

Abstract
Nanoparticles containing DNA complexed with the cationic polymer polyethylenimine (PEI)2 are efficient vehicles to transduce DNA into cells and organisms. DNA/PEI nanoparticles (DNPs) also elicit rapid and systemic release of pro-inflammatory cytokines that promote anti-tumor immunity. Here we report that DNPs possess previously unrecognized immunomodulatory attributes due to rapid up-regulation of IDO enzyme activity in lymphoid tissues of mice. IDO induction in response to DNP treatment caused dendritic cells (DCs) and regulatory T cells (Tregs) to acquire potent regulatory phenotypes. As expected, DNP treatment stimulated rapid increase in serum levels of IFN type I (IFNαβ) and II (IFNγ), which are both potent IDO inducers. IDO-mediated Treg activation was dependent on IFN type I receptor signaling, while IFNγ receptor signaling was not essential for this response. Moreover, systemic IFNγ release was caused by TLR9-dependent activation of Natural Killer cells, while TLR9 signaling was not required for IFNαβ release. Accordingly, DNPs lacking immunostimulatory TLR9 ligands in DNA stimulated IFNαβ production, induced IDO and promoted regulatory outcomes, but did not stimulate potentially toxic, systemic release of IFNγ. DNP treatment to induce IDO and activate Tregs blocked antigen-specific T cell responses elicited in vivo following immunization, and suppressed joint pathology in a model of immune-mediated arthritis. Thus, DNPs lacking TLR9 ligands may be safe and effective reagents to protect healthy tissues from immune-mediated destruction in clinical hyper-immune syndromes.
Introduction
Nanoparticles containing the cationic polyamine polyethylenimine (PEI) are efficient vehicles to transduce nucleic acids into cells and tissues (1–3). Previous studies on DNA/PEI nanoparticles (DNPs) focused on elucidating factors that influence the efficiency and stability of gene expression following DNA transduction. Relatively few studies focused on potentially toxic pro-inflammatory and immune stimulatory responses to DNP treatment, a key consideration when developing novel reagents for clinical applications. Several reports have described rapid, systemic release of pro-inflammatory cytokines such as IL-12, IFNγ and TNFα following DNP treatment in rodents. IL-12 released after DNP treatment mediated potent anti-tumor effects in mice bearing tumors, generating interest in exploiting such innate immunostimulatory responses to DNPs to boost anti-cancer therapy (4, 5). However sustained, systemic release of pro-inflammatory cytokines may provoke unacceptable toxicities that preclude chronic DNP treatments needed to achieve clinical efficacy.
Regulatory CD4 T cells of the Foxp3-lineage (Tregs) manifest potent immune regulatory phenotypes that may be exploited to treat and prevent hyper-immune syndromes such as autoimmunity and allograft rejection (6). However, the paucity of reliable methods to activate Tregs while not co-activating effector T cells, and the innate potential for Tregs to undergo functional re-programming in some settings of inflammation are formidable barriers to successful immunotherapy using Tregs (7). Previously, we reported that resting Tregs underwent rapid activation to acquire potent regulatory phenotypes in mice treated systemically with relatively high doses of TLR9 ligands (CpG oligonucleotides) due to induction of IDO enzyme activity in a rare subset of CD19+ DCs (8, 9). Tregs with stable regulatory phenotypes were also found in tumor-draining lymph nodes, and IDO activity in CD19+ DCs was essential to maintain Treg regulatory phenotypes (10, 11). In both inflammatory settings IDO-activated Tregs blocked production of pro-inflammatory cytokines by innate immune cells, prevented clonal expansion of antigen-activated effector T cells, and blocked Treg functional re-programming to become helper/effector T cells in response to TLR9-mediated activation signals (9, 11, 12). Thus, reagents that stimulate APCs to express IDO may constitute a novel class of immunomodulatory drugs potentially able to suppress immune-mediated tissue destruction by selective induction and maintenance of Treg regulatory phenotypes in patients with hyper-immune syndromes such as autoimmunity, allergies and transplanted allografts (13–16).
Several reagents that induce IDO enzyme activity in APCs have been described, including IFNs, reagents that stimulate IFN release such as TLR ligands (e.g. the TLR4 and TLR9 ligands LPS and CpGs, respectively), histone de-acetylase inhibitors, and engineered immunomodulatory reagents such as soluble CTLA4 (CTLA4Ig), some forms of which are approved for clinical use (e.g. Orencia™) to treat hyper-immune syndromes (17). IFNs, TLR ligands and histone de-acetylase inhibitors also elicit well-documented pro-inflammatory responses at doses when IDO is not induced, essentially precluding the use of such reagents in clinical settings of hyper-immunity. Moreover, IDO induction in APCs following CTLA4Ig treatment is critically dependent on poorly defined functional modalities in the immunoglobulin (Ig) domain that may be absent in CTLA4Ig isoforms developed for clinical applications (17).
Here we report that DNA/PEI nanoparticles (DNPs) possess potent and previously unrecognized immunomodulatory attributes. Immunomodulatory responses to DNPs overcame the immune stimulatory effects of induced pro-inflammatory cytokines by stimulating DCs and Tregs to acquire potent IDO-dependent regulatory phenotypes, which blocked T cell responses to immunization and ameliorated hyper-immunity that caused pathologic joint injury.
Materials and Methods
Mice
Mice were bred in a specific pathogen-free facility. The local (GHSU) Institutional Animal Care and Use Committee approved all procedures involving mice. TCR transgenic mice used as sources of responder T cells in suppression assays were described previously (8, 9).
DNA/PEI nanoparticle (DNP) treatment
Bacterial pDNA (pEGFPN1, Clontech) was prepared using an endotoxin-free Kit (Qiagen, Valencia, CA). Poly dA:dT (pAT) was purchased from Invivogen (San Diego, CA). DNPs were prepared using PEI or tetramethyl-rhodamine-conjugated PEI (Invivo-JetPEI™, Polyplus/VWR, Suwanee, GA) according to manufacturer's instructions. Mice were injected (i/v) with 30 µg pDNA or 21 µg pAT mixed with PEI (N:P =10 & 16.7, respectively).
Kynurenine and IDO enzyme activity detection in tissues
Snap frozen tissues were homogenized in PBS at 100 mg/ml (spleen) and 50 mg/ml (lymph nodes). Kynurenine and tryptophan were measured by HPLC after de-proteination using a C18 reverse phase column (18). IDO enzyme activities in cell-free tissue homogenates were measured as described (19). IDO activity was calculated as pmol kynurenine/hr/mg tissue.
IFN Assays
Serum IFNγ was measured by ELISA (eBioscience, San Diego, CA). Serum IFN type I was measured using an IFN bioactivity assay (20). Briefly, NCTC929 cells were pretreated with serial diluted serum samples overnight in the presence of 2 µg/ml of anti-IFNγ antibody (clone XMG1.2, BD Biosciences), and then infected with VSV. After 3 days, viable cells were stained with crystal violet and quantified by light absorption at 595 nm using a plate reader.
1-methyl-[D]-tryptophan (1MT)
1MT (a generous gift of NewLink Genetics Inc.) was prepared as a 20 mM stock solution in 0.1M NaOH, adjusted to pH 7.4, and stored in the dark. For in vitro use, 1MT was added at a final concentration of 100 µM. For in vivo treatment mice were provided with 1MT (2 mg/ml) in drinking water with sweetener (Nutrasweet) to enhance palatability (21).
Analytical flow cytometry
Cells were stained with the following antibodies; anti-CD4 (clone RM4–5), anti-Thy1.1 (clone OX-7), anti-NK1.1 (clone PK136), anti-CD49b (clone DX5), anti-IFNγ (clone XMG1.2) from Pharmingen-BD-Biosciences (San Jose, CA), and analyzed using a LSR2 flow cytometer (Becton-Dickinson). PBS-57 loaded CD1d tetramer and control tetramer were acquired from the NIH Tetramer Core facility. CFSE was purchased from Invitrogen. To detect intracellular IFNγ, cells were stained with anti-NK1.1 and anti-CD49b (DX5) mAbs, fixed (Cytofix/Cytoperm, BD Bioscience), and stained with anti-IFNγ. Some cells were incubated with brefeldin (1 µM, 3 hrs., BD Bioscience) to accumulate cytokines without further stimulation ex vivo. To detect IFNγ in OT-2 T cells, dLN cells were stimulated with PMA/ionomycin for 4 hrs. with brefeldin, stained with anti-CD4 and anti-Thy1.1 then fixed and stained to detect IFNγ.
