

Preface
Access to genetic data across studies is an important aspect of identifying new genetic associations through genome-wide association studies (GWAS). Meta-analysis across multiple GWAS with combined cohort sizes of tens of thousands of individuals often uncovers many more genome-wide associated loci than the original individual studies, which emphasizes the importance of tools and mechanisms for data sharing. However, even sharing summary-level data, such as allele frequencies, inherently carries some degree of privacy risk to study participants. Here we discuss mechanisms and resources for sharing data from GWAS, particularly focusing on approaches for assessing and quantifying privacy risks to participants from sharing of summary-level data.
Introduction
Population-based genetic studies have the potential to unlock biological mechanisms of disease and reveal their genetic underpinnings. In particular, genome-wide association studies (GWAS) using hundreds of thousands to millions of Single Nucleotide Polymorphisms (SNPs) have emerged over recent years as a particularly fruitful study design for identifying common variants with subtle genetic effects in complex disorders1. While a few initial studies made substantial findings by studying only a few hundred individuals, such as in age-related macular degeneration2, more often the small effect size of associated SNPs requires genotyping thousands of individuals when studying complex diseases across a population. Within the past two years there has been a trend towards including tens to hundreds of thousands of individual participants in GWAS3. Examples include: a meta-analysis of 5,539 cases and 17,231 controls for rheumatoid arthritis and 4,533 cases and 10,750 controls for celiac disease, which collectively identified 7 shared loci for these diseases4; 6,688 cases and 13,685 controls for Alzheimer’s disease, which identified 5 new genome-wide significant associations 5; meta-analysis of 22,233 individuals with coronary artery disease and 64,762 controls taken from 14 GWAS, which identified 13 new susceptibility loci 6; and meta-analysis of >100,000 individuals, which identified 59 newly associated loci with cholesterol and blood lipid levels7. These studies emphasize how increasing study sizes of tens or hundreds of thousands of individuals is enabling discovery of multiple genome-wide significant associations, rather than a single or a few loci as was frequently the case in early studies. Often these large-scale GWAS result from meta-analysis of many previous studies and are inherently enabled by sharing genetic data.
A consideration for any genetic study is the need to protect individual participants from the risk of re-identification, and maintaining privacy becomes more complex when data is shared beyond the original study within which the individual agreed to participate. We address these considerations by first discussing frameworks and resources for sharing data from GWAS, and then highlighting some of the risks associated with common modes of sharing data. Data can be shared either as information about the individual or as population-level data; we focus particularly on privacy challenges when sharing population-level data such as allele frequencies to a large audience, which until recently was regarded as relatively ‘safe’ in terms of privacy. We describe quantitative approaches and additional considerations in assessing risk to the privacy of individual participants at varying levels of sharing aggregate data. We consider quantitative approaches in the most depth, as there is currently much deliberation in the research community regarding how risk can be assessed and taken into account when planning studies.
What level of data can be shared?
Sharing individual-level data
Generally, the most comprehensive data sharing from GWAS is distribution of full phenotypic information accompanied by individual-level genotype data for each participant. Phenotypic information could be tied to a set of full medical records, such as has been conducted by the eMERGE Network in linking genotype data to various conditions including dementia, lipid levels, and type 2 diabetes8, or could be limited to a dichotomous trait, such as a case/control status. At a simple level this could be genotype calls (e.g., AA/AG/GG) for >500,000 SNPs, though it could include the raw array data used for calling genotypes or identifying copy number variants.
Access to the individual-level data has several advantages for analysis across datasets. First, access to individual-level data allows for joint analysis across all samples, giving greater power to detect associations than meta-analysis of summary-level statistics9. Second, access to individual-level data ensures a uniform analysis across all datasets, both in terms of application of quality control filters (such as SNP missingness) as well as higher-level analysis such as imputation. For example, imputation is often used to combine datasets genotyped on platforms and predict untyped markers10. However, imputation can vary by method, such as Beagle, Mach, and Impute, or it can the training set, such as using the 1000 Genomes datasets to identify indirectly typed variants11. Third, sharing of individual-level data can allow assessment of multiple variants in combination within a single individual (such as SNPxSNP interactions) for calculating combined effects of multiple associated variants; for example, a cumulative effect of 5 variants was identified in a meta-analysis of prostate cancer GWAS12. Finally, access to the individual-level, raw array probe-level data can be used to ascertain evidence for copy number variants associated with disease; recently, enrichment of duplications of VIPR2 were observed to be associated with schizophrenia utilizing multiple mental health GWAS13.
Clearly, the most important challenge with sharing individual-level genomics data is protecting privacy of individual participants. As discussed by Heeney and colleagues, individual genetic data from GWAS are not only uniquely identifying, they can predict risk to disease, and it is possible for consumers outside the scientific community to generate genetic profiles on individuals (such as through 23andMe)14,15. In addition to privacy issues, sharing individual-level data has challenges arising from the size and variability of files associated with array-based genotype data. For example, sharing of probe-level data for CNVs or genotype-level data involves file sizes exceeding 100 megabytes per sample and often contains highly specific formatting/referencing requirements essential for avoiding strand and genome-build inconsistencies. Informatically, summary-level data are often preferred over individual level data when the researcher’s goal is only rapid acquisition of allele-frequencies, p-values, or other summary-level data for sets of SNPs for exploratory or confirmatory purposes.
