

Abstract
The efficacy, safety, and tolerability of Lu AA21004 vs. placebo using venlafaxine XR as active reference in patients with DSM-IV-TR major depressive disorder (MDD) were evaluated. Lu AA21004 is a novel antidepressant that is a 5-HT3 and 5-HT7 receptor antagonist, 5-HT1A receptor agonist, 5-HT1B receptor partial agonist and inhibitor of the 5-HT transporter in recombinant cell lines. In this 6-wk, multi-site study, 429 patients were randomly assigned (1:1:1:1) to 5 or 10 mg Lu AA21004, placebo or 225 mg venlafaxine XR. All patients had a baseline Montgomery–Åsberg Depression Rating Scale (MADRS) total score ⩾30. The primary efficacy analysis was based on the MADRS total score adjusting for multiplicity using a hierarchical testing procedure starting with the highest dose vs. placebo. Lu AA21004 was statistically significantly superior to placebo (n=105) in mean change from baseline in MADRS total score at week 6 (p<0.0001, last observation carried forward), with a mean treatment difference vs. placebo of 5.9 (5 mg, n=108), and 5.7 (10 mg, n=100) points. Venlafaxine XR (n=112) was also significantly superior to placebo at week 6 (p<0.0001). In total, 30 patients withdrew due to adverse events (AEs) – placebo: four (4%); 5 mg Lu AA21004: three (3%); 10 mg Lu AA21004: seven (7%); and venlafaxine: 16 (14%). The most common AEs were nausea, headache, hyperhidrosis, and dry mouth. No clinically relevant changes over time were seen in the clinical laboratory results, vital signs, weight, or ECG parameters. In this study, treatment with 5 mg and 10 mg Lu AA21004 for 6 wk was efficacious and well tolerated in patients with MDD.
Keywords: Depression, Lu AA21004, MADRS, serotonin receptor affinity, venlafaxine
Introduction
Lu AA21004 (1-[2-(2,4-dimethyl-phenylsulfanyl)-phenyl]-piperazine) is a novel compound under development as an antidepressant (Bang-Andersen et al. 2011) with affinity for the human 5-HT1A, 5-HT1B, 5-HT3 and 5-HT7 receptors and the 5-HT transporter (SERT) (Moore et al. 2008). Based on preclinical data, these affinities are considered to be of clinical relevance and involved in the mechanism of action at therapeutic doses. In vivo, Lu AA21004 increases the extracellular levels of serotonin (5-HT), noradrenaline, dopamine, acetylcholine and histamine in rat prefrontal cortex and hippocampus (Moore et al. 2008).
Lu AA21004 is extensively metabolized in the liver and at least five cytochrome P450 isoenzymes appear to be involved. The metabolism of Lu AA21004 to its major metabolite (pharmacologically inactive) is mediated primarily by CYP2D6. In addition, Lu AA21004 does not seem to be a clinically relevant inhibitor or inducer of cytochrome P450 isoenzymes. The terminal elimination half-life after multiple doses is estimated at ~60–70 h. The exposure (Cmax and area under curve) increased linearly with dose (2.5–60 mg). The absorption of Lu AA21004 is independent of food intake (Wang et al. 2009) and maximum plasma concentrations are reached 3–16 h after dosing. The rationale for choosing the Lu AA21004 doses (5 and 10 mg) in this proof-of-concept study was based on non-clinical and phase I data. Approximately 60–80% occupancy of the human SERT is required to achieve a therapeutic effect with selective serotonin reuptake inhibitors (SSRIs) or serotonin noradrenaline reuptake inhibitors (SNRIs) (Meyer, 2007). In contrast, an occupancy level of 41% with Lu AA20004 in rats led to a significant increase in extracellular levels of 5-HT, perhaps due to the additional pharmacological activities of Lu AA21004, which may counteract negative feedback mechanisms operating at cellular and network levels. The dose of 5 mg/d corresponds to a SERT occupancy of ~40% in human brain and was, therefore, expected to be an effective dose (Areberg et al. 2009).
The aim of this phase II clinical study was to investigate the efficacy, safety, and tolerability of two fixed doses (5 and 10 mg/d) of Lu AA21004 vs. that of placebo after 6 wk treatment in adult patients with major depressive disorder (MDD). Venlafaxine XR (225 mg/d) was used as the active reference.
Method
This randomized, double-blind, fixed-dose, placebo-controlled, active reference study recruited 429 randomized patients from 49 psychiatric settings in 11 countries (Australia, Austria, Canada, Czech Republic, Finland, France, Italy, Malaysia, Slovakia, Spain, Sweden). Outpatients with MDD were recruited from psychiatric settings from August 2006 to August 2007. Advertisements were used in Australia, Austria, Canada, Finland, Malaysia, and Sweden. The study was conducted in accordance with the principles of Good Clinical Practice (ICH, 1996) and the Declaration of Helsinki (WMA, 1964). Local ethics committees approved the study design and eligible patients gave their written informed consent before participating.
Eligible patients were randomized equally (1:1:1:1) to one of the four treatment arms for a 6-wk double-blind treatment period. Randomized patients were given 1-wk wallet cards at each visit and were instructed to take two capsules per day, orally, at the same time every day (preferably in the morning). Lu AA21004 was dosed at 5 or 10 mg/d for 6 wk and venlafaxine at 75 mg/d for 4 d, 150 mg/d for the following 3 d, and 225 mg/d for the remainder of the treatment period. Efficacy and tolerability were assessed at screening, baseline and after 1, 2, 3, 4, 5, and 6 wk. Patients who completed the 6-wk double-blind treatment period entered a 2-wk double-blind taper period. During this period, patients on 5 mg/d Lu AA21004 switched to placebo; patients on 10 mg/d Lu AA21004 received 5 mg/d Lu AA21004 for the first week (week 7) and placebo for the second week (week 8); patients on placebo remained on placebo; patients on venlafaxine received 150 mg/d venlafaxine for the first week (week 7) and 75 mg/d for the second week (week 8). Patients were contacted for a safety follow-up 4 wk after the completion visit. Down-taper medication was also offered to patients who withdrew.
