

Abstract
The present study is the first to utilize bacterial cocaine esterase (CocE) to increase elimination of a lethal dose of cocaine and evaluate its cardioprotective effects. Rats received one of 5 treatments: CocE 1 min after saline; CocE 1 min after a lethal i.p. dose of cocaine; saline 1 min after a lethal i.p. dose of cocaine; CocE immediately after observing a cocaine-induced convulsion; and CocE 1 min after observing a cocaine-induced convulsion. Measures were taken of ECG, blood pressure, and cardiac troponin I (cTnI). The specificity of CocE against cocaine was determined by evaluating its actions against the cocaine analogue, WIN-35,065-2, which lacks an ester attack point for CocE. In addition, CocE’s effects were compared with those of midazolam, a benzodiazepine often used to manage cocaine overdose. Whereas CocE alone had negligible cardiovascular effects, it blocked or reversed cocaine-induced QRS complex widening, increased QTc interval, ST elevation, bradycardia, and hypertension. When administered 1 min after cocaine, CocE inhibited myocardial damage; however, administered 1 min after a cocaine-induced convulsion (approximately 40 s before cocaine-induced death), CocE did not block cTnI release, but did restore cardiac function. Midazolam blocked convulsions, but exhibited inadequate protection against cocaine-induced cardiotoxicity. The majority of rats given cocaine plus midazolam died. CocE did not prevent the lethal cardiovascular effects of WIN-35,065-2. In all likelihood, CocE rapidly and specifically reduced the body burden of cocaine and inhibited or reversed the cardiovascular consequences of high-dose cocaine. These results support CocE as a potential therapeutic avenue in cocaine overdose.
Keywords: Cardiovascular effect, ECG (electrocardiograph), Heart rate, Blood pressure, Cocaine toxicity, Cardiac troponin
1. Introduction
Over the past three decades, cocaine abuse has reached epidemic proportions in areas of the world. For example, increased cocaine use in the United States has resulted in a 47% increase in cocaine-related emergency room visits between 1999 and 2002 (SAMHSA, 2003). Among the numerous consequences of cocaine use, cardiovascular and cerebrovascular complications appear most life-threatening (Afonso et al., 2007; Kramer et al., 1990; Tseng et al., 1992). However, despite the fact that cocaine has been abused for more than 100 years, surprisingly little progress has been made in novel treatments of acute cocaine overdose (e.g., McCord et al., 2008).
Cocaine is a monoamine reuptake inhibitor acting at dopamine, norepinephrine, and serotonin transporters. It is absorbed quickly through mucous membranes and readily crosses the blood brain barrier, thereby rapidly increasing postsynaptic neurotransmitter levels via these different transporters. In addition, cocaine serves as a Na+ channel blocker, reducing electrical conduction in myocardial cells (Crumb and Clarkson, 1990). Current cocaine overdose treatments focus on managing the various symptoms of cocaine toxicity (McCord et al., 2008; Zimmerman, 2003). However, the pathogenesis of cocaine-mediated ischemia can be a result of increased oxygen demand, vasoconstriction, and/or enhanced platelet aggregation, making symptom-based treatment problematic. In addition to treating the cardiovascular response to cocaine overdose, seizures, metabolic acidosis, and respiratory alkalosis must also be controlled, further complicating the treatment strategy of a cocaine overdose patient.
An alternative strategy of controlling cocaine toxicity is to increase the rate of cocaine elimination. Over the past 30 years, human plasma butyrylcholinestrase (BChE) has been recognized for its ability to hydrolyze cocaine to non-toxic products (Inaba et al., 1978; Stewart et al., 1977). The most efficient mutations of BChE reverse the hemodynamic response to a non-lethal dose of cocaine (Gao and Brimijoin, 2004), and BChE has shown some promise indecreasing cocaine lethality in rats and mice (Hoffman et al., 1996). Recently, a bacterial cocaine esterase (CocE) found living in the soil surrounding the coca plant has been shown to hydrolyze cocaine with a catalytic efficiency that is nearly three orders of magnitude greater than that of endogenous esterases (Larsen et al., 2002; Turner et al., 2002). This action is highly specific: CocE does not metabolize WIN-35,065-2, a cocaine derivative that lacks cocaine’s benzoyl ester bond (Cooper et al., 2006).
Given CocE’s robust cocaine-hydrolyzing effect, it has some potential as a lead toward an antidote against cocaine-induced toxicity. Although our lab has shown that CocE protects against general cocaine toxicity and lethality in rats and mice as well as inhibiting cocaine-induced epileptogenic activity in rats (Cooper et al., 2006; Jutkiewicz et al., 2009; Ko et al., 2007), its specific ability to prevent the deleterious cardiovascular effects of cocaine toxicity has yet to be reported. The following studies utilized ECG and blood pressure telemetry in freely moving rats to determine CocE’s ability to prevent or reverse the cardiac and hemodynamic alterations induced by cocaine overdose. Furthermore, the extent to which cocaine esterase prevented myocardial injury in the presence of a lethal dose of cocaine was assessed using a biomarker of cardiac injury, cardiac troponin I (cTnI). The selectivity of CocE for cocaine was explored by evaluating its effects on cardiotoxicity produced by WIN-35,065-2. In addition, because benzodiazepine treatment has been effective clinically to reduce cocaine-induced seizures (Spivey and Euerle, 1990) and cardiotoxicity (Baumann et al., 2000), we compared the effects of midazolam pretreatment with those of CocE.
2. Methods
2.1. Drugs
(−)-Cocaine hydrochloride obtained from NIDA (Bethesda, MD, USA) was dis-solved in sterile water for injection to 180 mg/ml. A dose of 180 mg/kg cocaine given i.p., which produces 100% lethality (Cooper et al., 2006), was used in these studies. WIN-35,065-2 ((−)-2β-carbomethoxy-3β-phenyltropane was provided by Dr.F. Ivy Carroll (NIDA, Research Triangle Institute, NC, USA), dissolved in sterile water and administered at 560 mg/kg (i.p.), also the smallest dose exhibiting an LD100. CocE (supplied by Drs. D. Narasimhan and R.K. Sunahara; for a detailed description of CocE purification and synthesis see (Cooper et al., 2006)) was diluted with phosphate-buffered saline (PBS) to 10 mg/ml. A dose of 1 mg (0.1 ml) administered as a bolus through the implanted intravenous catheter, was used for all studies. Midazolam was also provided by NIDA (Bethesda, MD, USA) and diluted in saline to 1 mg/ml.
