

Abstract
It has become increasingly evident that protein degradation via the ubiquitin proteasome system plays a fundamental role in the development, maintenance and remodeling of synaptic connections in the CNS. We and others have recently described the activity-dependent regulation of proteasome activity and recruitment of proteasomes into spine compartments involving the phosphorylation of the 19S ATPase subunit, Rpt6, by the plasticity kinase Ca2+/calmodulin-dependent protein kinase II α (CaMKIIα) (Bingol and Schuman, 2006; Djakovic et al., 2009; Bingol et al, 2010). Here, we investigated the role of Rpt6 phosphorylation on proteasome function and synaptic strength. Utilizing a phospho-specific antibody we verified that Rpt6 is phosphorylated at Serine 120 (S120) by CaMKIIα. In addition, we found that Rpt6 is phosphorylated by CaMKIIα in an activity-dependent manner. Furthermore, we showed that a serine 120 to aspartic acid phospho-mimetic mutant of Rpt6 (S120D) increases its resistance to detergent extraction in rat hippocampal dendrites, indicating phosphorylated Rpt6 may promote the tethering of proteasomes to scaffolds and cytoskeletal components. Expression of Rpt6 S120D decreased miniature EPSC (mEPSC) amplitude, while expression of a phospho-dead mutant (S120A) increased mEPSC amplitude. Surprisingly, homeostatic scaling of mEPSC amplitude produced by chronic application of bicuculline or tetrodotoxin is both mimicked and occluded by altered Rpt6 phosphorylation. Together, these data suggest that CaMKII-dependent phosphorylation of Rpt6 at S120 may be an important regulatory mechanism for proteasome-dependent control of synaptic remodeling in slow homeostatic plasticity.
Introduction
The inherent turnover rate (half-life) for any given protein is determined by a combination of its synthesis and degradation. Importantly, cell biological signals are capable of altering rates of synthesis, degradation, or both. In neurons, this may contribute to the dynamic nature of the overall protein stoichiometry of functionally relevant microdomains such as synapses. The ubiquitin proteasome system (UPS) is a major pathway for protein turnover in eukaryotic cells. The selective degradation of proteins via the UPS involves the recognition and modification of target proteins with ubiquitin chains by ubiquitin ligases, and delivery of the ubiquitin-modified protein to the 26S proteasome, a large energy-dependent protease consisting of a proteolytic 20S core particle (CP) and a 19S regulatory particle (RP), where they are subsequently degraded (Hershko and Ciechanover, 1998).
The UPS is known to target several key synaptic proteins and has been shown to play an essential role in the development, maintenance, and remodeling of synaptic connections (for review, see Patrick, 2006; Yi and Ehlers, 2007). Furthermore, large cohorts of synaptic proteins are degraded bidirectionally in response to chronic activity blockade or upregulation (Ehlers, 2003). Yet the molecular mechanisms that regulate activity-dependent synaptic protein degradation by the UPS remain unknown.
We have recently described the activity-dependent regulation of proteasome activity involving the phosphorylation of the 19S ATPase subunit, Rpt6, by Ca2+/calmodulin-dependent protein kinase II α (CaMKIIα) (Djakovic et al., 2009). Rpt6, also known as psmc5, is a 45 kDa ATPase subunit in the 19S regulatory particle of the proteasome. Rpt6 together with Rpt1–5 form a hexameric ring, known as the “base” of the 19S. All six Rpt proteins have two main functional domains: an N-terminus coiled-coil domain important for formation of the base; and a C-terminus ATPase domain that is involved in ATP-dependent substrate unfolding and 20S CP opening (Marques et al., 2009). Studies on Archaea proteasomes have identified an additional functional domain in Rpt proteins, known as the OB fold, which has ATP-independent chaperone activity (Zhang et al., 2009b).
Here, we investigated the role of Rpt6 phosphorylation on proteasome function and synaptic strength. Using a phospho-specific antibody, we demonstrate that CaMKIIα phosphorylates Rpt6 on serine 120 (S120) in an activity-dependent manner. While expression of a phospho-mimetic mutant of Rpt6 (S120D) alone in heterologous cells is not sufficient to increase proteasome activity, expression of a phospho-dead mutant (S120A) blocks CaMKIIα-dependent stimulation of the proteasome. In addition, in hippocampal neurons, mimicking Rpt6 phosphorylation at S120 increases its association with scaffolds and/or cytoskeletal components. We find that mimicking or blocking phosphorylation produces opposite effects on synaptic strength. Strikingly, we find that homeostatic scaling of miniature EPSC (mEPSC) amplitude produced by chronic application of bicuculline (BIC) or tetrodotoxin (TTX) is both mimicked and occluded by altered Rpt6 phosphorylation. Together, these data suggest that CaMKII-dependent phosphorylation of Rpt6 at S120 may be an important regulatory mechanism for proteasome-dependent control of synaptic remodeling in slow homeostatic plasticity.
