

Abstract
Objectives:
To determine if there are in vivo differences in γ-aminobutyric acid (GABA) in the motor cortex and subcortical white matter of patients with amyotrophic lateral sclerosis (ALS) compared with healthy controls using proton magnetic resonance spectroscopy (1H-MRS).
Methods:
In this cross-sectional study, 10 patients with ALS and 9 age- and sex-matched healthy controls (HCs) underwent 3T edited 1H-MRS to quantify GABA centered on the motor cortex and the subcortical white matter.
Results:
Compared with healthy controls, patients with ALS had significantly lower levels of GABA in the left motor cortex (1.42 ± 0.27 arbitrary institutional units vs 1.70 ± 0.24 arbitrary institutional units, p = 0.038). There was no significant difference in GABA levels between groups in the subcortical white matter (p > 0.05).
Conclusion:
Decreased levels of GABA are present in the motor cortex of patients with ALS compared to HCs. Findings are consistent with prior reports of alterations in GABA receptors in the motor cortex as well as increased cortical excitability in the context of ALS. Larger, longitudinal studies are needed to confirm these findings and to further our understanding of the role of GABA in the pathogenesis of ALS.
Amyotrophic lateral sclerosis (ALS) is a fatal progressive neurodegenerative disease involving the spinal anterior horn cells, the corticospinal tract, and the motor cortex with a mean survival of 3–5 years.1 Clinical presentation of ALS is heterogeneous, making an accurate and timely diagnosis challenging in many cases. Whereas the detection of lower motor neuron (LMN) signs can be facilitated with EMG, there is a need for reliable assessment of upper motor neuron (UMN) involvement to supplement the clinical examination.2 As a result, there has been interest in developing UMN markers to aid ALS diagnosis and further our understanding of disease pathophysiology.
To address these issues, proton magnetic resonance spectroscopy (1H-MRS) has been used in a number of studies to investigate brain metabolism in ALS. Significantly higher levels of Glx (combined measure of glutamate and glutamine) as determined by MRS have been reported in the medulla of patients with ALS,3 adding credibility to the hypothesis that glutamate excitotoxicity plays a role in ALS pathophysiology. Riluzole is an antiglutamatergic drug; however, riluzole may also partially modulate the GABAergic system.4 One possibility is that decreased inhibition through GABAergic activity (in addition to increased excitation through glutamatergic activity) contributes to neuronal damage.
Conventional 1H-MRS techniques are unable to resolve GABA due to signal overlap and low signal intensity; recent advances in MRS editing techniques now allow for direct quantification of GABA at 3 Tesla field strength.5 Our study aimed at assessing differences in GABA levels in the motor cortex and the subcortical white matter between patients with ALS and healthy controls (HC).
METHODS
Participants.
We recruited 10 patients with ALS from the University of Michigan ALS clinic; we also recruited 9 HC. All ALS participants met the El Escorial criteria for probable or probable, laboratory-supported ALS. ALS and HC participants were between 18 and 80 years of age and right handed. Subjects were excluded if they had active substance abuse; psychiatric disease; history of CNS infection, head injury, or cerebrovascular disease; family history of motor neuron disease; or contraindication for MRI. We graded UMN scores based on a scale combining the Ashworth Spasticity Scale, presence of pathologic reflexes, and the Pseudobulbar Affect Scoring (scale range 0–33, with a higher score indicating higher disease burden). We determined LMN scores based on presence of atrophy, fasciculations, or decreased reflexes in the bulbar, cervical, and lumbosacral segments (range 0–6).
Standard protocol approvals, registrations, and patient consents.
The University of Michigan Institutional Review Board approved all study protocols (HUM00037485). All subjects provided informed written consent.
Magnetic resonance spectroscopy.