Sharing summary-level data
An alternative to sharing individual-level data is sharing aggregate data or summary statistics. Such statistics include genotype counts, allele frequencies, p-values, odds ratios, and other measures of effect size. In dichotomy analysis (case/control), researchers can use genotype counts to calculate summary-level statistics (p-value, odds ratio) under different genetic models (dominant, additive, recessive or co-dominant). When study conditions are carefully matched, the counts from different data sets can be used directly in meta-analysis if individual genotypes are inaccessible. Also, in the context of a publication, sharing allele frequencies, p-values and odds ratios is routine and essential for future studies to replicate the most associated SNPs. As highlighted earlier, many large scale GWAS have been enabled by meta-analyses that can only be conducted with access to the many associated markers with low-effect size that are generally not included in the tables or supplementary data of an individual study’s primary report. Data sharing is essential to this process. Increasingly, large studies are carefully managed efforts involving multiple consortia where each member contributes summary statistics that are independently prepared in a consistent manner and then combined for meta-analysis7,16,17.
From the perspective of risk to the privacy of individual participants, aggregating data into summary statistics provides some level of privacy protection. However, as discussed in later sections, some degree of residual identifying information remains in the cumulative analysis of large numbers of SNPs.
Mechanisms for data sharing
Study consortia
Meta-analysis in GWAS with tens to hundreds of thousands of individuals are often carried out by multi-center consortia that have set up mechanisms whereby either summary-level or individual-level data are gathered into a central database. The Coronary Artery Disease Genome-Wide Replication And Meta-analysis (CARDIoGRAM) consortium published one such example in which a steering committee provided oversight to several coordinated analysis groups who submitted carefully constructed summary-analysis results to a centralized database where the combined meta-analysis was completed16. Another current example is the Gene Environment Association Studies (GENEVA) consortium, which consists of 14 independent GWAS for various phenotypes and includes over 80,000 study participants. Consistent quality control and the use of centralized data deposition to the database of Genotypes and Phenotypes (dbGaP) for individual-and summary-level data are essential to GENEVA18. International efforts that use centralized computing also include the Psychiatric Genetics Consortium19, in which individual-level data are uploaded to a common server for structured analysis across a series of psychiatric disorders including bipolar, schizophrenia, autism and other mood disorders.
Databases for broader distribution
The consortia described above represent major efforts at coordinated and controlled approaches for data analysis. It is expected that the individual-level data and summary-level data will have utility in future studies, and so additional long-term data-sharing mechanisms are essential. For example, individual researchers might be looking to validate associations of a specific gene or variant not listed in the primary publications. Beyond individual researchers, new consortia may wish to rely on historical data; for example, the Population Reference Sample (POPRES)20 study created a common resource of controls useful for adding power to future case/control GWAS. An important aspect of enabling and realizing the future value for truly large-scale GWAS using thousands of samples has been the emergence of common repositories for distributing both individual-level and summary-level data. These databases provide a centralized resource for sharing data while providing mechanisms to protect privacy of individual study participants.
Two notable resources include samples genotyped through the International HapMap21 and 1000 Genomes projects,11 which are broadly distributed through multiple databases including dbSNP22. These resources typically are utilized to observe the variability of population-specific allele frequencies and generally are not used to generate significance of association signals. A number of other databases allow for controlled or restricted access to individual-level data and/or summarylevel data. For NIH-funded studies, dbGaP23 currently holds individual- and/or summary-level data (available via a controlled-access process) for approximately 1,900 datasets covering more than 257,000 individuals. Other sources of individual-level data include: the Genome Medicine Database of Japan (GeMDBJ), developed by the Study Groups of Japan's Millennium Genome Project (MGP) and maintains over 570,000 SNPs on 2,000 patients in three disease groups; the Wellcome-Trust Case Control Consortium (WTCCC), which maintains over 500,000 SNPs from approximately 14,000 individuals; and the EBI European Genome-Phenome Archive, which distributes data from WTCCC phase 1, 2, and 3 and 20 other study-specific providers. PheGeni24, HuGE Navigator25, GWAS Central26, JSNP27, and dbSNP provide individual- and summary-level data for several hundred thousand SNPs across diseases and several thousand samples with multiple levels of controlled access.
An important aspect of databases such as dbGaP is their ability to provide study-specific levels of controlled access to individual- and summary-level data. At one extreme, completely unrestricted access is strongly desired for at least a subset of variants, such as a list of hundreds to thousands of associated SNPs for tables within a publication, summary measures for individual phenotypic measures, and study protocols and data collection forms. A more constrained approach is to approve institutional users that have the ability to download more extensive summary-level data (that is, more than hundreds to thousands of SNPs) for studies without additional approvals. In the case of dbGaP, open access is available for broad release of non-sensitive data. For example, the Phenotype-Genotype Integrator allows one to query p-values across studies for limited number of SNPs in an open-access manner28. Controlled access through dbGaP is available when additional oversight is required for sensitive data sets involving individual-level genotype data, individual genome or exome sequences, or comprehensive GWAS for a published study, including allelic direction of effect. Access is controlled through an application process reviewed by one or more NIH Data Access Committees (DAC) that oversee the dataset(s) of interest, with terms of use conveyed through a Date Use Certification (DUC) agreement.
