

Abstract
Human apolipoprotein L1 (ApoL1) possesses both extra- and intra-cellular functions crucial in host defense and cellular homeostatic mechanisms. Alterations in ApoL1 function due to genetic, environmental, and lifestyle factors have been associated with African sleeping sickness, atherosclerosis, lipid disorders, obesity, schizophrenia, cancer, and chronic kidney disease (CKD). Importantly, two alleles of APOL1 carrying three coding-sequence variants have been linked to CKD, particularly in Sub-Saharan Africans and African Americans. Intracellularly, elevated ApoL1 can induce autophagy and autophagy-associated cell death, which may be critical in the maintenance of cellular homeostasis in the kidney. Similarly, ApoL1 may protect kidney cells against renal cell carcinoma (RCC). We summarize the role of ApoL1 in RCC and CKD, highlighting the critical function of ApoL1 in autophagy.
Keywords: Apolipoprotein L (ApoL), ApoL1, ApoL6, ApoA1, Autophagy, Apoptosis, ESKD, FSGS, Genetics, GSGS, HIVAN, Kidney transplantation
Since the identification and characterization of human apolipoprotein L1 (ApoL1) as an interacting protein of apolipoprotein A1 (ApoA1) and a minor component of high density lipoprotein (HDL) particles in 1997 [1], the human ApoL protein family was thought to be predominantly involved in lipid transport and metabolism [1–6]. In fact, several lines of evidence indicate that ApoLs are associated with hyperlipidemia, obesity and atherosclerosis [2, 4–6]. However, recent studies have identified novel roles for ApoL proteins in host innate immunity [7–13], apoptosis [14,15] and autophagy [16,17], which can be attributed, in part, to the structural similarities between ApoLs and bacterial colicins [10,18], other human apolipoproteins, and Bcl-2 family proteins [14,17–19]. The human APOL family consists of six genes localized in chromosome 22q12.3 to 13.1 [3, 18–21], a genomic hotspot for a number of diseases, including schizophrenia [22–28], prostate cancer [29,30], and liver and breast cancer [31,32]. More recently, two alleles of APOL1 harboring three coding-sequence variants have been discovered in kidney disease patients of African ancestry and linked to the pathogenesis of non diabetic chronic kidney disease (CKD), including idiopathic focal and segmental glomerulosclerosis (FSGS), hypertension-attributed kidney disease, and HIV-1-associated nephropathy (HIVAN), under a recessive inheritance pattern [33–39]. The expression of APOL1 has been detected in renal proximal tubular epithelial cells [19], podocytes and pre-glomerular vascular structures [40], suggesting that ApoL1 could play a role in the pathogenesis of renal diseases that affects the structure and/or function of these cells. Moreover, these APOL1 variants have been considered potentially risk factors for predicting the histopathology and progression of HIVAN [41] as well as kidney transplant rejection [42,43]. Similarly, given the results of previous studies showing a potential role of ApoL1 in tumorigenesis, and its expression in renal proximal tubular cells, we speculate that these APOL1 variants, by affecting the process of macroautophagy (hereafter referred to as autophagy), may also play a role in the pathogenesis of renal cell carcinoma (RCC). For all the above reasons, this review will summarize and discuss the role of ApoL1 in RCC and CKD, highlighting the critical function of ApoL1 in autophagy.
ApoL1 in autophagy and autophagy-associated cell death
We have recently shown that ApoL1 is a novel BH3-only protein and, when overexpressed intracellularly, induces autophagy and autophagy-associated cell death in a variety of cancer cell types [16,17]. However, the intracellular role of ApoL1 in vivo has not been investigated. Interestingly, lack of functional autophagy has been shown to correlate with the outcome of various diseases, including cancer, cardiomyopathies, inflammatory bowel disease, obesity, neurodegenerative diseases, and CKD [44–48]. Autophagy is a conserved, lysosomal-dependent mechanism that involves the sequestration and degradation of intracellular constituents. In this process, a fraction of cytosolic proteins and organelles is first enwrapped within a double-membrane organelle, referred to as the autophagosome. Subsequently, the autophagosome fuses with endosomes and lysosomes to form two other types of autophagic vesicles, amphisomes and autolysosomes in mammalian cells. This process results in the degradation of the sequestered materials by lysosomal enzymes in the autolysosome (Figure 1) [46]. Autophagy is a catabolic active process that generates amino acids, fatty acids, glucose and nucleotides, which can be recycled to produce energy and building blocks for the survival of cells [47]. In this manner, low physiological levels of autophagy is necessary to preserve the homeostasis and function of cells, and facilitate the turnover of proteins, organelles and lipid droplets. In addition, autophagy can also function as a survival mechanism during times of acute or chronic stress through the elimination and recycling of damaged/aggregated proteins and organelles. It has been demonstrated in mammalian cells that the induction of autophagy, for example in response to starvation, is mediated in part via inactivation of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1), by p53 and/or other factors [48–50]. Interestingly, two of the p53 downstream targets, Bnip3 and DRAM, have been shown to directly regulate autophagy (Figure 1). Upon autophagy induction, mTORC1 is inactivated leading to the activation of ULK1 complex (including ULK1, Atg13, FIP200, and Atg101), that in turn activates the class III phosphatidylinositol 3-kinase complex, including Vps34, Vps15, Beclin 1, Barkor/Atg14L, and Ambra1 [46,47]. Beclin 1, the mammalian homologue of yeast Atg6, is a BH3-only protein required for autophagy and is regulated by Bcl-2 family members and other interacting proteins [51,52]. The elongation of phagophore membranes into autophagosomes is regulated by three ubiquitination-like reactions. First, the ubiquitin-like molecule Atg12 is conjugated to Atg5 by Atg7, which acts like an E1 ubiquitin-activating enzyme, and by Atg10, which is similar to an E2 ubiquitin-conjugating enzyme. The Atg5-Atg12 