FACS-sorted DCs
CD19+ and CD19neg DCs were sorted from spleens of untreated and DNP-treated B6 mice using a Dako Cytomation MoFlo cell sorter as described (22). Sorted DCs for cytospin analyses were collected onto microscope slides and DCs for RT-PCR analyses were sorted into tubes containing RNA protect reagent (Qiagen) before preparing RNA samples for quantitative RT-PCR analyses as described (23, 24).
Immunohistochemistry
Paraffin-embedded tissue sections (5 µm) and cytospin preparations were stained with custom made polyclonal rabbit anti-murine IDO antibody (Southern Biotechnology, Birmingham, AL) as described (25). For immunofluorescence staining frozen sections (7 µm) were fixed (methanol), blocked with 1% non-fat milk in PBS, incubated with primary (1.5 hrs) and dye-conjugated secondary (1 hr) antibodies, and mounted in anti-fade media with DAPI, (Invitrogen, Carlsbad, CA). CD11c and B220 mAbs were from BioLegend (San Diego, CA). Mouse absorbed Cy3 conjugated goat-anti-rabbit antibody, Dylight 488 conjugated goat-anti-armenian hamster antibody and AMCA-conjugated donkey-anti-rat antibody were from Jackson ImmunoResearch (West Grove, PA). PEI/Rhodamine was from Polyplus/VWR. A confocal microscope (Zeiss LSM510M) was used to generate some immunofluorescence images.
DC and Treg suppression assays
MACS-enriched splenic DCs (CD11c+) and Tregs (CD4+CD25+) were isolated according to manufacturer’s instructions (Miltenyi Biotec Inc., Auburn, CA), except that cells were incubated with beads at RT. T cell stimulatory activity of DCs was assessed by culturing graded numbers of DCs (6–50×103/well) with responder (MACS-enriched) CD8+ T cells from OT-1 (+SIINFEKL peptide) TCR-Tg mice +/−1MT (22). Suppressor activity of Tregs was assessed by culturing graded numbers of Tregs (1.25–20×103/well) with responder A1 (H-Y-specific) T cells, APCs (female CBA) and male (H-Y) peptide; a cocktail of anti-PD-1, anti-PD-L1, and anti-PD-L2 mAbs was added to some cultures to block PD-1-PD-L interactions (9, 11). Occasionally, IDO-independent T cell suppression manifests (as no effect of adding 1MT) at higher numbers of DCs (Fig. 2E) or Tregs for technical reasons probably related to mouse health status, age, and strain.
Figure 2. DNP treatment induces splenic DCs and Tregs to acquire regulatory phenotypes via IDO.
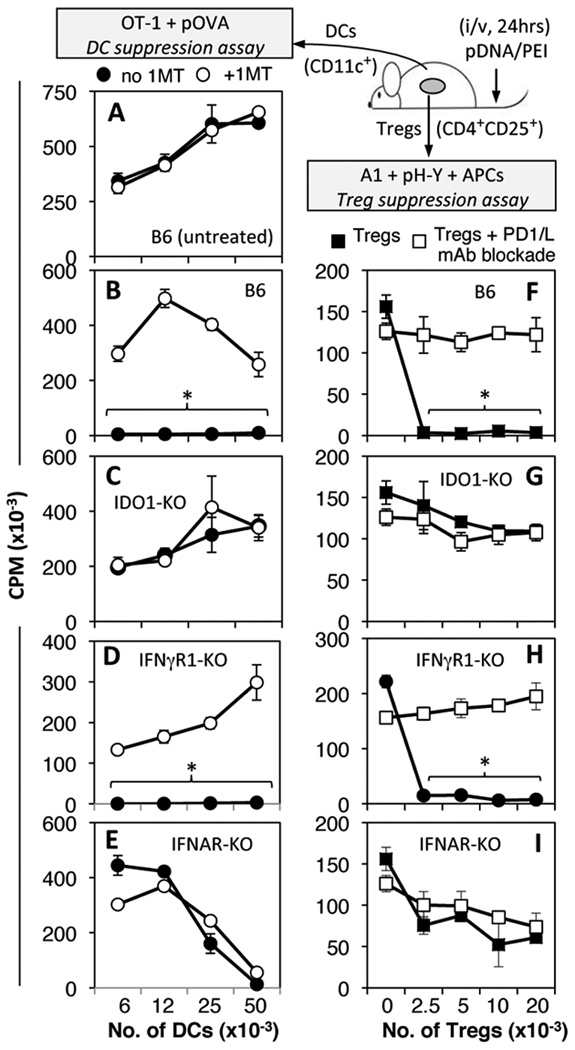
A–E. Graded numbers of MACS-enriched splenic CD11c+ DCs from untreated B6 (A) or pDNA/PEI-treated (i/v, 24 hrs.) mice were cultured with responder OT-1 T cells and OVA peptide. Parallel cultures contained IDO inhibitor (1MT, 200 µM). F–I. Graded numbers of MACs-enriched CD4+CD25+ Tregs were cultured with A1 T cells & APCs and H-Y peptide. Parallel cultures contained a cocktail of anti-PD-1 and anti-PD-L1/L2 blocking mAbs. T cell proliferation was assessed by measuring thymidine incorporation (72 hrs). Data are the means (+/− 1sd) from triplicate cultures. Asterisks highlight significant IDO-dependent suppression mediated by DCs or Tregs. Data are representative of experiments performed with 2 or more DNP-treated mice.
In vivo suppression assays
CFSE-labeled OVA-specific (Thy1.1) T cells from OT-1 or OT-2 donor mice were injected (i/v) into recipient B6 (Thy1.2) mice 1 day before immunization. CFSE labeling was performed by incubating MACS-enriched splenic CD8+ (OT-1) or CD4+ (OT-2) T cells at ~ 10×106 cells/ml in PBS with 2 µM CFSE (37°C, 15 mins). Mice were then immunized with 106 erythrocyte-free spleen cells from Act-mOVA transgenic mice (s/c), and treated with DNPs.
mBSA-induced arthritis model
B6 mice were sensitized with methylated BSA (mBSA, s/c, 500 µg in CFA, day 0), and booster injections (mBSA/IFA days 7, 14) Arthritis was induced by intra-articular challenge with mBSA (10 µg in PBS, day 21). Arthritis severity was evaluated by measuring joint swelling, neutrophil infiltration, and histological analysis as described (26, 27). To assess joint injury knee joints were dissected, fixed, and embedded in paraffin. Sagittal joint sections (5 mm) were stained with Safranin-O and counterstained with fast green/iron hematoxylin. Popliteal and inguinal LN cells draining inflamed joints were pooled, cultured (106 cells/well) +/− mBSA (100 µg/ml) for 36 hrs., and IL-17 and IL-6 concentrations were measured using a multiplex bead system (Luminex™) according to manufacturer's instructions.
Statistical Analysis
The unpaired Student’s t test (Graphpad Prism) was used for statistical analyses.
Results
DNPs induce rapid increase in IDO enzyme activity in mice
Elevated IDO protein levels were detected in mucosal and lymphoid tissues of mice following systemic DNP treatment to transduce bacterial plasmid DNA (pDNA) into mouse tissues (data not shown). To investigate this unanticipated response to DNP treatment levels of kynurenine (Kyn) - a tryptophan metabolite made by cells expressing IDO - were assessed in homogenates prepared from lymphoid tissues of DNP-treated mice (Table I). Significantly higher Kyn levels were detected in spleen and peripheral lymph nodes (LNs, ~10-fold and ~4-fold increase, respectively) relative to basal Kyn levels in tissues from untreated mice (Table I). Increased IDO enzyme activity was also detected in cell-free lysates from spleens of DNP-treated mice (Table I), measured as Kyn produced ex vivo in the presence of IDO enzyme substrate cocktail (19). However, elevated IDO enzyme activity was not detected in spleens from mice treated with PEI or pDNA alone (Table I). Thus DNP treatment stimulated rapid up-regulation of IDO enzyme activity in lymphoid tissues.