2.2. Subjects
Male Sprague-Dawley rats weighing 300–350 g (Harlan, Indianapolis, IN, USA) were singly housed following surgery and maintained in a 12-h light/dark, climate controlled room. Food and water were available ad libitum. All studies were approved by the Committee on the Use and Care for Animals at the University of Michigan Medical School and conformed to the guidelines set forth by the NIH Guide for the Use of Laboratory Animals.
2.3. Surgical procedures
Rats were anesthetized using ketamine (100 mg/kg; i.m.) and xylazine (10 mg/kg; i.m.), and all surgical conditions were aseptic.
2.3.1. Intravenous catheter surgery
All rats were implanted with an indwelling venous catheter using Micro-Renathane tubing (Braintree Scientific Inc., Braintree, MA, USA) inserted 3 cm into the jugular vein. Rats used in the cardiac troponin I studies were also implanted with a femoral vein catheter for blood sampling. Catheters were flushed daily with 0.3 ml of heparinized saline (50 U/ml).
2.3.2. Telemeter implantation surgery
The telemetric BP and ECG transmitters (Models TA11PA-C40 and TA11CTA-F40, Data Sciences, Transoma Medical, Inc., St. Paul, MN, USA) were implanted subcutaneously in the rat and secured to the abdominal wall. The catheter extending from the base of the BP transmitter was placed 3 cm into the left femoral artery. One electrode from the bottom of the ECG transmitter was sutured to the muscle above the xiphoid process and the second electrode was sutured to the right of the clavicle. ECG and BP transmitters were implanted in separate sets of rats. All rats were singly housed and allowed 6–8 days to recover before testing.
2.4. Cardiovascular data acquisition
The telemetry system consisted of battery operated ECG or pressure transmitters and receivers (Data Sciences, Transoma Medical, St. Paul, MN, USA). The ECG analysis program 4.02 from Data Sciences (St. Paul, MN, USA) was used to collect QRS, QTc (Bazette’s correction), HR, and ST elevation in 5 s intervals. Mean arterial pressure (MAP) was acquired using Dataquest A.R.T. 3.01.
2.5. Cardiac troponin I quantification. Blood (300 μl) was drawn from an indwelling femoral catheter 1, 2, and 4 h after treatment and centrifuged at 4100 r.p.m. Plasma was stored at −80 °C until it was quantified using an ELISA for cTnI (Life Diagnostics, Inc., West Chester, PA, USA).
2.5. Experimental design
2.5.1. Justification for dosages selected for cocaine, WIN-35,065-2, and cocE
Experimental justification of the doses used in this study can be obtained from a previous publication from our lab (Cooper et al., 2006). Briefly, 180 mg/kg cocaine and 560 mg/kg WIN-35,065-2 were selected for use in this study because they are the smallest doses (in quarter logarithmic increments) exhibiting an LD100. Furthermore, given the pharmacokinetic activity of cocaine in vivo (Sun et al., 2002), the peak plasma concentration of this dose would achieve toxic levels. In our previous publication, dose–effect curves were also conducted for cocE, which revealed that 1.0 mg (i.v.) was the smallest dose of cocE effectively inhibiting fatality in 100% of rats treated with a toxic dose of cocaine.
2.5.2. Cardioprotective effects of cocaine esterase: telemetry studies
To determine whether CocE prevented cocaine-induced cardiovascular alterations, we recorded ECG and MAP measurements in separate sets of animals in five randomly assigned treatment groups shown as individual conditions (at left in table under each measure) [5-7 rats per group, each used once], shown in Table 1. The first two groups served as controls. One received saline (i.p.) followed 1 min later by 1 mg CocE (i.v.; Group 1: saline + CocE); the other received cocaine (180 mg/kg i.p.) followed 1 min later by PBS vehicle (i.v.; Group 2: cocaine + PBS). The remaining three groups of rats received an injection of cocaine followed by CocE administration at one of three times: 1 min after the cocaine injection (Group 3: cocaine + CocE 1 min after cocaine); immediately after a cocaine-induced convulsion (Group 4: cocaine + CocE after convulsion); or 1 min after a cocaine-induced convulsion (Group 5: cocaine + CocE 1 min after convulsion). Electrocardiograph and blood pressure traces were recorded continuously for 2 h following cocaine injection or until the animal died. All i.v. injections were administered to freely moving rats in their home cage.
Table 1.
Cardioprotective effects of cocaine esterase; changes in QRS, QT, ST elevation, heart rate and mean arterial pressure.
| Baseline | 1 min before convulsion | 1–2min after convulsion | 4min after cocaine/saline |
20min after cocaine/saline |
|
|---|---|---|---|---|---|
| QRS duration (ms) | |||||
| Saline+CocE | 18.1 (2.4) | NA | NA | 17.5 (0.73) | 17.5 (1.2) |
| Cocaine+PBS | 17.2 (1.1) | 23.9 (2.3)*** | 26.8 (1.9)*** | NA | NA |
| Cocaine+CocE 1min after cocaine | 17.4 (1.2) | NA | NA | 18.4 (1.3) | 18.6 (1.5) |
| Cocaine+CocE after convulsion | 17.5 (1.2) | 24.9 (2.6)*** | 19.1 (2.0) | NA | 18.4 (1.3) |
| Cocaine+CocE 1min after convulsion | 18.8 (1.9) | 24.9 (2.7)*** | 19.4 (1.4) | NA | 19.1 (1.0) |
| QTc interval (ms) | |||||
| Saline+CocE | 150.8 (17) | NA | NA | 147.1 (22) | 141.5 (11) |
| Cocaine+PBS | 146 (12) | 163.1 (20) | 180.6 (24)† | NA | NA |
| Cocaine+CocE 1min after cocaine | 157.5 (12) | NA | NA | 165.5 (11) | 166.6 (15) |
| Cocaine+CocE after convulsion | 158.2 (4.8) | 170.6 (14) | 174.2 (17) | NA | 176 (15) |
| Cocaine+CocE 1min after convulsion | 157.5 (22) | 170 (4.0) | 178.7 (29) | NA | 173 (29) |
| ΔST elevation/depression (mV) | |||||
| Saline+CocE | 0 | NA | NA | (−) 0.7 (5.6) | 1.3 (15) |
| Cocaine+PBS | 0 | 61.3 (53) | (−) 193.3 (134)** | NA | NA |
| Cocaine+CocE 1min after cocaine | 0 | NA | NA | 1.8 (17) | 11.5 (8.6) |
| Cocaine+CocE after convulsion | 0 | 50.4 (28)** | 23.1 (20) | NA | 22.8 (25) |
| Cocaine+CocE 1min after convulsion | 0 | 70.2 (51) | (−) 42.5 (75) | NA | 24.8 (22) |
| Heart rate (bpm) | |||||
| Saline+CocE | 399.2 (14) | NA | NA | 418.3 (17) | 401.2 (21) |
| Cocaine+PBS | 401.5 (17) | 339.2 (11)* | 241.3 (54)*** | NA | NA |
| Cocaine+CocE 1min after cocaine | 403.4 (15) | NA | NA | 416.6 (44) | 414.2 (47) |
| Cocaine+CocE after convulsion | 405.7 (11) | 328.6 (15)** | 367.5 (62) | NA | 374.7 (39) |
| Cocaine+CocE 1min after convulsion | 403.3 (8.8) | 265.2 (26) | 313.5 (66)* | NA | 366.4 (42) |
| Mean arterial pressure (mmHg) | |||||
| Saline+CocE | 108.1 (9) | NA | NA | 105.0 (10) | 107.5 (9) |
| Cocaine+PBS | 106.4 (11) | 151.5 (14)* | 86.9 (31) | NA | NA |
| Cocaine+CocE 1min after cocaine | 111.7 (11) | NA | NA | 119.9 (16) | 118.9 (14) |
| Cocaine+CocE after convulsion | 112.6 (8) | 150.3 (20)** | 130.7 (16) | NA | 116.3 (15) |
| Cocaine+CocE 1min after convulsion | 118.0 (7) | 168.9 (14)** | 161.2 (19)** | NA | 121.8 (5) |
Data are presented as mean (±SD); all treatments groups contain n = 5–7 rats. One way ANOVA, Bonferroni post hoc results.