complex then interacts non-covalently with Atg16L1 and this resulting the Atg12-Atg5-Atg16L1 complex associates with phagophores but dissociates from mature autophagosomes [53]. The second ubiquitin-like reaction involves the conjugation of mammalian LC3-II, GABARAP and GATE-16, homologues of yeast Atg8 [54], to phosphatidylethanolamine (PE) by Atg7 and Atg3 (another E2-like), resulting in autophagosome-associated LC3-II. The Atg12-Atg5 complex may be able to enhance LC3-II conjugation to PE by acting in an E3-like fashion. In this way, the Atg12-Atg5-Atg16L1 complex may determine the sites of autophagosome synthesis by regulating the targeting of LC3-II to Atg12-Atg5-associated membranes. Interestingly, LC3-II remains associated with autophagosomes until after their fusion with lysosomes. The LC3-II inside the autolysosomes is degraded, while the LC3-II on the cytoplasmic surface can be delipidated and recycled. Thus, Atg12-Atg5-Atg16L1-positive and LC3-positive structures can be considered to be phagophores and Atg12-Atg5-Atg16L1-negative and LC3-positive vesicles can be regarded as mature autophagosomes [55,56]. P62, also known as sequestosome 1/SQSTM1, is an ubiquitously expressed protein and one of the best characterized substrates of selective autophagy. p62 has been shown to directly interacts with LC3 on the phagophore through the LC3-interacting region. Subsequently, p62 is incorporated into the autophagosome and then degraded (Figure 1). Impairment of autophagy is accompanied by accumulation of p62. This leads to the formation of large aggregates, which include p62 and ubiquitin. P62 also has been implicated in the recognition process of autophagy-mediated degradation of damaged Mitochondria, known as mitophagy. However, the precise mechanism is still under investigation [46, 57]. Notably, ApoL1, which its expression is inducible by p53, is found highly expressed in cells undergoing elevated levels of autophagy including renal tubular epithelial cell and pancreatic exocrine granular cells ([19] and The Human Protein Atlas websitehttp://www.proteinatlas.org/ENSG00000100342). Moreover, previous studies using immunoblot analysis have shown a time-dependent accumulation of LC3-II, and an increase in the LC3-II to LC3-I ratio in cells overexpressing ApoL1. These findings suggest that overexpression of ApoL1 induces autophagy, which is partially inhibited in murine Atg5−/− and Atg7−/− embryonic fibroblast cells. In addition, 3-methyladenine and wortmannin, two inhibitors of PI3KCIII and thus autophagy, also can reduce ApoL1-induced cell death [16]. Thus, one mechanism of ApoL1-induced autophagy may involve, directly or indirectly, activation of the translocation of LC3-II to the autophagosomes (Figure 1). Interestingly, we recently observed that ApoL1-EGFP fusion protein was localized in the cytosol and lysosomes (Figure 2A), suggesting that ApoL1-regulated autophagy, for survival or for death, may involve lysosomes. Nevertheless, as autophagy is a bona fide stress responsive mechanism and usually activated during cell death progression, the role of autophagy in autophagy-associated cell death is still under intensive investigation and there are numerous lines of evidence support either cells died with autophagy [58–62] or died of autophagy [62–66].
Figure 1.

The role and subcellular localization of ApoL1 in autophagy in human cells. A wealth of literature has shown that expression of APOL1 can be induced by the signaling pathways of IFN-γ, TNF-α and p53. Intracellularly, ApoL1 is lysosomal and cytosolic (see Figure 2A), and can induce (macro)autophagy and autophagy-associated cell death when overexpressed. Interestingly, another lysosomal protein, DRAM, also a p53 downstream target, can induce autophagy and apoptosis. Autophagy, a lysosome-dependent, self-eating, and catabolic mechanism, can be initiated by various stresses (for example, starvation, inflammatory cytokines, and pathogens). Upon autophagy induction, mTORC1 is inactivated leading to the activation of ULK1 complex (including ULK1, Atg13, FIP200, and Atg101), that in turn activates the class III phosphatidylinositol 3-kinase complex, including at least Vps34, Vps15, and Beclin 1. The elongation of phagophore membranes into autophagosomes is regulated by two ubiquitination-like reactions. First, the ubiquitin-like molecule Atg12 is conjugated to Atg5 by Atg7, which acts like an E1 ubiquitin-activating enzyme, and by Atg10, which is similar to an E2 ubiquitin-conjugating enzyme. The Atg5-Atg12 complex then interacts non-covalently with Atg16L1 and this resulting the Atg12-Atg5-Atg16L1 complex associates with phagophores but dissociates from mature autophagosomes. The second ubiquitin-like reaction involves the conjugation of mammalian LC3-II, a homologue of yeast Atg8, to phosphatidylethanolamine (PE) by Atg7 and Atg3 (another E2-like), resulting in autophagosome-associated LC3-II. The Atg12-Atg5 complex may be able to enhance LC3-II conjugation to PE by acting in an E3-like fashion. In this way, the Atg12-Atg5-Atg16L1 complex may determine the sites of autophagosome synthesis by regulating the targeting of LC3-II to Atg12-Atg5-associated membranes. Interestingly, LC3-II remains associated with autophagosomes until after their fusion with lysosomes. The LC3-II inside the autolysosomes is degraded, while the LC3-II on the cytoplasmic surface can be delipidated and recycled. Subsequently, the autophagosome fuses with endosomes and lysosomes to form two other types of autophagic vesicles, amphisomes and autolysosomes in human cells. This process results in the degradation of the sequestered materials by lysosomal hydrolases in the autolysosome. P62, an ubiquitously expressed protein, is one of the best characterized substrates of selective autophagy and mitophagy. p62 has been shown to bind polyubiquinated proteins and altered mitochondria and directly interact with LC3II on the phagophore through the LC3-interacting region. Subsequently, p62-cargo complex is incorporated into the autophagosome and then degraded. Mitophagy has been shown to be regulated by Bnip3, a BH3-only protein and a p53 downstream target. Extracellularly, ApoL1 has been shown to be an interacting protein of ApoA1 in lipoprotein complexes.
Figure 2.
