

Abstract
Objective:
Recommendations for the diagnosis of preclinical Alzheimer disease (AD) have been formulated by a workgroup of the National Institute on Aging and Alzheimer's Association. Three stages of preclinical AD were described. Stage 1 is characterized by abnormal levels of β-amyloid. Stage 2 represents abnormal levels of β-amyloid and evidence of brain neurodegeneration. Stage 3 includes the features of stage 2 plus subtle cognitive changes. Stage 0, not explicitly defined in the criteria, represents subjects with normal biomarkers and normal cognition. The ability of the recommended criteria to predict progression to cognitive impairment is the crux of their validity.
Methods:
Using previously developed operational definitions of the 3 stages of preclinical AD, we examined the outcomes of subjects from the Mayo Clinic Study of Aging diagnosed as cognitively normal who underwent brain MRI or [18F]fluorodeoxyglucose and Pittsburgh compound B PET, had global cognitive test scores, and were followed for at least 1 year.
Results:
Of the 296 initially normal subjects, 31 (10%) progressed to a diagnosis of mild cognitive impairment (MCI) or dementia (27 amnestic MCI, 2 nonamnestic MCI, and 2 non-AD dementias) within 1 year. The proportion of subjects who progressed to MCI or dementia increased with advancing stage (stage 0, 5%; stage 1, 11%; stage 2, 21%; stage 3, 43%; test for trend, p < 0.001).
Conclusions:
Despite the short follow-up period, our operationalization of the new preclinical AD recommendations confirmed that advancing preclinical stage led to higher proportions of subjects who progressed to MCI or dementia.
Guidelines for the diagnosis of Alzheimer disease (AD) have recently been revised by workgroups of the National Institute on Aging and Alzheimer's Association (NIA-AA). An asymptomatic or latent form of the disease, dubbed preclinical AD, was proposed, in which a cognitively normal (CN) person has evidence of AD pathophysiologic changes.1
The new NIA-AA criteria for preclinical AD are conceptualized as having 3 stages.1 Stage 1 is characterized by abnormal levels of β-amyloid. Abnormal levels of β-amyloid can be demonstrated by PET amyloid imaging or CSF β-amyloid levels. Stage 2 represents abnormal levels of β-amyloid and, in addition, brain neurodegeneration as evidenced by brain atrophy on structural MRI, abnormalities on [18F]fluorodeoxyglucose (FDG) PET, or elevated levels of CSF tau. Stage 3 includes the features of stage 2 (i.e., β-amyloidosis and neurodegeneration) as well as subtle cognitive changes. The order of the NIA-AA stages is meant to imply that the risk for cognitive impairment due to AD increases progressively across successive stages.
We have developed an operational approach to the new criteria and evaluated the distribution of preclinical AD stages among CN subjects.2 Although recent studies have begun to investigate the risk for cognitive decline in persons with abnormal β-amyloidosis (preclinical AD, stages 1–3 in aggregate),3,4 there are no studies, to our knowledge, that have examined the other stages individually as defined in the new NIA-AA criteria. In the present analysis, we prospectively examined the utility of our operationalization of the criteria to predict progression to cognitive impairment or dementia in CN subjects from the Mayo Clinic Study of Aging (MCSA).
METHODS
Subjects.
The MCSA is a population-based study of cognitive aging that was established in Olmsted County, Minnesota, in October 2004.5 All subjects are reevaluated every 15 months. Since 2004, the MCSA enrolled 2,454 subjects who proved to be CN. The study design,5 the prevalence of mild cognitive impairment (MCI)6 and dementia,7 and the incidence of MCI8 have been reported. In 2005, subjects were invited to undergo brain MRI. Beginning in 2006, both newly and previously enrolled subjects were offered the opportunity to undergo PET imaging. The only exclusion criteria were specific contraindications to MRI.
Standard protocol approvals and patient consents.
All study protocols were approved by the Mayo and Olmsted Medical Center Institutional Review Boards, and all subjects provided signed informed consent to participate in the study and in the imaging protocols.