Main entry criteria
Patients with MDD presenting with a current major depressive episode according to DSM-IV-TR criteria (APA, 1994) were included in the study if they were an outpatient of either sex, aged from 18 yr to 65 yr, with a Montgomery–Åsberg Depression Rating Scale (MADRS) (Montgomery & Åsberg, 1979) total score ⩾30 at the baseline visit.
Patients were excluded if they had any current psychiatric disorder other than MDD as defined in DSM-IV-TR [assessed using the Mini International Neuropsychiatric Interview (MINI; Sheehan et al. 1998)], or if they had a current or past history of manic or hypomanic episode, schizophrenia or any other psychotic disorder, including major depression with psychotic features, mental retardation, organic mental disorders, or mental disorders due to a general medical condition, any substance abuse disorder within the previous 6 months, presence or history of a clinically significant neurological disorder (including epilepsy), any neurodegenerative disorder, or any Axis II disorder that might compromise the study.
Patients at serious risk of suicide, based on the investigator's clinical judgement, or who had a score of ⩾5 on item 10 of the MADRS scale (suicidal thoughts) were also excluded, as were those receiving formal behaviour therapy or systematic psychotherapy, or were pregnant or breastfeeding, had a known hypersensitivity or were non-response to venlafaxine, or whose current depressive symptoms were considered by the investigator to have been resistant to two adequate antidepressant treatments of at least 6 wk duration, or had previously been exposed to Lu AA21004.
Patients were also excluded if they were taking the following psychotropic drugs within 2 wk prior to baseline or during the study: Reversible or irreversible monoamine oxidase inhibitors, SSRIs (fluoxetine within 5 wk), SNRIs, tricyclic antidepressants, psychoactive herbal remedies, any drug used for augmentation of antidepressant action or any other antidepressant drugs, oral antipsychotic and anti-manic drugs, or dopamine antagonists, any anxiolytics (including benzodiazepines); and any anticonvulsant drug, serotonergic agonists, narcotic analgesics or cough agents, anti-arrhythmics, oral anticoagulants, proton pump inhibitors, steroids, cisapride, macrolide antibiotics, antifungal agents, antihypertensives, all anti-inflammatory agents, anti-migraine agents, pseudoephedrine, hypolipidaemics, and episodic use of insulin. Occasional use of zolpidem, zopiclone and zaleplon for insomnia was allowed.
Patients were withdrawn if they became pregnant during the study, if the investigator considered it to be in the best interest of the patient for safety/efficacy reasons, if laboratory values were outside normal ranges and clinically significant, if they were considered to be at significant risk of suicide, if they scored ⩾5 points on item 10 (suicidal thoughts) of the MADRS, if the randomization code for a patient was broken, if consent to participate was withdrawn, if they did not take study medication for more than 6 consecutive days, or if the patient was lost to follow-up. The patient could be withdrawn from the study if a serious adverse event (SAE) occurred. If adverse events (AEs) were contributory to withdrawal, they were always regarded as the primary reason for withdrawal.
Efficacy rating
Patients were evaluated using the MADRS from baseline to week 6. Rater training was undertaken to increase inter-rater reliability, and was chaired by an experienced investigator. Only those investigators who had actively participated in rater training sessions prior to inclusion of patients into the study and had received rater certification were allowed to rate patients. Patient ratings were assessed by the same investigator at each visit, whenever possible.
Allocation to treatment
The medication was given as capsules of identical appearance. Patients who met the selection criteria at the baseline visit were assigned to double-blind treatment according to a computer-generated randomization list. The details of the randomization series were unknown to any of the investigators and were contained in a set of sealed opaque envelopes. At each study site, sequentially enrolled patients were assigned the lowest randomization number available in blocks of four. All investigators, study personnel and participants were blinded to treatment assignment for the duration of the entire study. The randomization code was broken for one patient (accidentally) who had completed the study before this was discovered, and was therefore not withdrawn from the study.
Analysis sets
All safety analyses were based on the all-patients-treated set (APTS), comprising all randomized patients who took at least one dose of study medication. All efficacy analyses were based on a modified intent-to-treat set (ITT) – the full-analysis set (FAS), comprising all patients in the APTS who had at least one valid post-baseline MADRS total score assessment.
Power and sample size calculations
It was planned to randomize a minimum of 384 patients with a DSM-IV-TR diagnosis of a major depressive episode (MDE) into the double-blind period of the study. With 96 patients in each treatment group and a standard deviation (s.d.) of 9 points, the power to detect a true treatment effect of 3.7 points on the MADRS total score at week 6, using last observation carried forward (LOCF), would be 80%.
Primary efficacy analysis
Four hypotheses were part of the primary efficacy analysis, which was fully adjusted for multiplicity using a hierarchical testing procedure at the 5% level of significance as long as the previous hypothesis was rejected. The order of testing was: no difference between the 10 mg dose vs. placebo at week 6, no difference between 5 mg vs. placebo at week 6, no difference between 10 mg dose vs. placebo at week 1, and finally no difference between 5 mg dose vs. placebo at week 1. The statistical model was an analysis of covariance (ANCOVA) of the change from baseline in MADRS total score (FAS, LOCF) with treatment and site as fixed factors and the baseline MADRS score as a covariate. The primary efficacy analysis was repeated on observed cases (OC) data, using both an ANCOVA and a mixed model for repeated measurements (MMRM).