p < 0.05 versus baseline.
p < 0.01 versus baseline.
p < 0.001 versus baseline.
A trend towards significance (p = 0.06).
2.5.3. Selective effects of cocaine esterase: telemetry studies
The effect of CocE on WIN-35,065-2-induced cardiotoxicity was evaluated to determine the specificity of CocE’s actions. Rats received an injection of WIN-35,065-2 (560 mg/kg, i.p.), followed 1 min later by an i.v. injection of either PBS or CocE. The same ECG and MAP measurements were collected as in the cocaine studies described above.
2.5.4. Protective effects of cocaine esterase: cTnI analysis
cTnI measurement was used to determine the extent to which CocE might be able to change this marker of cocaine-induced cardiac damage. Administration regimens identical to Groups 1, 3, and 5 (described above) were used. For all groups, blood was drawn 1, 2, and 4 h after cocaine or saline injection.
2.5.5. Effect of benzodiazepine treatment against cocaine-induced cardiotoxicity
As a comparison with cocaine esterase, the protective effects of midazolam were evaluated in rats treated with a lethal dose of cocaine. Midazolam (1 mg/kg) was administered 15 min prior to 180 mg/kg cocaine. The same ECG and pressure parameters were collected as described above.
2.6. Data and statistical analysis
Baseline cardiovascular parameters were collected for 20 min prior to experimental treatment. Each parameter [QRS duration, QTc interval, ΔST elevation (change from baseline), heart rate, and MAP] was compared at baseline among treatment groups using a one-way ANOVA with Bonferroni post hoc tests. Within each treatment group, one-way ANOVAs were used to compare baseline measurements with various post-treatment time points depending on treatment regimen (see Table 1). Individual subjects given cocaine alone convulsed; they differed in the latency to convulsion and for comparison purposes, time points were set to the initiation of the convulsion for each subject. Thus, in the figures where convulsions are indicated (convulsions were 25–30 s in duration), time after and before the cocaine convulsion (or in the case of WIN 35-065, the initial convulsion) serve as loci for displaying temporal relations. In the saline + CocE (Group 1) and cocaine + CocE 1 min after cocaine (Group 3), baseline measures were compared with 4 and 20 min after saline or cocaine, respectively; in the cocaine + PBS (Group 2), baseline measures were compared with those taken 1 min before and 1 min after a cocaine-induced convulsion; in the cocaine + CocE after convulsion (Group 4), baseline measures were compared with those taken 1 min before and 1 and 20 min after cocaine-induced convulsion; and in the cocaine + CocE 1 min after convulsion (Group 5), baseline measures were compared with those obtained 1 min before, and 2 and 20 min after convulsion. Rats given cocaine in the absence of the esterase convulsed in an average of 4 min. Therefore, in Group 3, in which no convulsion occurred because of early CocE administration, we evaluated the cardiovascular parameters 4 min after cocaine administration. In addition, rats treated with cocaine + PBS convulsed 140 (±71) s before death. Therefore, in the group treated with midazolam prior to cocaine (where rats died in the absence of a convulsion), we calculated each rat’s ECG and MAP measurements 140 s prior to death. We compared these measures to baseline using a paired t-test. p-Values of <0.05 were considered significant.
A two-way ANOVA followed by Bonferroni post hoc analysis was used to identify time- and treatment-dependent differences in cTnI levels between Group 1 and Groups 3 and 5.
3. Results
3.1. Percent convulsions and survival for ECG and MAP studies
Group 1 (saline + cocE), as expected, exhibited no convulsions and all survived, whereas all members of Group 2 (cocaine + PBS) convulsed and none survived. Time to death was 333 s (±35SE). CocE administered 1 min after cocaine (Group 3) completely blocked cocaine-induced convulsions, and all rats survived. CocE administered immediately following cocaine-induced convulsions (Group 4) also inhibited the lethal effects of cocaine (100% survival). CocE given 1 min after the end of the convulsion (approximately 80 s prior to death, Group 5) saved 80% of rats.
All rats treated with WIN-35-065-2, followed by either PBS or CocE exhibited convulsions, and none survived. Time to death was 563 s (±134SE) for WIN-35-065-2, and 541 s (±26SE) for WIN-35-065-2 and CocE. Midazolam blocked completely cocaine-induced convulsions; however only 20% of the rats in this group survived.