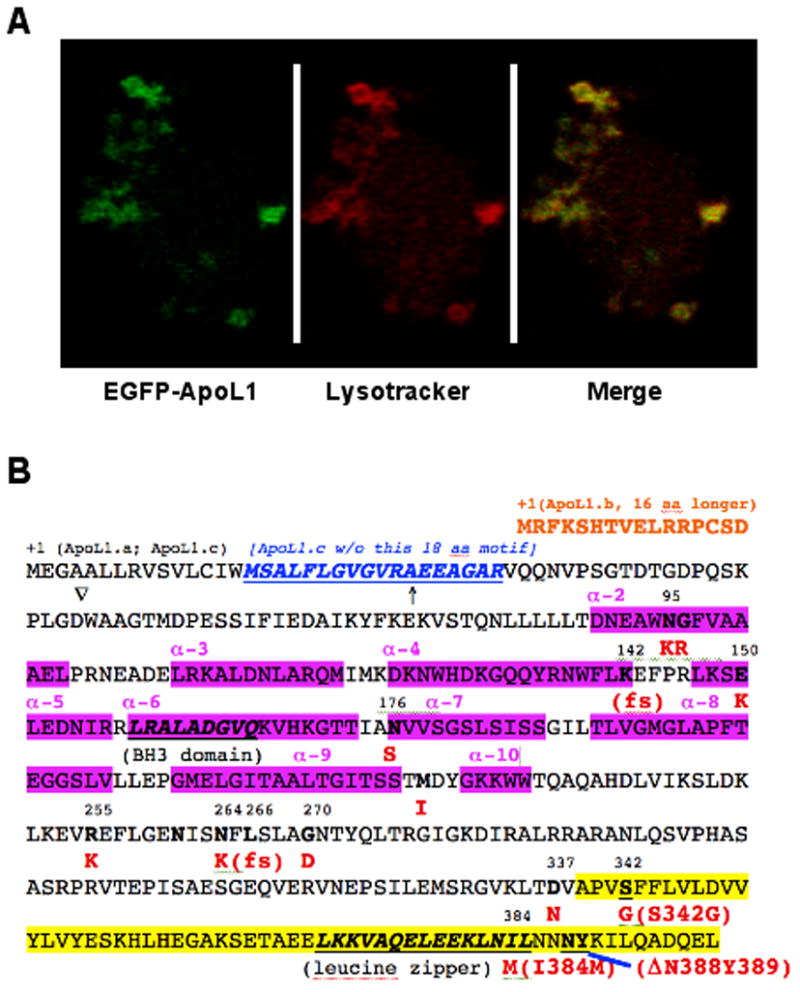
(A). Human ApoL1 localizes in the cytosol and lysosomes. Confocal microscopy analysis showed that EGFP-ApoL1 fusion protein (in Green) and the lysotracker (in Red) were co-localized (in yellow) almost perfectly in human fibroblast cells, suggesting that human ApoL1 is mainly lysosomal. (B). Non-synonymous SNPs, domains and motifs of human ApoL1 isoforms. Three ApoL1 isoforms can be generated by alternative splicing. ApoL1.a, a 398 amino-acid (aa) polypeptide, is shown in its complete sequence. ApoL1.b is 16 aa N-ter longer than ApoL1.a (Shown in orange), while ApoL1.c is missing a 18 aa motif (shown in blue and underlined) close to the N-ter of ApoL1.a. Interestingly, fourteen naturally occurring, non-synonymous variant/alleles of APOL1 have been identified thus far, based on the residue numbers of ApoL1.a, N95K, G96R, K142fs, E150K, N176S, M218I, R255K, N264K, L266fs, G270D, D337N, S342G, I384M and DN388Y389 (amino acid changes are in red). The BH3 domain (codons 158-166) and the leucine zipper domain (codons 365-392) are in bold and underlined. The two risk alleles of APOL1 in ESKD in African Americans, S342G/I384M and DN388Y389, are in the C-ter domain (highlighted in yellow; codons 342 to 398), which interacts with trypanosomal SRA (see text). Interestingly, I384M mutation is present in the leucine zipper domain, which may affect binding of ApoL1 with other proteins in human cells. K142fs and L266fs alleles were identified in an Indian patient whose serum lacked the TLF activity [101].↑, indicates the putative signal peptide, an N-ter 27 aa leader sequence of ApoL1.a); ∇, putative proteolytic cleavage site (between D and W) to generate 39 kDa form of ApoL1. N261, a potential N-glycosylation site. The alpha helixes (α-2 to 10) in the protein are also indicated (in pink).
ApoL1, autophagy, and cancer
As stated above, intracellular accumulation of ApoL1 has been linked to the processes of autophagy and cell death in the context of cancer. Others have shown that genetic, biochemical or pharmacological inhibition of autophagy results in increased free radicals, aggregated macromolecules, elevated DNA damage and metabolic deficiencies that lead to global cellular damage and apoptosis [67,68]. For all these reasons, alterations in proteins that impair the process of apoptosis and autophagy have been linked to the pathogenesis of different types of cancers. It is worth noting however, that the process of cell death in mammalian cells is often preceded or accompanied by autophagic vacuolization [58–62]. This finding, raises the controversial issue of whether autophagy per se, directly induces cell death, or alternatively, it is just an epiphenomenon seen in cells under stress that are destined to die. This notion is supported by the results of mouse studies, in which knockouts of essential autophagy genes showed that several chemotherapy drugs prevented the growth of tumor cells without affecting the process of “autophagic cell death” [61]. Nevertheless, even, assuming the autophagy may be a secondary phenomenon seen in cells subjected to severe stress destined to die, many studies have suggested that it can play a role in the pathogenesis of several human cancers, including glioblastomas, hepatocellular carcinomas, tumors associated with tuberous sclerosis, and pancreatic cancer [58,60,62]. For example, pancreatic cancers have been shown to depend on autophagy for survival. More specifically, pancreatic ductal adenocarcinoma cells (PDAC) exhibit constitutive upregulated autophagy under basal conditions, most likely due to the hypoxic environment present within tumors [69]. In turn, acquired alterations in autophagy may allow PDAC cells to survive under high stress conditions. Further, inhibition or targeted regulation of autophagy within these cells may provide a therapeutic treatment for cancers that require elevated autophagic activity. It is worth noting that mice with systemic mosaic deletion of Atg5 and liver-specific Atg7−/− developed benign liver adenomas [70]. These tumor cells originated autophagy-deficient liver cells and showed mitochondrial swelling, p62 accumulation, oxidative stress and DNA damage responses. The size of the Atg7−/− liver tumors is reduced by simultaneous deletion of p62. Together, these results suggest that autophagy is important for the suppression of spontaneous tumorigenesis through a cell-intrinsic mechanism in the liver, and that p62 accumulation contributes to tumor progression [70]. In contrast, the role of autophagy in kidney pathology was recognized just recently. In 2007, Chien and colleagues showed the induction of Beclin 1 and LC3 during kidney ischemia-reperfusion in rats, and both autophagy and apoptosis were suppressed by the expression of Bcl-XL and Bcl-2 [63]. Suzuki and colleagues further demonstrated the formation of autophagosome-like structures in renal tubular cells during ischemia-reperfusion in mice and, notably, in transplanted kidneys in humans [64]. In cultured tubular cells, autophagy inhibitors could suppress H2O2-induced cell death, suggesting that autophagy might contribute to cell death under the condition [64]. Very recently, Gozuacik and colleagues showed autophagy in renal tubular cells following tunicamycin-induced ER stress in mice, and autophagy may serve as a second cell-killing mechanism, or type II programmed cell death, that acts in concert with apoptosis during ER stress [65]. Taken together, it is evident that either too much or too little autophagic activity can affect the regulation of cell growth in a significant manner.