Secondary efficacy analysis
Prospectively defined secondary clinician-rated variables were: MADRS total score, 24-item Hamilton Depression (HAMD24) total score (Hamilton, 1960), Clinical Global Impression – Improvement (CGI-I) and Clinical Global Impression – Severity (CGI-S) scores (Guy, 1976), Hamilton Anxiety (HAMA) total score (Hamilton, 1959), remission [defined as MADRS ⩽10, 17-item HAMD (HAMD17) ⩽7 or as a CGI-S score ⩽2] and response (defined as ⩾50% decrease from baseline in MADRS or HAMD24 total score, or a CGI-I score ⩽2) at all time points.
The change from baseline to each visit in all the secondary efficacy variables, except response and remission, was analysed using an ANCOVA, adjusting for baseline score, site, and treatment, using both OC and LOCF data. For CGI-I, the baseline CGI-S score was used for adjustment. The change from baseline to each visit in all the secondary efficacy variables, except response and remission, was also analysed using MMRM to compare the treatment groups over all assessment points simultaneously using OC data.
Response and remission rates for each visit were evaluated using Fisher's exact test. The CGI-S and CGI-I scores were analysed at the last visit (OC and LOCF) using ANCOVA. Unless otherwise stated, the terms ‘significant’ and ‘significantly’ refer to statistical significance at the 5% level, two-sided. Efficacy analyses that were not multiplicity-controlled were considered secondary. The principal statistical software used was SAS® version 9.1 (SAS Institute Inc., USA).
Tolerability assessments
Each patient was asked a non-leading question (such as, ‘how do you feel?’) at each visit, starting at baseline. All AEs (including any change in concurrent illnesses or new illnesses) either observed by the investigator or reported spontaneously by the patient were recorded. AEs were coded using the lowest level term according to the Medical Dictionary for Regulatory Activities, version 10.0. The time to withdrawal due to AEs was analysed using the Cox model. The incidences of individual AEs were compared between the treatment groups using Fisher's exact test.
As a post-hoc analysis, the safety database was searched at preferred-term and verbatim-term level for possible suicide-related AEs, as described by the FDA (Laughren, 2006).
Results
Patient baseline characteristics
The APTS comprised 426 patients (placebo, 105; venlafaxine, 113; 5 mg Lu AA21004, 108; 10 mg Lu AA21004, 100) (Fig. 1). Slightly more patients than planned were enrolled in the study, raising the power from 80% to 84%. There were no clinically relevant or statistically significant differences between the treatment groups in patient demographics or clinical characteristics at baseline (Table 1). Patients had a mean age (±s.d.) of 43.3±11.5 yr, 62.7% were women, and 92.0% were Caucasian.
Fig. 1.

Flow chart of patient disposition. AE, Adverse events, ITT, intention to treat; LoE, lack of efficacy; MADRS, Montgomery–Åsberg Depression Rating Scale; PBO, placebo; Ven 225, venlafaxine XR 225 mg.
Table 1.
Baseline patient characteristics
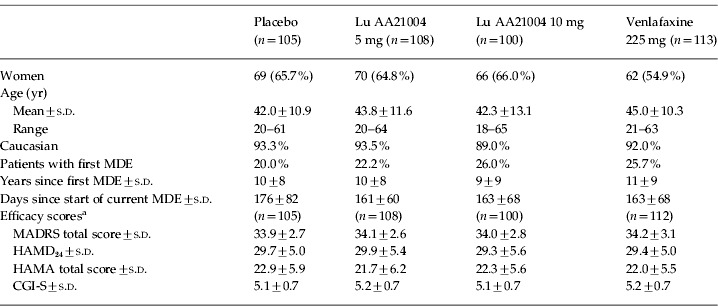
Based on the full-analysis set: CGI-S, Clinical Global Impression – Severity; HAMA, Hamilton Rating Scale for Anxiety; HAMD24, Hamilton Rating Scale for Depression (24 items); MADRS, Montgomery–Åsberg Depression Rating Scale; MDE, major depressive episode; s.d., standard deviation.
The mean baseline MADRS total score was 34.0, indicating a severely depressed patient population, consistent with the mean CGI-S score of 5.1. Patients were diagnosed with their first MDE ~10 yr prior to enrolment. Between 74% and 80% of the patients in each treatment group had had a previous MDE and their current episode had started about 5 months prior to enrolment (Table 1). There was a substantial level of anxiety symptoms, as indicated by a mean baseline HAMA total score of 22.2. About 40% (range 36–41%) of the patients in each treatment group had a concurrent medical condition. The number of patients taking zolpidem, zopiclone, or zaleplon prescribed episodically for insomnia was similar for placebo (n=3), venlafaxine (n=6), 5 mg Lu AA21005 (n=3), and 10 mg Lu AA21005 (n=3). Between 21% and 33% of the patients took concomitant medication that they continued with, and 26–29% commenced concomitant medication during the study.
Withdrawals from the study
The withdrawal rate due to all reasons during the entire study was 15% (Fig. 1), ranging from 9% (5 mg Lu AA21004) to 18% (venlafaxine and 10 mg Lu AA21004). More than 80% of the patients in each treatment group completed the study (Fig. 1). There was a slightly larger proportion of patients who completed the study in the 5 mg Lu AA21004 group than in the placebo, 10 mg Lu AA21004, or venlafaxine groups. The proportions of patients who withdrew due to AEs was statistically significantly different between venlafaxine and placebo, but not between the Lu AA21004 groups and placebo. There was an even distribution of withdrawals for any reason over time and no statistically significant differences between the treatment groups, between men and women, or between patients aged ⩽50 or >50 yr. The median compliance with study medication was 98%.