All MCSA subjects undergo a clinical and cognitive assessment every 15 months that includes 9 neuropsychological tests.5,6 The evaluations of all subjects were reviewed by a consensus panel consisting of physicians (neurologists and geriatricians), neuropsychologists, and study nurses. Subjects in the present study were diagnosed by the consensus panel as being CN, based on the clinical assessments including mental status examinations, informant interviews and a neuropsychological testing battery described below.5 Imaging findings were not used in forming a clinical diagnosis.
The neuropsychological battery was constructed as described previously.5 Domain-specific measures are formulated from the Wechsler Adult Intelligence Scale–Revised (WAIS-R), Wechsler Memory Scale-Revised (WMS-R), Auditory Verbal Learning Test (AVLT), Trail Making Test (TMT), category fluency test, and Boston Naming Test (BNT). Four cognitive domains are assessed: Executive (TMT: Part B, WAIS-R Digit Symbol); Language (BNT, category fluency); Memory (WMS-R Logical Memory-II, delayed recall percent retention, WMS-R Visual Reproduction-II delayed recall percent retention, AVLT delayed recall percent retention); and Visuospatial (WAIS-R Picture Completion, WAIS-R Block Design).
Subjects were considered to be CN if they performed within the normative range and did not meet criteria for MCI or dementia.5,6 A diagnosis of MCI was defined according to published criteria9 as cognitive concern by subject, informant, nurse, or physician; impairment in one or more of the 4 cognitive domains; essentially normal functional activities; and absence of dementia, according to the DSM-IV criteria.10 Subjects with MCI were classified as having amnestic MCI if the memory domain was impaired or nonamnestic MCI if the memory domain was not impaired. A diagnosis of dementia was based on the DSM-IV criteria.10
Although the MCSA uses age-corrected and (as appropriate) education-corrected Mayo Older Adult Normative Scores in forming a clinical diagnosis,11 we analyzed unadjusted z scores using means and SDs from a reference sample that was composed of 1,624 CN subjects from the initial MCSA enrollment visit. This large sample of subjects provided test-level, domain-level, and global-level reference means and SDs.
Our approach to obtaining domain-level z scores was as follows. First, individual test scores were converted to z scores using the test's reference mean and SD. Second, a raw domain score was calculated by taking the average of the component z scores. Third, the resulting raw domain score was converted to a z score using the domain-level reference mean and SD. A global cognitive summary score was formed by summing the 4 individual domain z scores, and this was converted to a z score using the global reference mean and SD. This global summary z score was used to assess cognitive impairment in our subjects.
We identified 529 MCSA CN subjects with cross-sectional MRI and PET imaging and global cognitive z score data. As of November 2011, 299 subjects (57%) had at least one follow-up examination in which their diagnostic status was determined. One subject with a follow-up visit less than 1 year after the baseline and 2 subjects missing the 15-month visit were subsequently excluded, leaving 296 subjects in our longitudinal analysis. Seventeen subjects (7%) among the 230 not included in the longitudinal analysis were lost to follow-up.
Imaging methods.
MRI was performed at 3 T with a 3-dimensional-magnetization-prepared rapid gradient echo sequence12 as described previously. Our primary MRI measure was hippocampal volume (measured with FreeSurfer software, version 4.5.013) adjusted for total intracranial volume (HVa).14 Total intracranial volume was measured by an algorithm developed by our laboratory.15 We calculated HVa as the residual from a linear regression of hippocampal volume (y) vs total intracranial volume (x).
PET images16 were acquired using a PET/CT scanner. A CT image was obtained for attenuation correction. The 11C-Pittsburgh compound B (PiB) PET scan consisting of 4 5-minute dynamic frames was acquired from 40 to 60 minutes after injection.17,18 [18F]FDG-PET images were obtained 1 hour after the PiB scan. Subjects were injected with [18F]FDG and imaged after 30–38 minutes, for an 8-minute image acquisition consisting of 42-minute dynamic frames.
Quantitative image analysis for both PiB and FDG was done using our in-house fully automated image processing pipeline.19 A global cortical PiB-PET retention ratio was formed by calculating the median uptake over voxels in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus regions of interest (ROIs) for each subject and dividing this by the median uptake over voxels in the cerebellar gray matter ROI of the atlas.20 FDG-PET scans were analyzed in a similar manner. For FDG-PET, we used the glucose metabolic rates from an AD signature set of ROIs consisting of angular gyrus, posterior cingulate, and inferior temporal cortical ROIs21 normalized to pons uptake.