Privacy for summary-level data
Risks of identification in shared summary-level datasets
As seen from many meta-analyses, summary-level data has great utility when combining multiple studies or even validating a small number of SNPs. It was originally assumed that summary-level data completely anonymized the participants and this type of data could be openly distributed for all SNPs. For example, if the genotypes of 10 individuals for a particular SNP were AA, AT, AT, AA, TT, AT, AA, AT, AA and AT, respectively, the allele frequency summary statistic is 65% A (13A’s of 20 total alleles). Intuitively one cannot determine much about the individuals when reporting only the allele frequency summary statistic of 65% for a single SNP. Initial views were that this would extend to larger numbers of SNPs, particularly when the average allele frequencies were for hundreds if not thousands of individuals. However, it was shown in 2008 by Homer et al29 that, using the marginal information in tens of thousands of SNPs, one could resolve whether an individual was a member of a cohort, even in cohort sizes exceeding 1,000 individuals, provided one had access to genotype data from that individual and access to genetic data from a reference population30.
This concept can be explained by considering a simple scenario. Let’s suppose we have a dataset of 10 SNPs where the minor allele frequency is 60% for the A allele for all 10 SNPs. Let’s now suppose we want to determine if a person is in this dataset with the additional knowledge that the this individual has an AA genotype for these 10 SNPs. If it was known that the allele frequency of these 10 SNPs was actually 50% A by having a reference dataset, we could construct a statistic that cumulatively accounts for the fact that the observed allele frequency in the dataset of interest is biased towards the A allele as compared to the reference set. Returning to the example, we see a shift from the expected 50% A allele frequency to an observed 60% A allele frequencies for 10 of 10 SNPs where the individual of interest is homozygous AA. Remarking a number of analytical approaches are possible, one could calculate a probability that the person is in a dataset by observing 10 of 10 SNPs shifted in their average allele frequency consistent with the allele found in the person of interest.
The publication by Homer et al. demonstrated an example cumulative test-statistic showing that one could determine membership in summary level allele frequency datasets by comparison to a reference dataset. The numbers of SNPs, their minor allele frequency, and to a small extent the accuracy of measuring allele frequency, were all found to influence the ability to improve the estimate of cohort membership. A series of subsequent studies investigated the implications and statistical aspects of estimating sample membership from aggregate data from GWAS compared to a reference population. Specifically, studies by Jacobs et al.31 and Sankaraman et al.30 formulated statistical frameworks based on likelihood ratios that optimize power to estimate membership, in part by leveraging the binomial distribution associated with sampling biallelic markers equimolar pooled across a defined number of samples. In fact, by the Neyman-Pearson Lemma32 these methods are an optimal solution. Additionally, Braun et al.33 showed that a high rate of false-positives can arise in the original formulation utilized by Homer et al., if effects of linkage disequilibrium are ignored and a normal distribution is assumed. Conversely, Zhou and colleagues showed that linkage disequilibrium can be leveraged for improving power in a statistical approach using multiple correlated markers within long haplotype blocks34. Linear-regression based frameworks have also been presented by Visscher et al35 while David Clayton has presented a Bayesian-based alternative to the frequentist approach36. Finally, Sampson and Zhao demonstrated methods to address aspects of unknown ancestry with the use of multiple reference populations37.
What are the implications?
The major realization from these papers was that, theoretically, there might be a risk that someone could determine if an individual were in a dataset even if only summary-level genotype frequencies were available, provided they had access to that person’s genotypes for those SNPs and had a sufficiently representative reference set of allele frequencies. Since most GWAS are studies of disease, this implies that there might be a path to determine medically relevant information about participants from summary-level data. Following publication of the Homer et al. paper30, the NIH addressed sharing of data from GWAS in a paper in Science38, and NIH and many other groups stopped openly distributing disease-specific summary-level datasets. The level of risk to participants and the appropriateness of this response have been intensely debated. Krawczak and colleagues39 have argued that current NIH policy is counterproductive due to the increased burden on international consortia to comply with NIH-based central repositories’ requirements. Some of this debate was published in a series of articles in PLOS Genetics, including one article that provided five views of balancing research with protecting privacy15. The authors generally agreed that some risk is inherent to genetic studies and that a balance between research and privacy is needed, though there was less agreement on where the balance lies. Interim models were suggested whereby credentials of individuals and institutions could be validated to allow access to full summary-level data, and these models are consistent with a study by Haga and O’Daniel showing individuals are more likely to participate in studies with some restrictions for online access40. Further research is on-going to try to further assess risks for study participants.