Materials and Methods
Antibodies and reagents
Antibodies.
αβ (20S) core proteasome [polyclonal antibody (pAb) and monoclonal antibody (mAb)] and Rpt6 (mAb) antibodies were from Enzo Life Sciences, PSD-95 (mAb; 7E3–1B8) was from Calbiochem, HA (mAb) antibody was from Covance, HA (pAb) antibody was from SCBT, and CaMKIIα (mAb; 6G9) antibody was from Abcam. Custom rabbit (pAb; Clone #07) anti-Rpt6 phospho-specific antibody for serine 120 (pS120) was generated commercially (ProSci) against a synthetic phosphorylated [NH2-ALRND(pS)YTLHK-OH].
Reagents.
Bicuculline and tetrodotoxin were from Tocris Bioscience, λ phosphatase was from NEB, and protease and phosphatase inhibitor cocktails were from Roche.
DNA and Sindbis constructs
RSV-CaMKIIα T286D was a generous gift from Anirvan Ghosh (University of California, San Diego, San Diego, CA). Ubiquitinated GFP (GFPu) was provided by Ron Kopito (Stanford University, Palo Alto, CA). Rat Rpt6 cDNA (Open Biosystems) was cloned into pRK5. An HA epitope tag was attached to the N terminus of Rat Rpt6 cDNA. Serine to alanine (S120A) phospho-dead or aspartic acid (S120D) phospho-mimetic mutants were created using quick change site-directed mutagenesis and verified by sequencing. HA-Rpt6 wild type (WT), S120A, and S120D were then cloned into pSinrep5. Sinrep5 pSG promoter was duplicated, and GFP was inserted downstream of the second promoter for coexpression. Production of recombinant Sindbis virions was performed as previously described (Djakovic et al., 2009).
Immunoprecipitation
Transfected HEK293 cells (polyethylenimine) or transduced neuronal cultures (Sindbis) were lysed in RIPA buffer with protease and phosphatase inhibitor cocktails. Anti-20S, pS120, HA, or IgG control antibodies was added to lysates, which were rocked at 4°C overnight, and the protein–antibody complexes were then captured by further incubation of lysates with Protein A agarose beads. The samples were then washed, boiled in sample buffer, and resolved by SDS-PAGE. The gels were then transferred to nitrocellulose membranes and probed in Western blot analysis with primary antibodies (see figure legends).
Neuronal cultures
Rat dissociated hippocampal or cortical neurons from postnatal day 1 pups of either sex were plated at a density of 45,000 cells/cm2 onto poly-d-lysine-coated coverslips or glass bottom 35 mm dishes (hippocampal cultures) (Mattek) or poly-d-lysine-coated 6-well dishes at ∼500,000 cells per well (cortical cultures) and were maintained in B27 supplemented Neurobasal media (Invitrogen) until ≥14 d, as previously described (Djakovic et al., 2009).
Immunostaining
Mature hippocampal neuron cultures (>14DIV) were infected with Sindbis HA-tagged Rpt6 (WT, S120A, or S120D variants) virus (coexpressing EGFP for identification) for 20 h. For colocalization experiments, neurons were fixed and stained with anti-HA (pAb) and PSD-95 (mAb) antibodies, washed, and subsequently stained with Alexa fluorescent secondary antibodies. Triton extraction was performed as previously described (Allison et al., 1998). Briefly, neurons were treated with 1% Triton X-100 and 4% polyethylene glycol (molecular weight, 40,000) in BRB80 buffer (80 mm PIPES, 1 mm MgCl2, and 1 mm EGTA) for 5 min and then rinsed in BRB80 buffer. Neurons were then fixed and stained with an anti-HA (mAb) antibody.