The median (interquartile range [IQR]) number days between the cognitive assessment and the MRI scan was 53 (39–66) days. The median (IQR) number of days between the MRI scan and the PET scan was 21 (8–35) days.
Operationalization of preclinical criteria.
We previously described our approach to the definition of imaging cutpoints for stages 1 and 2 of the preclinical criteria.2 Using subjects with clinically diagnosed AD dementia from the Mayo Alzheimer's Disease Research Center, we chose the values for each imaging biomarker that corresponded to 90% sensitivity. For abnormal brain β-amyloidosis, a requirement for all stages of the preclinical criteria, we used the cutpoint for the PiB-PET global cortical ratio of 1.5. For the markers of neurodegenerative changes required for stages 2 and 3, subjects were classified as having neurodegeneration if they had abnormal hippocampal atrophy or abnormal FDG-PET hypometabolism. The 90% sensitivity cutpoint for HVa was −0.70. For the FDG-PET hypometabolism ratio of the AD signature/cerebellar regions, the cutpoint value was 1.31.
For the subtle cognitive change required for stage 3,1 we defined the cognitive cutpoint based on the 10th percentile on the global neuropsychological composite z score from the baseline assessments of the 450 CN subjects who were part of the cross-sectional group with imaging biomarker assessments.2 In a secondary analysis we used the 10th percentile from the memory domain z score. The 10th percentile on our global cognitive composite corresponded to a z score of −0.85, whereas a value of −1.04 corresponded to the 10th percentile on the memory domain.
Subjects who were normal for the β-amyloid, neurodegenerative, or cognitive criteria were labeled as stage 0 to indicate that they are not currently on the AD pathophysiology pathway.
It was further necessary to define another group that did not conform to the 3 defined stages of the NIA-AA preclinical criteria. This group included subjects with abnormal neurodegeneration biomarkers but normal β-amyloid imaging. We designated this group as representing a suspected non-Alzheimer pathway (SNAP).2 We left unclassified 2 other sets of subjects: those with normal biomarkers but abnormal cognition and those with abnormal β-amyloid imaging and abnormal cognition but no neurodegeneration. This latter group was small, but we suspect these subjects may be misclassified and belong somewhere in NIA-AA preclinical stages 1–3.
Statistical analysis.
We summarized data for descriptive purposes using the median (IQR) for the continuous variables and counts (percent) for the categorical variables. We tested for differences in the continuous variables between groups using the Wilcoxon rank sum test, and χ2 tests were used for categorical variables. Because of the short follow-up period and variable duration of follow-up across the cohort, we focused on comparing the proportion of subjects that progressed to MCI or dementia by their 15-month follow-up visit. Because of the importance of keeping follow-up times the same across subjects, all subjects who were stable through 15 months were considered nonprogressors in our analysis, even if they were found to subsequently progress. We used a χ2 trend test to test for an increase in proportions from stage 0 to stage 3 and χ2 tests for 2 × 2 tables using the n − 1 method.22 All p values reported are 2-sided, and we did not adjust for multiple comparisons.
RESULTS
The characteristics of the 296 CN subjects in the longitudinal study group and the 2 larger groups (all MCSA CN subjects and all MCSA CN subjects with baseline imaging biomarkers and global cognitive z score) from which they were drawn are shown in table 1. The proportion of subjects falling into each stage of the preclinical criteria was similar to that for the larger group of 529 CN subjects who had imaging biomarkers measured. The demographics of the subjects in the longitudinal group, broken down by preclinical stage, are shown in table 2. Of 296 subjects, 31 (10%) progressed (27 amnestic MCI, 2 nonamnestic MCI, 1 vascular dementia, and 1 dementia with Lewy bodies). None developed AD dementia within 1 year of follow-up. Both patients who progressed to non-AD dementia fell into preclinical AD stage 2. Their PiB-PET global cortical ratios were 2.5 and 2.6; both met the FDG-PET criteria for abnormal glucose metabolism in AD signature regions.
Table 1.