Table I.
Kynurenine and IDO activity in lymphoid tissues
| DNA nanoparticles pDNA |
Kynurenine | IDO activity | ||||
|---|---|---|---|---|---|---|
| Mouse | CpG | CpG-free | pAT | Tissue | (pmol/mg)a | (pmol/mg/hr)a |
| B6 | − | − | − | spleen | 0.7 ± 0.3 | 19.1 ± 2.7 |
| B6 | + | − | − | spleen | 7.0 ± 1.2b | 77.1 ± 17.8b |
| B6 | − | − | − | LNs | 2.6 ± 1.2 | nt |
| B6 | + | − | − | LNs | 10.2 ± 2.2b | nt |
| B6 | PEI only | spleen | nd | 15.3 ± 2.6 | ||
| B6 | pDNA only | spleen | nd | 16.8 ± 1.5 | ||
| B6 | − | + | − | spleen | 4.8 ± 2.5b | 54.9 ± 28.5c |
| B6 | − | − | + | spleen | 5.7 ± 3.1b | 74.0 ± 36.4c |
| TLR9-KO | − | − | − | spleen | 4.9 | 20.2 |
| TLR9-KO | + | − | − | spleen | 12.7 | 53.2 |
| TLR9-KO | − | + | − | spleen | 12.1 | 57.0 |
Notes: nd, not detected (below background); nt, not tested
a mean ± 1sd; p <0.002 – 0.0001b, p<0.013c (treated vs untreated)
bold numbers: IDO activity above basal levels (untreated mice)
DNPs induce IDO in peri-follicular regions of lymphoid tissues
To detect cells induced to express IDO after DNP treatment B6 mice were treated with pDNA/PEI (i/v, 24 hrs), and lymphoid tissues were stained to detect IDO. Dispersed clusters of cells expressing IDO were detected in peri-follicular regions of spleen and peripheral LNs, and stained cells displayed plasmacytoid morphologies (Fig. 1A, B). This pattern resembled staining patterns observed in mice treated with CTLA4Ig or high-doses of oligonucleotides with CpG motifs to ligate B7 and TLR9, respectively, which induced IDO in rare, but distinctive CD19+ DCs with attributes of both DCs and B cells (23, 28). To test if DNP treatment induced selective IDO expression in CD19+ DCs we sorted CD19+ and CD19neg DCs from spleens of DNP-treated B6 mice and stained sorted cells to detect IDO. Most CD19+ DCs expressed IDO (Fig. 1C) while almost no CD19neg DCs expressed IDO (Fig. 1D), indicating that DNP treatment stimulated selective IDO expression in CD19+ DCs. Though most IDO+ cells in lymphoid tissues co-expressed CD11c some IDO+ cells did not express CD11c (Fig. 1E, F), indicating that DNPs induced IDO in CD19+ DCs and some non-DCs in peri-follicular regions.
Figure 1. DNP treatment induces selective IDO expression in lymphoid tissues.
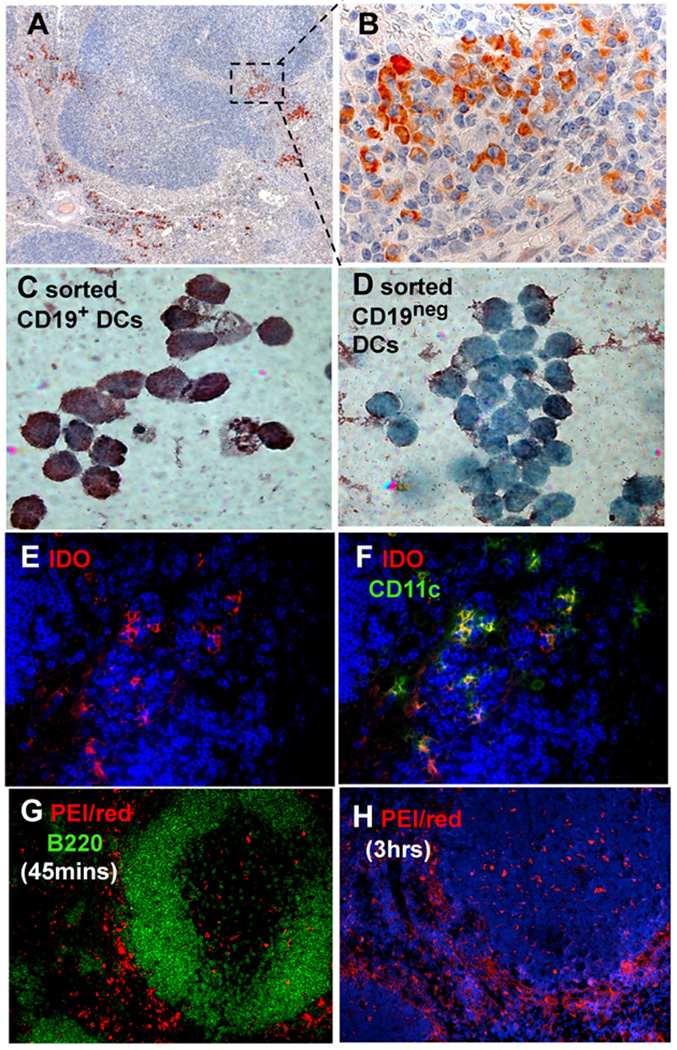
A–F. B6 mice were treated with pDNA/PEI (30 µg/6 µl, i/v, 24 hrs). Spleen sections (A, B) and FACS-sorted CD19+ (C) and CD19neg (D) DCs were stained to detect IDO (A–D) and counterstained with hematoxylin. E, F. Inguinal LNs were stained to detect IDO (red), CD11c (green) and B220 (blue). G, H. B6 mice were treated with DNPs containing rhodamine-conjugated PEI (red) and pDNA for 45 mins. (G) or 3 hrs. (H) and PEI was detected in spleen sections. Original magnifications; A, G, H (×200), B–D, (×1000); (E, F, ×400). Data are representative of experiments with at least 3 mice.
To evaluate where DNPs accumulated in lymphoid tissues B6 mice were treated with DNPs containing rhodamine-conjugated PEI. Only 45 minutes after DNP treatment dye-conjugated DNPs (red) had accumulated in peri-follicular regions adjacent to splenic B cell areas (B220+, Fig. 1G). After 3 hours DNPs had accumulated in marginal zones, and a few cells within lymphoid follicles also stained intensely (Fig. 1H, and data not shown), suggesting that DNPs were rapidly and selectively ingested by cells located in peri-follicular/marginal zones.
DNP treatment induces DCs to acquire potent regulatory phenotypes via IDO
To assess if DNP treatment induced DCs to acquire regulatory phenotypes dependent on IDO graded numbers of splenic CD11c+ DCs from DNP-treated mice (i/v, 24 hrs) were cultured with OVA-specific OT-1 T cells and cognate peptide (pOVA), SIINFEKL (Fig. 2, left). In this assay DCs from untreated mice exhibited T cell stimulatory activity ex vivo, while DCs from mice treated with other IDO inducers (CpGs, CTLA4Ig) did not stimulate T cell proliferation, unless the IDO inhibitor 1-methyl-[D]-tryptophan (1MT) was added to cultures (22, 28). As expected, DCs from untreated mice stimulated robust OT-1 proliferation that was not enhanced by adding 1MT (Fig. 2A, closed and open symbols, respectively). In contrast, DCs from DNP-treated B6 mice did not stimulate OT-1 T cell proliferation, unless 1MT was present in cultures and irrespective of how many DCs were added to cultures (Fig. 1B). DCs from DNP-treated mice lacking intact IDO1 genes (IDO1-KO mice) stimulated robust OT-1 proliferative responses that were not enhanced by adding 1MT (Fig. 2C). To assess IFN signaling requirements to induce functional IDO, mice lacking intact genes encoding IFNγ (IFNγR1-KO) and IFN type I (IFNAR-KO) receptors were treated with DNPs. IFNγR1-KO mice were used rather than IFNγR2-KO mice as the IFNγRα subunit (encoded by IFNγR1) is essential to bind IFNγ while the IFNγRβ subunit (encoded by IFNγR2) is required only for optimal IFNγ signaling; thus IFNγR1 gene ablation eliminates IFNγ signaling while IFNγR2 ablation may not eliminate IFNγ signaling completely (29). DCs from DNP-treated IFNγR1-KO mice mediated IDO-dependent T cell suppression comparable with B6 mice (Fig. 2D), while DCs from DNP-treated IFNAR1-KO mice did not mediate suppression via IDO (Fig. 2E); loss of T cell stimulatory properties at high DC numbers may be linked to the genetic defect in these mice or to non-specific suppression that occasionally manifests in these assays for technical reasons. Thus DCs acquired potent IDO-dependent T cell regulatory phenotypes after DNP treatment and IFN type I, but not IFNγ signaling was essential for regulatory responses to manifest.