Confocal microscopy
For all imaging purposes, we used a Leica DMI6000 inverted microscope outfitted with a Yokogawa Spinning disk confocal head, an Orca ER High Resolution CCD camera (6.45 μm/pixel at 1×) (Hamamatsu), Plan Apochromat 40×/1.25 na and 63×/1.4 na objective, and a Melles Griot argon/krypton 100 mW air-cooled laser with 488/568/647 nm excitations or a PerkinElmer solid-state laser with 488/561/647 nm excitations. Confocal z-stacks were acquired in all experiments. All imaging was acquired in the dynamic range of 8 bit or 12 bit acquisition (0–255 and 0–1024 pixel intensity units, respectively) with Simple PCI (C Imaging) or Volocity (PerkinElmer) imaging software.
Florescence intensity quantitation and colocalization
Maximum projected confocal z-stack images were thresholded above background, and mean fluorescence was measured. For Triton extraction experiments, dendrites from untreated and extracted neurons were imaged. Mean fluorescent intensity of Triton-extracted dendrites was divided by the average mean fluorescent intensity of untreated dendrites to determine the percentage of Rpt6 that was extracted. All values were normalized to control (WT Rpt6) levels. For GFPu experiments, fixed cells were imaged and mean GFP fluorescence of individual cells was measured. Mean fluorescence was averaged for each experimental group and normalized to the control. All data analysis was performed with NIH ImageJ and statistical analysis was performed with Prism (GraphPad). For colocalization: the extent of colocalization was determined by deriving the Pearson's correlation coefficient for red and green images using NIH ImageJ Colocalization Indices plugin (with default values). Representative images showing for overlapping immunofluorescent signals in Figure 3B, bottom, Coloc, was generated using NIH ImageJ Colocalization plugin (with default values).
Figure 3.
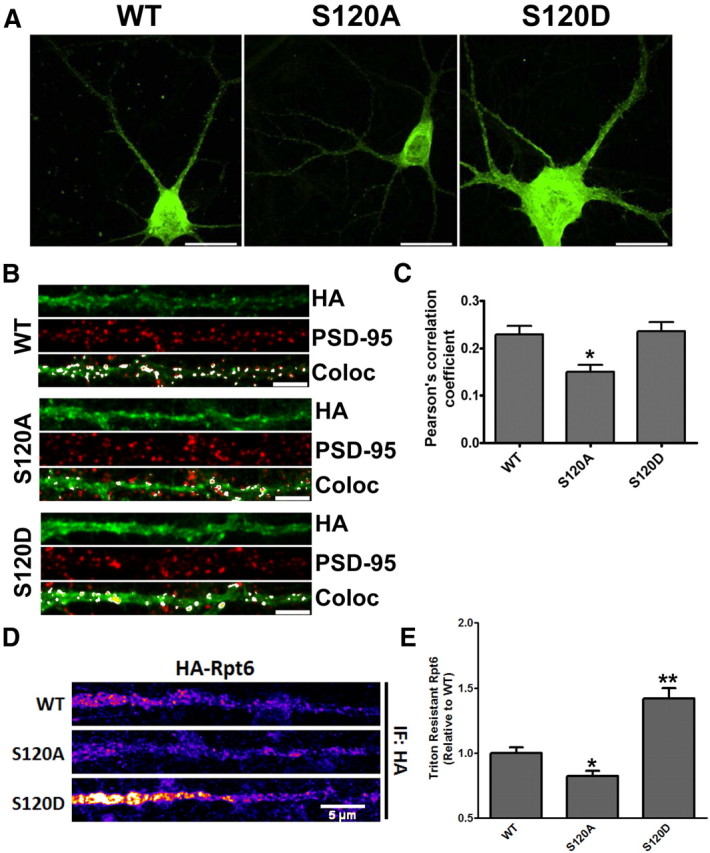
Phosphorylation affects the distribution and detergent-resistant extraction of Rpt6 in hippocampal dendrites. A, Representative images of rat hippocampal neurons transduced with HA-Rpt6 WT, S120A, or S120D Sindbis virus (20–24 h) and immunostained with anti-HA antibodies. Arrows indicate straightened dendrites in B. Scale bars, 20 μm. B, Representative straightened dendrites showing immunofluorescent signals for HA-Rpt6 (green), PSD-95 (red), and overlapping signals [white: colocalized (Coloc)]. Scale bars, 5 μm. C, S120A has decreased overlap with PSD-95 compared with WT and S120D. Bar graph shows the average (±SEM) Pearson's coefficient for WT/PSD-95, S120A/PSD-95, and S120D/PSD95. One-way ANOVA with post hoc Bonferroni's multiple-comparison test, *p < 0.05; n = 40 dendrites (one dendrite per cell) over two separate experiments. D, Straightened dendrites of HA-Rpt6-infected neurons that were Triton-X extracted and stained for HA-Rpt6 (HA antibody). S120D Rpt6 was resistant to Triton extraction, while S120A was more readily extracted compared with Rpt6 WT. E, Bar graph shows the average (±SEM) dendritic HA-Rpt6 fluorescent signal after detergent extraction. n = 3; 20–30 dendrites/experiment. **p < 0.001; *p < 0.05, ANOVA with post hoc Dunnett's multiple-comparison test.