3.2. CocE’s protective effect against cocaine-induced bradycardia
CocE alone had no effect on HR (F(2,13) = 1.9; p = 0.2), QTc interval (F(2,12) = 0.3; p = 0.8), QRS duration (F(2,20) = 0.4; p = 0.7) or ST elevation (F(2,13) = 0.06; p = 0.95) (n = 6). It did exhibit a robust ability to inhibit the cardiotoxic effects of cocaine (see Table 1 and Figs. 1 and 2). Cocaine in the absence of the esterase produced significant bradycardia following the initial tachycardic response to the injection (Fig. 1a and Table 1; F(2,16) = 29; p < 0.001), such that 1 min prior to convulsions, rats exhibited a reduced HR (p < 0.05; n = 6). Bradycardia was exacerbated by the cocaine-induced convulsion (p < 0.001), and HR continued decreasing until death, determined by respiratory cessation and lack of heart beat. CocE inhibited or reversed cocaine-induced HR changes when administered either 1 min after cocaine (Table 1 and Fig. 1b; F(2,16) = 0.17; p = 0.8; n = 6), or immediately following the convulsion (Table 1 and Fig. 1c, F(3,26) = 4.4; p = 0.013; n = 7), respectively. When CocE was administered 1 min after the convulsion (convulsions were 25–35 s in duration), HR returned to resting rates more slowly (F(3,21) = 9.8; p = 0.0005; n = 6); 2 min after convulsion (~1 min post-CocE), HR remained reduced (p < 0.01), returning to baseline approximately 4 min after convulsion (3 min post-CocE; Fig. 1d).
Fig. 1.

Heart rate and QTc interval following cocaine: cardioprotective effects of CocE. Cocaine (180 mg/kg, i.p.) produced robust reductions in heart rate (a) and QTc interval elongation (e) as compared with baseline (B). CocE (1 mg, i.v.) was able to block or largely reverse these effects when administered 1 min after cocaine (b, f), immediately following convulsion (c, g) or 1 min after convulsion (d, h). Asterisk indicates time of death for each rat in the group (if applicable).
Fig. 2.

QRS duration and ST elevation/depression following cocaine: cardioprotective effects of CocE. Cocaine (180 mg/kg, i.p.) + PBS produced QRS widening (a) and ST elevation prior to cocaine-induce convulsion, followed by transient ST depression after convulsion (e) as compared with baseline (B). CocE (1 mg, i.v.) blocked or largely reversed the effects on QRS duration and ST segment when administered 1 min after cocaine (b, f), immediately following cocaine-induced convulsion (c, g) or 1 min after convulsion (d, h). Asterisk indicates time of death for each rat in the group (if applicable).
3.3. CocE’s protective effect against cocaine-induced QTc interval prolongation
The QTc interval of the ECG was also affected by cocaine administration (Table 1 and Fig. 1e-h); QTc interval prolongation occurred 1 min before (trend) and after (p < 0.05) cocaine-induced convulsions (F(2,15) = 3.8; p = 0.05). In addition, the QTc interval shortened rapidly 30–60 s prior to death (Fig. 1e). CocE, given either immediately after cocaine (Table 1 and Fig. 1f; F(3,22) = 1.4; p = 0.27) or immediately after a cocaine-induced convulsion (Fig. 1 g; F(3,19) = 0.6; p = 0.6), prevented the development of QTc prolongation. When CocE was given 1 min after a cocaine-induced convulsion, it prevented the development of an exaggerated QTc prolongation (F(3,19) = 0.6; p = 0.6); nevertheless, under these conditions, a non-significant lengthening of the QTc interval was observed 2 min after cocaine-induced convulsions (Table 1 and Fig. 1h).
3.4. CocE’s protective effect against cocaine-induced QRS widening
QRS widening was observed following cocaine + PBS (Table 1, F(2,15) = 30; p < 0.0001) both 1 min before and after cocaine-induced convulsion (p < 0.001) and continued to widen until death (Fig. 2a). This effect was completely blocked in rats that were given CocE 1 min after cocaine (Table 1 and Fig. 2b; F(2,16) = 0.8; p = 0.5). CocE also reversed the QRS widening when administered immediately (Table 1 and Fig. 2c; F(3,26) = 22; p < 0.0001) or 1 min (Table 1 and Fig. 2d; F(3,22) = 15; p < 0.0001) after the cocaine-induced convulsion.
3.5. CocE’s protective effect against cocaine-induced ST elevation
ST elevation was observed prior to cocaine-induced convulsion (Table 1 and Fig. 2e; F(2,17) = 16.5; p = 0.0002). After the convulsion, and 1-2 min prior to death, ST depression was evident, most likely as a result of the escalating ischemia. Minimal ST elevation was observed when CocE was administered 1 min after cocaine (Table 1 and Fig. 2f; F(2,17) = 1.8; p = 0.2). CocE also inhibited the transient ST depression observed after convulsions when it was administered immediately following the convulsion (Table 1 and Fig. 2 g; F(3,24) = 5.5; p = 0.006). Alternatively, when cocE was administered 1 min after the cocaine-induced convulsion, ST depression was observed post-convulsion (Table 1 and Fig. 2h; F(3,22) = 5.7; p = 0.006). However, 2 min after the convulsion, ST depression was nearly 41/2 fold less severe than when cocaine was administered in the absence of cocE (Table 1, cocaine + PBS).
Representative ECG traces are depicted in Fig. 3. The time frames selected for each treatment regimen display CocE’s cardioprotective effect compared with high-dose cocaine in the absence of CocE.
Fig. 3.

Representative ECG traces: cardioprotective effects of CocE. Cocaine (180 mg/kg, i.p.) produced cardiac electrical abnormalities. When CocE (1 mg, i.v.) was administered either 1 min after cocaine, immediately or 1 min after convulsions (conv), this electrical instability was alleviated. Each ECG trace represents 100 ms. Calibration bar at the top of the figure denotes 0.5 mV.
3.6. CocE’s effect on WIN-35,065-2-induced ECG changes
Similar to toxic doses of cocaine, WIN-35,065-2 (n = 5) produced bradycardia (Fig. 4a; F(2,13) = 6.8; p = 0.02), QTc interval prolongation (Fig. 4b; F(2,14) = 5.5; p = 0.02), QRS widening (Fig. 4c; F(2,14) = 11.4; p = 0.002) and slight ST elevation followed by depression (Fig. 4d; F(2,15) = 0.9; p = 0.4). Interestingly, WIN-35,065-2’s cardiovascular effects (except for QTc interval prolongation) were not present until the drug-induced convulsion. Although time to convulsion (average ± SD) did not differ between cocaine (198 ± 93 s) and WIN-35,065-2 (184 ± 64 s), WIN-35,065-2 administration was accompanied by multiple convulsions prior to death compared to a single cocaine-induced convulsion. Surprisingly, CocE administration 1 min after WIN-35,065-2 (n = 6) increased the time to convulsion as compared with PBS administration (281 ± 71 s vs. 184 ± 64 s respectively; t(10) = 2.5; p = 0.03). Nevertheless, WIN-35,065-2-induced bradycardia (Fig. 4e; F(2,16) = 3.9; p = 0.046), QTc interval prolongation (Fig. 4f; F(2,14) = 7.7; p = 0.006), QRS widening (Fig. 4g; F(2,16) = 6.7; p = 0.009), and slight ST depression (Fig. 4h; F(2,17) = 1.5; p = 0.25), were unchanged by CocE administration.