ApoL1, autophagy and renal cell carcinoma (RCC)
RCC is among the ten leading causes of cancer related deaths, and accounts for 2% of all cancers [71–73]. RCC occurs in a sporadic and a hereditary form, and both forms are frequently associated with structural alterations of the short arm of chromosome 3 (i.e., 3p). Genetic studies of the families at high risk for developing RCC led to the cloning of genes whose alteration results in tumor formation. These genes are either tumor suppressors (e.g., VHL, TSC) or oncogenes (e.g., MET, RAS). RCC originates in proximal renal tubular epithelial cells, which have, among other functions, an important role in the absorption and transport of water, glucose, salt, and several nutrients [74,75]. These processes require a significant amount of energy and therefore proximal tubules have a well developed lysosomal system to participate in the reabsorption and degradation of albumin and other low molecular weight plasma proteins that are filtered by the kidney. In addition, rapidly proliferating cells consume energy and require an active lysosomal system to degrade and recycle proteins, lipids and amino acids that are needed for the process of cell growth [47]. For all these reasons, it is tempting to speculate that autophagy may play a critical role modulating the growth, regeneration, and survival of proximal tubular cells. We have found high levels of ApoL1 protein expressed in renal proximal tubular epithelial cells [19], and these findings also suggest that ApoL1 may play an active role in these processes. In support of this notion, autophagy has been shown to be renal protective during acute ischemic injury [76–78] and cisplatin-induced kidney injury in mice [79–81]. More specifically, these studies showed that cisplatin can induce the formation of autophagic vesicles in renal proximal tubular epithelial cells. These changes appear to be mediated by p53 and modulated by anti-death proteins, like Bcl-2 (Figure 1) [80,81]. Thus, given the fact that renal proximal tubular epithelial cells show significant p53-associated autophagic activity under stress and express considerable amounts of ApoL1 [19], it is worth discussing the potential interactions that p53, ApoL1, and autophagy may have in the pathogenesis of RCC.
RCC usually maintains a wild type p53 tumor suppressor gene, however, the function of p53 could be modulated by other mechanisms including alterations in the RAS, PI3K/AKT/mTOR and/or NF-κB pathways in RCC [71,73,82]. In addition, it is well known that activation of p53 by various stresses can induce autophagy in cancer cells [49,83,84]. Interestingly, we observed a time-dependent expression of APOL1 in a number of cell lines overexpressing wildtype nuclear p53. Thus, one pathway by which p53 induces autophagy could be through the intracellular accumulation of ApoL1 that leads to activation and translocation of LC3-II and/or changing the homeostasis of phospholipids [17]. In addition, inhibitors of PI3KCIII and therefore autophagy also reduced ApoL1-induced cell death. Furthermore, we recently observed downregulated expression of ApoL1 in RCC tissues and cell lines (Hu et al., unpublished observations). These findings suggest that ApoL1 could be an important signaling protein that is targeted to elimination by RCC cells for their survival advantage. It should be noted, however, that mice with Atg5 targeted deletion in the proximal tubules did not develop kidney tumors but exhibited accumulation of misfolded proteins and deformed organelles, and susceptible to ischemia-reperfusion injury [76]. These mice only showed hypertrophic changes in renal proximal tubular cells, suggesting that, unlike the liver-specific autophagy-deficient mice that develop liver tumors, suppression of autophagy in kidney proximal tubular epithelial cells do not cause kidney tumors. These findings are consistent with the notion that other alterations in tumor suppressor genes and/or oncogenes are required for the development of RCC. Unfortunately, since rodents do not carry APOL1 gene, it is difficult to study the potential interactions between autophagy and ApoL1 in the pathogenesis of renal tumors or RCC in mice.
ApoL1 and CKD
Chronic kidney disease (CKD) encompasses a group of renal lesions caused by a variety of etiological factors that destroy the renal parenchyma and decrease the renal function. For example, the term Focal and Segmental Glomerulosclerosis (FSGS), which defines a renal glomerular lesion associated with CKD, represents a common and final injury pathway, used by different primary or secondary renal diseases, hypertension, toxins, drugs, and viral infections [85]. The National Kidney Foundation classifies CKD in five stages based on the extent of renal damage and pathological events including glomerular filtration rate, renal excretory function, plasma creatinine, proteinuria levels, and percent of functionally active nephrons [86]. Although diabetes and hypertension are the leading causes of stage 5 kidney disease, also referred to as End Stage Kidney Disease (ESKD), many other environmental, genetic, and lifestyle factors have been associated with CKD.
The kidney filtration barrier comprises a complex framework of renal structures that include glomerular endothelial cells, extracellular matrix proteins that constitute the basement membrane, and podocytes. Podocytes are terminally differentiated cells surrounding the glomerular capillaries. The long processes or “foot projections” of the podocytes wrap around capillaries interdigitate with one another forming filtration slits that are highly permeable to water, small solutes and ions, but restrict the passage of albumin, large molecules, and cells into the glomerular filtrate [86]. In addition, injured mesangial cells, which are specialized contractile smooth muscle cells that provide structural support to renal glomeruli, can decrease the glomerular filtration rate (GFR). In CKD, the kidney’s ability to maintain the GFR and other renal functions is impaired, preventing the clearance of waste products and toxic metabolites from the blood and urinary filtrate. Ultimately, these changes further compromise the structure of renal glomeruli and tubules and affect the homeostasis of the whole body [86]. In essence, CKD can be the result of many pathological insults and processes that affect different glomerular and tubulo-interstitial cells. In recent years podocyte injury has been considered a major culprit for the development of FSGS. Moreover, the chronic loss of podocytes in the urine is considered an important factor leading to FSGS and CKD. Inflammation, stress-induced damage, changes in lipid profiles, viral infections, hypertension, and alterations of survival and autophagic pathways have also been shown to contribute to the pathogenesis of glomerular and tubulointerstitial injury leading to CKD [85,86]. Interestingly, recent studies have suggested several mechanisms by which reduced autophagy can affect the function of podocytes, proximal renal tubular epithelial cells, and the pathophysiology of different renal diseases [62,79]. In addition, APOL1 is detected in normal podocytes, and its expression appears to be reduced in patients with FSGS and HIVAN [40]. Taken together, these findings suggest that the APOL1 risk variants may modulate the outcome of these kidney diseases by affecting the function of podocytes. For example, mice with podocyte-specific deletion of Atg5 showed proteinuria and a progressive decline in renal function [87]. After 20 months, mice displayed significant Global and Segmental Glomerulosclerosis (GSGS) or FSGS caused by puromycin aminonucleoside and adriamycin, which normally upregulated autophagic activity in intact podocytes [87]. As podocytes are the primary target for toxic aggregates and oxidative stress, it could be hypothesized that accumulation protein aggregates and damaged organelles in autophagy-deficient podocytes, may overwhelm the intracellular degradation pathway of ubiquitinated proteins and induce CKD. Interestingly, 22-month-old Atg5−/− mice presented a significant reduction in the number of podocytes in non-sclerosed glomeruli, suggesting that podocyte loss precedes the development of FSGS [87]. In addition, studies have found increased levels of Atg3 mRNA in podocytes from patients with FSGS relative to control renal samples [88]. Moreover, genetic alterations in several proteins expressed by podocytes, including nephrin, podocin, and CD2, are also associated with the development of Glomerulosclerosis [89]. Since podocytes appear to require and exhibit high levels of autophagy [90], they may rely on this mechanism to fight stressful situations. Consequently, autophagic balance of podocytes appears to be essential mechanism to preserve the renal function, and loss of autophagic capabilities may be an important contributing factor for the development of ESKD.