Electrophysiology
Hippocampal neurons expressing Rpt6 WT, S120A, or S120D Sindbis virus (20–24 h) were identified by GFP expression. Whole-cell patch-clamp recordings were made from cultured hippocampal neurons bathed in HBS (containing 119 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 30 mm glucose, and 10 mm HEPES [pH 7.4]), plus 1 μm TTX and 10 μm BIC, with an Axopatch 200B amplifier. Pipettes were filled with internal solutions containing 100 mm cesium gluconate, 0.2 mm EGTA, 5 mm MgCl2, 2 mm adenosine triphosphate, 0.3 mm guanosine triphosphate, and 40 mm HEPES, pH 7.2. Pipette resistances ranged from 3 to 6 MΩ. For infected neurons additionally treated with BIC (50 μm, 48 h) or TTX (2 μm, 24 h): neurons were infected 20 h and 4 h after addition of BIC or TTX, respectively. Cultured neurons with a pyramidal-like morphology were voltage-clamped at −70 mV and series resistance was left uncompensated. mEPSCs were analyzed off-line using Synaptosoft mini analysis software. Statistical differences between experimental conditions were determined by either unpaired t tests (two groups) or by ANOVA and indicated post hoc multiple-comparison test (>2 experimental conditions).
Results
Rpt6 is phosphorylated at serine 120 by CaMKIIα
We have previously demonstrated that proteasome function is regulated by synaptic activity involving the key plasticity kinase CaMKIIα. Furthermore, we showed that Rpt6, a 19S RP subunit, is phosphorylated by CaMKIIα (Djakovic et al., 2009). To gain further insight into the functional relevance of Rpt6 phosphorylation by CaMKIIα, we first sought to identify and verify the site of CaMKII phosphorylation on Rpt6. Using phosphorylation site prediction software (ScanSite), we identified serine 120 (S120) as a candidate site for CaMKII. Indeed, a recent study showed that S120 of Rpt6 can be phosphorylated by CaMKIIα in in vitro kinase reactions (Bingol et al., 2010). To demonstrate the phosphorylation of Rpt6 at S120 by CaMKII, we generated a polyclonal phospho-specific-antibody against S120 (pS120). When transfected in HEK293 cells, we find that wild-type (WT) Rpt6 was recognized by the pS120 antibody (Fig. 1A). Coexpression with constitutively active CaMKIIα T286D led to a dramatic increase in Rpt6 phosphorylation as detected by our phospho-specific antibody (approximately threefold increase) (Fig. 1A,B). In contrast, we observed no pS120 immunoreactivity in cells expressing a phospho-dead mutant variant of Rpt6 (S120A) either alone or coexpressing CaMKIIα T286D (Fig. 1C). Furthermore, treating the nitrocellulose membranes with λ-phosphatase removed the majority of pS120 immunoreactivity (Fig. 1D). To determine whether the same effect could be seen with endogenous Rpt6, we enriched for endogenous phosphorylated Rpt6 by immunoprecipitation with our pS120 antibody from lysates of HEK293 cells and neuronal cultures and then probed the blots for endogenous Rpt6. We observed that expression of CaMKIIα T286D increased phosphorylation of endogenous Rpt6 at S120 in both HEK293 cells and dissociated rat cortical neurons (Fig. 1E). Interestingly, CaMKIIα was also coimmunoprecipitated with phosphorylated Rpt6 (Fig. 1E).
Figure 1.