Fig. 4.

Cardiotoxic effects of WIN-35,065-2: insensitivity to CocE. 560 mg/kg WIN-35,065-2 (i.p.) was administered 1 min prior to intravenous PBS (a-d) or CocE (e and f). WIN-35,065-2 administration (+PBS) elicited bradycardia (a), elongated QTc interval (b), QRS widening (c) and ST depression (d) compared with baseline (B). Although CocE (1 mg i.v.) administered 1 min after WIN-35,065-2 delayed the first convulsion compared with WIN + PBS, cardiac electrical abnormalities were unaffected by the esterase treatment (e-h). Asterisk indicates time of death for each rat in the group.
3.7. CocE’s effect on cocaine- and WIN-35,065-2-induced hypertension
Cocaine + PBS (n = 6) produced a robust pressor response within 1 min of i.p. administration (Fig. 5a and Table 1; F(2,14) = 12.9; p = 0.001). Mean arterial pressure (MAP) peaked immediately before a convulsion (p < 0.05). Following cocaine-induced convulsions, respiratory depression was evident, bradycardia worsened, and MAP decreased precipitously until death. CocE alone had no effect on MAP (see Table 1), and when CocE was administered 1 min after cocaine (n = 6), no appreciable cocaine-induced MAP changes were observed (Table 1 and Fig. 5b; F(2,17) = 0.6; p = 0.5). CocE given immediately following convulsions (n = 5) reversed cocaine-induced hypertension (p < 0.05) within 2 min (1 min after CocE infusion) (Table 1 and Fig. 5c; F(3,19) = 6.3; p = 0.005). Rats given CocE 1 min after convulsion (n = 6) developed cocaine-induced hypertension (p < 0.01) which did not quite return to baseline within 2 min (1 min post-CocE). However, 3 min after esterase administration, MAP had returned to resting levels (Table 1 and Fig. 5d; F(3,12) = 15.9; p = 0.0006). WIN-35,065-2 also produced a pressor response (Fig. 6a; F(2,16) = 57; p < 0.0001, n = 6), and CocE was ineffective in blocking this hemodynamic response (Fig. 6b; n = 5, F(2,14) = 26; p < 0.0001).
Fig. 5.

Hemodynamic instability following cocaine: efficacy of the esterase antidote. 180 mg/kg cocaine (i.p.) + PBS (i.v.) produced a robust pressor response, followed by rapid hypotension after the convulsion (a) as compared with baseline (B). CocE given 1 min after cocaine completely blocked cocaine’s pressor response (b). CocE administered immediately following cocaine-induced convulsion (c) or 1 min after convulsion (d) rapidly reversed cocaine’s pressor response. Asterisk indicates time of death for each rat in the group (if applicable).
Fig. 6.

Hemodynamic instability following WIN-35,065-2: insensitivity to CocE. 560 mg/kg WIN (i.p.) + PBS (i.v.) treatment resulted in a pressor response prior to the first convulsion (a) compared with baseline (B). CocE administration 1 min after WIN delayed the first convulsion; however it did not inhibit WIN’s pressor response (b). Asterisk indicates time of death for each rat in the group.
3.8. Effect of benzodiazepine treatment on the cardiotoxic effects of cocaine (data not shown)
Although midazolam demonstrated some protection against the toxic effects of cocaine, 80% of rats died, regardless of inhibiting the convulsion. These rats exhibited duration of time to lethality comparable to rats treated with cocaine + PBS; midazolam + cocaine produced death within 466 (±128) s in ECG implanted rats and 440 (±106) s for rats implanted with pressure-transmitters (t(8) = 2.0; p = 0.08 and t(10) = 0.9; p = 0.4, respectively vs. cocaine + PBS).
Cardiovascular alterations following midazolam + cocaine were similar to cocaine + PBS treatment. The average time from convulsion to death in cocaine + PBS rats was 140 71 s. Therefore base-line cardiovascular measures in rats given midazolam + cocaine were compared with measures collected 140 s prior to cocaine-induced lethality, the average time at which, in the absence of mida-zolam, the rat would convulse. QRS duration increased 140 s prior to death (17.5 (±1.3) ms vs. 24.9 (±1.6) ms; t(4) = 6.7; p = 0.003), similar to cocaine treatment alone. Additionally, midazolam-treated rats were susceptible to cocaine-induced bradycardia (401 (±31) bpm vs. 289 (±66) bpm; t(4) = 5.6; p = 0.005), and hypertension (118.5 (±6) mmHg vs. 140 (±6) mmHg; t(5) = 7.6; p = 0.0006). ST elevation was observed almost immediately following cocaine injection and was sustained until 178 ± 22 s before death when ST depression ensued. To identify ST segment alterations (change from baseline) that occurred 1 min before and after the time at which a cocaine-treated rat would normally convulse, baseline measures were compared with measures taken 240 and 30 s prior to cocaine-induced death (F(2,14) = 26.2; p < 0.0001); ST elevation occurred at the earlier time point whereas ST depression ensued 30 s prior to death (117 ± 85 and −387 ± 180 mV, respectively). In contrast to cocaine-treated rats, midazolam pretreated rats did not exhibit significant QTc prolongation 240 s prior to cocaine-induced death compared to post-midazolam baseline (163 (±12) ms vs. 168 (±20) ms; t(4) = 0.15; p = 0.89).
3.9. Ability of CocE to inhibit myocardial damage (cTnI)
Cocaine injection followed 1 min later by CocE inhibited cTnI release similar to saline injection (Fig. 7), further indicating CocE’s potential cardioprotective capability. Alternatively, when CocE was administered 1 min after cocaine-induced convulsions (approximately 40 s prior to when death would have occurred), increases in cTnI levels were observed 1 and 2 h after cocaine injection (F(2,30) = 12.5; p = 0.0001), and returned towards normal at 4 h.
Fig. 7.