Efficacy
Primary endpoint
On the pre-defined primary efficacy endpoint, both doses of Lu AA21004 were statistically significantly (p<0.0001) superior to placebo in mean change from baseline in MADRS total score at week 6 (FAS, LOCF), with mean treatment differences to placebo of 5.9 (5 mg) and 5.7 (10 mg) points (Table 2) in a multiplicity-controlled analysis. These differences to placebo correspond to a standardized effect size (Cohen's d) of 0.56 (5 mg) and 0.54 (10 mg). Venlafaxine was also statistically significantly (p<0.0001) superior to placebo at week 6, with a mean treatment difference to placebo of 6.4 points (LOCF). The estimated treatment differences and nominal p values at week 6 obtained from an analysis using MMRM were similar to those obtained in the ANCOVA analyses [5.6±1.3 (5 mg Lu AA21004), 7.2±1.4 (10 mg Lu AA21004), 7.6±1.3 (venlafaxine), all p<0.0001] (Table 2). As a sensitivity analysis, the non-parametric Kruskal–Wallis test showed a statistically significant difference between the active treatments and placebo. The assumption of homogeneity of variances across treatment groups was confirmed using Bartlett's test (p=0.90). At week 1, with a difference from placebo in the MADRS total score of 0.8 for 10 mg (p=0.2377) and 0.2 for 5 mg (p=0.7489), none of the active treatments separated significantly from placebo.
Table 2.
Change from baseline in MADRS total score at week 6 (FAS)
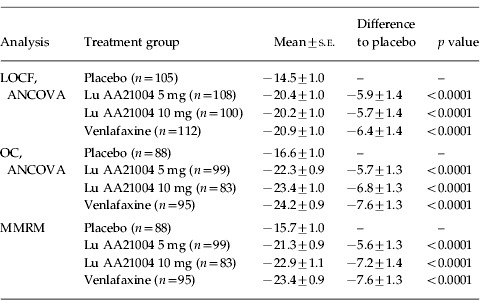
FAS, Full-analysis set; LOCF, last observation carried forward; MADRS, Montgomery–Åsberg Depression Rating Scale; MMRM, mixed model repeated measures; OC, observed cases; s.e., standard error of the mean.
Secondary efficacy analyses
MADRS
The mean MADRS total score decreased in all active treatment groups from 34.1 at baseline to ~13.4 in the LOCF analysis and to ~10.9 in the OC analysis at week 6. For Lu AA21004, a statistically significant difference compared to placebo in the change from baseline in MADRS total score, in favour of Lu AA21004, was seen from week 2 (10 mg) or week 3 (5 mg) onwards (LOCF and OC). For venlafaxine, a statistically significant difference to placebo was seen from week 2 (OC) or week 3 (LOCF) onwards (Fig. 2). At week 6, the proportion of MADRS responders (patients with ⩾50% decrease in MADRS total score) and remitters (MADRS score ⩽10) was statistically significantly higher in all active treatment groups than placebo (LOCF and OC) (Table 3). Single item analysis at week 6 showed a statistically significant advantage for both doses of Lu AA21004 for 9 out of the 10 items (except for ‘concentration difficulties’) relative to placebo.
Fig. 2.

Mean change from baseline in Montgomery–Åsberg Depression Rating Scale (MADRS) total scores (ANCOVA, FAS, OC, over time) and LOCF (week 6). * p<0.05, ** p<0.01, *** p<0.001 vs. placebo. FAS, Full-analysis set; LOCF, last observation carried forward; OC, observed cases.
Table 3.
Proportion (%) of responders and remitters at week 6 (FAS, mean)
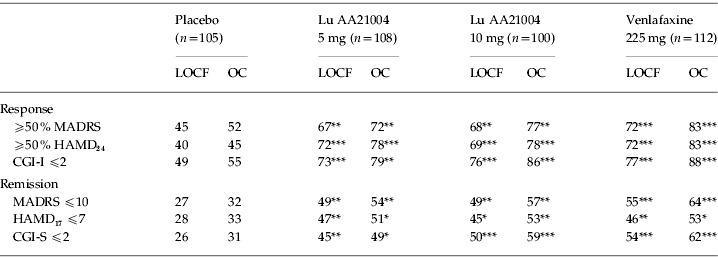
CGI-S, Clinical Global Impression – Severity; CGI-I, Clinical Global Impression – Improvement; HAMD17, Hamilton Rating Scale for Depression (17 items); HAMD24, Hamilton Rating Scale for Depression (24 items); FAS, full-analysis set; LOCF, last observation carried forward; MADRS, Montgomery–Åsberg Depression Rating Scale; OC, observed cases.
p<0.05, ** p<0.01, *** p<0.001 vs. placebo.
HAMD24
The mean HAMD24 total score decreased in all active treatment groups from 29.5 at baseline to ~11.7 in the LOCF analysis and ~9.7 in the OC analysis at week 6 (Table 4). For Lu AA21004 (5 mg and 10 mg), a statistically significant difference to placebo was seen from week 1 onwards. For venlafaxine, a statistically significant difference to placebo was seen from week 2 (OC) or week 3 (LOCF) onwards. At week 6, the proportion of HAMD24 responders (patients with ⩾50% decrease in HAMD24 total score) and remitters (HAMD17 score ⩽7) was statistically significantly higher in all active treatment groups compared to placebo (LOCF and OC) (Table 3).
Table 4.
Mean change from baseline in efficacy variables at week 6, difference to placebo (FAS)
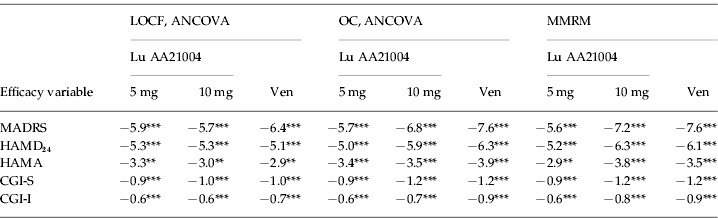
CGI-S, Clinical Global Impression – Severity; CGI-I, Clinical Global Impression – Improvement; FAS, full-analysis set; HAMA, Hamilton Rating Scale for Anxiety; HAMD24, Hamilton Rating Scale for Depression (24 items); LOCF, last observation carried forward; MADRS, Montgomery–Åsberg Depression Rating Scale; OC, observed cases; MMRM, mixed model repeated measures; Ven, venlafaxine.