Subjects were imaged on a Philips Achieva 3T system (Best, Netherlands) using an 8-channel receive head coil. We used T1-weighted 3-dimensional magnetization-prepared rapid gradient echo imaging to place 3.0 cm × 2.0 cm × 3.0 cm voxels in the left motor cortex and the left subcortical white matter located caudally to the motor cortex (figure 1). These 2 anatomic locations were chosen based on prior MRS ALS study results. Single-voxel point resolved spectroscopy (PRESS) spectra (repetition time [TR]/echo time [TE] = 2,000/35 msec) were acquired using VAPOR water suppression with 32 averages. A MEGA-PRESS experiment for editing GABA was performed with the following parameters: TE = 68 msec (TE1 = 15 msec, TE2 = 53 msec); TR = 1.8 s; 256 transients of 2k data points; spectral width = 2 kHz; frequency selective editing pulses (14 msec) applied at 1.9 ppm (“on”) and 7.46 ppm (“off”). Slice-selective refocusing was performed using amplitude-modulated pulse GTST1203 (length = 7 msec, bandwidth = 1.2 kHz). Conventional PRESS spectroscopy was analyzed using LC Model. MEGA-PRESS spectroscopy was analyzed using in-house postprocessing software in MATLAB with Gaussian curve fitting to the GABA and inverted N-acetylaspartate (NAA) peaks. We measured GABA relative to the NAA signal in the edited spectra.6 The GABA:NAA ratio was then multiplied by the NAA concentration determined from LC Model analysis of a short-TE PRESS spectrum of the same voxel which provides a concentration of NAA relative to water (in AIU). CSF correction was performed for each voxel using statistical parametric mapping (SPM, Wellcome Trust Centre for Neuroimaging). Metabolite concentrations were only used for statistical analysis from LC Model if the Cramér-Rao bounds were less than 20%.
Figure 1. Voxel placement and resulting spectrum.

T1-weighted images show single voxel placement centered on the left motor cortex in the sagittal (A), axial (B), and coronal projections (C) and on the left subcortical white matter located caudal to the motor cortex in the sagittal (D), axial (E), and coronal projections (F). Representative magnetic resonance spectroscopy spectrum from the motor cortex using MEGA-point resolved spectroscopy editing technique (G). Combined measure of glutamine and glutamate (Glx) is resolved at 3.8 ppm, γ-aminobutyric acid (GABA) at 3.0 ppm with an inverted N-acetylaspartate (NAA) peak at 2.0 ppm.
Statistical analyses.
We performed 2-tailed independent sample t tests using Stata v.11 (College Station, TX) to determine differences in GABA levels and Spearman correlations for associations between GABA levels and UMN scores. Significance was set a priori at a p value of 0.05.
RESULTS
Participants.
Characteristics of the patients with ALS are presented in the table. Mean age of the patients with ALS was 61.5 ± 7.7 years and mean age of the HC was 58.6 ± 6.9 years (p = 0.63). Mean duration of symptoms was 29.2 ± 14.4 months and the mean revised Amyotrophic Lateral Sclerosis Functional Rating Scale value was 38.5 ± 6.7.
Table.
Patient characteristics
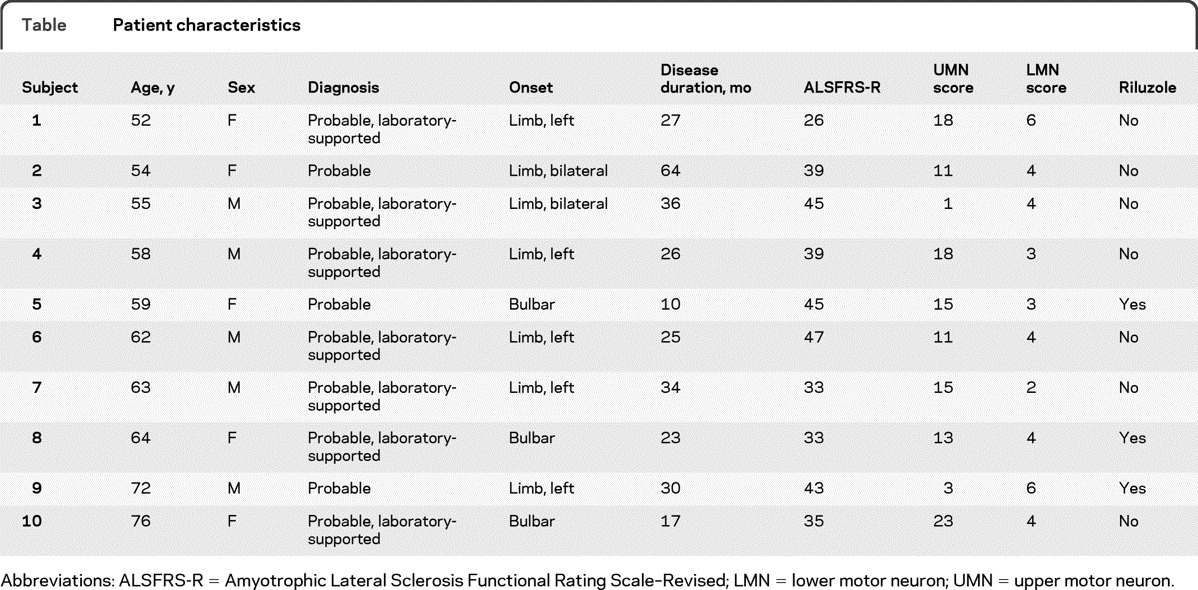
Abbreviations: ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; LMN = lower motor neuron; UMN = upper motor neuron.
