

Abstract
Lantibiotics are a large family of antibacterial peptide natural products containing multiple post-translational modifications, including the thioether structures lanthionine and methyllanthionine. Efforts to probe structure–activity relationships and engineer improved pharmacological properties have driven the development of new methods to produce non-natural analogues of these compounds. In this study, solid-supported chemical synthesis was used to produce analogues of the potent lantibiotic epilancin 15X, in order to assess the importance of several N-terminal post-translational modifications for biological activity. Surprisingly, substitution of these moieties, including the unusual N-terminal d-lactyl moiety, resulted in relatively small changes in the antimicrobial activity and pore-forming ability of the peptides.
Lantipeptides are a diverse and growing class of ribosomally synthesized, polycyclic natural products, classified by the presence of the thioether-containing residues meso-lanthionine (Lan) and (2S,3S,6R)-3-methyllanthionine (MeLan) (Figure 1).1 These moieties are installed post-translationally into a linear precursor peptide via enzymatic dehydration of serine and threonine to the α,β-unsaturated residues 2,3-didehydroalanine (Dha) and (Z)-2,3-didehydrobutyrine (Dhb), respectively, followed by intramolecular Michael-type addition of cysteine thiols to yield the thioether cross-links. Lantipeptides that have antimicrobial activities are called lantibiotics, and many of these exert their activity via binding to lipid II to inhibit peptidoglycan biosynthesis and/or forming pores in the cell membrane.2 As members of this family display substantial activity against pathogenic bacteria,3 several lantibiotics are currently in development for the treatment of bacterial infections.4 Fueled by this therapeutic potential, in vitro and in vivo engineering efforts have sought to improve the pharmacological properties of these compounds using their natural biosynthetic machinery;5,6 such approaches include recent methods to incorporate non-proteinogenic amino acids.7 As a complementary strategy to bioengineering, chemical synthesis presents the opportunity to widen further the chemical space accessible for the production of lantibiotics and other cyclic peptides,8 including the design of non-natural cross-links.9 The successful construction of lactocin S10 and both peptides of lacticin 314711 by solid-phase peptide synthesis (SPPS) highlight the growing applicability of chemical approaches to produce complex lantibiotics and analogues.
Figure 1.

Sequences and ring topologies of nisin and epilancin 15X (1) and chemical structures of their post-translational modifications.
Epilancin 15X (1, Figure 1), a cationic lantibiotic first isolated from the clinical strain Staphylococcus epidermidis 15X154, currently ranks among the most active lantibiotics against several clinically relevant bacterial pathogens, including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus.12 Despite this potent activity, the mode of action of this compound is currently unknown. While its C-terminal ring system resembles the pore-forming portion of the lantibiotic nisin (Figure 1),2,6,131 lacks nisin’s A- and B-rings that target lipid II;14 indeed, 1(15) and the structurally related lantibiotic epilancin K716 do not bind to lipid II. The combination of potent activity and lack of lipid II binding presents the intriguing possibility that 1 and related lantibiotics may bind to a different biological target.16,17 The N-terminus of 1 is adorned with an unusual d-lactyl (DLac) cap, which is installed by the oxidoreductase ElxO via stereospecific reduction of a pyruvyl (Pyr) moiety generated from hydrolysis of an N-terminal Dha.18 Additional post-translational modifications beyond Lan/MeLan are often critical for the biological activity of lantibiotics,19 but the importance of the DLac group has not been assessed in this regard. Previous attempts to reconstitute the biosynthesis of 1in vitro or in a heterologous host were unsuccessful,18 and no study has yet demonstrated biosynthetic manipulation in the producing organism. Hence, at present no analogues of this lantibiotic have been reported. We therefore employed SPPS to construct analogues of 1 to probe the importance of its N-terminal cluster of post-translational modifications for bioactivity.