Genomics, molecular genetics, and protein isoforms of human APOL1
It has been well known for years that various kidney diseases that cause FSGS are clustered among populations of African descent. Recent linkage and mapping genetic analyses have demonstrated that two alleles containing variants in the last exon of the APOL1 gene appear to explain the striking racial disparities in the frequency of idiopathic FSGS, hypertension-attributed ESKD, and HIVAN between African Americans and European Americans [91]. These studies have localized a region in chromosome 22q12-13 that harbors the MYH9 and APOL1 genes, which possess risk variants associated with FSGS, ESKD, and HIVAN in people of African ancestry [33–43]. As stated above, ApoL1 is essential for its primate-specific role in autophagy, the N-terminal (N-ter) and C-terminal (C-ter) contain domains/motifs vital to its localization, translocation and immunity effector functions. In fact, there are three protein isoforms, ApoL1.a, ApoL1.b and ApoL1.c, encoded by four putative human APOL1 mRNA variants [19]. ApoL1.a, a 398 amino-acid (aa) polypeptide, is the most abundant and relatively well-studied protein. Resulting from alternatively splicing, ApoL1.b, 414 aa, is 16 aa N-ter longer than ApoL1.a, while ApoL1.c, 380 aa, is missing a 18 aa motif close to the N-ter of ApoL1.a due to alternative splicing (Figure 2B). Using ApoL1.a as an example, besides the BH3 domain (codons 158-166), there is a leucine zipper domain (LZD; codons 365-392) in ApoL1. The N-ter of ApoL1 isoforms consist of an anionic pore-forming domain thought to be involved in anion channel formation and cell death in trypanosomes. Adjacent to the pore-forming domain is the membrane–addressing domain, which consists of pH-sensitive α–helices and facilitates the association with HDL particles at a neutral pH or intracellular regions at an acidic pH. Moreover, C-ter of ApoL1 possesses an amphipathic α-helix containing the LZD embedded in the serum resistance-associated protein (SRA) interacting domain (see below). Mounting evidence has suggested a vital role for the LZD in protein-protein interaction [92,93]. Interestingly, it has been shown that the LZD may also bind lipid species and DNA to promote signal transduction. For example, Yamamoto and colleagues have revealed that the LZD of ApoE3 confers stability to the N-ter domain in an inactive state. When the LZD interacted with surface binding sites of lipoprotein complexes, it resulted in the anchoring of ApoE3 to the lipid particle surface, which facilitates a conformational change of the N-ter helices [93]. In turn, it can be speculated that binding of the LZD of ApoL1 to other components of lipoprotein particles and/or membrane receptors promotes structural changes of ApoL1, allowing for the functional properties of the N-ter and the BH3 domains of ApoL1.
With regard to the link of ApoL1 and CKD, there are two haplotypes of APOL1, harboring three coding sequence mutations, have been identified as risk variants in this region. The first one, termed G1, is a two non-synonymous SNP (NSNP) haplotype (rs73885319 (A→G; S342G) and rs60910145 (G→T; I384M). The second one, termed G2, is a two codon deletion haplotype rs71785313 (6-bp in frame deletion; ΔN388Y389). Interestingly, the frequency of the APOL1 G1 haplotype distributed in African populations is consistent with the pattern of African ancestry risk of developing ESKD [34]. In fact, based on odds ratios and p values APOL1 risk variants are more strongly associated with ESKD than the leading MYH9 risk variants. Aside from the 5 NSNPs mentioned above, other naturally occurring, non-synonymous variant/alleles of APOL1 have been identified. According to the residue numbers of ApoL1.a, they are N95K, G96R, E150K, N176S, M218I, R255K, N264K, G270D, and D337N, suggesting that ApoL1 is very polymorphic (Figure 2B). It is logical to speculate that protein products of some of these NSNPs may have altered activity, stability or subcellular localization of ApoL1.