Rpt6 is phosphorylated at S120 by CaMKIIα. A, B, Lysates from HEK293 cells expressing either HA-Rpt6 WT alone or HA-Rpt6 WT plus CaMKIIα T286D showed an approximately threefold increase in Rpt6 pS120 immunoreactive signal in coexpressing cells (n = 4; p < 0.001, unpaired Student's t test). C, When expressed in HEK293 cells alone or together with CaMKIIα, Rpt6 S120A has no pS120 immunoreactivity. D, λ-Phosphatase treatment of nitrocellulose membranes efficiently removed pS120 immunoreactivity. E, Expression of CaMKIIα T286D in HEK293 cells (transfection) and neurons (viral transduction) increased phosphorylation of endogenous Rpt6. Representative blot from three experiments. F, G, Increased phosphorylation of Rpt6 at S120 and enhanced association of Rpt6 and CaMKIIα in neurons treated with BIC (20 μm, 24 h), but not in cells treated with BIC and the CaMKII inhibitor AIP (2 μm). Immunoprecipitates (using Rpt6 pS120 antibody) from lysates of treated neuronal cultures were analyzed by SDS-PAGE. The blots were then probed with Rpt6 (mAb) and CaMKIIa (mAb) antibodies. n = 3–6 individual experiments; one-way ANOVA with post hoc Bonferroni's multiple-comparison test, *p < 0.05).
We have previously shown that increasing neuronal activity by blocking inhibitory synaptic transmission with BIC, leads to a significant stimulation of proteasome activity (Djakovic et al., 2009; Jakawich et al., 2010). To determine whether such treatments would regulate endogenous Rpt6 phosphorylation, we treated neurons with BIC (20 μm) for 24 h, and then immunoprecipitated phosphorylated Rpt6 with the pS120 antibody from the resulting cell lysates. We found that treatment of neurons with BIC led to a significant (approximately threefold) increase in endogenous Rpt6 phosphorylation at S120 (Fig. 1F,G). In contrast, coapplication of the specific CaMKII inhibitor, Autocamtide-2 Related Inhibitory Peptide (AIP) (2 μm), completely blocked this BIC-induced phosphorylation of Rpt6 at S120 (Fig. 1F,G). Once again, we observed a dramatically increased association between Rpt6 and CaMKIIα in BIC-treated neurons. Together these results provide strong evidence that Rpt6 is phosphorylated at S120 in an activity and CaMKIIα-dependent manner.
Phosphorylation of Rpt6 S120 is required for CaMKIIα-dependent stimulation of the proteasome
Since we previously demonstrated that CaMKIIα stimulates activity of the proteasome (Djakovic et al., 2009), we hypothesized that CaMKIIα-dependent activation of the proteasome may require S120 phosphorylation. When expressed in HEK293 cells, Rpt6 WT, S120A and a phospho-mimetic variant S120D coimmunoprecipitated with the 20S CP at similar levels, suggesting that S120 phosphorylation may not be required for incorporation of Rpt6 into the proteasome (Fig. 2A). Interestingly, expression of Rpt6 S120A or S120D alone had no significant effect on the degradation of the GFPu proteasome degradation reporter compared with WT Rpt6 in HEK293 cells (Fig. 2B). However, coexpression of Rpt6 S120A with CaMKIIα T286D blocked CaMKII-dependent stimulation of GFPu degradation (Fig. 2C). This is supported by a recent study that reported that expression of Rpt6 S120A blocks activation of the proteasome following addition of purified CaMKIIα to cell lysates (Bingol et al., 2010). Together, these data suggest that phosphorylation of Rpt6 at S120 is necessary, but not sufficient, for CaMKIIα-mediated activation of the proteasome.
Figure 2.