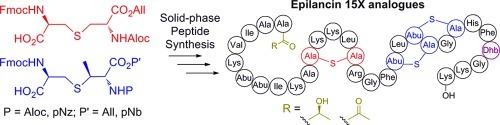
We envisioned the solid-supported total synthesis to utilize orthogonally protected Lan/MeLan building blocks,10,11,20−22 which allow selective installation of each cross-link into the growing peptide backbone via amide-forming cyclization between the free carboxylate of the Lan/MeLan residue and the N-terminus of the peptide. As 1 contains one Lan ring and a pair of overlapping MeLan rings, three such building blocks were necessary. Allyl-protected Lan building block 2 (Scheme 1) was accessed by the procedure of Pattabiraman et al.20 However, attempting to apply a similar scheme to MeLan building blocks 3 and 4, featuring allyl and p-nitrobenzyl protection, respectively, resulted in low yields and partial loss of stereochemical integrity. Therefore, an alternative scheme was developed using protected β-methyl-d-cysteine 8, which was prepared from d-threonine (7) in five steps (Scheme 1).21 We envisioned that 8 might be utilized as a common intermediate in the syntheses of both 3 and 4, thereby streamlining the overall approach. Following protecting group manipulation of 8, the corresponding p-methoxybenzyl (Mob)-protected thiols 9 and 11 were deprotected and condensed with l-bromoalanine 6 under phase-transfer conditions, resulting in 10 and 12 with full preservation of stereochemistry. tert-Butyl ester removal yielded MeLan building blocks 3 and 4, which were used subsequently for SPPS.
Scheme 1. Structure of Lan Building Block 2 and Synthesis of MeLan Building Blocks 3 and 4.

Reagents and conditions: (a) tert-butyl 2,2,2-trichloroacetimidate, EtOAc, cyclohexane, 93%; (b) PPh3, CBr4, CH2Cl2, 79%; (c) AlocOSu, iPr2NEt, CH2Cl2, 97%; (d) Hg(OAc)2, PhOMe, CF3CO2H, then dithiothreitol; (e) 6, NaHCO3, Bu4NBr, Bu3P, EtOAc, H2O, 64% (two steps); (f) CF3CO2H, PhSiH3, CH2Cl2, 96%; (g) pNzCl, iPr2NEt, CH2Cl2, 96%; (h) Pd(PPh3)4, PhNHMe, THF; (i) pNbBr, NaHCO3, DMF, 93% (two steps); (j) 6, NaHCO3, Bu4NBr, Bu3P, EtOAc, H2O, 85% (two steps); (k) CF3CO2H, PhSiH3, CH2Cl2, 95%. Aloc, allyloxycarbonyl; pNz, p-nitrobenzyloxycarbonyl; pNb, p-nitrobenzyl.
With these building blocks in hand, syntheses of epilancin 15X analogues were performed. Given the structural similarities between nisin and 1 in the C-terminal ring system, we decided to investigate the importance of the unusual N-terminal segment of 1 for antimicrobial activity. 9-Fluorenylmethoxycarbonyl (Fmoc)-based SPPS was commenced using preloaded Wang resin (15, Scheme 2) with a substitution of 0.1 mmol/g to prevent intermolecular side reactions during cyclization.20N,N′-Diisopropylcarbodiimide (DIC) and 1-hydroxybenzotriazole (HOBt) or 1-hydroxy-7-azabenzotriazole (HOAt) were used for coupling reactions to minimize side products observed with other activating agents. Fragment 13 (Figure 2), representing Phe27-Dhb28 of 1, and building blocks 2, 3, and 4 were incorporated successfully using this chemistry. The thioether rings were installed by selective deprotection of the appropriate Lan/MeLan residue followed by cyclization, which was complete in all cases within 4 h. Nitrobenzyl-based protecting groups were removed from the resin-bound intermediate 16 (Scheme 2) by treatment with 6 M stannous chloride and 5 mM HCl; allyl-based protecting groups were not affected by these conditions. Subsequent cyclization promoted by (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP), HOAt, and 2,4,6-collidine yielded ring C (17). During the installation of ring B (18) and later ring A (20), allyl-based protecting groups were removed with Pd(PPh3)4 and phenylsilane. After successful construction of ring A, the desired absolute stereochemistry of the thioether rings was confirmed by peptide hydrolysis, derivatization of the resulting amino acids, and analysis by gas chromatography–mass spectrometry (GC-MS) with a chiral stationary phase (see Supporting Information).