HAMA
The level of anxiety symptoms, as assessed by the mean HAMA total score decreased in all active treatment groups from ~22 at baseline to ~10.1 in the LOCF analysis (Table 4) and ~8.4 in the OC analysis at week 6. For Lu AA21004, a statistically significant difference to placebo was seen in change from baseline in HAMA total score from week 2 (10 mg, OC) or week 3 (LOCF and OC) onwards (Fig. 3). For venlafaxine, a statistically significant difference to placebo was seen from week 3 (OC) or week 4 (LOCF) onwards.
Fig. 3.

Mean change from baseline in Hamilton Rating Scale for Anxiety (HAMA) total scores (ANCOVA, FAS, OC, over time) and LOCF (week 6). * p<0.05, ** p<0.01, *** p<0.001 vs. placebo. FAS, Full-analysis set; LOCF, last observation carried forward; OC, observed cases. Some patients were excluded due to the use of a non-validated scale in France.
CGI
The mean CGI-S score decreased in all active treatment groups from ~5.2 at baseline to ~2.6 in the LOCF analysis (Table 4) and ~2.3 in the OC analysis at week 6. The mean CGI-I score improved in all active treatment groups to ~2.0 at week 6 (LOCF, Table 4). For Lu AA21004, a statistically significant difference to placebo was seen in mean CGI-I score from week 1 (10 mg) or week 2 (5 mg) onwards (LOCF). For venlafaxine, a statistically significant difference to placebo was seen from week 3 onwards (LOCF). At week 6, the proportion of CGI responders (CGI-I ⩽2) and CGI remitters (CGI-S ⩽2) was statistically significantly higher in all active treatment groups than placebo (LOCF and OC) (Table 3).
Tolerability and safety
AEs
Since Lu AA21004 is a compound with a new mode of action, its safety and tolerability profile is described in some detail below. During the 6-wk treatment period, approximately three-fifths of patients in the placebo (61%) and 5 mg Lu AA21004 (68%) groups and approximately three-quarters of the patients in the 10 mg Lu AA21004 (74%) and venlafaxine (75%) groups had one or more AE. A total of 30 (7%) patients withdrew due to AEs: four (4%) in the placebo group, three (3%) in the 5 mg Lu AA21004 group, seven (7%) in the 10 mg Lu AA21004 group, and 16 (14%) in the venlafaxine group. Only in the venlafaxine group, did statistically significantly more patients withdraw due to AEs than in the placebo group (p=0.009). Seven patients withdrew from the study due to nausea: three (3%) in the 10 mg Lu AA21004 group and four (4%) in the venlafaxine group.
AEs reported by ⩾5% of patients during the 6-wk treatment period are shown in Table 5. The most common AEs reported in the active treatment groups were nausea, headache, hyperhidrosis, and dry mouth. For Lu AA21004, nausea (5 and 10 mg), hyperhidrosis (10 mg), and vomiting (10 mg) were the only AEs reported with an incidence statistically significantly higher than placebo. For the majority of patients reporting nausea, it was transient and mild or moderate in intensity. In addition to nausea and hyperhidrosis, the incidence of dry mouth, constipation, and anorgasmia were statistically significantly higher in the venlafaxine group than placebo group.
Table 5.
Adverse events (AEs) with an incidence of ⩾5% in any group in the 6-wk double-blind treatment period (APTS)
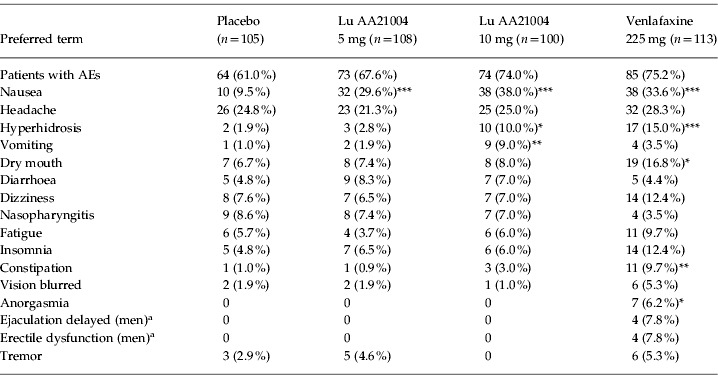
APTS, All-patients-treated set.
Number of men: n=36 (placebo), n=38 (5 mg), n=35 (10 mg), n=51 (venlafaxine).
p<0.05, ** p<0.01, *** p<0.001 vs. placebo.
In all treatment groups, the majority of patients who had AEs, had mild or moderate AEs. The incidence of severe AEs was 4% in the placebo group, 6% in the Lu AA21004 groups, and significantly higher at 12% in the venlafaxine group (p=0.026, Fisher's exact). Severe AEs reported by at least two patients in any Lu AA21004 treatment group included: severe headache by three patients (3%) in the 10 mg Lu AA21004 group and two patients (1.9%) in the placebo group, and two patients (1.9%) in the 5 mg Lu AA21004 group had severe fatigue. In addition, severe AEs reported by at least two patients in the venlafaxine group were: severe nausea and severe vomiting, each reported by two patients (1.8%), severe insomnia in four patients (3.5%), severe dizziness in three patients (2.7%), and severe hyperhidrosis in two patients (1.8%).
For patients treated with both Lu AA21004 doses, the incidence of AEs related to sexual dysfunction (anorgasmia, delayed ejaculation, erectile dysfunction, decreased libido, impotence, abnormal organism, abnormal sexual function) was at placebo level [1.9% (5 mg) and 1.0% (10 mg) vs. 1.9% (placebo)]. In total, 23 AEs related to sexual dysfunction were reported by 18 patients, comprising seven women and 11 men. Of the women (n=267), two were in the placebo group, one from each of the Lu AA21004 groups, and three from the venlafaxine group. Of the men (n=159), all 11 were from the venlafaxine group, in which the incidence of AEs related to sexual dysfunction was statistically significantly higher than placebo (12.4% vs. 1.9%, p=0.0033, Fisher's exact test). Two patients withdrew due to AEs related to sexual dysfunction; one due to anorgasmia and 1 due to delayed ejaculation, both from the venlafaxine group.