DNP treatment induces Tregs to acquire potent regulatory phenotypes via IDO
Previously, we reported that IDO activity induced in CD19+ DCs stabilized Treg regulatory phenotypes, and prevented Treg functional re-programming at sites of tumor growth and following TLR9 ligation (8, 9, 11, 12). To evaluate if DNP treatment activated Tregs via IDO, graded numbers of splenic CD4+CD25+ cells from DNP-treated B6 mice (i/v, 24 hrs) were added to Treg suppression assays containing responder male (H-Y) antigen-specific CD4+ T cells from A1 TCR-Tg mice, cognate H-Y peptide, and APCs from CBA female mice (Fig. 2, right). Because these cultures contained no Treg mitogens, and Tregs (B6) were genetically mismatched with responder T cells and APCs, Tregs cannot activate during culture and regulatory phenotypes must be generated in vivo (9, 11). Adding as few as 2,500 Tregs (Tregs:Responders = 1:20) from DNP-treated B6 mice suppressed A1 T cell proliferation completely (Fig. 2F, closed symbols). Robust A1 T cell proliferation was restored in the presence of a cocktail of mAbs that block interactions between programmed death ligand-1 (PD-1), and its ligands, PD-L1 and PD-L2 (Fig. 2F, open symbols), a hallmark feature of IDO-activated Tregs (9, 11). Consistent with the interpretation that Treg activation was IDO-dependent, Tregs from DNP-treated IDO1-KO mice had no significant regulatory activity, and PD-1/PD-L mAb blockade did not enhance A1 T cell proliferation (Fig. 2G). Tregs from DNP-treated IFNγR-KO mice mediated potent PD-1/PD-L-dependent suppression, comparable with Tregs from DNP-treated B6 mice (Fig. 2H), while Tregs from DNP-treated IFNAR-KO mice did not mediate potent suppression, and PD-1/PD-L blockade had no effect on A1 T cell proliferation (Fig. 2I). These outcomes were consistent with data from DC suppression assays, and support the hypothesis that DNP treatment induced DCs and Tregs to acquire regulatory phenotypes dependent on induction of IDO in DCs via IFN type I receptor signaling.
IDO and IFN type I induction following DNP treatment is not TLR9-dependent
Bacterial pDNA contains immunostimulatory (un-methylated CpG) motifs that ligate TLR9 in innate immune cells to trigger release of pro-inflammatory cytokines, including IFN type I. As TLR9 ligands also induced IDO via IFN type I and activated Tregs (9, 22), we tested if DNPs lacking TLR9 ligands (CpG motifs) induced IDO activity. Spleens from B6 mice treated with DNPs containing CpG-free pDNA or synthetic poly dA:dT polymers (pAT) exhibited increased IDO activity levels comparable with levels induced by DNPs containing TLR9 ligands (Table I). Consistent with these findings, DNP-induced IDO activity levels were comparable in spleens of TLR9-deficient (TLR9-KO) and B6 mice (Table I). Thus IDO induction following DNP treatment was not dependent on TLR9 signaling.
To further evaluate innate immune responses to DNP treatment serum IFN type I and II levels were assessed in mice treated with DNPs containing or lacking TLR9 ligands. Consistent with previous reports (1, 5), treating B6 mice with DNPs containing TLR9 ligands (pDNA/PEI) induced substantial increases in serum IFNγ levels 24 hours after DNP administration (Fig. 3A). In contrast, DNP treatment in mice with defective genes encoding TLR9 or MyD88 - an adaptor molecule required for signaling via several TLR dependent pathways in innate immune cells – induced no detectable increases in serum IFNγ levels (Fig. 3A). Moreover, treating wild-type (B6) mice with DNPs lacking TLR9 ligands (CpG-free pDNA/PEI, pAT/PEI) elicited no detectable increases in serum IFNγ (Fig. 3B). In contrast, DNP treatment stimulated comparable increases in serum IFN type I levels, irrespective of the presence or absence of TLR9 ligands in DNPs (Fig. 3C). Thus systemic IFNγ release induced by DNPs was dependent on TLR9/MyD88 signaling in innate immune cells, while IFN type I release was not dependent on this pathway.
Figure 3. DNPs induce systemic interferon release.
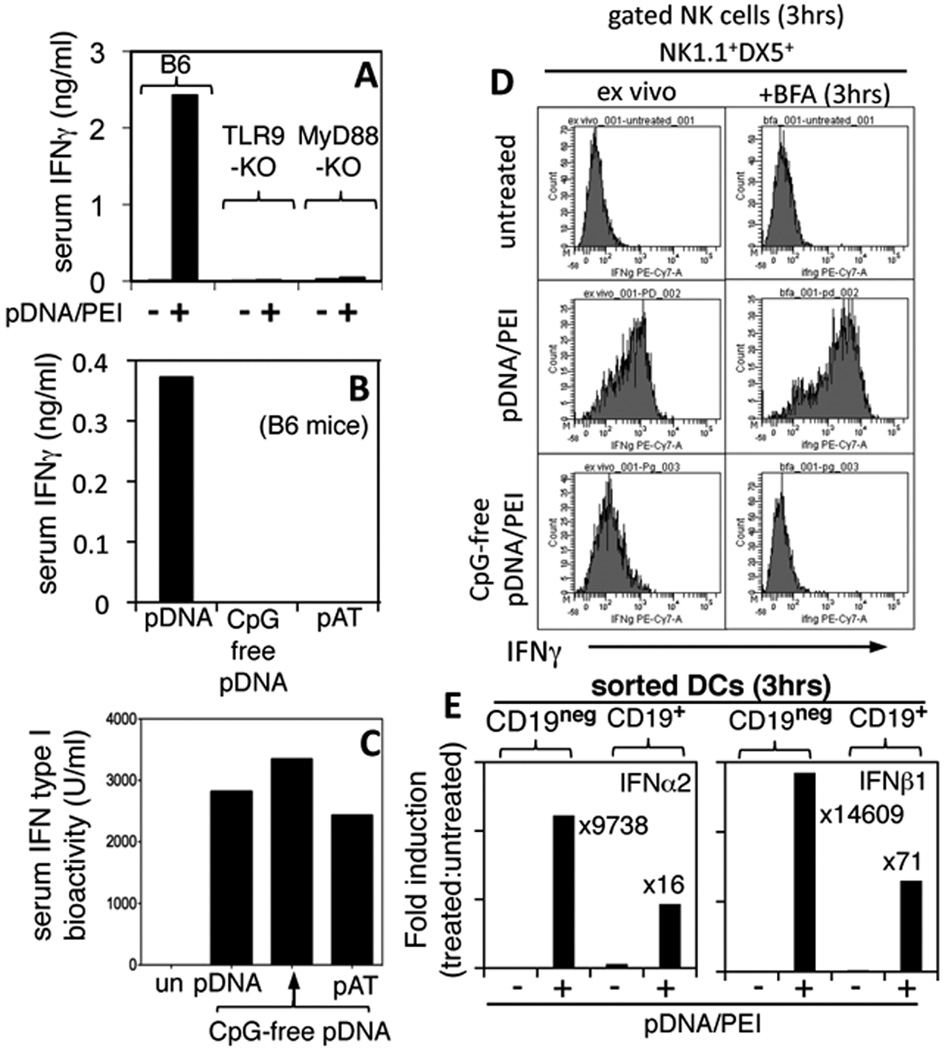
A, B. Mice were treated with DNA/PEI nanoparticles containing pDNA, CpG-free pDNA, or pAT and serum IFNγ levels were assessed after 24 hrs. (ELISA). C. Serum IFN type I levels were assessed by adding serum from DNP-treated mice to IFN bioassays (see Methods). D. Mice were treated with DNPs and after 3 hrs. splenocytes were stained with NK markers (NK1.1, DX5) and intracellular IFNγ directly (ex vivo) or after culture for 3 hr. with GolgiPlug (BFA). E. RNA from FACS-sorted CD19neg and CD19+ DCs from DNP-treated mice (3 hrs) were analyzed (qPCR) to detect IFNα2 and IFNβ1 transcripts. Data were normalized to β-actin levels in each sample and expressed as fold induction over basal levels (treated vs. untreated). Data are representative of 2 or more experiments.