Lastly, we remark that estimating membership in a dataset requires access to genetic data from an individual or relative. Clearly, meeting this requirement assumes some loss of privacy already, such as disease risks or ancestry. Indeed, Malin and colleagues describe determining membership from aggregate SNP data as reidentification since some aspects of a person’s identity are available in a publication recommending policies for minimizing identification risks in clinical research data41. Still, access to genetic data of an individual doesn’t render privacy expectations moot. First, participation as a phenotyped case in a study more accurately reflects being diagnosed with a disease then disease risk predictions from SNP data due to the modest effect size of most associations. Even in strongly associated examples such as an >5 odds ratio for the APOE-ε4 allele with Alzheimer’s, there is only modest predictive value for individuals already mildly cognitive impaired developing Alzheimer’s disease42. Second, the original PLOS Genetics publication and subsequent publications showed that one could also learn about immediate relatives of the genotyped individual due to shared genetics, such as a child about a parent, without knowing the exact regions or variants they share29,30. Even in the cases of related individuals with shared genetics there exist important expectations of privacy.
Assessing risk for summary data
As noted above, in practice some level of open distribution of aggregate data is necessary to communicate results in the literature. Assessing privacy risk is an important aspect of disseminating findings from GWAS that reach significance and those that don’t. A dilemma that has been faced by many researchers is what balance should be struck between releasing summary-level data during publication or through searchable databases and minimizing the risk to the privacy of study participants. For example, how do researchers determine the number of SNPs that should be placed on the web or in a supplementary table? Is releasing summary-level data from 1,000 or 5,000 SNPs reasonable? Managing and assessing the risk when sharing summary-level data should balance multiple factors - both quantitative and non-quantitative - as well as have a clear deliberation process.
Non-quantitative risk assessment should include consideration of the potential consequences of someone in a particular cohort being identified as a participant. For example, identification of participants in studies of readily observable common traits, such as obesity or hair color, would be less concerning than identification of individuals in studies of alcohol dependence, illegal behavior, or psychiatric conditions. These types of non-quantitative risk considerations are often study specific and higher-level restrictions on access may be warranted for higher-risk studies. Within databases such as dbGAP, there is ability to define restriction on access through the Data Use Certification agreement. For example, some datasets require applicants to obtain IRB approval for access, while many other datasets allow for general use access following institutional and user agreement to standard sharing and reporting policies for GWAS.
Quantifying the risk of making summary-level data broadly available is an essential part of the risk assessment process, and one that lends itself to more traditional approaches for risk assessment. Box 1 introduces several key concepts in risk assessment, such as sensitivity, specificity and positive predictive value (PPV). Each of these metrics gives insight into a specific type of risk. Beyond these metrics, software tools also exist for quantifying risk associated with summary-level data from GWAS. Notably, Sankaraman and colleagues 30 published a method and software tool called SecureGenome, which utilizes an input genotype set and a reference set and determines the number of highly ranked SNPs that can be safely exposed from the upper bounds of the optimally solved likelihood ratio test.
Box 1. Risk assessment definitions applied to sharing GWAS aggregate datasets.
In order to consider risk-assessment definitions, it is useful to first think of standard ‘ability of a test to detect a disease’ measures of sensitivity, specificity, positive and negative predictive values, as shown in the upper table. Each of these can be converted to an ‘ability to classify an individual as being in a genome-wide association study (GWAS) data set’, as shown in the lower table.

The risk-assessment definitions in the context of GWAS data sets are listed below.
Type II error. The proportion of times that someone who is actually in the data set is not identified as being in the data set. For example, with 20% type II error, there is a 20% chance of failing to determine that someone is in a data set.
Type I error. The proportion of times that someone is predicted to be in the data set when they are not. For example, with 5% type I error, there is a 5% chance of determining that someone is in the data set when they are not.
Sensitivity. The ability to detect true positives (that is, the correct classification of people in the data set). In both cases, this would be (a) / (a + c). For example, with a sensitivity of 30%, only 30% of test individuals in the data set will be correctly classified as being in the data set; 70% of those actually in the data set will be missed.
Specificity. The proportion of those people that are not in the data set who are correctly classified as not being in the data set (that is, true negatives). From the table, this would be (d) / (b + d). For example, with a specificity of 40%, only 40% of test individuals will be correctly classified as not being in the data set; 60% of those classified as being in the data set actually are not.
Power. The proportion of times that an individual who is actually in the data set will be correctly classified as being in the data set. For example, with 80% power, there is an 80% chance of correctly classifying someone as being in the data set.
Positive predictive value. The positive predictive value (PPV) is defined as the number of true positives divided by the total number of all positives ((a) / (a + b)). This measure is frequently used for rare disorders. Similarly, most individuals from a population would not actually be in a GWAS data set. PPV is the proportion of all individuals predicted to be positive from a population that are truly in a data set. With 20% PPV, only 20% of those identified as being in the cohort actually will be; 80% will not (and hence the ratio of false positives to true positives would be 4:1).