No possibly suicide-related AEs were found in the database search during the entire study. A decrease in MADRS item 10 score (suicidal thoughts) from baseline was seen in all treatment groups at all weeks. A numerical superiority over placebo was seen in all active treatment groups from week 2 onwards.
SAEs
No deaths occurred during the study. Three patients had SAEs: two in the 10 mg Lu AA21004 group (one patient with worsening of MDD, and one patient with Varicella zoster infection) and one in the venlafaxine group (brain tumour).
Vital signs, weight, clinical laboratory values, ECGs
No consistent trends were observed for vital signs, weight, clinical laboratory values or ECG in the active treatment groups, and there were no marked differences between patients receiving active treatment and those receiving placebo. The incidence of potentially clinically significant (PCS) values was generally low and evenly distributed among the treatment groups for vital signs, weight or clinical laboratory values, and no patients withdrew due to a PCS value.
All mean vital signs were within the reference ranges and the mean changes from screening were generally small [⩽2 mmHg (supine diastolic blood pressure), ⩽5 mmHg (supine systolic blood pressure), or ⩽4 bpm (supine pulse)].
The mean weight change from baseline to week 6 was ⩽±0.3 kg in the Lu AA21004 and placebo groups and −0.8 kg in the venlafaxine group, which was not considered to be clinically relevant. Weight gain (⩾7%) was recorded for one placebo patient and three patients in the 10 mg Lu AA21004 group, whereas weight loss (⩾7%) was recorded for two patients in the 10 mg Lu AA21004 group, and one patient in the venlafaxine group. No patients withdrew due to weight change.
The mean changes in clinical laboratory values were small and similar between treatment groups and the incidence of PCS values was generally <2% in any treatment group for any laboratory test. No clinically relevant abnormalities in ECG values were found after administration of Lu AA21004.
Discussion
This is the first double-blind, randomized, placebo-controlled study to evaluate the efficacy, safety and tolerability of Lu AA21004 in patients with MDD. The active reference, venlafaxine XR (225 mg), was included with the purpose of validating the study methodology and patient population, and was effective on the primary efficacy analysis. Both doses of Lu AA21004 resulted in a significant improvement compared to placebo on the primary efficacy analysis. It has been suggested (Moncrieff & Kirsch, 2005) that the difference in total scores for an active treatment vs. placebo can be driven by a few single individual items in a rating scale. However, this is not the case in the present study, in which both doses of Lu AA21004 showed significantly greater efficacy than placebo on nine of the 10 MADRS items.
There is a large difference to placebo for all active treatment groups of about 5–6 points on the HAMD24 (which translates to ~4 points on the HAMD17), which is more than the ~2 points on the HAMD17 seen in FDA pivotal antidepressant studies (Kirsch et al. 2002). This also confirms the assay sensitivity of the studied population, who were not only severely depressed, but also had a substantial level of anxiety symptoms at baseline. At week 6, the proportion of MADRS responders (patients with ⩾50% decrease in MADRS total score) and remitters (MADRS score ⩽10) was statistically significantly greater in all active treatment groups than in placebo (LOCF and OC). The difference between active treatment and placebo of ~6 points on the MADRS translates into a clinically relevant difference in response rates of between 22% and 32% units, compared to an average of 16% units for antidepressants approved by the competent European authorities (Melander et al. 2008). The robustness of the results was also confirmed by the significantly better outcome than placebo on HAMD24, HAMA, CGI-I and CGI-S.
Several pharmacological mechanisms are likely to account for the multimodal antidepressant action of Lu AA21004. It has been estimated that an 80% occupancy of the human SERT is achieved at standard doses of SSRIs or SNRIs (Meyer, 2007). However, 5 mg Lu AA21004 occupies ~40% of SERT sites, suggesting the existence of additional mechanisms involved in its therapeutic activity. Hence, SERT blockade by SSRIs evokes a series of negative feedback mechanisms that attenuate the increase in extracellular (synaptic) concentration of 5-HT, including the activation of 5-HT1A and 5-HT1B autoreceptors on serotonergic neurons (Artigas et al. 1996, 2001). The partial agonist activity of Lu AA21004 at 5-HT1B receptors may therefore help counteract the inhibition of terminal 5-HT synthesis and release evoked by 5-HT1B receptor activation. Likewise, its full agonist activity at human 5-HT1A receptors expressed in cell lines is predicted to evoke a rapid desensitization of 5-HT1A autoreceptors (Haddjeri et al. 2009), thereby normalizing serotonergic cell firing and 5-HT release. On the other hand, given the presence of excitatory 5-HT3 receptors in GABAergic interneurons in cortical and limbic areas (Morales et al. 1996; Puig et al. 2004), their activation by 5-HT may induce a GABA-mediated inhibition of neurotransmitter release. In support of this view, the 5-HT3 receptor antagonist ondansetron augments the increase of extracellular 5-HT in the ventral hippocampus induced by the SSRI paroxetine (Mørk et al. 2009). Moreover, blockade of 5-HT7 receptors has been shown to produce rapid antidepressant-like effects in the rat in behavioural and electrophysiological experimental paradigms (Mnie-Filali et al. 2011). In addition to these effects on the 5-HT system, the systemic administration of Lu AA21004 increases the extracellular concentration of dopamine, noradrenaline and acetylcholine (Haddjeri et al. 2009), an effect probably contributing to its antidepressant activity.