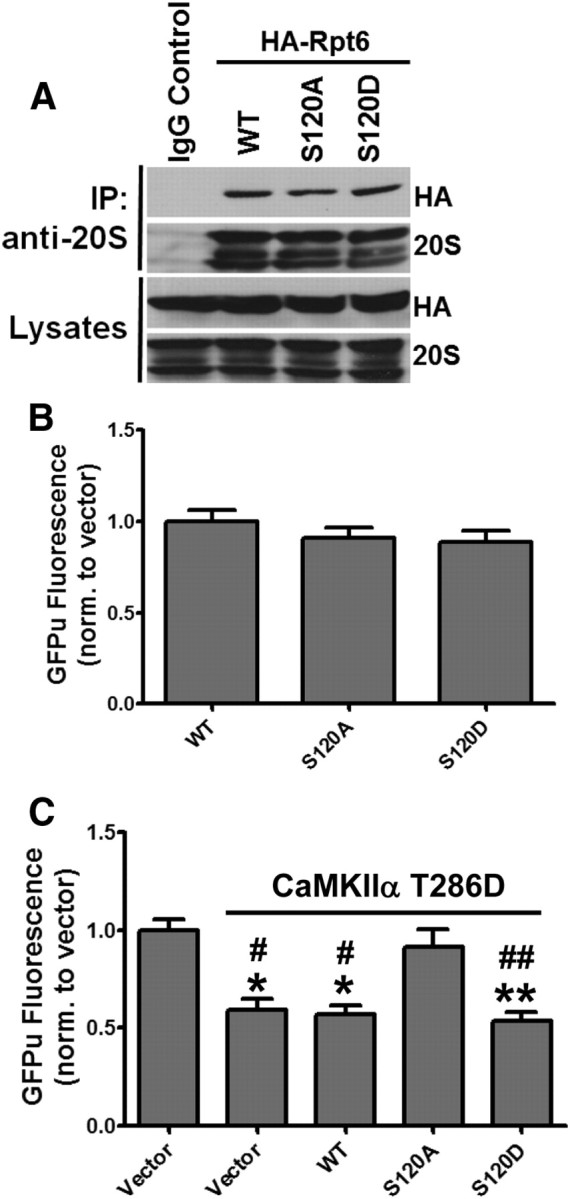
Phosphorylation of Rpt6 S120 is required for CaMKIIα-dependent stimulation of the proteasome. A, Ectopically expressed Rpt6 is efficiently incorporated into endogenous proteasomes. Proteasomes were immunoprecipitated from HEK293 cells expressing Rpt6 WT, S120A, or S120D. Immunoprecipitates and lysates were immunoblotted for Rpt6 (HA) or 20S proteasome subunits. B, HEK293 cells were coexpressed with GFPu and Rpt6 WT, S120A, or S120D. GFPu fluorescence was imaged after 18 h of expression. No significant differences were observed across the three Rpt6 constructs. C, HEK293 cells were cotransfected with β-gal (control vector) or CaMKIIα T286D and Rpt6 (WT, S120A, S120D) along with the GFPu proteasome reporter. S120A-attenuated CaMKIIα-dependent GFPu degradation. n >116 cells from three independent experiments. Compared with control: **p < 0.001; *p < 0.01; compared with CaMKIIα + S120A: ##p < 0.001; #p < 0.05; one-way ANOVA with post hoc Bonferroni's multiple-comparison test.
Phosphorylation affects the distribution and detergent-resistant extraction of Rpt6 in hippocampal dendrites
To further explore the effects of Rpt6 phosphorylation on proteasome function, we set out to determine whether phosphorylation of Rpt6 affected its distribution in neuronal dendrites. We first asked whether there were any differences in the distribution of Rpt6 S120A and S120D in hippocampal dendrites. We expressed HA-Rpt6 WT, S120A, and S120D in mature dissociated rat hippocampal neurons and immunostained for both Rpt6 (HA) and the postsynaptic protein PSD-95 (Fig. 3A,B). All three were distributed throughout the soma and dendrites in both punctate and diffuse pattern (Fig. 3A,B). We compared the average (±SEM) Pearson's coefficient for Rpt6 and PSD-95 in WT, S120A-, and S120D-expressing cells, and found there to be no significant differences between Rpt6 WT and S120D. However, Rpt6 S120A had a significantly decreased Pearson's coefficient compared with WT or S120D (p < 0.05; 0.23 ± 0.02, 0.15 ± 0.01, 0.24 ± 0.02, respectively, for WT, S120A, and S120D) (Fig. 3C). This suggests that the inability to phosphorylate Rpt6 at S120 decreases its accumulation at synapses.
Translocation to, and sequestration of proteasomes in, dendritic spines following neuronal stimulation involves CaMKIIα (Bingol and Schuman, 2006; Bingol et al., 2010), which may account for why we observed no differences between WT and S120D. Synaptic proteins can be tethered to postsynaptic sites by association with scaffold proteins and the actin cytoskeleton, often rendering them resistant to detergent extraction (Allison et al., 1998). We therefore asked whether phosphorylation of Rpt6 at S120 increases its detergent resistance in hippocampal dendrites. Hippocampal neurons expressing Rpt6 WT, S120A, or S120D were detergent extracted with Triton X-100 as previously described (Allison et al., 1998). We found that S120D had significantly increased detergent resistance relative to WT, while S120A was more readily extracted by the Triton X-100 treatment (p < 0.05; 1.00 ± 0.04, 0.82 ± 0.04, 1.42 ± 0.08, respectively, for WT, S120A and S120D) (Fig. 3D,E). This suggests that proteasomes may be tethered in spine compartments in part by phosphorylation of Rpt6.