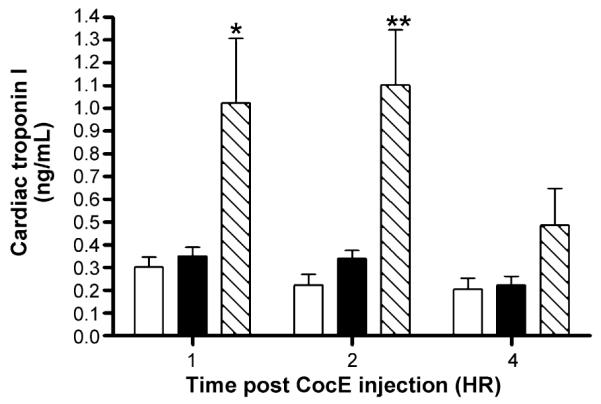
Biochemical analysis of myocardial injury: cardiac troponin I. CocE (1 mg; i.v.) inhibited the release of cTnI (ng/ml) when administered 1 min after 180 mg/kg cocaine (closed bars) and therefore exhibited cTnI levels 1, 2, and 4 h after treatment similar to that of saline (i.p.) + CocE (open bars). When CocE treatment was delayed until 1 min after cocaine-induced convulsion (hatched bars), cTnI release was evident 1 and 2 h after treatment, returning to control levels at 4 h. *p < 0.05, **p < 0.01 vs. saline + CocE treatment group.
4. Discussion
This study is the first to report the promising protective cardiovascular effects of cocaine esterase treatment using a rat model of cocaine toxicity. These experiments indicate that increasing the rate of cocaine degradation by administering a cocaine esterase intravenously has potential as a lead to a therapeutic antidote for cocaine overdose. Previous HPLC-MS studies from our lab revealed that cocE (an equimolar concentration compared to the in vivo dose used in the current studies) added to human plasma spiked with 300 μM cocaine (a dose exceeding toxic levels in both rats and humans) decreases the cocaine concentration to 2 μM within 1 min (Cooper et al., 2006). In the current studies, not only did CocE prevent death in 100% of rats treated 1 min after a lethal injection of cocaine or immediately following cocaine-induced convulsions, similar to previous reports (Cooper et al., 2006), it rapidly returned cocaine-induced electrocardiographic alterations towards baseline. Furthermore, administration of CocE 1 min following the cessation of cocaine-induced convulsions rescued 80% of rats from the lethal effects of cocaine overdose, and returned ECG parameters towards normal within 2-4 min. Importantly, cocE was also benign when administered alone at active anti-cocaine dose levels.
Large doses of cocaine depress ventricular function, inhibit repolarization, and slow electrical conduction in the heart via its Na+ channel blocking properties (Kerns et al., 1997; Magnano et al., 2006; Perera et al., 1997). These effects were observed here following a lethal dose of cocaine as indicated by QRS widening, QTc interval prolongation and bradycardia, respectively. In addition, cocaine produced marked hypertension. These changes suggest that, whereas electrical conduction was hampered in the heart, coronary vasoconstriction, and hypertension (both documented responses to cocaine (Foltin et al., 2003; Isner and Chokshi, 1991)) reduced myocardial oxygen supply and increased oxygen demand, respectively. Rats also exhibited an electrical abnormality, ST elevation, prior to convulsion, followed by ST depression emerging 1-3 min prior to cocaine-induced death. Rapid and robust inhibition or reversal of these potentially devastating cardiovascular consequences of cocaine toxicity was achieved following cocaine esterase infusion in a time-dependent manner. CocE administered 1 min after a lethal dose of cocaine blocked the development of cocaine-induced QRS widening, QTc interval prolongation, and bradycardia. In addition, CocE inhibited the development of the pressor response to cocaine. In addition, a major cardiovascular consequence that occurs in a small portion of human cocaine toxicity cases is aortic dissection (Hsue et al., 2002). Since cocE only halts the further progression of cocaine-induced cardiovascular dysfunction by reducing plasma cocaine concentrations to non-toxic levels, careful examination of the conditions for producing cocaine-induced dissection and the course of treatment by CocE under these circumstances will be necessary.
Currently, the only studies attempting to accelerate cocaine degradation as a means to alleviate cocaine-induced cardiovascular alterations have utilized natural or mutant versions of butyrlcholinesterase (BChE), the enzyme that degrades cocaine in mammals. A large dose of BChE did not elicit as much cardio pro-tective effect following a sub-lethal dose of cocaine as we observed with CocE after toxic cocaine doses.
In addition to blocking the development of cocaine-induced cardiovascular effects, CocE also reversed the cocaine-induced electrical alterations and pressor response. As a result, when CocE was administered approximately 40 s before death (1 min after convulsion), 80% of rats survived an otherwise lethal dose of cocaine. Although cardiac electrical activity returned towards normal shortly after CocE administration and was maintained throughout the 2 h of data collection, it will be necessary to gain a better understanding of the potential pathological electrical disturbances that may occur when the esterase treatment is delayed until seconds before cocaine-induced death. This study was designed to identify the acute cardioprotective effects of cocE in the face of cocaine toxicity, and since cocaine was most likely still on board for the duration of ECG recordings, this study cannot identify the extent of enduring cardiac electrical damage when esterase treatment was delayed.
It should also be noted that these studies utilized cocaine-naïve rats and therefore no preexisting cardiac damage was present. Future studies should identify whether cocE exhibits similar life-saving capacity during cocaine toxicity when the cardiovascular system has been damaged by prior cocaine abuse. Furthermore, the esterase has been shown to increase anti-CocE antibody titers following repeated administration, thereby decreasing the effectiveness of CocE (Ko et al., 2007). The immunogenic properties of bacterial CocE should be considered when determining CocE’s therapeutic value. CocE also has a short half-life (~13 min), so in its current form it is not a practical prophylactic treatment for cocaine dependency; however, strategic amino acid mutations renders esterases that are longer acting. Despite the need for additional studies, these results strongly support its potential as a lead for cocaine toxicity in emergency settings.
Currently, a number of therapeutics are recommended to manage the cardiovascular symptoms of cocaine-associated myocardial ischemia. For example, similar to benzodiazepines, nitroglycerin has also been shown to reverse cocaine-induced vasoconstriction and relieve cocaine-associated chest pain (Baumann et al., 2000; Brogan et al., 1991). [Phentolamine, an α-adrenergic antagonist, and calcium channel blockers have been reported to relieve cocaine-induced coronary constriction, and thus decrease ischemia. In addition, cocaine increases platelet aggregation, and as a result anti-platelet and anti-thrombin agents are also recommended.] Although these therapeutics and others are no doubt useful in symptom management, cocaine esterase is able to rapidly reduce or reverse all cocaine-associated cardiovascular alterations. Therefore, CocE may represent a viable first treatment for cocaine-related emergency patients, removing the source of the cardiovascular complications, in lieu of, or as a compliment to treating the symptoms.