Scheme 2. Synthesis of Epilancin Analogues 21, 22, and 23.

Reagents and conditions: (a) SnCl2, HCl, DMF; (b) piperidine, DMF; (c) PyAOP, HOAt, 2,4,6-collidine, DMF; (d) Fmoc-Leu-OH, DIC, HOBt, DMF; (e) Pd(PPh3)4, PhSiH3, DMF, CH2Cl2. Prior to cleavage from resin, all residues contained appropriate side-chain protecting groups for Fmoc-SPPS: tert-butyloxycarbonyl (Boc) for Lys, 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) for Arg, trityl (Trt) for His. Green residues are those modified from the natural product.
Figure 2.

Structures of Dha/Dhb-containing fragments prepared by solution-phase chemistry (see Supporting Information).
Following synthesis of resin-bound intermediate 20, analogues of 1 were prepared in a divergent fashion. In the first analogue (21, Scheme 2), the importance of the three N-terminal α,β-unsaturated residues (Dha3, Dhb7, Dhb8) was probed by replacement with their reduced counterparts l-alanine and l-2-aminobutyric acid (Abu), respectively, (Dha3Ala/Dhb7Abu/Dhb8Abu). To probe the importance of the last step in epilancin biosynthesis, the reduction of an N-terminal pyruvyl moiety to DLac,18 a second analogue (22) was prepared that also contained Pyr in place of DLac. To construct this analogue, fragment 14 (Figure 2) was coupled to the resin-bound 29-mer; the Boc-Dha moiety spontaneously hydrolyzed to the desired ketone upon tert-butyloxycarbonyl (Boc) removal. Based on the biological activities of these peptides (see below), a third analogue (23, Scheme 2) was prepared in which the eight N-terminal residues were truncated and an acetyl cap was added. All peptides were cleaved from the resin and globally deprotected with trifluoroacetic acid using triisopropylsilane (for 21 and 23) or anisole (for 22) as the primary scavenging agent, as triisopropylsilane is known to (partially) reduce peptide ketones.23 After reversed-phase HPLC purification, these peptides were isolated in overall yields of 1.6–1.9%, with an average yield of 92–93% per step.
Liquid culture antimicrobial assays using a standard two-fold dilution protocol were performed with authentic 1 and 21–23 against the indicator strain Staphylococcus carnosus TM300 to assess quantitatively the effect of each set of alterations on antimicrobial activity. Interestingly, both full-length analogues 21 and 22 largely maintained the nanomolar activity of parent peptide 1 (Figure 3). Substitution of the three N-terminal α,β-unsaturated residues of 1 with the corresponding l-amino acids in 21 gave only a modest increase in the IC50 value from 95 ± 9.6 to 270 ± 23 nM (minimum inhibitory concentration (MIC) values of 250 and 625 nM, respectively). This increase may potentially be due to loss of the planarity of these unsaturated structures, but a covalent mechanism of action involving these residues can be ruled out. Replacement of the DLac moiety of 21 with Pyr in 22 yielded a slight additional increase in IC50 to 354 ± 8 nM (MIC 1.25 μM). This observation suggests that the N-terminal acyl group plays only a minor role in activity and more likely serves to improve the stability of the peptide. While both Pyr and Lac moieties are known to protect lantibiotics from aminopeptidases,18 enzymatic formation of DLac from Pyr by ElxO may further stabilize 1 by removing the ketone, a potential site of nucleophilic attack in vivo. Analogue 23, in which the eight N-terminal residues of 1 were removed, still possessed activity against S. carnosus (MIC 12.5 μM) but diminished by nearly 2 orders of magnitude compared to 1. This N-terminal tail region, which contains primarily nonpolar residues, may therefore be important for proper binding interactions between epilancin 15X and its putative biological target, or alternatively for the molecule to span the membrane in pore-forming activity.