Phosphorylation of Rpt6 regulates synaptic strength
Having shown that CaMKII phosphorylates Rpt6 in an activity-dependent manner, we examined whether changes to Rpt6 phosphorylation in turn had any effect on synaptic function. mEPSCs were recorded from cultured rat hippocampal neurons expressing Rpt6 WT, S120A, or S120D. We found that expression of Rpt6 S120A and S120D produced opposite effects on synaptic strength (Fig. 4A,B). Expression of S120D for 24 h caused a significant decrease in mEPSC amplitude (11.37 ± 0.63 pA), while expression of S120A led to a significant increase in mEPSC amplitude (17.90 ± 0.76 pA) compared with WT infected neurons (14.73 ± 1.16 pA) (n = 8–12 cells per condition; p < 0.05) (Fig. 4B). No significant differences in mEPSC frequency were seen, suggesting the observed effect was likely to be postsynaptic in origin (Fig. 4C). This scaling of mEPSC amplitude following Rpt6 S120A or S120D expression mimics changes in mEPSC amplitude observed following chronic blockade or upregulation of synaptic activity. Therefore, we decided to investigate whether Rpt6 phosphorylation altered homeostatic scaling of mEPSC amplitude produced by prolonged TTX or BIC treatments. Treating hippocampal neurons with TTX (2 μm, 24 h) or BIC (50 μm, 48 h) alone, resulted in an increase and a decrease in mEPSC amplitude, respectively (13.42 ± 0.63, 19.06 ± 1.29, and 10.96 ± 0.62 pA, respectively, for untreated, TTX treated. and BIC treated) (n = 15–29 cells per condition; p < 0.05) (Fig. 4D). Strikingly, we found that expression of Rpt6 S120A mimics TTX-induced and occludes BIC-induced scaling of mEPSC amplitude (S120A plus TTX: 18.08 ± 1.83 pA; S120A plus BIC: 18.13 ± 1.99 pA) (Fig. 4D). Conversely, expression of Rpt6 S120D mimics BIC and occludes TTX-induced scaling of mEPSC amplitude (S120D plus BIC: 12.49 ± 1.19 pA; S120D plus TTX: 13.98 ± 1.11 pA) (n = 14–19 cells per condition; no significance: p > 0.05 for S120A and S120D bracketed groups, respectively) (Fig. 4D). No significant differences in mEPSC frequency were observed (data not shown). Together, these data suggest that changes in proteasome function via phosphorylation of Rpt6 at S120 may be involved in homeostatic synaptic plasticity.
Figure 4.

Homeostatic scaling of mEPSC amplitude produced by chronic application of bicuculline or tetrodotoxin is both mimicked and occluded by altered Rpt6 phosphorylation. A, Example traces of mEPSCs recorded from hippocampal neurons expressing Rpt6 WT, S120A, or S120D. Calibration: 10 pA, 250 ms. B, S120A Rpt6 increases while S120D decreases mEPSC amplitude relative to WT, respectively. C, No significant change to mEPSC frequency was observed. n = 8, 12, and 9, respectively, for WT, S120A, and S120D; *p < 0.05, ANOVA with post hoc Fisher's least significant difference (LSD) test. D, Rpt6 S120A mimics and occludes scaling of mEPSC amplitude produced by chronic application of TTX (2 μm, 24 h) and BIC (50 μm, 48 h), respectively. Conversely, Rpt6 S120D mimics and occludes scaling of mEPSC amplitude produced by chronic application of BIC and TTX, respectively. n = 14–29 cells per condition. For comparison of untreated, TTX, and BIC: *p < 0.05, ANOVA and post hoc Fisher's LSD test. For comparison of S120A or S120D with or without TTX and BIC, respectively: p > 0.05, ANOVA. NS, No significant difference in bracketed conditions. Note: S120A and S120D alone conditions in D are taken from B for comparison purposes.