It is worthy noting that extracellular ApoL1 is a crucial component of human trypanolytic factors (TLF) that provide resistance against trypanosomes, including Trypanosoma brucei brucei. There are a number of excellent papers already documenting this particular important area of research [7–13, 94–100]. While two types of TLF, TLF1 and TLF2, have been identified thus far, TLF1 is relatively well studied, shown to be resided in a minor subclass of serum HDL, which contains two primate specific proteins, ApoL1 and haptoglobin-related protein (Hpr), as well as ApoAI. These protein complexes provide an important defense mechanism against the flagellated parasitic protozoa trypanosome. ApoL1 alone has been shown to be sufficient to confer trypanolytic activity in vitro and in vivo [94–97]. Regarding TLF1-resistant trypanosomes, they produce the SRA, which binds specifically to ApoL1 of TLF1. Microscopic analysis demonstrated that ApoL1 uptaken by trypanosomes is targeted to the lysosome, leading to the formation of anion channels, triggering the depolarization of the lysosomal membrane, and causing an influx of chloride that leads to the osmotic swelling of the lysosome until the parasite dies [9,10,13]. Formation of the SRA-ApoL1 complex prevents killing and allows parasite growth in humans. It has been shown that SRA directly interacts with the C-ter α-helical region of ApoL1 (Figure 2B). We have reported that APOL1 expression can be induced by circulating inflammatory cytokines, including interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), further supporting the role of ApoL1 in extracellular innate immunity [16,17,19]. Importantly, variants of G1 and G2 alleles are located in the last exon resulting in codon changes in both SRA-binding and the LZD in the C-ter of ApoL1 isoforms. Consequently, protein products of the G1 and G2 alleles can effectively lyze serum-resistant Trypanosoma brucei rhodesiense, and the protein product of the G2 allele lose binding affinity with SRA, suggesting a positive selection and inheritance of both alleles in populations that once encountered trypanosomal infection threat [68, 70]. These two variants in APOL1 may be beneficial to individuals living in African regions with T.b. rhodesiense and, possibly, T.b. gambiense, enabling them to resist African sleeping sickness. Consistent with this hypothesis, a case report documented an Indian patient whose serum lacked functional ApoL1 and lost trypanolytic activity. The patient was infected by a serum-sensitive T. evansi, a strain that would normally not infect humans [101]. Mutation analysis showed that the patient had two frameshift mutations, K142fs and L266fs, resulting in loss of detectable ApoL1 protein in the serum (Figure 2B). Thus, with apparent autosomal recessive inheritance, one copy of an APOL1 G1 or G2 risk variant provides protection from African sleeping sickness, whereas possession of two APOL1 risk variants, leads to FSGS, HIVAN and hypertensive nephrosclerosis.
ApoL1 in hypertensive nephrosclerosis, FSGS, HIVAN, and kidney transplantation
As discussed above, a spectrum of three renal diseases, hypertensive nephrosclerosis (arteriolar nephrosclerosis and hypertension-attributed ESKD), FSGS, and HIVAN have been linked to the presence of the APOL1 risk alleles. A remarkable observation is that the link between nephrosclerosis and hypertension in African American patients (labeled hypertensive nephrosclerosis), could be now explained, at least in part, by the APOL1 risk variants [38]. Nephrosclerosis is the result of scarring of the renal parenchyma, and is associated with pathological changes occurring in the preglomerular microvasculature and the secondary involvement of glomeruli and interstitium. These patients typically present with advanced disease and it is difficult to determine the original process that initiated the renal disease. Therefore, nephrologists usually assume that hypertension is responsible for the development of nephrosclerosis in approximately 25% of the patients with ESKD. Recent studies have documented the expression of ApoL1 protein in preglomerular vessels, a key renal structure involved in the pathogenesis of hypertension-attributed nephrosclerosis Moreover, the family clustering of ESKD attributed to hypertension in African Americans can now be explained by the APOL1 risk alleles. Therefore, the APOL1 genetic variation is associated with more than a seven-fold increase in risk for hypertension-attributed ESKD [102]. These findings also suggest that mild to moderate elevations in blood pressure per se, might not be the primary cause responsible for the development of nephrosclerosis.
HIVAN is a renal disease characterized by severe proteinuria associated with FSGS, frequently presenting the collapsing phenotype, and microcystic tubular dilatation leading to renal enlargement and rapid progression to ESKD [103,104]. At the cellular level, HIVAN is characterized by the loss, hypertrophy, and proliferation of glomerular and tubular epithelial cells. Histological studies have shown HIVAN to be the predominant lesion in the 2.3 million HIV-1 infected patients living in Sub-Saharan Africa. Interestingly, the United States Renal Disease System reported that HIV-1 infected African Americans have a 50 times higher risk of developing ESKD than HIV-1-infected whites. HIVAN is considered the most prominent form of nondiabetic kidney disease. Interestingly, the G1 and G2 haplotypes of APOL1 have also been identified as risk variants for HIVAN [39,41,103,104]. Changes in the differentiation and proliferation status of glomerular podocytes, parietal epithelial cells, and tubular epithelial cells are known to play a key role in the pathogenesis of HIVAN. As stated above, podocytes require high levels of autophagic homeostasis for cellular integrity and survival [90], and autophagy-deficient podocytes exhibited glomerulopathy in aging mice, suggesting that autophagy is a major protective means to maintain functional podocytes and therefore kidney. It is conceivable that if ApoL1 plays an important role in regulating autophagy in kidney cells, both gain-of-function and loss-of-function mutations of APOL1 may disrupt ApoL1-dependent cellular homeostasis, leading to apoptosis and necrosis. Furthermore, patients possessing two APOL1 risk alleles have the highest risk for developing ESKD when infected with HIV-1 [37,39]. Up to date, however, the exact mechanisms by which HIV-1 induces glomerular epithelial and tubular injury are not clearly understood. We speculate that cytokines released by HIV-1-infected cells may play a critical role in this process as well. Certain cytokines released by HIV-infected cells can upregulate the expression of ApoL1 in renal epithelial cells and induce apoptotic and/or proliferative changes in these cells. Overall, these changes may impair autophagy and disrupt the balance of intracellular protein degradation and recycling, increasing the susceptibility of these cells to detach, de-differentiate, and/or proliferate. Thus, elucidation of the dynamic signaling pathways and ApoL1 interactome may prove useful in designing specific therapeutic strategies for many different kidney diseases, and increasing of ApoL1 autophagic signals may provide a treatment for RCC.
Finally, coding-sequence variants in the APOL1 gene appear to predict the outcome of kidney transplants. A recent study found that kidneys from African American deceased donors harboring two APOL1 risk variants failed more rapidly after renal transplantation than those with zero or one risk variant [43]. These studies, however, has several weaknesses including the small sample size, lack of genotyping of APOL1 in the recipients, and few post-implantation biopsies. Therefore, although these findings need to be replicated in a larger number of patients, APOL1 genotyping could improve the donor selection process and the long term outcome of kidney allograft survival [42].