Positive predictive value
In this section we discuss a metric that can be used in quantitative risk assessments in the context of sharing data that specifically accounts for the size of the sampled population and the fact that most individuals from a population are actually not in the dataset. In a concept highlighted by Braun et al31, false positives depend on the number of participants from the population, and PPV as a metric can quantify the risk of correctly identifying an individual as being included within a dataset given that most individuals from the population are not actually included in the dataset. Calculating PPV requires determining the proportion of the ‘at-risk’ population that is in the genome-wide association study. For example, let’s assume someone wished to determine whether a person was within a dataset of 1,000 European ancestry individuals as part of the Framingham study (and they had genotype data for this person). Given an estimated 65,000 individuals in Framingham with an approximate 75% European ancestry population, the ‘at-risk’ population is approximately 50,000 individuals. The prevalence is thus 1,000/50,000=0.02. Without any data, the risk of positively identifying a person who is actually in the dataset is 2%. Thus the prevalence allows estimation of the positive predictive value given this prior knowledge. Prevalence of participants within a study may be quite low in large-scale studies, or may become reasonably high in small ‘at-risk’ populations such as a GWAS of the Native Hawaiian populations or Old Order Amish. The influence of prevalence on risk assessment through PPV is illustrated in Figure 1, in which a simulation with a high prevalence is compared with a simulation with a low prevalence. With low prevalence, the risk of resolving membership of a cohort is greatly reduced. Therefore, the strength of PPV as a measure is that it inherently accounts for the prior probability that a person selected at random is actually in the dataset and inherently accounts for key aspects of the population as part of risk assessment29,30.
Figure 1. Sharing 5,000 SNPs at different prevalence or prior probabilities.
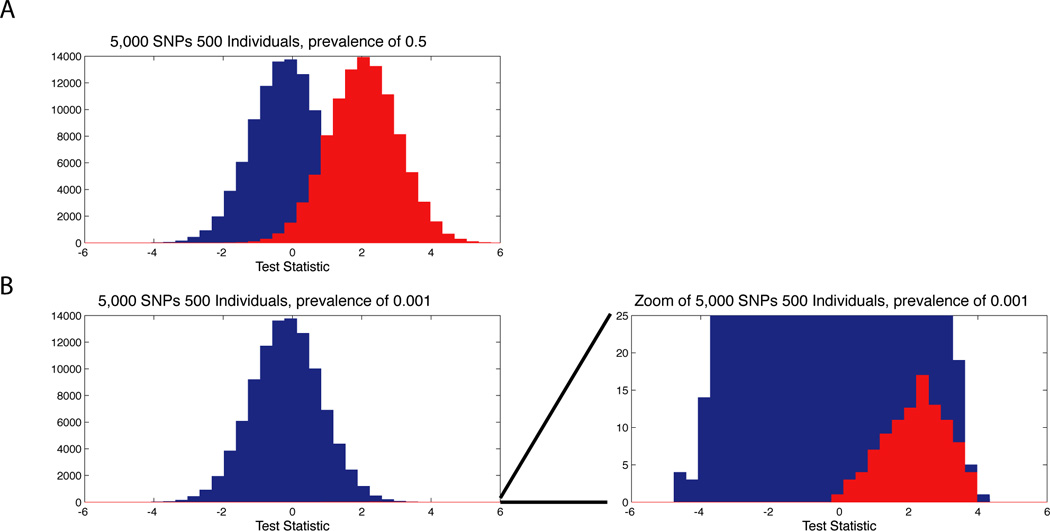
In the plots, we use simulations to show how the prior probability of being in a dataset impacts the ability to resolve if a person within a population using summary level allele frequencies from 5,000 SNPs on datasets of 500 individuals. In (a) we show a histogram of test-statistics based on the approach of Jacobs et al31 for resolving membership in 100,000 simulations when the person tested is actually within a dataset (red) and 100,000 simulations when the person tested is not within a dataset (blue). Since the simulations of being in a dataset and not within a dataset are equal, the prevalence or prior probability of being in the dataset is 0.5. In (b) we show 100,000 simulations when the person is not within the dataset (blue) and 100 simulations when they are within the dataset, equivalent to a prevalence or prior probability of being in the dataset of 0.001. The figures is zoomed to the right showing how a large number of tests of individuals not in the dataset can obscure the ability to distinguish true positive and false-positives. Describing risk as PPV allows one to consider prevalence for being in a dataset as a prior, thus increasing the accuracy in assessing the risk of a person within a dataset being correctly identified.
As explained above, researchers are often faced with the question of how many SNPs should be included in the summary data that they release. The PPV is one way to obtain a quantitative risk assessment for different numbers of SNPs and different study sizes; Table 1 provides several examples of PPV as a risk assessment in simulations of releasing between a few hundred and few thousand most associated SNPs by p-value from a study with different prevalence settings. In these simulations we used a prevalence of 0.01, which could be similar to a study of cardiovascular traits in a Framingham population, and 0.001, which could be similar to a study of 1,000 individuals with Major Depression sampled from a population defined to include all U.S. persons of European ancestry. The results of these simulations show the importance of considering prevalence; for 5,000 SNPs and a cohort size of 500 the PPV is 29.2% for a prevalence of 0.01 and 7.5% for a prevalence of 0.001, both with a discrimination threshold of 0.001. Further results from Table 1 suggest that sharing 1,000 SNPs for datasets with > 500 individuals generally lead to a low PPV, regardless of the population size. Taken together, the process of assessing risk with PPV and/or other statistical metrics can be used to inform discussions of non-quantitative risks.
