conditions or multi-step sequences, or both, and they lack in generality and efficiency. Faced with such difficulties in our total synthesis approach toward some of these natural products (i.e. 1 and 2, Figure 1),[10] we opted to explore the use of elemental sulfur in the presence of NaHMDS. As we describe below, these explorations led to a general and practical sulfenylation method of 2,5-diketopiperazines that is simple to perform at ambient temperature in common organic solvents.
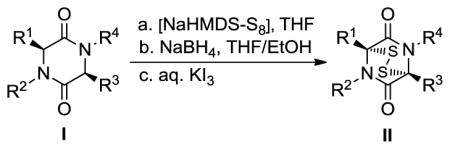
The new sulfenylation method involves three sequential steps from 2,5-diketopiperazine substrates (I, Table 1) to epidithiodiketopiperazines (II, Table 1) or bis-methylthiodiketopiperazines (III, Table 2) with no purification in between steps. Thus 2,5-diketopiperazine I was added to a freshly prepared solution of elemental sulfur and NaHMDS in THF at room temperature. After a short period of stirring, additional NaHMDS was added and the reaction mixture was stirred at the same temperature until the sulfenylation was complete, at which time the reaction was quenched with aq. NH4Cl. The crude product was transferred to a THF:EtOH (1:1) solution through extraction with CH2Cl2, drying, evaporation and dissolution, and then reduced with NaBH4 to the corresponding dithiolate, which was oxidized with KI3 to afford epidithiodiketopiperazines (II), or methylated with MeI to give the corresponding bis-methyldithiodiketopiperazines (III).
Table 1.
Preparation of epidithiodiketopiperazines from 3,6-diketopiperazines (DKPs).[a]
 | |||
|---|---|---|---|
| Entry | Substrate | Product[b] | Overall yield (%)[c] |
| 1 |
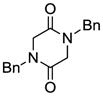 3 |
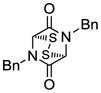 13 |
40 |
| 2 |
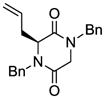 4 |
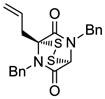 14 |
63 |
| 3 |
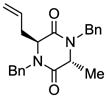 5 |
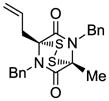 15 |
70 |
| 4 |
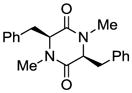 6 |
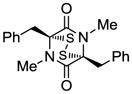 16 |
69 |
| 5 |
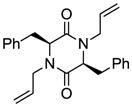 7 |
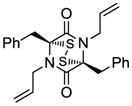 17 |
65 |
| 6 |
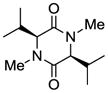 8 |
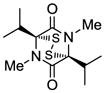 18 |
45 |
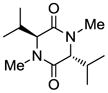
| 7 |
 9 |
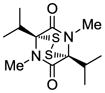 18 |
47 |
| 8 |
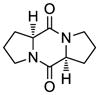 10 |
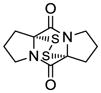 19 |
65 |
| 9 |
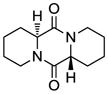 11 |
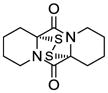 20 |
68 |
| 10 |
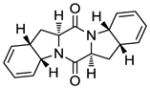 12 |
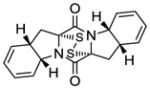 21 |
55[d] |
Reactions were performed on 100 mg scale of DKP.
Racemic mixture unless otherwise stated.
Isolated yield after flash column chromatography.
ca. 1.4:1 dr.
Table 2.
Preparation of bis-methylthiodiketopiperazines from 3,6-diketopiperazines (DKPs).[a]
 | |||
|---|---|---|---|
| Entry | Substrate | Product[b] | Overall yield (%)[c] |
| 1 |
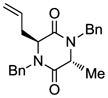 5 |
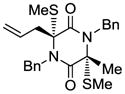 22 |
70 |
| 2 |
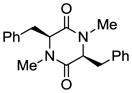 6 |
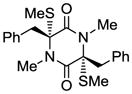 23 |
72 |
| 3 |
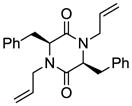 7 |
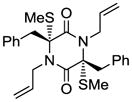 24 |
63 |
| 4 |
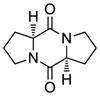 10 |
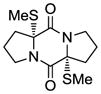 25 |
64 |
| 5 |
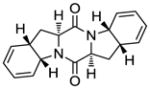 12 |
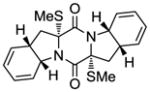 26 |
58[d] |
Reactions were performed on 100 mg scale of DKP.