To identify cells expressing IFNs after DNP (pDNA/PEI) treatment flow cytometric and quantitative RT-PCR analyses were performed to detect intracellular IFNγ and IFN type I (IFNα2, IFNβ1) expression, respectively. Splenocytes expressing intracellular IFNγ were detected soon after DNP treatment (3 hrs.), and phenotypic analysis revealed that splenic Natural Killer (NK) cells expressing NK1.1 and DX5 accounted for almost all the cells expressing IFNγ at this early phase in the innate response to DNPs (Fig. 3D, and data not shown). Gated NK1.1+DX5+ cells from pDNA/PEI-treated mice expressed uniformly high levels of intracellular IFNγ relative to NK cells from untreated mice when cells were analyzed directly (ex vivo), or after culture (for 3 hrs.) in the presence of brefeldin (BFA) to block protein secretion (Fig. 3D, center and upper panels, respectively). In contrast, gated NK cells from mice treated with DNPs lacking TLR9 ligands (CpG-free pDNA/PEI) expressed much lower levels of IFNγ only marginally higher than levels in NK cells from untreated mice, and IFNγ was undetectable after ex vivo stimulation with BFA (Fig. 3D, lower and upper panels, respectively). Invariant NK T (iNKT) cells - gated CD1d-tetramer+TCRβ+NK1.1+ cells - from pDNA/PEI-treated mice did not express IFNγ (data not shown). RT-PCR analyses of RNA from FACS-sorted CD19+ and CD19neg DCs from B6 untreated and pDNA/PEI-treated mice (3 hrs after DNP treatment) revealed substantial increases in IFNα2 and IFNβ1 transcripts in both DC subsets (Fig. 3E). After normalizing results to β-actin levels, IFNα2 and IFNβ1 transcript levels were increased ~3 orders of magnitude in CD19neg DCs, and between 1–2 orders of magnitude in CD19+ DCs, indicating that splenic DCs made copious amounts of IFN type I soon after DNP treatment, and that IFNα2 production, unlike IDO expression at later times (Fig. 1), was not restricted to the CD19+ DC subset. Thus, DNPs stimulated rapid and uniform NK cell activation leading to systemic IFNγ and IFNαβ release, and IFNγ but not IFNαβ release was TLR9-dependent as DNPs lacking TLR9 ligands did not activate NK cells.
DNP treatment suppresses antigen-specific T cell responses elicited in vivo
To test if DNP treatment suppressed T cell responses elicited in vivo after OVA immunization, OVA-specific T cell responses were monitored in B6 (Thy1.2) mice harboring marked cohorts of CD4 T cells transferred from OT-2 TCR (Thy1.1) transgenic donor mice and labeled with the dye CFSE. As depicted (Fig. 4A), CFSE-labeled OT-2 T cells were injected into recipients one day before mice were immunized (s/c) with OVA+ splenocytes from Act-mOVA transgenic mice (30). Flow cytometric analyses were performed five days after OVA immunization to detect gated donor (CD4+Thy1.1) OT-2 T cells in inguinal LNs draining immunization sites. As expected, OT-2 T cells proliferated extensively in OVA-immunized B6 mice (Fig. 4B, second left panel) such that total numbers of OT-2 and OT-2 Th1 (expressing intracellular IFNγ) T cells increased >20-fold and >30-fold relative to non-immunized controls (Fig. 4C, 4D, respectively).. In contrast, DNP treatment blocked OT-2 T cell clonal expansion (Fig. 4B, second right panel) and differentiation such that total numbers of OT-2 and Th1 OT-2 T cells were even lower than in non-immunized controls (Fig. 4C, D). Continuous oral dosing with 1MT starting 2 days before OVA immunization restored robust OT-2 T cell proliferation in OVA-immunized, DNP-treated mice (Figs. 4B, right panel), revealing that IDO activity was essential for the regulatory effects of DNP treatment to manifest in vivo. Comparable outcomes were obtained in experiments using OVA-specific OT-1 (CD8) T cells instead of OT-2 T cells (data not shown). Comparable outcomes were also obtained when TLR9-deficient mice were used as OT-1 recipients and treated with DNPs containing TLR9 ligands, and when B6 mice were treated with DNPs lacking TLR9 ligands (data not shown). Thus, DNP treatment blocked in vivo T cell responses to exogenous antigen, and IDO activity, but not TLR9 signaling was essential for T cell regulatory effects to manifest after DNP treatment.
Figure 4. DNP treatment suppresses OVA-specific T cell responses via IDO.
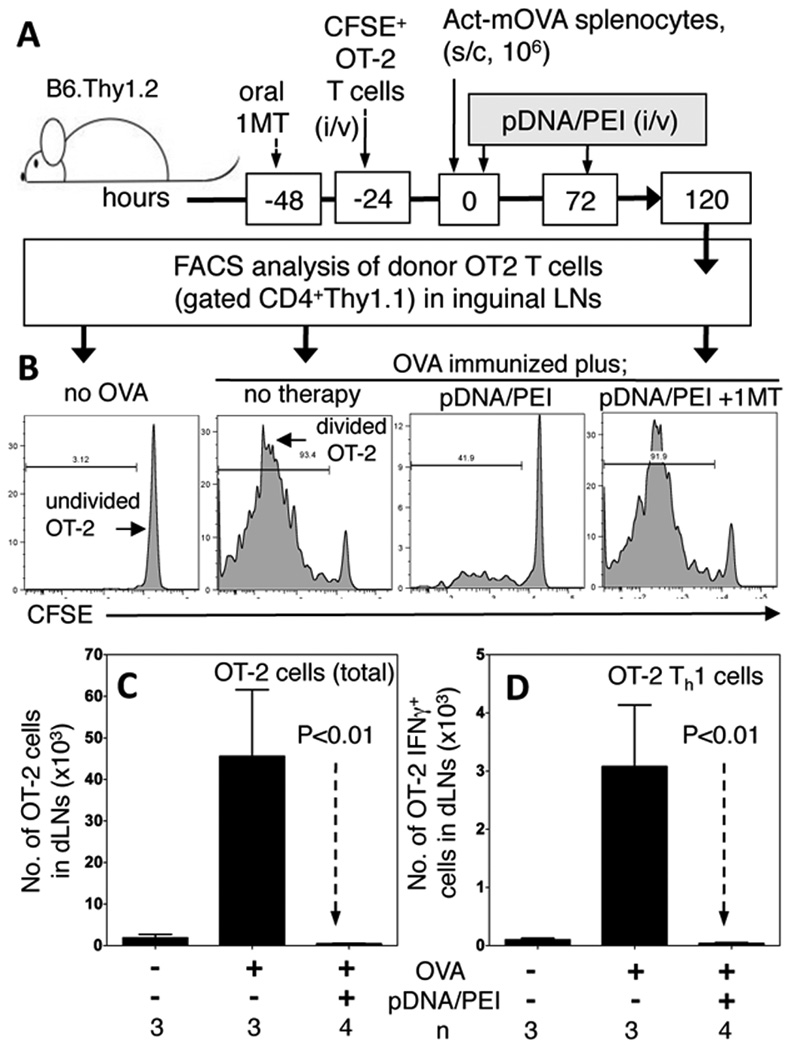
A. CFSE-labeled OT-2 (CD4+Thy1.1) T cells were injected into B6 (Thy1.2) mice. Mice were immunized with Act-mOVA splenocytes and treated twice with pDNA/PEI nanoparticles. After 120 hrs. CFSE and intracellular IFNγ levels on gated dLN OT-2 T cells were assessed. B. Histograms showing CFSE levels on gated OT-2 T cells from representative mice treated as indicted. C. Total numbers of OT-2 T cells in inguinal dLNs. D. Numbers of OT-2 dLN T cells expressing IFNγ. Data are representative of 3 independent experiments.