Descriptive characteristics of MCSA CN participants
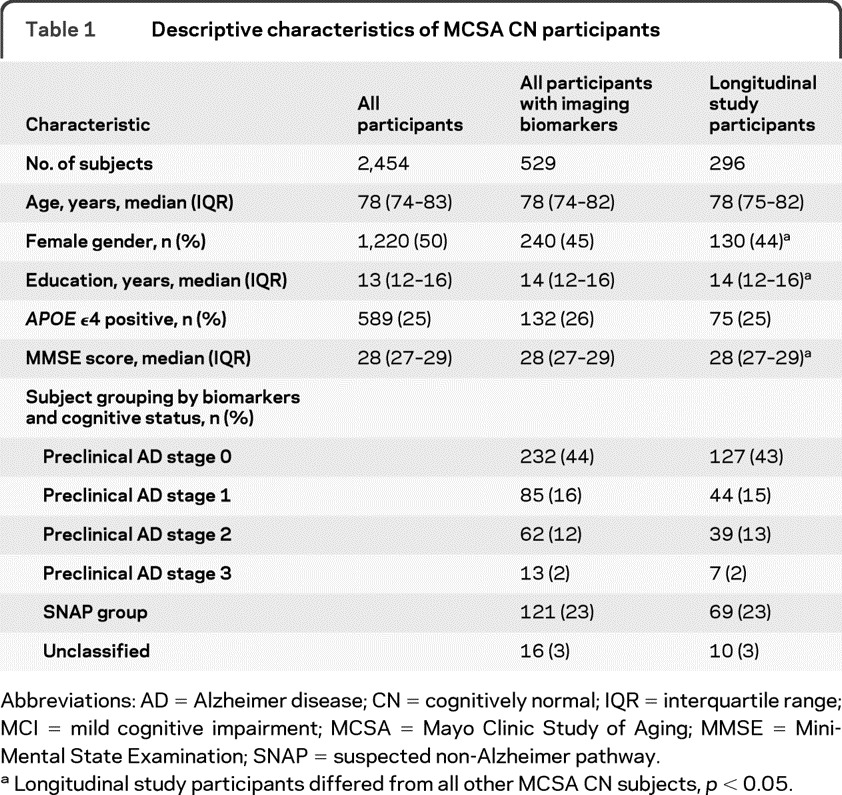
Abbreviations: AD = Alzheimer disease; CN = cognitively normal; IQR = interquartile range; MCI = mild cognitive impairment; MCSA = Mayo Clinic Study of Aging; MMSE = Mini-Mental State Examination; SNAP = suspected non-Alzheimer pathway.
Longitudinal study participants differed from all other MCSA CN subjects, p < 0.05.
Table 2.
Descriptive characteristics of participants by preclinical AD stage

Abbreviations: AD = Alzheimer disease; CN = cognitively normal; FDG = fluorodeoxyglucose; IQR = interquartile range; MCI = mild cognitive impairment; MCSA = Mayo Clinic Study of Aging; MMSE = Mini-Mental State Examination; PiB = Pittsburgh compound B.
Stage 0, all biomarkers normal; stage 1, abnormal PiB-PET; stage 2, abnormal PiB-PET and falling abnormal on at least one neurodegeneration biomarker; stage 3, abnormal PiB-PET, neurodegeneration and cognitive z score; SNAP, suspected non-Alzheimer pathway, where at least one neurodegeneration biomarker is abnormal with normal PiB-PET with or without abnormal cognition.
The median global cognitive z scores are >0 for the study group, which reflects their volunteer nature and the fact that about three-fourths of subjects had had one or more cognitive testing sessions before the PET baseline visit.
The proportion of subjects with MCI or dementia at the follow-up visit by preclinical stage (using the global cognitive z score to inform cognitive abnormality) is shown in table 2. Different groupings of stages are compared in table 3. A larger proportion of subjects progressed as stage increased from 0 to 3 (p value for trend <0.001). There were no significant differences between stages 1 and 2 nor between 2 and 3, although the sample sizes are small. There was no significant difference between the SNAP group and stages 1–3 (10% vs 18%, p = 0.18), although the absolute proportion of subjects who progressed to MCI or dementia was higher in stages 1–3. We repeated the analyses using the memory composite z score and found similar results (stage 0, 4%; stage 1, 10%; stage 2, 19%; stage 3, 44%; p value for trend <0.001; stage 0 [4%] vs stages 1–3 [17%], p = 0.002).