Figure 3.

Growth inhibition activity of authentic epilancin 15X (1) and analogues 21–23 against Staphylococcus carnosus TM300.
To test the latter possibility, we used flow cytometry to examine the ability of analogues 21 and 23 to alter the membrane potential and permeability of S. carnosus cells. Despite removal of the N-terminal segment, 23 still depolarized the membrane (Figure 4a) and disrupted membrane integrity (Figure 4b). Higher concentrations of 23 were needed than for full-length analogue 21 to elicit these responses, and these observations correlated well to the relative differences in antibiotic activity (Figure 3). These findings are consistent with the importance of the nisin-like C-terminal portion of 1 for pore-forming activity and suggest that the N-terminal segment may be involved in binding to a putative non-lipid II biological target to increase potency. The pore-forming activity of nisin is enhanced about 103-fold by binding to lipid II.26 In comparison, the effect of the N-terminus of epilancin 15X is more modest, with its MIC values increasing only 2 orders of magnitude upon removal of the eight N-terminal residues.
Figure 4.

Flow cytometry analysis of the pore-forming abilities of 21 and 23 against S. carnosus TM300. (a) Membrane depolarization activity measured by DiOC2(3) mean fluorescence intensity (MFI).24 (b) Membrane permeability activity measured by propidium iodide (PI) uptake.25
The work presented here extends the application of solid-supported chemical synthesis for the production of lantibiotic peptides. Analogues of epilancin 15X, a lantibiotic for which variants have not been made using biosynthetic machinery reconstituted in vitro or in bacterial hosts, were successfully produced. Interestingly, analogues 21 and 22 largely maintained the potent activity of the natural product. In addition, the roles of different portions of epilancin 15X for biological activity were investigated: the nisin-like C-terminal ring system allows for pore-forming activity; the linear N-terminal region increases potency, perhaps via binding to an as yet unidentified biological target; and the N-terminal DLac moiety likely plays more of a role in improving peptide stability than in antimicrobial activity. Further studies will focus on identifying this putative non-lipid II biological target of epilancin 15X, which may represent further diversification of lantibiotic modes of action.
Acknowledgments
We thank Weixin Tang (University of Illinois) for providing Lan/MeLan standards for GC-MS analysis and Dr. Juan Velásquez (University of Illinois) for providing authentic epilancin 15X. This work was supported by the National Institutes of Health (GM58822 to W.A.V.), the Robert C. and Carolyn J. Springborn Endowment (to P.J.K.), and an American Heart Association Midwest Affiliate predoctoral fellowship (11PRE7620039 to P.J.K.).
Supporting Information Available
Experimental procedures, characterization of new molecules, and GC-MS results. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Knerr P. J.; van der Donk W. A.. Annu. Rev. Biochem. 2012, DOI: 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- a Bierbaum G.; Sahl H.-G. Curr. Pharm. Biotechnol. 2009, 10, 2–18. [DOI] [PubMed] [Google Scholar]; b Breukink E.; de Kruijff B. Nat. Rev. Drug Discov. 2006, 5, 321–323. [DOI] [PubMed] [Google Scholar]