Table 1. Risk assessment with different prevalence parameters.
The table shows sensitivity, specificity and positive predictive value (PPV) for sharing <5,000 SNPs for <5,000 individuals, assuming a prevalence of 0.001 (upper part of the table) or 0.01 (lower part of the table), based on simulated Framingham SNP Health Association Resource (SHARe) genomewide association data. In these simulations, summary-level allele frequency data sets were created by randomly selecting a fixed number of individuals from the Framingham SHARe data set into two data sets. From these data sets, SNPs that failed Hardy–Weinberg equilibrium (<10-6), minor allele frequency (<0.01), missingness (<0.01) and call rate (<0.97) were removed using the PLINK analysis tool set43. Association statistics were calculated for all SNPs, but sharing of allele-frequency data was only assumed for the most associated SNPs by P value (5,000 SNPs in the examples shown in the table). Individuals in and not in the data set were evaluated at a defined prevalence with a significance threshold of 0.005, with the entire process repeated until 100,000 simulations were completed.
| Prevalence = 0.001 | ||||
|---|---|---|---|---|
| SNPs | Cohort Size |
Sensitivity | Specificity | Positive Predictive Value |
| 100 | 100 | 0.05 | 0.99 | 0.010 |
| 100 | 500 | 0.04 | 0.99 | 0.004 |
| 100 | 1000 | 0.01 | 0.99 | 0.004 |
| 500 | 500 | 0.19 | 0.98 | 0.011 |
| 500 | 1000 | 0.12 | 0.98 | 0.008 |
| 1000 | 500 | 0.36 | 0.97 | 0.012 |
| 1000 | 1000 | 0.21 | 0.97 | 0.007 |
| 5000 | 500 | 0.83 | 0.99 | 0.075 |
| 5000 | 1000 | 0.51 | 0.99 | 0.038 |
| Prevalence = 0.01 | ||||
| SNPs |
Cohort Size |
Sensitivity | Specificity |
Positive Predictive Value |
| 100 | 100 | 0.05 | 0.99 | 0.080 |
| 100 | 500 | 0.06 | 0.99 | 0.067 |
| 100 | 1000 | 0.04 | 0.99 | 0.034 |
| 500 | 500 | 0.28 | 0.96 | 0.061 |
| 500 | 1000 | 0.17 | 0.97 | 0.049 |
| 1000 | 500 | 0.44 | 0.95 | 0.076 |
| 1000 | 1000 | 0.27 | 0.95 | 0.056 |
| 5000 | 500 | 0.89 | 0.98 | 0.292 |
| 5000 | 1000 | 0.63 | 0.98 | 0.275 |
Summary and implications
The path for future GWAS will benefit from and depend on data sharing. Recent large-scale efforts show that careful, coordinated efforts of sharing summary-level data leads to the discovery of many new genome-wide significant associations. With hindsight, these associations are often apparent in the original studies, though not at levels to merit follow-up sequencing. Clearly, the sharing of data and the ability to access summary-level data will be an important part of identifying new associations in future studies. Protecting the privacy of participants is an important part of this process. Quantitatively assessing privacy risks using PPV incorporates population size and can inform discussion of non-quantitative factors such as the impact of an individual being identified within studies. Therefore, it is our opinion that quantitative tools should play a useful part in assessing the risk of determining that an individual is in a dataset from release of aggregate SNP genome-wide genotyping datasets and subsets of these datasets.
Acknowledgements
This manuscript represents the views and opinions of its authors and does not necessarily represent the views or policies of the National Institutes of Health (NIH) or the U.S. Department of Health and Human Services. This research was supported in part by the Intramural Research Program of the NIH, National Library of Medicine. DWC would like to acknowledge support from NHLBI award U01 HL086528. We thank Ian Marpuri and Lin Gyi for their support coordinating the development of this work.
Glossary
- Allele frequency
The frequency of the less-common allele of a polymorphism. It has a value between 0 and 0.5 and can vary between populations.
- Bayesian
A statistical framework for evaluating a hypothesis. The Bayesian approach assesses the probability of a hypothesis being correct by incorporating the prior probability of the hypothesis.
- Discrimination threshold
The significance threshold for rejecting the null hypothesis in a statistical test.
- Frequentist
A statistical framework for evaluating a hypothesis. The frequentist approach tests a hypothesis as being correct given the strength of a data set.
- Imputation
A method for inferring untyped variants from neighboring variants, based on linkage disequilibrium and haplotype structure.
- Linear regression
The estimation of a first-order relationship between two variables, which involves fitting a line of best fit to the data.
- Missingness
The percentage of samples that do not receive a genotype call for a SNP in a genome-wide association study.