Table 3.
Proportion of participants who progressed to MCI/AD within 15 months by stage
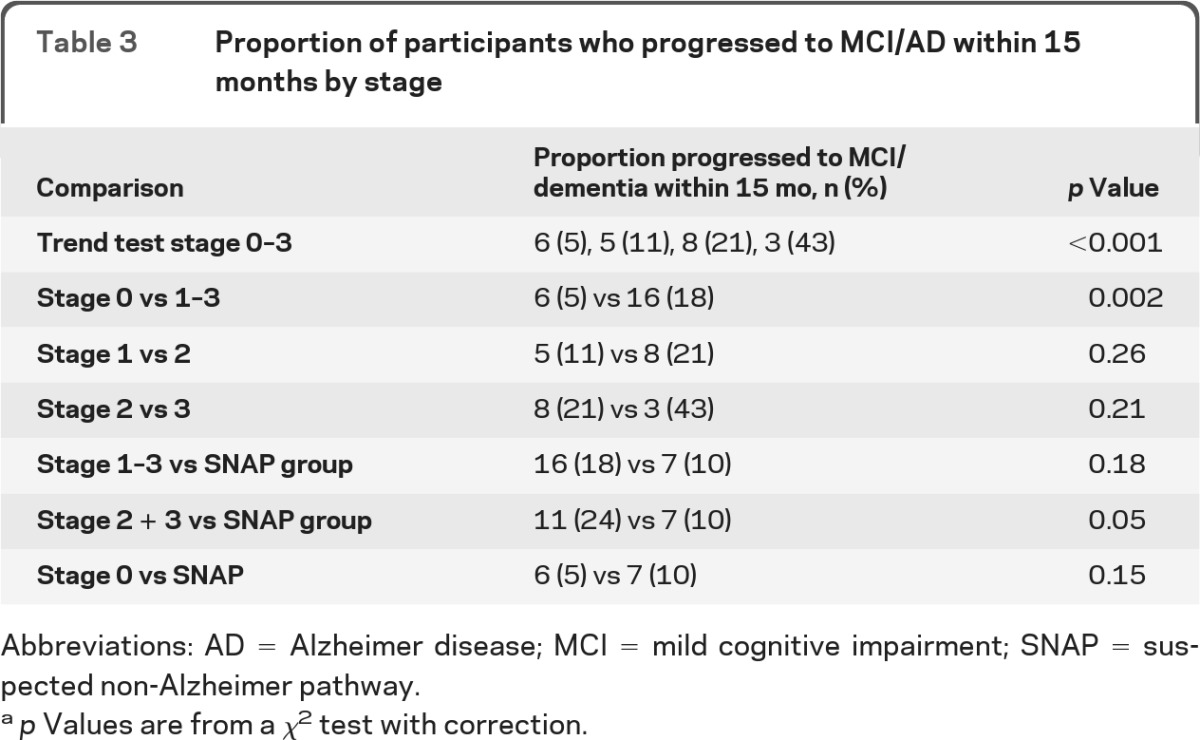
Abbreviations: AD = Alzheimer disease; MCI = mild cognitive impairment; SNAP = suspected non-Alzheimer pathway.
p Values are from a χ2 test with correction.
DISCUSSION
The best measure of the validity of the recently proposed criteria for preclinical AD ought to be their predictive accuracy for progression to mild cognitive impairment or dementia. Our analyses offer preliminary support for their predictive validity. The proportion of subjects who developed MCI or dementia over 1 year of follow-up increased across stages. Subjects with β-amyloidosis (stages 1–3) were at greater risk for progression to MCI or dementia than those in stage 0. Although our participants were virtually all of European descent, they were drawn from a population-based sample of elderly participants.