Due to the profile of Lu AA21004, a hierarchical procedure was used to test for onset of action at week 1. Although not significant on the MADRS, Lu AA21004 displayed onset of antidepressant action, with significant improvement vs. placebo at week 1 onwards for both doses on HAMD24, and for the 10 mg dose on CGI-I.
The proportion of patient withdrawals has been used in recent years as an indirect index of drug effectiveness in the real world (Kahn et al. 2008; Lieberman et al. 2005; Trivedi et al. 2006). The analysis of withdrawal rates in patients treated with Lu AA21004 indicates a better tolerability profile compared to the active reference, venlafaxine. Compared to placebo, significantly more patients withdrew due to AEs only in the venlafaxine group.
The most common AEs reported in the active treatment groups were nausea, headache, hyperhidrosis, and dry mouth. No possibly suicide-related AEs were found. No consistent trends were observed for vital signs, weight, clinical laboratory values or ECG in the active treatment groups, and there were no marked differences between patients receiving active treatment and those receiving placebo.
Sexual dysfunction during antidepressant treatment is one of the main reasons for the lack of compliance (Kennedy & Rizvi, 2009). According to the present data, the incidence of spontaneously reported AEs related to sexual dysfunction was similar to placebo in patients treated with either dose of Lu AA21004. In the venlafaxine group, the incidence of AEs related to sexual dysfunction was significantly higher than that of placebo (12.4% vs. 1.9%). Unlike SSRIs, Lu AA21004 also displays moderate to high affinity for 5-HT1A, 5-HT1B, 5-HT3 and 5-HT7 receptors (see above). There is limited information on the role of these receptors on sexual drive, although the increase in plasma testosterone levels evoked in male rats by the proximity of female rats is further enhanced by the selective 5-HT3 antagonist ondansetron, which suggests that 5-HT3 receptor blockade may lead to an enhanced sexual drive (Amstislavskaya & Popova, 2004).
The generalizability of results from this study to the broad population of depressed patients, like most randomized controlled trials, is limited by the inclusion and exclusion criteria. Patients aged <18 yr or >65 yr were not included, nor were patients with specified psychiatric or medical comorbidities, or patients at risk of suicidal behaviour, nor those with treatment-resistant depression or with mild to moderate depression. The titration of venlafaxine XR, from 75 mg to 225 mg over 7 d, was according to the manufacturer's instructions. Only one patient treated with venlafaxine withdrew in the first week of treatment, indicating that there was no bias due to early withdrawals in this treatment arm.
In conclusion, treatment with 5 mg and 10 mg Lu AA21004 for 6 wk in this proof-of-concept study was well tolerated and efficacious in reducing depressive and anxious symptoms in patients with MDD.
Acknowledgements
This study was sponsored by H. Lundbeck A/S. The authors gratefully acknowledge the participation of the following investigators at the psychiatric sites in this study. Australia: Thomas George, Michael Theodoros, Graham Burrows. Austria: Siegfried Kasper, Margot Schmitz, Harald Schubert. Canada: Javed Ali, David Bakish, Martin Tremblay, Raymond Matte, Marie-Josée Filteau. Czech Republic: Petr Roček, Jiri Bilik, Jan Drahozal, Juraj Rektor, Michaela Klabusayova, Erik Herman, Zdenek Solle. Finland: Antti Ahokas, Anna Savela, Hannu Koponen, Riitta Jokinen, Ulla Lepola, Anneli Timonen, Marko Sorvaniemi, Markku Timonen. France: Joël Gailledreau, Francis Gheysen, Pierre Le Goubey, Marcel Zins-Ritter, Paule Khalifa, Christian Gaussares, Daniel Bonnaffoux. Italy: Giovanni Battista Cassano, Pietro Bria. Malaysia: Teck-Hoe Yen, Sulaiman Ahmad Hatim. Slovakia: Marek Zelman, Viera Korinkova, Eva Janikova, Peter Korcsog, Peter Molcan. Sweden: Ingemar Sjödin, Eva Scheutz, Kurt Wahlstedt, Anders Elverfors, Maj-Liz Persson, Angela Marré-Lippitz. The authors thank D. J. Simpson (H. Lundbeck A/S) for technical assistance in the preparation of the manuscript. The authors are entirely responsible for the scientific content of this article.
[Trial Registration: www.clinicaltrials.gov identifier: NCT00839423.]
Statement of Interest
Over the past 2 years, E. Álvarez has received consulting and educational honoraria from Eli Lilly, Sanofi-Aventis, Lundbeck, Pfizer; and has participated in clinical trials sponsored by Eli Lilly, Bristol–Myers, Sanofi-Aventis, Servier and Lundbeck. F. Artigas declares having received lecture fees from CSC Pharmaceuticals and Lilly on antidepressant drugs. Over the past 2 years, V. Perez has received consulting and educational honoraria from Eli Lilly, AstraZeneca, Sanofi-Aventis, Lundbeck, Pfizer; and has participated in clinical trials sponsored by Eli Lilly, Bristol-Meyers, AstraZeneca and Lundbeck. H. Loft (biostatistician) and M. Dragheim are employees of H. Lundbeck A/S, Denmark. The Editor-in-Chief of this Journal, Dr Alan Frazer, serves on an advisory board for Lundbeck that deals with the mechanism of action of Lu AA21004. Consequently, he removed himself from the review process, which was handled exclusively by an appropriate Field Editor.