- Neyman–Pearson lemma
A theorem that assures the optimality of a likelihood ratio test between simple hypotheses at a given threshold.
- Prevalence
The prior probability that a person is in a data set of interest. Alternatively, the term can refer to the fraction of individuals in a data set out of the total number of individuals that could be in the data set.
- Reference data set
A data set of samples from individuals who are from the same population that was sampled in the summary-level data set of interest.
Biographies
Dr. David W. Craig is an Associate Professor at the Translational Genomics Research Institute. Dr. Craig earned his PhD. In Bioengineering from the University of Washington studying the impact protein sequence on structure and function. He research group at the TGen is focused on development and application high-throughput genomics software tools for improving diagnosis and treatment of disease.
Dr. Manolio is Director of the Office of Population Genomics of the National Human Genome Research Institute, NIH. She received her M.D. from the University of Maryland in 1980 and her Ph.D. in human genetics/genetic epidemiology from Johns Hopkins University in 2001. She joined the National Heart, Lung, and Blood Institute in 1987 and moved to NHGRI in 2005 to lead efforts in applying genomic technologies to population research, including the Electronic Medical Records and Genetics (eMERGE) Network and the NHGRI Genome-Wide Association Catalog. She has authored over 200 original research papers and has research interests in genome-wide association studies.
Dr, Zhenyuan Wang earned a Ph.D from the University of Illinois at Urbana-Champaign. He participated in the human genome project. As a member of dbGaP team, Dr. Wang is working on collecting, displaying and redistributing individual genotypes and GWAS results.
Dr. Ostell is the Chief of the Information Engineering Branch (IEB) of the National Center for Biotechnology Information (NCBI). Dr. Ostell earned a Ph.D. in molecular biology from Harvard University, developed commercial software for biotechnology, then helped create NCBI in 1988. As IEB Chief, Dr. Ostell has been responsible for designing, developing, building, and deploying the production resources at NCBI from its beginning including PubMed, GenBank, BLAST, Entrez, RefSeq, dbSNP, PubMed Central, dbGaP, and many others. In 2007 Dr. Ostell was inducted into the United States National Academies, Institute of Medicine, and made an NIH Distinguished Investigator in 2011.
Robert M. Goor, Ph.D., is a Staff Scientist in the Information Engineering Branch of the National Center for Biotechnology Information (NCBI). He earned a doctorate in Mathematics from the University of Michigan for work in mathematical analysis and optimization. He has taught mathematics, engineering and computer science at the college level. In addition, he has engaged in applied mathematical research in the private sector, primarily with The General Motors Research Labs, focusing on advanced robotics. Dr. Goor joined NIH in January, 2006, and has built a mathematically-based, computerized quality assurance tool for assessing and interpreting measurements of short tandem repeat (STR) markers.
Dr. Sherry is the Reference Collection Section Chief within the Information Engineering Branch (IEB) of the National Center for Biotechnology Information (NCBI). He earned his Ph.D. from the Pennsylvania State University in 1996, and post-doc'd at the LSU Medical Center until joining NCBI in 1998. His group develops genetic variation-related archives and information systems for basic research and medical genetics including dbSNP, dbGaP, SRA, PheGenI, and ClinVar. He conducts research on the architecture of population genetic information to ensure NCBI information systems are both useful to researchers and respectful to study participants.
Michael Feolo is a staff scientist at the NIH’s National Center for Biotechnology Information, and since 2007 has been the team lead for the database of Genotype and Phenotype. He received an M.S. from the Department of Biomedical Informatics, University of Utah in 2000. Michael is also the team lead for the dbMHC, a web interface and permanent public archive data about the HLA genes. Prior to leading the dbGaP team, Michael worked in the database of Single Nucleotide Polymorphisms from 2000 to 2007 and participated as a member of the planning and analysis committees for the International HapMap Project.