Although the risk increased significantly across stages, we were not able to demonstrate a significant difference in outcomes between stages 1 and 2 or 2 and 3, in part because of the few subjects in each group. However, the observed percentages suggest a possible increase in proportion of subjects who progressed to MCI or dementia in stage 3 (43%) compared with stage 2 (21%). Beyond these initial steps toward validating the criteria, the results also support a model of sequential pathophysiologic alterations that may lead to clinical AD dementia.23 Studies of persons with MCI at baseline have shown that the combination of abnormal β-amyloid biomarkers and neurodegenerative biomarkers conveys an increased risk for incident dementia compared with possession of only one abnormal biomarker type.24–27
In addition to the short follow-up and modest number of subjects who developed MCI or dementia, our findings should be considered preliminary because of other limitations of our analyses. First, there are several ways that the NIA-AA preclinical AD criteria could be operationalized.1 For imaging cutpoints, we chose values that were based on a level of abnormality that captured 90% of our group of patients with clinically diagnosed AD dementia.2 Sensitivity analyses showed that a less abnormal cutpoint included more subjects in stages 1–3 and the SNAP group and more abnormal cutpoints included fewer subjects.2 Among imaging features to define the neurodegenerative criteria, we allowed either hippocampal volume loss or FDG-PET hypometabolism rather than just one of these because each captures a different aspect of neurodegeneration. However, a requirement for abnormalities in both of them would have been more conservative. We also could have used alternative ROIs for volumetric MRI that included the AD signature regions28 similar to what we used in FDG-PET,26 or we could have chosen a smaller ROI such as the posterior cingulate gyrus alone for FDG-PET. Given the preliminary nature of the current analysis, we were unable to perform comparisons of different ROIs and cutpoints until we have studied more subjects for a longer period of time. Second, we did not use CSF results, because in the MCSA, the number of subjects with both CSF and longitudinal follow-up was much smaller than the current imaging-based cohort. The correct cutpoints and the correct imaging or biofluid modality will be the ones that, on future longitudinal analyses, offer the best combination of specificity and sensitivity.
We used only PiB-PET imaging to define brain β-amyloidosis. As expected, about one-third of our elderly subjects had abnormal levels of PiB retention.19,29–32 In our study group, amyloid positivity alone was associated with progression at 1 year (17% vs 8%, p = 0.02). The relationship between CSF β-amyloid and PiB-PET tracer retention levels is very strong.33,34 We would expect, therefore, that use of CSF β-amyloid levels for the definition of abnormal amyloidosis would yield similar results. The increased risk of MCI or dementia in subjects in stages 1–3 compared with stage 0 was consistent with prior studies that have shown that cognitively normal subjects with evidence of β-amyloidosis are at greater risk for cognitive decline.3,4 There are far more studies showing that persons with MCI who have abnormal β-amyloid biomarkers are more likely to progress to dementia than those lacking such biomarkers.4,35–38 Current models of AD biomarkers predict that β-amyloidosis should have the same predictive relationship for progression along the spectrum of cognitive impairment in both the cognitively normal and MCI phases of the process.23
The subtle cognitive decline feature of stage 3 of the preclinical AD criteria1 has been a challenge to operationalize and conceptualize. We chose to use a cross-sectionally derived global cognitive score for practical reasons, because it allowed us to include more subjects in analyses. Persons who eventually become demented have lower cognition than persons who do not progress to dementia.39 That was the case with our subjects: those in the lowest 10th percentile of global cognition had a greater risk for MCI or dementia than the rest of the group (32% vs 8%, p < 0.001). Conceptually, low cognitive functioning should be viewed as a measure of the outcome itself and not as a risk factor for cognitive decline to avoid circularity. In the specific context of the assessment of preclinical AD and not preclinical dementia in general, low cognitive functioning has meaning only in subjects who have evidence of β-amyloid accumulation and neurodegeneration.
One-quarter of our subjects had abnormal neurodegeneration biomarkers but normal β- amyloid imaging, a proportion similar to that of our cross-sectional group.2 We designated this group by the acronym SNAP. With 15 of 31 progressors (48%) below the cutpoint for PiB retention, our observations called attention to a non–β-amyloid preclinical state of dementia that might not be due to AD pathophysiology. Although our MRI and FDG-PET imaging biomarkers were based on AD pathophysiology, we believe that the SNAP group may have non-AD pathophysiologic processes such as cerebrovascular disease, synucleinopathies, non-AD tauopathies, or other neurodegenerative pathologic conditions.40 The SNAP subjects had a short-term prognosis that was not demonstrably different from preclinical AD stages 0 or stages 1–3 combined. To be sure, our analysis lacked power to detect subtle group differences.