DNP treatment attenuates joint injury in a model of immune-mediated arthritis
To test the hypothesis that DNP treatment attenuates immune-mediated tissue pathology a model of antigen-induced rheumatoid arthritis was employed (26). B6 mice were sensitized with methylated BSA (mBSA/CFA, day 0), boosted twice with mBSA/IFA (days 7, 14), and local joint arthritis was induced by intra-articular injection of mBSA (challenge) 21 days after initial immunization. Groups of mBSA-immunized mice were treated with DNPs lacking TLR9 ligands (pAT/PEI) or vehicle (PEI) starting one day before mBSA challenge (day 20), and continuing after local mBSA challenge (days 21, 22, 24, 26). DNP treatment reduced joint (knee) swelling significantly (measured cumulatively 1–7 days after mBSA challenge), relative to controls that received vehicle only (Fig. 5A). Therapeutic effects of DNP treatment on joint swelling were abrogated in mice lacking intact IDO1 genes (IDO1-KO), and in B6 mice given oral 1MT continuously from two days before initial mBSA immunization until experimental endpoints, 7 days after mBSA challenge (Fig. 5A). Significant therapeutic effects of DNP treatment were also observed for other measures of arthritis severity. Neutrophil infiltration into joints one day after mBSA challenge was reduced significantly in DNP-treated, mBSA-immunized B6 mice relative to immunized B6 mice, and DNP-treated, immunized mice lacking functional IDO (Fig. 5B). Similarly, levels of the pro-inflammatory cytokines IL-6 (Fig. 5C) and IL-17 (Fig. 5D) produced ex vivo by cells from inflamed inguinal and popliteal LNs draining sites of joint inflammation were reduced significantly in an IDO-dependent fashion. Moreover, DNP treatment prevented loss of sulfated proteoglycans in articular joint cartilage - a key indicator of joint injury (compare Figs. 6A, B, respectively) - when analyzed 7 days after local mBSA challenge (Fig. 6C). Oral 1MT (Fig. 6D) and IDO1 gene ablation (Fig. 6E) also eliminated therapeutic effects of DNP treatment by this measure. Thus, DNP treatment reduced joint inflammation and attenuated destructive immunity driving joint injury significantly, and these therapeutic effects were IDO dependent in this model of immune-mediated rheumatoid arthritis.
Figure 5. DNP treatment suppresses joint inflammation via IDO in a model of immune-mediated arthritis.
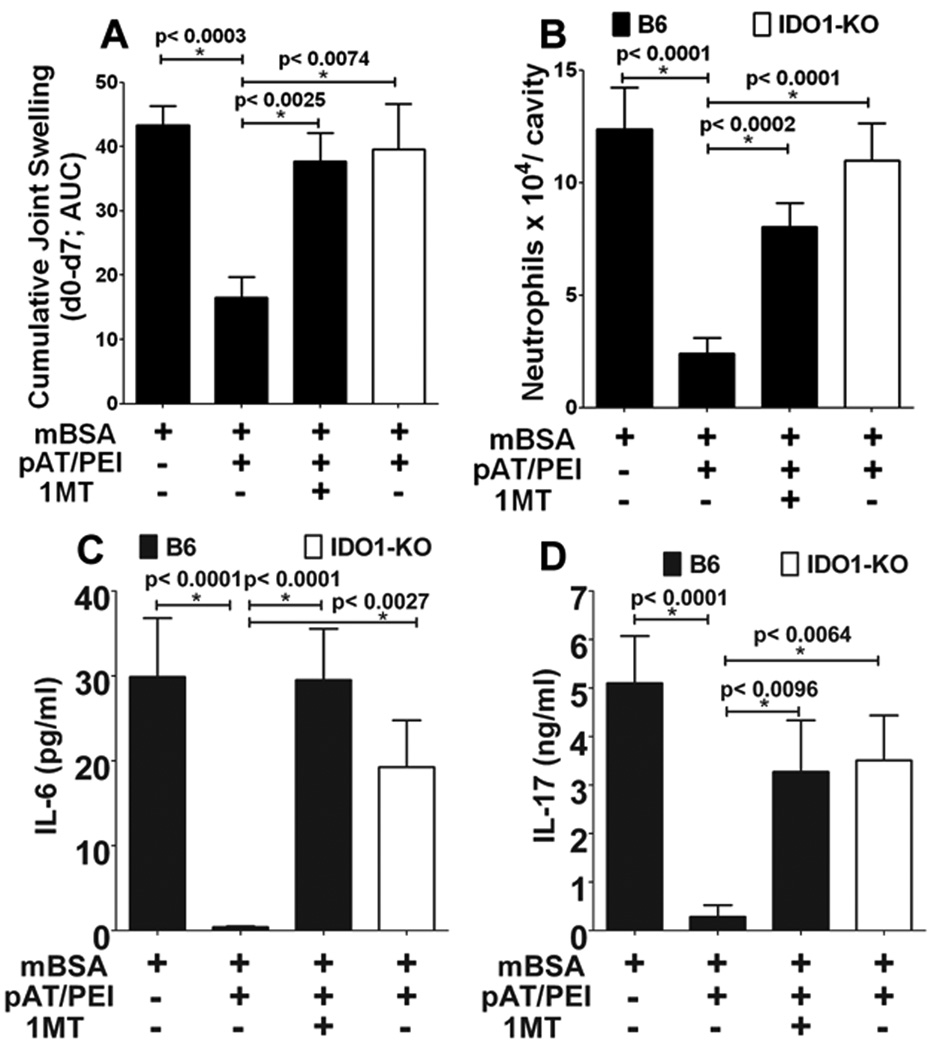
B6 and IDO1-KO mice (n=5) were immunized and boosted with mBSA, and arthritis was induced after 21 days by intra-articular mBSA injection (see Methods). pAT/PEI nanoparticles were administered five times (i/v, 20, 21, 22, 24 26 days after initial mBSA immunization). Some pAT/PEI-treated B6 mice also received oral 1MT starting two days before initial mBSA immunization. Arthritis progression was monitored by assessing cumulative joint swelling 7 days after mBSA challenge (A) represented as area under curve (AUC), and neutrophil infiltration into joints 1 day after local mBSA challenge (B). IL-6 (C) and IL-17 (D) cytokine levels in inflamed dLN cells were assessed by multiplex analysis. Data are representative of two independent experiments with 3–5 mice/group; statistical significance (*) was estimated using Student’s t test by combining both experiments.
Figure 6. DNP treatment protects joints from immune–mediated arthritis.