Footnotes
Weblinks
NCBI dbGaP: http://www.ncbi.nlm.nih.gov/dbgap
NIH GWAS Page (includes policy): http://gwas.nih.gov/
GWAS Central: https://www.gwascentral.org/
Public Population Project in Genomics (P3G): http://p3g.org
NHGRI catalog: http://www.genome.gov/gwastudies/
The European Genome-phenome Archive (EGA): http://www.ebi.ac.uk/ega/
SecureGenome: securegenome.icsi.berkeley.edu/securegenome/
Bibliography
- 1.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nature reviews. Genetics. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 2.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhernakova A, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS genetics. 2011;7:e1002004. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollingworth P, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature genetics. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nature genetics. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kho AN, et al. Electronic medical records for genetic research: results of the eMERGE consortium. Science translational medicine. 2011;3 doi: 10.1126/scitranslmed.3001807. 79re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nature genetics. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 10.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nature reviews. Genetics. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 11.Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng SL, et al. Cumulative association of five genetic variants with prostate cancer. The New England journal of medicine. 2008;358:910–919. doi: 10.1056/NEJMoa075819. [DOI] [PubMed] [Google Scholar]
- 13.Vacic V, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heeney C, Hawkins N, de Vries J, Boddington P, Kaye J. Assessing the privacy risks of data sharing in genomics. Public health genomics. 2011;14:17–25. doi: 10.1159/000294150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Church G, et al. Public access to genome-wide data: five views on balancing research with privacy and protection. PLoS genetics. 2009;5:e1000665. doi: 10.1371/journal.pgen.1000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preuss M, et al. Design of the Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circulation. Cardiovascular genetics. 2010;3:475–483. doi: 10.1161/CIRCGENETICS.109.899443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nature genetics. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelis MC, et al. The Gene, Environment Association Studies consortium (GENEVA): maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genetic epidemiology. 2010;34:364–372. doi: 10.1002/gepi.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Committee PGCS. A framework for interpreting genome-wide association studies of psychiatric disorders. Molecular psychiatry. 2009;14:10–17. doi: 10.1038/mp.2008.126. [DOI] [PubMed] [Google Scholar]
- 20.Nelson MR, et al. The Population Reference Sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am J Hum Genet. 2008;83:347–358. doi: 10.1016/j.ajhg.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 22.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic acids research. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mailman MD, et al. The NCBI dbGaP database of genotypes and phenotypes. Nature genetics. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leinonen R, et al. The European Nucleotide Archive. Nucleic acids research. 2011;39:D28–D31. doi: 10.1093/nar/gkq967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. A navigator for human genome epidemiology. Nature genetics. 2008;40:124–125. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]
- 26.Thorisson GA, et al. HGVbaseG2P: a central genetic association database. Nucleic acids research. 2009;37:D797–D802. doi: 10.1093/nar/gkn748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirakawa M, et al. JSNP: a database of common gene variations in the Japanese population. Nucleic acids research. 2002;30:158–162. doi: 10.1093/nar/30.1.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hindorff LA, et al. 2011 AMIA Summit on Translational Bioinformatics. San Francisco: 2011. PheGenI: an Integrated Resource for Browsing Genetic Association Data. [Google Scholar]
- 29.Homer N, et al. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genet. 2008;4:e1000167. doi: 10.1371/journal.pgen.1000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sankararaman S, Obozinski G, Jordan MI, Halperin E. Genomic privacy and limits of individual detection in a pool. Nat Genet. 2009;41:965–967. doi: 10.1038/ng.436. [DOI] [PubMed] [Google Scholar]
- 31.Jacobs KB, et al. A new statistic and its power to infer membership in a genome-wide association study using genotype frequencies. Nature genetics. 2009;41:1253–1257. doi: 10.1038/ng.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neyman J, Pearson E. On the Problem of the Most Efficient Tests of Statistical Hypotheses. Philosophical Transactions of the Royal Society of London. Series. A, Containing Papers of a Mathematical or Physical Character. 1933;231:289–337. [Google Scholar]
- 33.Braun R, Rowe W, Schaefer C, Zhang J, Buetow K. Needles in the haystack: identifying individuals present in pooled genomic data. PLoS Genet. 2009;5:e1000668. doi: 10.1371/journal.pgen.1000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang R, Li YF, Wang X, Tang H, Zhou X. Learning your identity and disease from research papers: information leaks in genome wide association study; Proceeding CCS '09 Proceedings of the 16th ACM conference on Computer and communications security; 2009. pp. 534–544. [Google Scholar]
- 35.Visscher PM, Hill WG. The limits of individual identification from sample allele frequencies: theory and statistical analysis. PLoS genetics. 2009;5:e1000628. doi: 10.1371/journal.pgen.1000628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clayton D. On inferring presence of an individual in a mixture: a Bayesian approach. Biostatistics. 2010;11:661–673. doi: 10.1093/biostatistics/kxq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampson J, Zhao H. Identifying individuals in a complex mixture of DNA with unknown ancestry. Statistical applications in genetics and molecular biology. 2009;8 doi: 10.2202/1544-6115.1469. Article 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zerhouni EA, Nabel EG. Protecting aggregate genomic data. Science. 2008;322:44. doi: 10.1126/science.322.5898.44b. [DOI] [PubMed] [Google Scholar]
- 39.Krawczak M, Goebel JW, Cooper DN. Is the NIH policy for sharing GWAS data running the risk of being counterproductive? Investigative genetics. 2010;1:3. doi: 10.1186/2041-2223-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haga SB, O'Daniel J. Public Perspectives Regarding Data-Sharing Practices in Genomics Research. Public health genomics. 2011 doi: 10.1159/000324705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malin B, Karp D, Scheuermann RH. Technical and policy approaches to balancing patient privacy and data sharing in clinical and translational research. Journal of investigative medicine : the official publication of the American Federation for Clinical Research. 2010;58:11–18. doi: 10.231/JIM.0b013e3181c9b2ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elias-Sonnenschein LS, Viechtbauer W, Ramakers IH, Verhey FR, Visser PJ. Predictive value of APOE-{varepsilon}4 allele for progression from MCI to AD-type dementia: a meta-analysis. Journal of neurology, neurosurgery, and psychiatry. 2011 doi: 10.1136/jnnp.2010.231555. [DOI] [PubMed] [Google Scholar]
- 43.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]