Of the 2 subjects who progressed to dementia, one received a diagnosis of vascular dementia because of extensive white matter hyperintensities and worsening cognitive impairment after a stroke. The other was diagnosed with dementia with Lewy bodies because of very prominent parkinsonism and profound apathy (see appendix e-1 on the Neurology® Web site at www.neurology.org). Even with imaging findings indicative of AD pathophysiology, non-AD processes may be present and relevant to the cognitive disorder.
This report represents a preliminary view of the utility of the preclinical AD criteria for assessing prognosis. The concept of preclinical AD enables a systematic approach to prevention of clinical cognitive impairment. We are encouraged that the model we tested showed promising predictive abilities.
Supplementary Material
GLOSSARY
- AA
Alzheimer's Association
- AD
Alzheimer disease
- AVLT
Auditory Verbal Learning Test
- BNT
Boston Naming Test
- CN
cognitively normal
- DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, 4th edition
- FDG
fluorodeoxyglucose
- HVa
hippocampal volume adjusted for total intracranial volume
- IQR
interquartile range
- MCI
mild cognitive impairment
- MCSA
Mayo Clinic Study of Aging
- NIA-AA
National Institute on Aging and Alzheimer's Association
- PiB
Pittsburgh compound B
- ROI
region of interest
- SNAP
suspected non-Alzheimer pathway
- TMT
Trail Making Test
- WAIS-R
Wechsler Adult Intelligence Scale–Revised
- WMS-R
Wechsler Memory Scale–Revised.
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Knopman took part in data collection, supervised analyses, generated the first and final drafts, and takes overall responsibility for the data and the manuscript. Dr. Jack took part in data collection, supervised analyses, and critically reviewed the manuscript. H.J. Wiste performed analyses and critically reviewed the manuscript. S.D. Weigand performed analyses and critically reviewed the manuscript. Dr. Vemuri performed analyses of imaging data and critically reviewed the manuscript. Dr. Lowe took part in data collection, performed analyses of imaging data, and critically reviewed the manuscript, Dr. Kantarci performed analyses of imaging data and critically reviewed the manuscript. Dr. Gunter performed analyses of imaging data. M.L. Senjem performed analyses of imaging data. Dr. Ivnik took part in data collection and critically reviewed the manuscript. Dr. Roberts critically reviewed the manuscript. Dr. Boeve took part in data collection and critically reviewed the manuscript. Dr. Petersen obtained funding, took part in data collection, and critically reviewed the manuscript.
DISCLOSURE
Dr. Knopman serves as Deputy Editor for Neurology®. Dr. Jack serves on scientific advisory boards for GE Healthcare, Bristol Meyers Squibb, and Eli Lilly. H.J. Wiste, S.D. Weigand, and Dr. Vemuri report no disclosures. Dr. Lowe serves on a scientific advisory board for Bayer Pharmaceuticals and receives research support from GE Healthcare, AVID Radiopharmaceuticals, and Siemens Molecular Imaging. Dr. Kantarci, Dr Gunter, M.L. Senjem, Dr. Ivnik, and Dr. Roberts report no disclosures. Dr. Boeve receives research support from GE Healthcare. Dr. Petersen serves on a scientific advisory board for GE Healthcare. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Sperling RA, Aisen P, Beckett L, et al. Towards defining the preclinical stage of Alzheimer's disease: recommendations from the National Institute on Aging and the Alzheimer's Association Workgroup. Alzheimers Dement 2011; 7: 280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR, Jr, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging-Alzheimer's Association criteria for preclinical Alzheimer disease. Ann Neurol Epub 2011 Sep 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roe CM, Fagan AM, Williams MM, et al. Improving CSF biomarker accuracy in predicting prevalent and incident Alzheimer disease. Neurology 2011; 76: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol 2011; 69: 181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roberts RO, Geda YE, Knopman D, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008; 30: 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petersen RC, Roberts RO, Knopman DS, et al. Prevalence of mild cognitive impairment is higher in men than in women: The Mayo Clinic Study of Aging. Neurology 2010; 75: 889–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knopman DS, Petersen RC, Rocca WA, Larson EB, Ganguli M. Passive case-finding for Alzheimer's disease and dementia in two U.S. communities. Alzheimers Dement 2011; 7: 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roberts RO, Geda YE, Knopman DS, et al. The incidence of MCI differs by subtype and is higher in men: The Mayo Clinic Study of Aging. Neurology 2012; 78: 342–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256: 183–194 [DOI] [PubMed] [Google Scholar]