Summary and future perspectives
ApoL1 and ApoL6 are crucial regulators of cellular homeostasis, death and survival, and disruption of functions of these proteins may present significant clinical complications from cancer to kidney failure. ApoL1, when overexpressed intracellularly, induces autophagy and autophagy-associated cell death in all cell types examined thus far including those originated from kidney normal and cancerous tissues. In contrast, cells stably transfected with ApoL6 showed mitochondrial-mediated cell death characterized by the release of cytochrome c and activation of caspases 9 and 8. ApoL6 overexpression also blocked Beclin 1-dependent autophagy. Hypothetically, an elevated ratio of ApoL6:ApoL1 could promote apoptosis through the inhibition of autophagic signals. Considering the role of ApoL1 in cellular homeostasis, gain- or loss-of-function mutations in ApoL1 could increase the risk of cell death leading to CKD. Furthermore, ApoL1-mediated lipid metabolism could also play a role in CKD. Moreover, HIV-1 infection and other stresses may act as a second trigger/hit to promote cell death, which may not be rescued by ApoL1-dependent autophagy in patients harboring two APOL1 risk alleles. Importantly, from the human ApoL1 interactome point of view, the I384M mutation is present in the LZD and therefore may disrupt interaction of ApoL1 with other critical proteins in kidney cells and in the circulation that lead to pathological consequences of CKD. Thus, understanding the biology and pathophysiology of ApoL1 is of great importance in the prevention and therapy of RCC and CKD. For the well-being of up to 40% of ESKD in African Americans as well as in Hispanic Americans and others with African ancestry around the world, future investigations should aim at understanding the molecular mechanisms of ApoL1-associated CKD states and the clinical implications of APOL1 genotype in the setting of proteinuria, FSGS, HIVAN, hypertension-attributed nephrosclerosis, and clinically “healthy” kidney donors.
Acknowledgments
We apologize to the scientists whose work could not be included in this review paper because of space limitations, and are grateful for the funding support from NIH and DOD, USA, NCI 5RO1CA106644, NIH P20RR016480 (NM-INBRE), and DOD PC040298 to CAH; and RO1DK49419 and R01HL55605 to PER.
List of abbreviations
- ApoL
apolipoprotein L
- CKD
chronic kidney disease
- ESKD
end stage kidney disease
- FSGS
Focal Segmental Glomerulosclerosis
- HIVAN
HIV-1 associated nephropathy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O’Connor PM, Malloy MJ, Kane JP. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem. 1997;272:25576–25582. doi: 10.1074/jbc.272.41.25576. [DOI] [PubMed] [Google Scholar]
- 2.Duchateau PN, Movsesyan I, Yamashita S, Sakai N, Hirano K, Schoenhaus SA, O’Connor-Kearns PM, Spencer SJ, Jaffe RB, Redberg RF, et al. Plasma apolipoprotein L concentrations correlate with plasma triglycerides and cholesterol levels in normolipidemic, hyperlipidemic, and diabetic subjects. J Lipid Res. 2000;41:1231–1236. [PubMed] [Google Scholar]
- 3.Duchateau PN, Pullinger CR, Cho MH, Eng C, Kane JP. Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J Lipid Res. 2001;42:620–630. [PubMed] [Google Scholar]
- 4.Albert TS, Duchateau PN, Deeb SS, Pullinger CR, Cho MH, Heilbron DC, Malloy MJ, Kane JP, Brown BG. Apolipoprotein L-I is positively associated with hyperglycemia and plasma triglycerides in CAD patients with low HDL. J Lipid Res. 2005;46:469–474. doi: 10.1194/jlr.M400304-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Horrevoets AJ, Fontijn RD, van Zonneveld AJ, de Vries CJ, ten Cate JW, Pannekoek H. Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood. 1999;93:3418–3431. [PubMed] [Google Scholar]
- 6.Li Q, Fan P, Bai H, Liu R, Huang Y, Wang X, Wu H, Liu Y, Liu B. Distribution and effect of apoL-I genotype on plasma lipid and apolipoprotein levels in Chinese normalipidemic and endogenous hypertriglyceridemic subjects. Clin Chim Acta. 2009;403:152–155. doi: 10.1016/j.cca.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Vanhollebeke B, Pays E. The trypanolytic factor of human serum: many ways to enter the parasite, a single way to kill. Mol Microbiol. 2010 May;76:806–814. doi: 10.1111/j.1365-2958.2010.07156.x. [DOI] [PubMed] [Google Scholar]
- 8.Pays E, Vanhollebeke B. Human innate immunity against African trypanosomes. Curr Opin Immunol. 2009;21:493–498. doi: 10.1016/j.coi.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 9.Pays E, Vanhollebeke B, Vanhamme L, Paturiaux-Hanocq F, Nolan DP, Perez-Morga D. The trypanolytic factor of human serum. Nat Rev Microbiol. 2006;4:477–486. doi: 10.1038/nrmicro1428. [DOI] [PubMed] [Google Scholar]
- 10.Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homble F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309:469–472. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 11.Shiflett AM, Bishop JR, Pahwa A, Hajduk SL. Human high density lipoproteins are platforms for the assembly of multi-component innate immune complexes. J Biol Chem. 2005;280:32578–32585. doi: 10.1074/jbc.M503510200. [DOI] [PubMed] [Google Scholar]
- 12.Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009;19:850–858. doi: 10.1101/gr.085647.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, Van Den Abbeele J, Pays A, Tebabi P, Van Xong H, Jacquet A, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422:83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Lu H, Jiang Z, Pastuszyn, Hu CA. Apolipoprotein L6, a Novel proapoptotic Bcl-2 Homology 3-Only protein, Induces Mitochondria-mediated Apoptosis in Cancer Cells. Mol Can Res. 2005;3:21–31. [PubMed] [Google Scholar]
- 15.Zhaorigetu S, Yang Z, Toma I, McCaffrey TA, Hu CA. Apolipoprotein L6, Induced in Atherosclerotic Lesions, Promotes Apoptosis and Blocks Beclin 1-dependent Autophagy in Atherosclerotic Cells. J Biol Chem. 2011;286:27389–27398. doi: 10.1074/jbc.M110.210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008:283. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–1082. doi: 10.4161/auto.7066. 7066 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanhollebeke B, Pays E. The function of apolipoproteins L. Cell Mol Life Sci. 2006;63:1937–1944. doi: 10.1007/s00018-006-6091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray PE, Hu C. Advances in our understanding of the pathogenesis of HIV-1 associated nephropathy in children. Future Virol. 2011;6:883–894. doi: 10.2217/fvl.11.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monajemi H, Fontijn RD, Pannekoek H, Horrevoets AJ. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics. 2002;79:539–546. doi: 10.1006/geno.2002.6729. [DOI] [PubMed] [Google Scholar]