- a Asaduzzaman S. M.; Sonomoto K. J. Biosci. Bioeng. 2009, 107, 475–487. [DOI] [PubMed] [Google Scholar]; b Piper C.; Cotter P. D.; Ross P. R.; Hill C. Curr. Drug Discovery Technol. 2009, 6, 1–18. [DOI] [PubMed] [Google Scholar]; c Cotter P. D.; Hill C.; Ross R. P. Curr. Protein Pept. Sci. 2005, 6, 61–75. [DOI] [PubMed] [Google Scholar]
- a Ghobrial O.; Derendorf H.; Hillman J. D. J. Pharm. Sci. 2010, 99, 2521–2528. [DOI] [PubMed] [Google Scholar]; b Donadio S.; Maffioli S.; Monciardini P.; Sosio M.; Jabes D. J. Antibiot. 2010, 63, 423–430. [DOI] [PubMed] [Google Scholar]
- a Ross A. C.; Vederas J. C. J. Antibiot. 2011, 64, 27–34. [DOI] [PubMed] [Google Scholar]; b Nagao J.; Nishie M.; Sonomoto K. Curr. Pharm. Biotechnol. 2011, 12, 1221–1230. [DOI] [PubMed] [Google Scholar]; c Field D.; Hill C.; Cotter P. D.; Ross R. P. Mol. Microbiol. 2010, 78, 1077–1087. [DOI] [PubMed] [Google Scholar]; d Cortés J.; Appleyard A. N.; Dawson M. J.; David A. H. Methods Enzymol. 2009, 458, 559–574. [DOI] [PubMed] [Google Scholar]
- Rink R.; Wierenga J.; Kuipers A.; Kluskens L. D.; Driessen A. J. M.; Kuipers O. P.; Moll G. N. Appl. Environ. Microbiol. 2007, 73, 5809–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Oldach F.; Al Toma R.; Kuthning A.; Caetano T.; Mendo S.; Budisa N.; Süssmuth R. D. Angew. Chem., Int. Ed. 2012, 51, 415–418. [DOI] [PubMed] [Google Scholar]; b Shi Y.; Yang X.; Garg N.; van der Donk W. A. J. Am. Chem. Soc. 2011, 133, 2338–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Levengood M. R.; Knerr P. J.; Oman T. J.; van der Donk W. A. J. Am. Chem. Soc. 2009, 131, 12024–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor A. B. Org. Biomol. Chem. 2011, 9, 7606–7628. [DOI] [PubMed] [Google Scholar]
- a Ross A. C.; McKinnie S. M. K.; Vederas J. C. J. Am. Chem. Soc. 2012, 134, 2008–2011. [DOI] [PubMed] [Google Scholar]; b Dekan Z.; Vetter I.; Daly N. L.; Craik D. J.; Lewis R. J.; Alewood P. F. J. Am. Chem. Soc. 2011, 133, 15866–15869. [DOI] [PubMed] [Google Scholar]; c Knerr P. J.; Tzekou A.; Ricklin D.; Qu H.; Chen H.; van der Donk W. A.; Lambris J. D. ACS Chem. Biol. 2011, 6, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Liu H.; Pattabiraman V. R.; Vederas J. C. Org. Lett. 2009, 11, 5574–5577. [DOI] [PubMed] [Google Scholar]; e Pattabiraman V. R.; Stymiest J. L.; Derksen D. J.; Martin N. I.; Vederas J. C. Org. Lett. 2007, 9, 699–702. [DOI] [PubMed] [Google Scholar]; f Ghalit N.; Reichwein J. F.; Hilbers H. W.; Breukink E.; Rijkers D. T. S.; Liskamp R. M. J. ChemBioChem 2007, 8, 1540–1554. [DOI] [PubMed] [Google Scholar]
- Ross A. C.; Liu H.; Pattabiraman V. R.; Vederas J. C. J. Am. Chem. Soc. 2010, 132, 462–463. [DOI] [PubMed] [Google Scholar]
- Liu W.; Chan A. S. H.; Liu H.; Cochrane S. A.; Vederas J. C. J. Am. Chem. Soc. 2011, 133, 14216–14219. [DOI] [PubMed] [Google Scholar]
- a Ekkelenkamp M. B.; Hanssen M.; Hsu S. T. D.; de Jong A.; Milatovic D.; Verhoef J.; van Nuland N. A. J. Febs Lett. 2005, 579, 1917–1922. [DOI] [PubMed] [Google Scholar]; b Verhoef J.; Milatovic D.; Ekkelenkamp M. B.. WO/2005/023852, March 17, 2005.