Racemic mixture unless otherwise stated.
Isolated yield after flash column chromatography.
ca.1.4:1 dr.
Table 1 demonstrates the generality and scope of the developed sulfenylation method for the preparation of a range of epidithiodiketopiperazines. Thus, 3,6-unsubstituted diketopiperazines (e.g. 3, entry 1) enter the reaction to provide the expected epidithiodiketopiperazine product, albeit in modest yield (40%). This result may be attributed to unhindered intermolecular reactions of the generated sulfur species, as supported by the higher yields obtained from 3,6-mono- (entry 2) and 3,6-disubstituted (entries 3–7) diketopiperazines. Furthermore, the present method accommodates equally well both syn (entries 4–6) and anti (entries 3 and 7) 3,6-disubstituted diketopiperazines, as well as polycyclic diketopiperazines (entries 8–10). It should be noted that sulfenylation through this protocol proceeds from the same side of the molecule even in the case of the anti diketopiperazines (entries 3 and 7). As a consequence of enolate formation, the epidithio products obtained in Table 1 are racemic. Product 21 is formed as a mixture of two diastereoisomers (ca. 1.4:1 dr) as previously demonstrated.[10]
With the generality and scope of the newly developed sulfenylation method for the synthesis of epidithiodiketopiperazines demonstrated, we then proceeded to explore its application to the preparation of bis-methylthiodiketopiperazines (III, Table 2) from diketopiperazine substrates (I). The protocol for the preparation of the latter compounds required modification of only the last step of the sequence employed for the preparation of the epidithiodiketopiperazine derivatives, namely methylation of the generated sulfenylated species after the sodium borohydride reduction with MeI. Table 2 summarizes the conditions used and shows a number of examples involving monocyclic, tricyclic and pentacyclic diketopiperazines as substrates. All bis-methylthiodiketopiperazines were obtained in good yields as single syn diastereoisomers (racemic mixtures) except for compound 26, which was isolated as a mixture of diastereoisomers (ca. 1.4:1 dr) by virtue of the additional stereogenic centers present in the starting substrate.[10] The absence of any anti products in these reactions provides support for an intramolecular attachment of the second sulfur moiety within the diketopiperazine scaffold and stands in contrast to the classical Schmidt method that provides mixtures of syn and anti products in much lower yields.[6]
Having developed this new sulfenylation method using the [NaHMDS-S8] reagent combination, we then sought to gain some mechanistic insight as to the chemistry involved. To this end, we attempted to analyze the products obtained by the initial mixing of elemental sulfur (S8) and NaHMDS as described above. High resolution mass spectrometry showed a major signal at m/e = C12H36N2O2S4H⊕ [M+H⊕] which corresponded to (TMS)2N-SSSS-N(TMS)2 (calcd: 449.0911; found: 449.0908).[11] Flash column chromatography with silica gel led to the isolation of this rather labile compound whose NMR spectroscopic data provided further support for its presumed structure noted above [1H NMR (CDCl3, 600 MHz) δ = 0.26 ppm; 13C NMR (CDCl3, 150 MHz) δ = 2.4 ppm]. A possible scenario for the formation of this species is depicted in Scheme 1. Thus, it is postulated that the first equivalent of NaHMDS opens the 8-membered ring sulfur cluster to an open-chain mono-TMS sulfenamide species (28) which suffers further attack from a second equivalent of NaHMDS preferentially at the middle of the sulfur chain (presumably due to electronic and steric repulsion at each end, respectively) to afford the observed N,N′-tetrathio-bis-TMS derivative (29). Indeed, freshly prepared and isolated species 29 served as a successful sulfenylating reagent of diketopiperazine 6 in the presence of 3.0 equiv of NaHMDS under the same conditions as those described in Table 1 to afford epidithiodiketopiperazine 16 in similar yield (57% yield) to the original protocol. The crude mixture [NaHMDS-S8] also exhibited mass spectrometric peaks corresponding to (TMS)2N-SSS-N(TMS)2 (calcd for C12H36N2O2S3H⊕ [M+H⊕]: 417.1190; found: 417.1186) and (TMS)2N-SSSSS-N(TMS)2 (calcd for C12H36N2O2S5H⊕ [M+H⊕]: 512.0280; found: 512.0241).