Comparison of brain GABA levels.
Due to inadequate signal to noise ratio, the GABA-edited spectra from 1 healthy control and 1 patient with ALS were not analyzed for the motor cortex. As shown in figure 2, subjects with ALS demonstrated significantly lower levels of GABA within the left motor cortex (1.42 ± 0.27 AIU) compared to HC (1.70 ± 0.24 AIU; p = 0.038). As seen in figure 2, there were no significant mean GABA level group differences in the left subcortical white matter between the subjects with ALS (1.42 ± 0.30 AIU) and the HC (1.53 ± 0.34 AIU; p = 0.45). There were no significant differences in the motor cortex mean GABA levels of the limb-onset subjects with ALS compared to the bulbar-onset subjects with ALS (p = 0.45) or between subjects taking riluzole and those not taking riluzole (p = 0.63). There were no significant correlations between GABA levels and UMN scores.
Figure 2. Decreased γ-aminobutyric acid (GABA) levels within the motor cortex of patients with amyotrophic lateral sclerosis (ALS).

Circles represent GABA levels in the left motor cortex (A) and subcortical white matter located caudal to the motor cortex (B) for individual patients with ALS and healthy controls (HC). Horizontal bars indicate the mean. Patients with ALS have reduced concentrations of GABA in the left motor cortex compared to healthy controls. There is no difference between ALS and healthy control GABA levels in the left subcortical white matter. AIU = arbitrary institutional units; SCWM = subcortical white matter.
DISCUSSION
Our results suggest that lower levels of GABA are present in the motor cortex of patients with ALS. Previous human and animal studies in ALS have reported alterations in cortical excitation or GABAergic function. A postmortem ALS study reported alterations in the α1 and β1 mRNA subunits of the GABAA receptor.7 Transcranial magnetic stimulation studies have demonstrated increased cortical excitability in patients with ALS.8,9 In addition, PET studies have also demonstrated cortical reductions in 11C-flumazinal, which binds to the GABAA receptor in patients with ALS, suggesting a reduction in inhibitory neurotransmission.9 Nieto-Gonzalez et al.10 reported additional evidence of the association between decreased GABAergic inhibition and cortical excitability using an animal model of ALS. To our knowledge, no previous studies have used MRS to investigate GABA alterations in ALS.
Although the number of subjects included in the study was relatively small, our cohort is comparable to other studies which apply relatively new neuroimaging techniques to study neurologic diseases. Given the small numbers, we were unable to adjust for potential confounders. We recognize that GABA levels measured by 1H-MRS do not differentiate between intracellular and extracellular components. Furthermore, the MEGA-PRESS editing technique used does not excite pure GABA signal, and the 3 ppm signal has a significant contribution from macromolecules with a similar coupling network to that of GABA (i.e., signal at 3 ppm coupled to a signal at 1.7 ppm). While it is possible that effects are driven by changes in the macromolecular component, our study is based upon prior evidence of GABAergic dysfunction in ALS, and we therefore interpret our observations as representing actual alterations in GABA levels. An additional limitation of our study is the relatively large size of the MRS voxels which is required to achieve adequate signal to noise ratio at 3T field strength.