References
- Amstislavskaya TG, Popova NK. The roles of different types of serotonin receptors in activation of the hypophyseal-testicular complex induced in mice by the presence of a female. Neuroscience and Behavioral Physiology. 2004;34:833–837. doi: 10.1023/b:neab.0000038136.27999.3d. [DOI] [PubMed] [Google Scholar]
- APA. Diagnostic and Statistical Manual of Mental Disorders. 4th edn. Washington, DC: American Psychiatric Association; 1994. Text Revision (DSM-IV-TR). [Google Scholar]
- Areberg J, Dragheim M, Brennum L, Stensbøl TB, San Francisco, CA, USA: 2009. ). Lu AA21004: efficacy at low serotonin transporter occupancy at clinically effective doses – from animal studies to depressed patients. Poster NR7-020 presented at the 162nd Annual Meeting of the American Psychiatric Association, 16–21 May 2009, . Accessed 9 March 2011. [Google Scholar]
- Artigas F, Celada P, Laruelle M, Adell A. How does pindolol improve antidepressant action? Trends in Pharmacological Sciences. 2001;22:224–228. doi: 10.1016/s0165-6147(00)01682-5. [DOI] [PubMed] [Google Scholar]
- Artigas F, Romero L, de Montigny C, Blier P. Acceleration of the effect of selected antidepressant drugs in major depression by 5-HT1A antagonists. Trends in Neuroscience. 1996;19:378–383. doi: 10.1016/S0166-2236(96)10037-0. [DOI] [PubMed] [Google Scholar]
- Bang-Andersen B, Ruhland T, Jørgensen M, Smith G. et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of mood and anxiety disorders. Journal of Medicinal Chemistry. 2011;54:3206–3221. doi: 10.1021/jm101459g. [DOI] [PubMed] [Google Scholar]
- Guy W. ECDEU Assessment Manual for Psychopharmacology. revised edn. Rockville, MD: National Institute of Mental Health; 1976. [Google Scholar]
- Haddjeri N, Etievant A, Moore N, Miller S. et al. Electrophysiological study of the effects of the novel antidepressant Lu AA21004 on the rat 5-HT neuronal activity. European Neuropsychopharmacology. 2009;19:S437. (Suppl. 3), [Google Scholar]
- Hamilton M. The assessment of anxiety states by rating. British Journal of Medical Psychology. 1959;32:50–55. doi: 10.1111/j.2044-8341.1959.tb00467.x. [DOI] [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICH. 1996. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073122.pdf) http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073122.pdf) ). Harmonised Tripartite Guideline E6: Guideline for Good Clinical Practice ( . Accessed 9 March 2011.
- Kahn RS, Fleischhacker WW, Boter H, Davidson M. et al. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371:1085–1097. doi: 10.1016/S0140-6736(08)60486-9. [DOI] [PubMed] [Google Scholar]
- Kennedy SH, Rizvi S. Sexual dysfunction, depression, and the impact of antidepressants. Journal of Clinical Psychopharmacology. 2009;29:157–164. doi: 10.1097/JCP.0b013e31819c76e9. [DOI] [PubMed] [Google Scholar]
- Kirsch I, Moore TJ, Scoboria A, Nicholls SS. The emperor's new drugs: an analysis of antidepressant medication data submitted to the U.S. Food and Drug Administration. Prevention and Treatment. 2002;5:1–11. , Article 23, pp. [Google Scholar]
- Laughren T. 2006. http://www.fda.gov/ohrms/dockets/ac/06/briefing/2006-4272b1-01-fda.pdf. http://www.fda.gov/ohrms/dockets/ac/06/briefing/2006-4272b1-01-fda.pdf ). Memorandum on Suicidality. ( ). Accessed 9 March 2011.
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS. et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Melander H, Salmonson T, Abadie E, van Zwieten-Boot B. A regulatory apologia – A review of placebo-controlled studies in regulatory submissions of new-generation antidepressants. European Neuropsychopharmacology. 2008;18:623–627. doi: 10.1016/j.euroneuro.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. Journal of Psychiatry and Neuroscience. 2007;32:86. [PMC free article] [PubMed] [Google Scholar]
- Mnie-Filali O, Faure C, Lambás-Señas L, Mansari ME. et al. Pharmacological blockade of 5-HT7 receptors as a putative fast acting antidepressant strategy. Neuropsychopharmacology. 2011;36:1275–1288. doi: 10.1038/npp.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncrieff J, Kirsch I. Efficacy of antidepressants in adults. British Medical Journal. 2005;331:155–157. doi: 10.1136/bmj.331.7509.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery S, Åsberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry. 1979;134:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- Moore NA, Bang-Andersen B, Brennum LT, Frederiksen K. et al. Lu AA21004: a novel potential treatment for mood disorders. European Neuropsychopharmacology. 2008;18:S321. (Suppl. 4), [Google Scholar]
- Morales M, Battenberg E, de Lecea L, Bloom FE. The type 3 serotonin receptor is expressed in a subpopulation of GABAergic neurons in the rat neocortex and hippocampus. Brain Research. 1996;731:199–202. doi: 10.1016/0006-8993(96)00557-4. [DOI] [PubMed] [Google Scholar]
- Mørk A, Brennum LT, Fallon SM, Lassen AB. et al. In vivo effects of the multi-target drug Lu AA21004: a novel potential treatment for mood disorders. Biological Psychiatry. 2009;65:73S. (8 Suppl. 1), [Google Scholar]
- Puig MV, Santana N, Celada P, Mengod G, Artigas F. In vivo excitation of GABA interneurons in the medial prefrontal cortex through 5-HT3 receptors. Cerebral Cortex. 2004;14:1365–1375. doi: 10.1093/cercor/bhh097. [DOI] [PubMed] [Google Scholar]
- Sheehan DV, Lecrubier Y, Sheenan KH, Amorim P. et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.) Journal of Clinical Psychiatry. 1998;59:22–33. (Suppl. 20), , quiz 34–57. [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA. et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. American Journal of Psychiatry. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wojtkowski T, Agyemang A, Homery M-C. et al. Effect of food on the pharmacokinetics of Lu AA21004 in healthy volunteers. Journal of Clinical Pharmacology. 2009;49:1115. [Google Scholar]
- WMA. World Medical Association; 1964. ). Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects ( . Accessed 9 March 2011. [Google Scholar]