- a Wiedemann I.; Benz R.; Sahl H.-G. J. Bacteriol. 2004, 186, 3259–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hasper H. E.; de Kruijff B.; Breukink E. Biochemistry 2004, 43, 11567–11575. [DOI] [PubMed] [Google Scholar]; c Wiedemann I.; Breukink E.; van Kraaij C.; Kuipers O. P.; Bierbaum G.; de Kruijff B.; Sahl H.-G. J. Biol. Chem. 2001, 276, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Hsu S.-T. D.; Breukink E.; Tischenko E.; Lutters M. A. G.; de Kruijff B.; Kaptein R.; Bonvin A. M. J. J.; van Nuland N. A. J. Nat. Struct. Mol. Biol. 2004, 11, 963–967. [DOI] [PubMed] [Google Scholar]
- Oman T. J.; Lupoli T. J.; Wang T.-S. A.; Kahne D.; Walker S.; van der Donk W. A. J. Am. Chem. Soc. 2011, 133, 17544–17547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brötz H.; Josten M.; Wiedemann I.; Schneider U.; Götz F.; Bierbaum G.; Sahl H.-G. Mol. Microbiol. 1998, 30, 317–327. [DOI] [PubMed] [Google Scholar]
- Hoffmann A.; Schneider T.; Pag U.; Sahl H.-G. Appl. Environ. Microbiol. 2004, 70, 3263–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velásquez J. E.; Zhang X.; van der Donk W. A. Chem. Biol. 2011, 18, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ökesli A.; Cooper L. E.; Fogle E. J.; van der Donk W. A. J. Am. Chem. Soc. 2011, 133, 13753–13760. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Castiglione F.; Lazzarini A.; Carrano L.; Corti E.; Ciciliato I.; Gastaldo L.; Candiani P.; Losi D.; Marinelli F.; Selva E.; Parenti F. Chem. Biol. 2008, 15, 22–31. [DOI] [PubMed] [Google Scholar]; c Cotter P. D.; O’Connor P. M.; Draper L. A.; Lawton E. M.; Deegan L. H.; Hill C.; Ross R. P. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18584–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hosoda K.; Ohya M.; Kohno T.; Maeda T.; Endo S.; Wakamatsu K. J. Biochem. 1996, 119, 226–230. [DOI] [PubMed] [Google Scholar]
- Pattabiraman V. R.; McKinnie S. M. K.; Vederas J. C. Angew. Chem., Int. Ed. 2008, 47, 9472–9475. [DOI] [PubMed] [Google Scholar]
- Narayan R. S.; VanNieuwenhze M. S. Org. Lett. 2005, 7, 2655–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Carrillo A. K.; VanNieuwenhze M. S. Org. Lett. 2012, 14, 1034–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mothia B.; Appleyard A. N.; Wadman S.; Tabor A. B. Org. Lett. 2011, 13, 4216–4219. [DOI] [PubMed] [Google Scholar]; c Martin N. I. J. Org. Chem. 2009, 74, 946–949. [DOI] [PubMed] [Google Scholar]; d Bregant S.; Tabor A. B. J. Org. Chem. 2005, 70, 2430–2438. [DOI] [PubMed] [Google Scholar]; e Mohd Mustapa M. F.; Harris R.; Bulic-Subanovic N.; Elliott S. L.; Bregant S.; Groussier M. F. A.; Mould J.; Schultz D.; Chubb N. A. L.; Gaffney P. R. J.; Driscoll P. C.; Tabor A. B. J. Org. Chem. 2003, 68, 8185–8192. [DOI] [PubMed] [Google Scholar]
- Marceau P.; Buré C.; Delmas A. F. Bioorg. Med. Chem. Lett. 2005, 15, 5442–5445. [DOI] [PubMed] [Google Scholar]
- Oman T. J.; van der Donk W. A. ACS Chem. Biol. 2009, 4, 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut I. M.; Blanke S. R.; van der Donk W. A. ACS Chem. Biol. 2011, 6, 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breukink E.; Wiedemann I.; Kraaij C. v.; Kuipers O. P.; Sahl H.-G.; de Kruijff B. Science 1999, 286, 2361–2364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.