In this study we also compared the cardioprotective effects of the benzodiazepine, midazolam, to CocE in the presence of a lethal dose of cocaine. Although midazolam pretreatment did inhibit the development of convulsions in all rats treated, it resulted in only 20% survival, as compared with 100% survival after CocE administration. Additionally, the remaining 80% of rats exhibited electrocardiographic and hemodynamic alterations similar to rats given cocaine + PBS. Therefore, inhibition of cocaine-induced convulsions was not sufficient to elicit cardioprotective effects comparable to those provided by CocE in our model. This suggests that cardiovascular effects of cocaine are a major contributor, even in the absence of convulsions, to lethality in this model.
In addition to cardiac electrophysiologic properties indicating cardiovascular dysfunction, cTnI plasma measurement is a reliable biomarker of myocardial injury (Fishbein et al., 2003; Ricchiuti et al., 1998) and a useful tool to identify irreversible myocardial damage in patients with cocaine-associated chest pain (Kontos et al., 2002). Although we confirmed that CocE blocked or largely reversed ST elevation/depression, an indicator of cocaine-induced transient cardiac electrical dysfunction, we sought to evaluate whether this was sufficient to abolish the development of more long-term myocardial injury. Despite CocE’s ability to prevent death when the enzyme was administered 1 min after cocaine-induced convulsions, it did not inhibit the release of cTnI, an excellent marker of cocaine-induced myocardial damage. Because seizures alone do not increase cTnI levels (Woodruff et al., 2003), these results indicate that cocaine-induced cardiac damage occur when there was a delay (in this case to 40 s before death) in CocE treatment following convulsions. Despite the observed cTnI release, electrical activity returned towards baseline within minutes, raising the question of the nature of the cocaine-induced myocardial damage. However, it should be noted that although there were no significant differences in ECG parameters 20 min after cocaine in all cocE-treated groups compared with saline + cocE, QTc prolongation and ST elevation are still evident at this time (Table 1), and may indicate enduring electrical changes. Further long term and histological studies of the cardiovascular effects of cocaine in the presence of delayed CocE treatment are necessary to determine the details of this potential cardiac injury.
The specificity of the action of CocE against cocaine was challenged using WIN-35,065-2, a cocaine derivative lacking the benzoyl ester bond present in cocaine, and the cleavage site for CocE. As expected, CocE did not protect against WIN-35,065-2-induced convulsions or lethality. In addition, CocE did not alter the cardiotoxic effects of WIN-35,065-2, further indicating the high level of specificity exhibited by CocE.
These studies are the first to report the capability of a bacterial cocaine esterase to prevent or protect against cocaine-associated cardiac death. Overall, these studies implicate bacterial cocaine esterase as being a highly efficient potential therapeutic for cocaine toxicity. Due to its ability to rapidly and specifically degrade cocaine (Cooper et al., 2006), CocE effectively inhibited or reversed the cardiovascular consequences of cocaine toxicity and lethality. It also inhibited myocardial damage when administered soon after cocaine administration. These results support the possible use of this bacterial esterase for the emergency management of cocaine overdose.
Acknowledgements
The authors would like to thank Yong-Gong Shi and Davina Barron for their technical support in this project. We also would like to thank Dr. B. Lucchesi for editorial advice on the manuscript.
Role of funding source
Funding for this study was provided by USPHS Grant DA 012416. The USPHS had no further role in the study design, collection of data, writing of the manuscript, or in the decision to submit this manuscript for publication.
Footnotes
Contributors
Susan K. Wood participated in creating the study design, collected and analyzed the data, and wrote the first draft of the manuscript. Diwa Narasimhan and Roger K. Sunahara purified and synthesized the cocaine esterase used in this study. Ziva Cooper contributed to creating the study design and conducted the initial dose-effect studies that identified the doses used in this study. James H. Woods obtained funding for the study and played an integral role in the development and evaluation of the project. All authors contributed to and have approved the final manuscript.
Conflict of interest
All authors declare that they have no conflict of interest.
References
- Afonso L, Mohammad T, Thatai D. Crack whips the heart: a review of the cardiovascular toxicity of cocaine. Am. J. Cardiol. 2007;100:1040–1043. doi: 10.1016/j.amjcard.2007.04.049. [DOI] [PubMed] [Google Scholar]
- Baumann BM, Perrone J, Hornig SE, Shofer FS, Hollander JE. Randomized, double-blind, placebo-controlled trial of diazepam, nitroglycerin, or both for treatment of patients with potential cocaine-associated acute coronary syndromes. Acad. Emerg. Med. 2000;7:878–885. doi: 10.1111/j.1553-2712.2000.tb02065.x. [DOI] [PubMed] [Google Scholar]
- Brogan WC, 3rd, Lange RA, Kim AS, Moliterno DJ, Hillis LD. Alleviation of cocaine-induced coronary vasoconstriction by nitroglycerin. J. Am. Coll. Cardiol. 1991;18:581–586. doi: 10.1016/0735-1097(91)90617-i. [DOI] [PubMed] [Google Scholar]
- Cooper ZD, Narasimhan D, Sunahara RK, Mierzejewski P, Jutkiewicz EM, Larsen NA, Wilson IA, Landry DW, Woods JH. Rapid and robust protection against cocaine-induced lethality in rats by the bacterial cocaine esterase. Mol. Pharmacol. 2006;70:1885–1891. doi: 10.1124/mol.106.025999. [DOI] [PubMed] [Google Scholar]