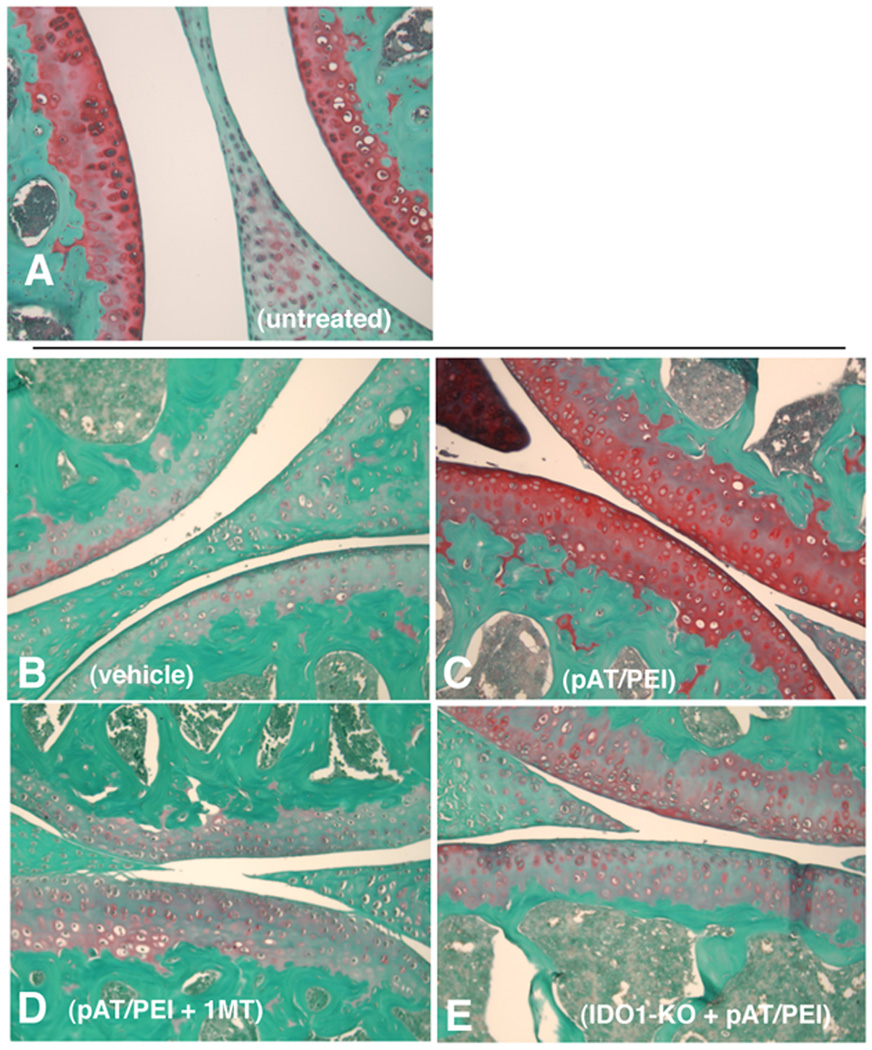
A–E. Immunohistochemical analyses of tissue sections of joints (2 µm) from untreated B6 mice (A), and mBSA immunized and challenged B6 (B–D) or IDO1-KO (E) mice treated with vehicle (B), DNPs (C, E) and DNPs plus oral 1MT (D) as described in Fig. 5. Sulfated proteoglycans in joints (red stain) were detected by staining affected joint cavities with Safranin-O seven days after mBSA challenge (day 28). Images are representative of mice analyzed in 2 separate experiments.
Discussion
In this study we show for the first time that nanoparticles containing DNA complexed with the cationic polyamine PEI (DNPs) exhibited potent and dominant immunomodulatory attributes that overcame the immune stimulatory effects of DNPs to suppress T cell responses and immune-mediated tissue injury. Intact IDO was essential for DNPs to induce immunomodulatory responses, and DNP treatment caused DCs and Tregs to acquire potent regulatory phenotypes via IDO. IDO induction in lymphoid tissues was dependent on IFN type I, but not IFNγ signaling. Moreover, TLR9/MyD88 signaling was essential for IFNγ release after DNP treatment, while IFN type I production was not dependent on TLR9/MyD88 signaling. Consequently, DNPs lacking immune stimulatory TLR9 ligands exhibited potent T cell regulatory attributes and attenuated immune-mediated joint pathology in a model of arthritis, but did not activate NK cells to produce IFNγ. These findings suggest that DNPs lacking TLR9 ligands may be effective and safe drugs to promote immune suppression and tolerance that prevents, ameliorates or reverses pathologic syndromes due to hyper-immunity in settings of autoimmunity, allergies and transplant rejection.
Previous studies on the use of PEI as a DNA delivery vehicle focused on optimizing the efficiency and stability of gene expression in cells following transduction (2, 3). Innate pro-inflammatory responses to DNP treatment were described in a murine lung tumor model, in which DNPs stimulated rapid TLR4-dependent, systemic IL-12 and IFNγ (a downstream IL-12 response gene) release that enhanced anti-tumor immunity (4, 5); however, DNPs also inhibited TLR4-mediated TNFα production by macrophages (4). Findings reported in the current study are consistent with these previous reports as serum levels of IFNγ and IFN type I were elevated rapidly in mice treated with DNPs containing bacterial pDNA that possesses un-methylated CpG motifs that ligate TLR9 to activate innate immune cells. Moreover, both IFNγ and IFN type I induce IDO gene expression in a range of cell types (17, 31), providing a rationale for rapid up-regulation of IDO enzyme activity observed in mucosal and lymphoid tissues of DNP-treated mice.
Cells ingest DNPs by phagocytosis or endocytosis, and dsDNA is soon released into cytoplasm because cationic polymers such as PEI disrupt endocytic vesicles via the ‘proton sponge’ effect (3). DNPs stimulated IDO expression by small populations of cells located in peri-follicular regions of spleen and peripheral LNs. Several immune cell types in these regions can acquire regulatory phenotypes, including macrophage, DC and B cell subsets. Treatments with B7 (CTLA4Ig) and TLR9 ligands (CpGs) induced selective IDO expression in rare, but distinctive CD19+ DCs with attributes of both DCs and B cells (22, 23, 28). DNP treatment also stimulated CD19+ DCs to uniformly express IDO, though a few other non-DC cell types also expressed IDO in peri-follicular regions. Prior to IDO induction DNPs accumulated in peri-follicular regions of lymphoid tissues, and CD19neg and CD19+ DCs up-regulated IFNαβ gene expression substantially a few hours after DNP treatment. The finding that IFNαβ signaling was essential to induce IDO and activate Tregs, while IFNγ signaling was not essential for regulatory responses via IDO, parallels our previous findings from studies using CTLA4Ig and CpGs as IDO inducers (8, 9, 22, 28). These findings support the hypothesis that DCs located in peri-follicular regions ingested DNPs rapidly to trigger IFN type I release that subsequently induced CD19+ DCs to express IDO via autocrine or paracrine pathways.
Our findings provide novel insights into early innate immune responses to DNPs that lead to dominant immune regulatory outcomes. Findings that PEI or dsDNA treatments alone did not up-regulate IDO activity, while DNPs containing dsDNA that lacked TLR9 ligands did induce IDO-mediated regulatory responses suggested that TLR9-independent mechanisms sense dsDNA in cells that ingested DNPs to trigger IFNαβ release. Most cells can sense cytosolic dsDNA via DDX41/STING, LRRFIP/β-catenin, AIM2/ASC pathways to induce rapid cytokine release (IFNαβ, IL-1β) after virus infection or ingestion of dying (apoptotic) cells (32–35). Our findings support the hypothesis that one (or more) of these DNA sensing pathways is required for innate immune responses to DNPs that lead to immune regulatory responses, though elucidating how dominant immune stimulatory or regulatory responses manifest via these pathways in response to particular insults and distinct settings will require further study. The finding that NK cells – which do not express TLR9 - were rapidly and uniformly activated in mice treated with DNPs containing TLR9 ligands suggested that other innate immune cells ingested DNPs and sensed TLR9 ligands to induce cytokines such as IL-12 and IL-18 that activate NK cells. The absence of this response in mice treated with DNPs lacking TLR9 ligands may reduce toxicities caused by sustained, systemic pro-inflammatory cytokine release that is not essential to induce IDO but potentiates immunity.
Treating mice with DNPs lacking TLR9 ligands reduced the severity of pre-established immune-mediated joint injury in an IDO-dependent fashion, revealing that regulatory responses to DNPs overcame immune-mediated joint destruction, and any toxicities caused by chronic DNP exposure. However, DNPs containing TLR9 ligands also suppressed OVA-specific T cell responses elicited in vivo, demonstrating that induced regulatory responses to DNP treatment also overcame the immune stimulatory effects of OVA-immunization and the collateral immune potentiating effects caused by DNP-induced systemic release of pro-inflammatory cytokines such as IFNγ, IL-12 and TNFα. This point notwithstanding, DNPs lacking TLR9 ligands are likely to be more effective and safer reagents to activate Tregs, and promote tolerogenic outcomes than DNPs containing TLR9 ligands in clinical settings of hyper-immunity. Indeed, some forms of PEI were approved for limited gene therapy applications in clinical settings despite the well documented pro-inflammatory responses elicited in response to DNPs (2, 3). Identifying novel and selective reagents to enhance the potent immune regulatory attributes of Tregs, while avoiding Treg functional re-programming and activation of effector T cells is a priority due to the need for more effective treatments to prevent autoimmune disease progression and to protect healthy tissue and organ allografts (6, 7). The findings reported in the current study justify further development and evaluation of DNPs to treat a broader range of clinical syndromes than hitherto considered.