Scheme 1.

Proposed mechanism for the formation of N,N′-tetrathio-bis-trimethylsilyl compound 29 from S8 (27) and NaHMDS.
Given the possibility of several sulfenylation agents within the reaction mixture, this sulfenylation reaction may be rather complex. However, using tetrasulfide species 29, the tentative mechanism shown in Scheme 2 for the case of diketopiperazine 6 as a substrate may be proposed. Thus, enolate formation[12] from 6 under the basic conditions employed may allow stepwise installation of sulfur at positions 3 and 6 through sequential inter- and intramolecular carbo–sulfur bond formations. This sequence furnishes intermediates 6b–6d as a mixture of oligosulfides from which the epitetrasulfide 31 was isolated as the major product (43% yield) together with epidisulfide 16 (8% yield), as well as several other unidentified products. An X-ray crystallographic analysis of 31 proved its tetrasulfide nature unambiguously (see X-ray derived ORTEP in Scheme 2).[13] Reduction of this oligosulfide mixture with NaBH4 then leads to the corresponding bis-thiolate species, whose oxidation with KI3 (after quenching with NH4Cl) furnish the epidithiodiketopiperazine product 16, whereas methylation affords bis-methylthiodiketopiperazine 23. Pure tetrasulfide 31 was also converted to 16 under the same NaBH4-KI3 conditions (94% yield).
Scheme 2.

A postulated mechanism for the formation of epidithiodiketopiperazine 16 and bis-methylthiodiketopiperazine 23 from diketopiperazine 6. Only one diastereoisomer of 6c and related compounds is shown.
In order to explore further the usefulness of this new sulfenylation procedure, we decided to cap the intermediate dithiolate species with other electrophiles. As shown in Scheme 3, quenching of the resulting dithiolate after NaBH4 reduction with pivaloyl chloride (PivCl) led to bis-pivalate 34 in 35% yield (syn isomer, unoptimized). Exposure of the latter to Mg(OMe)2, followed by bubbling of oxygen through the solution led to the formation of epidithiodiketopiperazine 35 (87% yield). On the other hand, employment of trimethylsilyl ethoxymethyl chloride (SEMCl) instead of PivCl to capture the dithiolate species generated from 33 and NaHMDS led to the formation of bis-SEM derivative 36 in 40% yield (syn isomer, unoptimized). Attempts to deprotect compound 36 with fluoride reagents failed; at best, only trace amounts of epidithiodiketopiperazine 35 were obtained upon oxidation of the derived mixture of products. However, the most interesting transformation of compound 36 was observed when this compound was treated with SnCl4 in THF at 25 °C in an attempt to cleave the SEM groups. Under these conditions, product 36, possessing the S-CH2-O bridge, was obtained in 93% yield. The assigned structure of the latter was consistent with its spectroscopic data and was unambiguously proven through X-ray crystallographic analysis (see X-ray derived ORTEP, Scheme 3).[14]
Scheme 3.

Synthesis of bis-sulfenylated diketopiperazine derivatives 34 and 36 and formation of O,S-acetal diketopiperazine 37. Reagents and conditions: a) NaHMDS (0.6 M in PhMe, 3.0 equiv), S8 (1.0 equiv), THF, 25 °C, 1 min; then 33 (1 M in THF, 1.0 equiv) 1 min; then NaHMDS (0.6 M in PhMe, 2.0 equiv), 25 °C, 0.5 h; b) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 0.75 h; c) PivCl (50 equiv), 25 °C, 15 h, 35%; d) Mg(OMe)2 (20 equiv), MeOH, 25 °C, 15 h, 87%; e) SEMCl (50 equiv), 25 °C, 15 h, 40%; f) SnCl4 (1.0 M in CH2Cl2, 14 equiv), CH2Cl2, 25 °C, 15 min, 93%.
A plausible mechanism for this unusual reaction is shown in Scheme 4. Thus, rapid cleavage of the tail end of the first SEM group may lead to trichlorotin alkoxide 36b,[15] whose decomposition to iminium species 36c by expulsion of a trichlorotinthio-SEM species may be facilitated by intramolecular activation of the departing S atom as shown on 36b. The latter species (36c) may then undergo ring closure to the observed product 37.