- Crumb WJ, Jr., Clarkson CW. Characterization of cocaine-induced block of cardiac sodium channels. Biophys. J. 1990;57:589–599. doi: 10.1016/S0006-3495(90)82574-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein MC, Wang T, Matijasevic M, Hong L, Apple FS. Myocardial tissue troponins T and I. An immunohistochemical study in experimental models of myocardial ischemia. Cardiovasc. Pathol. 2003;12:65–71. doi: 10.1016/s1054-8807(02)00188-6. [DOI] [PubMed] [Google Scholar]
- Foltin RW, Ward AS, Haney M, Hart CL, Collins ED. The effects of escalating doses of smoked cocaine in humans. Drug Alcohol Depend. 2003;70:149–157. doi: 10.1016/s0376-8716(02)00343-5. [DOI] [PubMed] [Google Scholar]
- Gao Y, Brimijoin S. An engineered cocaine hydrolase blunts and reverses cardiovascular responses to cocaine in rats. J. Pharmacol. Exp. Ther. 2004;310:1046–1052. doi: 10.1124/jpet.104.068122. [DOI] [PubMed] [Google Scholar]
- Hoffman RS, Morasco R, Goldfrank LR. Administration of purified human plasma cholinesterase protects against cocaine toxicity in mice. J. Toxicol. Clin. Toxicol. 1996;34:259–266. doi: 10.3109/15563659609013786. [DOI] [PubMed] [Google Scholar]
- Hsue PY, Salinas CL, Bolger AF, Benowitz NL, Waters DD. Acute aortic dissection related to crack cocaine. Circulation. 2002 Apr;105(13):1592–1595. doi: 10.1161/01.cir.0000012524.44897.3a. [DOI] [PubMed] [Google Scholar]
- Inaba T, Stewart DJ, Kalow W. Metabolism of cocaine in man. Clin. Pharmacol. Ther. 1978;23:547–552. doi: 10.1002/cpt1978235547. [DOI] [PubMed] [Google Scholar]
- Isner JM, Chokshi SK. Cardiovascular complications of cocaine. Curr. Probl. Cardiol. 1991;16:89–123. doi: 10.1016/0146-2806(91)90013-z. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM, Baladi MG, Cooper ZD, Narasimhan D, Sunahara RK, Woods JH. A bacterial cocaine esterase protects against cocaine-induced epilep-togenic activity and lethality. Ann. Emerg. Med. 2009;54(3):409–420. doi: 10.1016/j.annemergmed.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns W, 2nd, Garvey L, Owens J. Cocaine-induced wide complex dysrhythmia. J. Emerg. Med. 1997;15:321–329. doi: 10.1016/s0736-4679(97)00003-6. [DOI] [PubMed] [Google Scholar]
- Ko MC, Bowen LD, Narasimhan D, Berlin AA, Lukacs NW, Sunahara RK, Cooper ZD, Woods JH. Cocaine esterase: interactions with cocaine and immune responses in mice. J. Pharmacol. Exp. Ther. 2007;320:926–933. doi: 10.1124/jpet.106.114223. [DOI] [PubMed] [Google Scholar]
- Kontos MC, Anderson FP, Ornato JP, Tatum JL, Jesse RL. Utility of troponin I in patients with cocaine-associated chest pain. Acad. Emerg. Med. 2002;9:1007–1013. doi: 10.1111/j.1553-2712.2002.tb02134.x. [DOI] [PubMed] [Google Scholar]
- Kramer LD, Locke GE, Ogunyemi A, Nelson L. Cocaine-related seizures in adults. Am. J. Drug Alcohol Abuse. 1990;16:307–317. [PubMed] [Google Scholar]
- Larsen NA, Turner JM, Stevens J, Rosser SJ, Basran A, Lerner RA, Bruce NC, Wilson IA. Crystal structure of a bacterial cocaine esterase. Nat. Struct. Biol. 2002;9:17–21. doi: 10.1038/nsb742. [DOI] [PubMed] [Google Scholar]
- Magnano AR, Talathoti NB, Hallur R, Jurus DT, Dizon J, Holleran S, Bloomfield DM, Collins E, Garan H. Effect of acute cocaine administration on the QTc interval of habitual users. Am. J. Cardiol. 2006;97:1244–1246. doi: 10.1016/j.amjcard.2005.11.046. [DOI] [PubMed] [Google Scholar]
- McCord J, Jneid H, Hollander JE, de Lemos JA, Cercek B, Hsue P, Gibler WB, Ohman EM, Drew B, Philippides G, Newby LK. Management of cocaine-associated chest pain and myocardial infarction. A scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897–1907. doi: 10.1161/CIRCULATIONAHA.107.188950. [DOI] [PubMed] [Google Scholar]
- Perera R, Kraebber A, Schwartz MJ. Prolonged QT interval and cocaine use. J. Electrocardiol. 1997;30:337–339. doi: 10.1016/s0022-0736(97)80047-7. [DOI] [PubMed] [Google Scholar]
- Ricchiuti V, Sharkey SW, Murakami MM, Voss EM, Apple FS. Cardiac troponin I and T alterations in dog hearts with myocardial infarction: correlation with infarct size. Am. J. Clin. Pathol. 1998;110:241–247. doi: 10.1093/ajcp/110.2.241. [DOI] [PubMed] [Google Scholar]
- SAMHSA . Emergency Department Trends from the Drug Abuse Warning Network, Final Estimates 1995-2002. DAWN Series: D-24, DHHS Publication No. (SMA) 03-3780. US Department of Health and Human Services; Rockville, MD: 2003. Substance Abuse and Mental Health Services Administration, Office of Applied Studies. [Google Scholar]
- Spivey WH, Euerle B. Neurologic complications of cocaine abuse. Ann. Emerg. Med. 1990;19:1422–1428. doi: 10.1016/s0196-0644(05)82612-5. [DOI] [PubMed] [Google Scholar]
- Stewart DJ, Inaba T, Tang BK, Kalow W. Hydrolysis of cocaine in human plasma by cholinesterase. Life Sci. 1977;20:1557–1563. doi: 10.1016/0024-3205(77)90448-9. [DOI] [PubMed] [Google Scholar]
- Sun H, Shen ML, Pang YP, Lockridge O, Brimijoin S. Cocaine metabolism accelerated by a re-engineered human butyrylcholinesterase. J. Pharmacol. Exp. Ther. 2002;302:710–716. doi: 10.1124/jpet.302.2.710. [DOI] [PubMed] [Google Scholar]
- Tseng CC, Derlet RW, Albertson TE. Cocaine-induced respiratory depression and seizures are synergistic mechanisms of cocaine-induced death in rats. Ann. Emerg. Med. 1992;21:486–493. doi: 10.1016/s0196-0644(05)82511-9. [DOI] [PubMed] [Google Scholar]
- Turner JM, Larsen NA, Basran A, Barbas CF, 3rd, Bruce NC, Wilson IA, Lerner RA. Biochemical characterization and structural analysis of a highly proficient cocaine esterase. Biochemistry. 2002;41:12297–12307. doi: 10.1021/bi026131p. [DOI] [PubMed] [Google Scholar]
- Woodruff BK, Britton JW, Tigaran S, Cascino GD, Burritt MF, McConnell JP, Ravkilde J, Molgaard H, Andreasen F, Dam M, Jaffe AS. Cardiac troponin levels following monitored epileptic seizures. Neurology. 2003;60:1690–1692. doi: 10.1212/01.wnl.0000065881.46964.4f. [DOI] [PubMed] [Google Scholar]
- Zimmerman JL. Poisonings and overdoses in the intensive care unit: general and specific management issues. Crit. Care Med. 2003;31:2794–2801. doi: 10.1097/01.CCM.0000100123.50896.F0. [DOI] [PubMed] [Google Scholar]