Acknowledgments
We thank Liesl Desevilla, Gabriella Pacholczyk and the late Doris McCool for managing the mouse colonies used in this study, and Diane Addis for expert technical assistance with HPLC analyses. We thank NewLink Genetics Inc. for the generous gift of 1MT.
Non-standard abbreviations used
- PEI
polyethylenimine
- DNPs
DNA/PEI nanoparticles
- Tregs
regulatory (Foxp3-lineage) CD4 T cells
- DCs
dendritic cells
- PD-1/PD-L
Programmed Death-1/ligand
- 1MT
1-methyl-[D]-tryptophan
- Kyn
kynurenine
Footnotes
This study was supported by grants from the Carlos and Marguerite Mason Trust, and the NIH (AI075165) to ALM.
References
- 1.Kawakami S, Ito Y, Charoensit P, Yamashita F, Hashida M. Evaluation of proinflammatory cytokine production induced by linear and branched polyethylenimine/plasmid DNA complexes in mice. J Pharmacol Exp Ther. 2006;317:1382–1390. doi: 10.1124/jpet.105.100669. [DOI] [PubMed] [Google Scholar]
- 2.Intra J, Salem AK. Characterization of the transgene expression generated by branched and linear polyethylenimine-plasmid DNA nanoparticles in vitro and after intraperitoneal injection in vivo. J Control Release. 2008;130:129–138. doi: 10.1016/j.jconrel.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunther M, Lipka J, Malek A, Gutsch D, Kreyling W, Aigner A. Polyethylenimines for RNAi-mediated gene targeting in vivo and siRNA delivery to the lung. Eur J Pharm Biopharm. 2011;77:438–449. doi: 10.1016/j.ejpb.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Li P, Yin Y, Cai X, Huang Z, Chen J, Dong L, Zhang J. The promotion of type 1 T helper cell responses to cationic polymers in vivo via toll-like receptor-4 mediated IL-12 secretion. Biomaterials. 2010;31:8172–8180. doi: 10.1016/j.biomaterials.2010.07.056. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigo-Garzon M, Berraondo P, Ochoa L, Zulueta JJ, Gonzalez-Aseguinolaza G. Antitumoral efficacy of DNA nanoparticles in murine models of lung cancer and pulmonary metastasis. Cancer Gene Ther. 2010;17:20–27. doi: 10.1038/cgt.2009.45. [DOI] [PubMed] [Google Scholar]
- 6.Leslie M. Immunology. Regulatory T cells get their chance to shine. Science. 2011;332:1020–1021. doi: 10.1126/science.332.6033.1020. [DOI] [PubMed] [Google Scholar]
- 7.Phillips B, Trucco M, Giannoukakis N. Current state of type 1 diabetes immunotherapy: incremental advances, huge leaps, or more of the same? Clin Dev Immunol. 2011;2011:432016. doi: 10.1155/2011/432016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baban B, Chandler PR, Johnson BA, 3rd, Huang L, Li M, Sharpe ML, Francisco LM, Sharpe AH, Blazar BR, Munn DH, Mellor AL. Physiologic Control of IDO Competence in Splenic Dendritic Cells. J Immunol. 2011;187:2329–2335. doi: 10.4049/jimmunol.1100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183:2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munn DH, Sharma MD, Hou D, Baban B, Lee J, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor AL. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–290. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR, Blazar BR, Mellor AL, Munn DH. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity. 2010;33:942–954. doi: 10.1016/j.immuni.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood. 2009;114:5062–5070. doi: 10.1182/blood-2009-06-227587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 15.Ciorba MA, Bettonville EE, McDonald KG, Metz R, Prendergast GC, Newberry RD, Stenson WF. Induction of IDO-1 by immunostimulatory DNA limits severity of experimental colitis. J Immunol. 2010;184:3907–3916. doi: 10.4049/jimmunol.0900291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, Liu L, Liu K, Bizargity P, Hancock WW, Visner GA. Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2,3-dioxygenase-dependent immune suppression. J Immunol. 2009;183:1022–1031. doi: 10.4049/jimmunol.0900408. [DOI] [PubMed] [Google Scholar]
- 17.Johnson BA, 3rd, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1:645–661. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laich A, Neurauter G, Widner B, Fuchs D. More rapid method for simultaneous measurement of tryptophan and kynurenine by HPLC. Clin Chem. 2002;48:579–581. [PubMed] [Google Scholar]
- 19.Hoshi M, Saito K, Hara A, Taguchi A, Ohtaki H, Tanaka R, Fujigaki H, Osawa Y, Takemura M, Matsunami H, Ito H, Seishima M. The absence of IDO upregulates type I IFN production, resulting in suppression of viral replication in the retrovirus-infected mouse. J Immunol. 2010;185:3305–3312. doi: 10.4049/jimmunol.0901150. [DOI] [PubMed] [Google Scholar]
- 20.Tyring S, Fleischmann WR, Jr, Baron S. A convenient microassay for cytolysis and cytostasis. Methods Enzymol. 1986;119:574–579. doi: 10.1016/0076-6879(86)19077-x. [DOI] [PubMed] [Google Scholar]
- 21.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 22.Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting Edge: CpG Oligonucleotides Induce Splenic CD19+ Dendritic Cells to Acquire Potent Indoleamine 2,3-Dioxygenase-Dependent T Cell Regulatory Functions via IFN Type 1 Signaling. J Immunol. 2005;175:5601–5605. doi: 10.4049/jimmunol.175.9.5601. [DOI] [PubMed] [Google Scholar]
- 23.Johnson BA, 3rd, Kahler DJ, Baban B, Chandler PR, Kang B, Shimoda M, Koni PA, Pihkala J, Vilagos B, Busslinger M, Munn DH, Mellor AL. B-lymphoid cells with attributes of dendritic cells regulate T cells via indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2010;107:10644–10648. doi: 10.1073/pnas.0914347107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGaha TL, Chen Y, Ravishankar B, van Rooijen N, Karlsson MC. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood. 2011;117:5403–5412. doi: 10.1182/blood-2010-11-320028. [DOI] [PubMed] [Google Scholar]
- 25.Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. 2004;61:67–77. doi: 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Lemos HP, Grespan R, Vieira SM, Cunha TM, Verri WA, Jr, Fernandes KS, Souto FO, McInnes IB, Ferreira SH, Liew FY, Cunha FQ. Prostaglandin mediates IL-23/IL-17-induced neutrophil migration in inflammation by inhibiting IL-12 and IFNgamma production. Proc Natl Acad Sci U S A. 2009;106:5954–5959. doi: 10.1073/pnas.0812782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teige A, Bockermann R, Hasan M, Olofsson KE, Liu Y, Issazadeh-Navikas S. CD1d-dependent NKT cells play a protective role in acute and chronic arthritis models by ameliorating antigen-specific Th1 responses. J Immunol. 2010;185:345–356. doi: 10.4049/jimmunol.0901693. [DOI] [PubMed] [Google Scholar]
- 28.Baban B, Hansen AM, Chandler PR, Manlapat A, Bingaman A, Kahler DJ, Munn DH, Mellor AL. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int Immunol. 2005;17:909–919. doi: 10.1093/intimm/dxh271. [DOI] [PubMed] [Google Scholar]
- 29.Pestka S, Kotenko SV, Muthukumaran G, Izotova LS, Cook JR, Garotta G. The interferon gamma (IFN-gamma) receptor: a paradigm for the multichain cytokine receptor. Cytokine Growth Factor Rev. 1997;8:189–206. doi: 10.1016/s1359-6101(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 30.Ehst BD, Ingulli E, Jenkins MK. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant. 2003;3:1355–1362. doi: 10.1046/j.1600-6135.2003.00246.x. [DOI] [PubMed] [Google Scholar]
- 31.Huang L, Baban B, Johnson BA, Mellor AL. Dendritic cells, indoleamine 2,3 dioxygenase and acquired immune privilege. International Reviews of Immunology. 2010;29:133–155. doi: 10.3109/08830180903349669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y, Cao X. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat Immunol. 2010;11:487–494. doi: 10.1038/ni.1876. [DOI] [PubMed] [Google Scholar]
- 34.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]