Scheme 4.

Postulated mechanism for the formation of mixed thioacetal 37 from bis-SEMthiodiketopiperazine 36. Reagents and conditions: a) SnCl4 (1.0 M in CH2Cl2, 14 equiv), CH2Cl2, 25 °C, 15 min, 93%.
The described chemistry offers a general, practical and simple method for the introduction of sulfur into cyclic 2,5-diketopiperazines under mild conditions. This method has already proven its value in the total synthesis of epicoccin G (1),[10] 8,8′-epi-ent-rostratin B[10] and acetylaranotin,[16] where it was proven superior to previously known methods, and is expected to facilitate future expeditions in similarly complex and challenging molecules containing the epidithiodiketopiperazine and bis-methylthiodiketopiperazine structural motifs.
Experimental Section
Preparation of epidithiodiketopiperazines
To a suspension of elemental sulfur (8.0 equiv) in THF (0.2 M) at 25 °C under argon was added NaHMDS (0.6 M in PhMe, 3.0 equiv; LiHMDS and KHMDS gave inferior yields) dropwise over a period of 2 min. During the addition, the insoluble yellow S8 quickly changed color, initially into a dark blue, then dark orange, and finally light orange solution. This solution was stirred for an additional 1 min, and DKP (1.0 equiv) dissolved in THF (0.2 M) was added dropwise at 25 °C over a 2 min period, at which time the reaction mixture turned light brown. The mixture was stirred for an additional 1 min, then additional NaHMDS (0.6 M in PhMe, 2.0 equiv) was added, and the resulting mixture was stirred for 0.5 h at 25 °C. The reaction mixture was quenched with sat. aq. NH4Cl solution and extracted with CH2Cl2. The combined organic layers were dried over MgSO4, filtered, and concentrated to afford a brownish residue which was taken to the next step without purification. The residue was dissolved in a mixture of degassed THF:EtOH (1:1, 0.05 M) at 0 °C, and to the stirred solution under argon was added NaBH4 (25 equiv) in small portions over a period of 1 min. The resulting mixture was stirred for 45 min while it was allowed to reach ambient temperature. After this time, the solution was cooled to 0 °C and quenched by careful addition of sat. aq. NH4Cl solution. The resulting mixture was extracted with EtOAc, and to the combined organic extracts was added an aq. solution of KI3 (1.4 M). This mixture was stirred for 10 min and then quenched with sat. aq. Na2S2O3 solution; the resulting mixture was extracted with EtOAc. The combined organic layers were dried (MgSO4), filtered, and concentrated to give an oily residue. The crude product was purified by flash silica gel column chromatography or preparative thin layer chromatography.
Preparation of bis-methylthiodiketopiperazines
The DKP was processed through steps a and b as described in Table 1. To the mixture obtained after NaBH4 reduction (step b) at 0 °C was added MeI (50 equiv), and the resulting mixture was stirred at 25 °C for 15 h. After this time, the solution was quenched by careful addition of sat. aq. NH4Cl solution and extracted with CH2Cl2. The combined organic layers were dried (MgSO4), filtered, and concentrated to give an oily residue. The crude product was purified by flash silica gel column chromatography or preparative thin layer chromatography.
Supplementary Material
Footnotes
We thank Drs. D. H. Huang and L. Pasternack for NMR spectroscopic assistance, Dr. R. Chadha for X-ray crystallographic assistance and Dr. G. Siuzdak for mass spectrometric assistance. This work was supported by the Skaggs Institute for Research and grants from the National Institutes of Health (USA) and National Science Foundation (USA), as well as a postdoctoral fellowship from Fonds de Recherche Québec (to D. G.).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Martins MB, Carvalho I. Tetrahedron. 2007;63:9923–9932. [Google Scholar]; b) Dinsmore CJ, Beshore DC. Tetrahedron. 2002;58:3297–3312. [Google Scholar]
- 2.a) Guo H, Sun B, Gao H, Chen X, Liu S, Yao X, Liu X, Che Y. J Nat Prod. 2009;72:2115–2119. doi: 10.1021/np900654a. [DOI] [PubMed] [Google Scholar]; b) Wang JM, Ding GZ, Fang L, Dai JG, Yu SS, Wang YH, Chen XG, Ma SG, Qu J, Xu S, Du D. J Nat Prod. 2010;73:1240–1249. doi: 10.1021/np1000895. [DOI] [PubMed] [Google Scholar]
- 3.Nagarajan R, Huckstep LL, Lively DH, DeLong DC, Marsh MM, Neuss N. J Am Chem Soc. 1968;90:2980–2982.Studies toward aranotin: Gross U, Nieger M, Brase S. Chem Eur J. 2010;16:11624–11631. doi: 10.1002/chem.201001169.