Lower GABA levels as measured by edited MRS are present in the motor cortex of patients with ALS compared to HC. Our findings are concordant with prior reports of increased cortical excitability and decreased cortical GABA receptor binding. It is therefore conceivable that GABAergic transmission alterations play a role in ALS pathophysiology. These studies need to be replicated in larger cohorts of patients to further elucidate the role of GABA in ALS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank MR senior research technologist Suzan Lowe for her assistance in imaging data acquisition.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- GABA
γ-aminobutyric acid
- 1H-MRS
proton magnetic resonance spectroscopy
- HC
healthy control
- LMN
lower motor neuron
- NAA
N-acetylaspartate
- PRESS
point resolved spectroscopy
- SCWM
subcortical white matter
- TE
echo time
- TR
repetition time
- UMN
upper motor neuron
Footnotes
Editorial, page 1544
AUTHOR CONTRIBUTIONS
Dr. Foerster contributed to the drafting/revising of the manuscript, study concept/design, analysis/interpretation of data, acquisition of the data, statistical analysis, study supervision/coordination, and obtaining funding. Dr. Callaghan contributed to the drafting/revising of the manuscript, study concept/design, and analysis/interpretation of data. Dr. Petrou contributed to the drafting/revising of the manuscript, study concept/design, analysis/interpretation of data, acquisition of the data, and study coordination. Dr. Edden contributed to the drafting/revising of the manuscript, acquisition of the data, and contribution of vital tools. Dr. Chenevert contributed to the drafting/revising of the manuscript, acquisition of the data, and contribution of vital tools. Dr. Feldman contributed to the drafting/revising of the manuscript, study concept/design, analysis/interpretation of data, study supervision, and obtaining funding.
DISCLOSURE
Dr. Foerster has received support from the American Roentgen Ray Fund. Dr. Callaghan has received support from a T32 training grant, ADA Junior Faculty Award and the Taubman Medical Research Institute. Dr. Petrou has received support from the Radiology Society of North American Scholar Fund. Dr. Edden reports no disclosures. Dr. Chenevert has served as a consultant for Philips Healthcare. Dr. Feldman reports no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Eisen A, Schulzer M, MacNeil M, Pant B, Mak E. Duration of amyotrophic lateral sclerosis is age dependent. Muscle Nerve 1993; 16: 27– 32 [DOI] [PubMed] [Google Scholar]
- 2. de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 2008; 119: 497– 503 [DOI] [PubMed] [Google Scholar]
- 3. Pioro EP, Majors AW, Mitsumoto H, Nelson DR, Ng TC. 1H-MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurology 1999; 53: 71– 79 [DOI] [PubMed] [Google Scholar]
- 4. Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem 2010; 17: 1942– 1199 [DOI] [PubMed] [Google Scholar]
- 5. Mescher M, Merkle H, Kirsch J, Garwood M, Gruetter R. Simultaneous in vivo spectral editing and water suppression. NMR Biomed 1998; 11: 266– 272 [DOI] [PubMed] [Google Scholar]
- 6. Stagg CJ, Best JG, Stephenson MC, et al. Polarity-sensitive modulation of cortical neurotransmitters by transcranial stimulation. J Neurosci 2009; 29: 5202– 5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petri S, Krampfl K, Hashemi F, et al. Distribution of GABAA receptor mRNA in the motor cortex of ALS patients. J Neuropathol Exp Neurol 2003; 62: 1041– 1051 [DOI] [PubMed] [Google Scholar]
- 8. Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 2006; 129: 2436– 2446 [DOI] [PubMed] [Google Scholar]
- 9. Turner MR, Osei-Lah AD, Hammers A, et al. Abnormal cortical excitability in sporadic but not homozygous D90A SOD1 ALS. J Neurol Neurosurg Psychiatry 2005; 76: 1279– 1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nieto-Gonzalez JL, Moser J, Lauritzen M, Schmitt-John T, Jensen K. Reduced GABAergic inhibition explains cortical hyperexcitability in the wobbler mouse model of ALS. Cereb Cortex 2011; 21: 625– 635 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.