Discussion
In this study, we have further evaluated the biological relevance of Rpt6 phosphorylation on proteasome and synaptic function. Rpt6 S120 phospho-specific antibodies demonstrated that CaMKIIα phosphorylates Rpt6 on serine 120 in an activity-dependent manner (Fig. 1). Interestingly, we found that overexpression of the Rpt6 phospho-mutants alone had no effect on proteasome activity in HEK293 cells (Fig. 2). However, expression of the phospho-dead variant of Rpt6 (S120A) blocked CaMKIIα-dependent stimulation of proteasome function (Fig. 2). Mechanistically, it is yet to be fully determined how CaMKII mediates proteasome assembly or activity and whether other phosphorylation sites on Rpt6 or other proteasome subunits are required. Moreover, as several proteasome-interacting proteins have been identified (Glickman and Raveh, 2005; Schmidt et al., 2005), one plausible hypothesis is that CaMKIIα regulates these interactions to control the activity of proteasomes in neurons, although direct interactions between Rpt6 and CaMKIIα may also be required. Indeed, it has been shown that interactions between CaMKII and Rpt6 are involved in trafficking of proteasomes to spine compartments (Bingol et al., 2010). Interestingly, we found that increased synaptic activity enhances the association between CaMKIIα and phosphorylated Rpt6 (Fig. 1F). Furthermore, Rpt6 S120A was found to show decreased resistance to detergent extraction, while the opposite was true for Rpt6 S120D, indicating that phosphorylation of Rpt6 may promote the tethering of proteasomes to scaffolds and/or cytoskeletal components. Together, these findings further support the notion that phosphorylation of Rpt6 at S120 by CaMKIIα is involved in the trafficking and sequestration of proteasomes in spine compartments.
Modifications in synaptic strength are accompanied by changes in the protein composition of synapses. This process is dependent on neuronal activity, can be bidirectional, and requires UPS function (Ehlers, 2003). In addition, proteasome function has been shown to be required for various synaptic plasticity- and behavior-related paradigms such as LTP, LTD, and fear memory consolidation (Fonseca et al., 2006; Karpova et al., 2006; Dong et al., 2008; Lee et al., 2008). We hypothesized that regulating proteasome function via Rpt6 phosphorylation may be important for synaptic strength. In this regard, we found that expression of Rpt6 S120A and S120D produced opposite effects on synaptic strength (Fig. 4A,B). To our knowledge, this is the first demonstration that the phosphorylation status of a single proteasome subunit can modulate synaptic strength bidirectionally. Moreover, to our surprise, we found that homeostatic scaling of mEPSC amplitude produced by chronic application of BIC or TTX is both mimicked and occluded by altered Rpt6 phosphorylation (Fig. 4D). Together, these data indicate that CaMKII-dependent phosphorylation of Rpt6 at S120 may be an important regulatory mechanism for proteasome-dependent control of synaptic remodeling and efficacy.
Footnotes
S.N.D. was supported by the University of California, San Diego, and a Neuroplasticity of Aging NIH Training Grant. The work was supported by National Institutes of Health Grants MH085798 (M.A.S.), NS054732 (G.N.P.), and NS060847 (G.N.P.), and Grant P50-GMO85764 from the Center for Systems Biology. M.A.S. is a Pew Biomedical Scholar. We thank Alysia Birkholz and David Pardo for help with experiments; and George DeMartino, Jeff Keil, and members of the Patrick laboratory for helpful discussion, advice, and critical review of the manuscript.
The author's declare no competing financial interests.
References
- Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Upadhya SC, Ding L, Smith TK, Hegde AN. Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn Mem. 2008;15:335–347. doi: 10.1101/lm.984508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nägerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006;52:239–245. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Raveh D. Proteasome plasticity. FEBS Lett. 2005;579:3214–3223. doi: 10.1016/j.febslet.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Jakawich SK, Neely RM, Djakovic SN, Patrick GN, Sutton MA. An essential postsynaptic role for the ubiquitin proteasome system in slow homeostatic synaptic plasticity in cultured hippocampal neurons. Neuroscience. 2010;171:1016–1031. doi: 10.1016/j.neuroscience.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knöpfel T, Behnisch T. Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J Neurosci. 2006;26:4949–4955. doi: 10.1523/JNEUROSCI.4573-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ. Catalytic mechanism and assembly of the proteasome. Chem Rev. 2009;109:1509–1536. doi: 10.1021/cr8004857. [DOI] [PubMed] [Google Scholar]
- Patrick GN. Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol. 2006;16:90–94. doi: 10.1016/j.conb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Hanna J, Elsasser S, Finley D. Proteasome-associated proteins: regulation of a proteolytic machine. Biol Chem. 2005;386:725–737. doi: 10.1515/BC.2005.085. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- Zhang F, Hu M, Tian G, Zhang P, Finley D, Jeffrey PD, Shi Y. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009b;34:473–484. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]