- 4.a) Gardiner MD, Waring P, Howlett BJ. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]; b) Rezanka T, Sobotka M, Spizek J, Sigler K. Anti-infect Agents Med Chem. 2006;5:187–224. [Google Scholar]; c) Greiner D, Bonaldi T, Eskeland R, Roemer R, Imhof A. Nat Chem Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]; d) Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bibblel KC. Blood. 2007;109:2579–2588. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwasa E, Hamashima Y, Sodeoka M. Isr J Chem. 2011;51:420–433. [Google Scholar]
- 6.Ohler E, Poisel H, Tataruch F, Schmidt U. Chem Ber. 1972;105:635–641. doi: 10.1002/cber.19721050229. [DOI] [PubMed] [Google Scholar]
- 7.a) Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J Am Chem Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p. [DOI] [PubMed] [Google Scholar]; b) Iwasa E, Hamashima Y, Fujishiro S, Hashizume D, Sodeoka M. Tetrahedron. 2011;67:6587–6599. [Google Scholar]; c) Kishi Y, Fukuyama T, Nakatsuka S. J Am Chem Soc. 1973;95:6492–6493. doi: 10.1021/ja00800a078. [DOI] [PubMed] [Google Scholar]; d) Fukuyama T, Kishi Y. J Am Chem Soc. 1976;98:6723–6724. doi: 10.1021/ja00437a063. [DOI] [PubMed] [Google Scholar]; e) Poisel H, Schmidt U. Chem Ber. 1971;104:1714–1721. [Google Scholar]
- 8.a) Trown PW. Biochem Biophys Res Commun. 1968;33:402–407. doi: 10.1016/0006-291x(68)90585-8. [DOI] [PubMed] [Google Scholar]; b) Yoshimura Y, Nakamura H, Matsunari K. Bull Chem Soc. 1975;48:605–609. [Google Scholar]
- 9.a) Kim J, Movassaghi M. J Am Chem Soc. 2010;132:14376–14378. doi: 10.1021/ja106869s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim J, Ashenhurst A, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Öhler E, Tataruch F, Schmidt U. Chem Ber. 1973;106:396–398. doi: 10.1002/cber.19731060205. [DOI] [PubMed] [Google Scholar]; d) Overman LE, Sato T. Org Lett. 2007;9:5267–5270. doi: 10.1021/ol702518t. [DOI] [PubMed] [Google Scholar]; e) DeLorbe JE, Jabri SY, Mennen SM, Overman LE, Zhang FL. J Am Chem Soc. 2011;133:6549–6552. doi: 10.1021/ja201789v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolaou KC, Totokotsopoulos S, Giguère D, Sun Y, Sarlah D. J Am Chem Soc. 2011;133:8150–8153. doi: 10.1021/ja2032635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Siivari J, Maaninen A, Haapaniemi E, Laitinen RS, Chivers T. Z Naturforsch B: Chem Sci. 1995;50:1575–1582. [Google Scholar]; b) Schmidt M, Scherer O. Naturwissenschaften. 1963;50:302–304. [Google Scholar]
- 12.Treatment of 2,5-diketopiperazine 6 with excess NaHMDS under the sulfenylation conditions followed by quenching with D2O led to incorporation of only one D into the molecule.
- 13.CCDC 851862 contains the supplementary crystallographic data for compound 31. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 14.CCDC 850759 contains the supplementary crystallographic data for compound 37. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 15.a) Chandrasekhar V, Nagendran S, Baskar V. Coord Chem Rev. 2002;235:1–52. [Google Scholar]; b) Wakamatsu K, Orita A, Otera J. Organomet. 2008;27:1092–1097. [Google Scholar]
- 16.Codelli JA, Puchlopek ALA, Reisman SE. J Am Chem Soc. 2011 doi: 10.1021/ja209354e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.