- 10. American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 11. Ivnik RJ, Malec JF, Smith GE. WAIS-R, WMS-R and AVLT norms for ages 56 through 97. Clin Neuropsychol 1992; 6(suppl): 83–104 [Google Scholar]
- 12. Jack CR, Jr, Bernstein MA, Borowski BJ, et al. Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimers Dement 2010; 6: 212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002; 33: 341–355 [DOI] [PubMed] [Google Scholar]
- 14. Jack CR, Jr, Twomey CK, Zinsmeister AR, Sharbrough FW, Petersen RC, Cascino GD. Anterior temporal lobes and hippocampal formations: normative volumetric measurements from MR images in young adults. Radiology 1989; 172: 549–554 [DOI] [PubMed] [Google Scholar]
- 15. Gunter JL, Bernstein MA, Borowski BJ, et al. Measurement of MRI scanner performance with the ADNI phantom. Med Phys 2009; 36: 2193–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-B. Ann Neurol 2004; 55: 306–319 [DOI] [PubMed] [Google Scholar]
- 17. Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh compound-B. J Cereb Blood Flow Metab 2005; 25: 1528–1547 [DOI] [PubMed] [Google Scholar]
- 18. McNamee RL, Yee SH, Price JC, et al. Consideration of optimal time window for Pittsburgh compound B PET summed uptake measurements. J Nucl Med 2009; 50: 348–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008; 131: 665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lopresti BJ, Klunk WE, Mathis CA, et al. Simplified quantification of Pittsburgh compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med 2005; 46: 1959–1972 [PubMed] [Google Scholar]
- 21. Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 2011; 32: 1207–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Campbell I. Chi-squared and Fisher-Irwin tests of two-by-two tables with small sample recommendations. Stat Med 2007; 26: 3661–3675 [DOI] [PubMed] [Google Scholar]
- 23. Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010; 9: 119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bouwman FH, Schoonenboom SN, van der Flier WM, et al. CSF biomarkers and medial temporal lobe atrophy predict dementia in mild cognitive impairment. Neurobiol Aging 2007; 28: 1070–1074 [DOI] [PubMed] [Google Scholar]
- 25. Vemuri P, Wiste HJ, Weigand SD, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology 2009; 73: 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Landau SM, Harvey D, Madison CM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010; 75: 230–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CR, Jr, Wiste HJ, Vemuri P, et al. Brain β-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain 2010; 133: 3336–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2008; 19: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pike KE, Savage G, Villemagne VL, et al. β-Amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain 2007; 130: 2837–2844 [DOI] [PubMed] [Google Scholar]
- 30. Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010; 31: 1275–1283 [DOI] [PubMed] [Google Scholar]
- 31. Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 2008; 65: 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rentz DM, Locascio JJ, Becker JA, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 2010; 67: 353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weigand SD, Vemuri P, Wiste HJ, et al. Transforming cerebrospinal fluid Aβ42 measures into calculated Pittsburgh compound B units of brain Aβ amyloid. Alzheimers Dement 2011; 7: 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann Neurol 2006; 59: 512–519 [DOI] [PubMed] [Google Scholar]
- 35. Wolk DA, Price JC, Saxton JA, et al. Amyloid imaging in mild cognitive impairment subtypes. Ann Neurol 2009; 65: 557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scheinin NM, Aalto S, Koikkalainen J, et al. Follow-up of [11C]PIB uptake and brain volume in patients with Alzheimer disease and controls. Neurology 2009; 73: 1186–1192 [DOI] [PubMed] [Google Scholar]
- 37. Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009; 132: 1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Okello A, Koivunen J, Edison P, et al. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology 2009; 73: 754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson RS, Leurgans SE, Boyle PA, Bennett DA. Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol 2011; 68: 351–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007; 69: 2197–2204 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.