- 21.Page NM, Butlin DJ, Lomthaisong K, Lowry PJ. The human apolipoprotein L gene cluster: identification, classification, and sites of distribution. Genomics. 2001;74:71–78. doi: 10.1006/geno.2001.6534. [DOI] [PubMed] [Google Scholar]
- 22.Carrera N, Arrojo M, Paz E, Ramos-Rios R, Agra S, Paramo M, Brenlla J, Costas J. Testing the antagonistic pleiotropy model of schizophrenia susceptibility by analysis of DAOA, PPP1R1B, and APOL1 genes. Psychiatry Res. 2010;179:126–129. doi: 10.1016/j.psychres.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 23.McGhee KA, Morris DW, Schwaiger S, Nangle JM, Donohoe G, Clarke S, Meagher D, Quinn J, Scully P, Waddington JL, et al. Investigation of the apolipoprotein-L (APOL) gene family and schizophrenia using a novel DNA pooling strategy for public database SNPs. Schizophr Res. 2005;76:231–238. doi: 10.1016/j.schres.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Mimmack ML, Ryan M, Baba H, Navarro-Ruiz J, Iritani S, Faull RL, McKenna PJ, Jones PB, Arai H, Starkey M, et al. Gene expression analysis in schizophrenia: reproducible up-regulation of several members of the apolipoprotein L family located in a high-susceptibility locus for schizophrenia on chromosome 22. Proc Natl Acad Sci USA. 2002;99:4680–4685. doi: 10.1073/pnas.032069099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutcliffe JG, Thomas EA. The neurobiology of apolipoproteins in psychiatric disorders. Mol Neurobiol. 2002;26:369–388. doi: 10.1385/mn:26:2-3:369. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi S, Cui YH, Han YH, Fagerness JA, Galloway B, Shen YC, Kojima T, Uchiyama M, Faraone SV, Tsuang MT. Association of SNPs and haplotypes in APOL1, 2 and 4 with schizophrenia. Schizophr Res. 2008;104:153–164. doi: 10.1016/j.schres.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomita Y, Ikeda M, Mutoh H, Inada T, Iwata N, Ozaki N, Honda H. Association study between Apolipoprotein L and schizophrenia by exhaustive and rule-based combination analysis for identification of multilocus interactions. J Biosci Bioeng. 2007;103:303–310. doi: 10.1263/jbb.103.303. [DOI] [PubMed] [Google Scholar]
- 28.Venkatasubramanian G, Arasappa R, Rao NP, Behere RV, Kalmady S, Gangadhar BN. Inverse relationship between serum high density lipoprotein and negative syndrome in antipsychotic-naive schizophrenia. Clin Chem Lab Med. 2010;48:95–98. doi: 10.1515/CCLM.2010.004. [DOI] [PubMed] [Google Scholar]
- 29.Johanneson B, McDonnell SK, Karyadi DM, Quignon P, McIntosh L, Riska SM, FitzGerald LM, Johnson G, Deutsch K, Williams G, et al. Family-based association analysis of 42 hereditary prostate cancer families identifies the Apolipoprotein L3 region on chromosome 22q12 as a risk locus. Hum Mol Genet. 2010;19:3852–3862. doi: 10.1093/hmg/ddq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christensen GB, Baffoe-Bonnie AB, George A, Powell I, Bailey-Wilson JE, Carpten JD, Giles GG, Hopper JL, Severi G, English DR, et al. Genome-wide linkage analysis of 1,233 prostate cancer pedigrees from the International Consortium for Prostate Cancer Genetics using novel sumLINK and sumLOD analyses. Prostate. 2010;70:735–744. doi: 10.1002/pros.21106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu GN, Zuo L, Zhou Q, Zhang SM, Zhu HQ, Gui SY, Wang Y. Loss of heterozygosity on chromosome 10q22–10q23 and 22q11.2–22q12.1 and p53 gene in primary hepatocellular carcinoma. World J Gastroenterol. 2004;10:1975–1978. doi: 10.3748/wjg.v10.i13.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartikainen JM, Tuhkanen H, Kataja V, Eskelinen M, Uusitupa M, Kosma VM, Mannermaa A. Refinement of the 22q12-q13 breast cancer--associated region: evidence of TMPRSS6 as a candidate gene in an eastern Finnish population. Clin Cancer Res. 2006;12:1454–1462. doi: 10.1158/1078-0432.CCR-05-1417. [DOI] [PubMed] [Google Scholar]
- 33.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genovese G, Tonna SJ, Knob AU, Appel GB, Katz A, Bernhardy AJ, Needham AW, Lazarus R, Pollak MR. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int. 2010;78:698–704. doi: 10.1038/ki.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-Based Risk Assessment of APOL1 on Renal Disease. J Am Soc Nephrol. 2011;22:2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, et al. APOL1 Genetic Variants in Focal Segmental Glomerulosclerosis and HIV-Associated Nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freedman BI, Kopp JB, Langefeld CD, Genovese G, Friedman DJ, Nelson GW, Winkler CA, Bowden DW, Pollak MR. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21:1422–1426. doi: 10.1681/ASN.2010070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papeta N, Kiryluk K, Patel A, Sterken R, Kacak N, Snyder HJ, Imus PH, Mhatre AN, Lawani AK, Julian BA, Wyatt RJ, Novak J, Wyatt CM, Ross MJ, Winston JA, Klotman ME, Cohen DJ, Appel GB, D’Agati VD, Klotman PE, Gharavi AG. APOL1 variants increase risk for FSGS and HIVAN but not IgA nephropathy. J Am Soc Nephrol. 2011;22:1991–1996. doi: 10.1681/ASN.2011040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol. 2011;22:2119–2128. doi: 10.1681/ASN.2011010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fine DM, Wasser WG, Estrella MM, Atta MG, Kuperman M, Shemer R, Rajasekaran A, Tzur S, Racusen LC, Skorecki K. APOL1 Risk Variants Predict Histopathology and Progression to ESRD in HIV-Related Kidney Disease. J Am Soc Nephrol. 2011 Dec 1; doi: 10.1681/ASN.2011060562. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen DM, Mittalhenkle A, Scott DL, Young CJ, Norman DJ. African American living-kidney donors should be screened for APOL1 risk alleles. Transplantation. 2011;92:722–725. doi: 10.1097/TP.0b013e31822eec39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reeves-Daniel AM, DePalma JA, Bleyer AJ, Rocco MV, Murea M, Adams PL, Langefeld CD, Bowden DW, Hicks PJ, Stratta RJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11:1025–1030. doi: 10.1111/j.1600-6143.2011.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 47.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang WB, Wang Z, Shu F, Jin YH, Liu HY, Wang QJ, Yang Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem. 2010;285:40461–40471. doi: 10.1074/jbc.M110.164046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M, Vandenabeele P. Beclin1: A role in membrane dynamics and beyond. Autophagy. 2012:8. doi: 10.4161/auto.8.1.16645. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 53.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859–864. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol. 2011;12:226. doi: 10.1186/gb-2011-12-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanida I. Autophagy basics. Microbiol Immunol. 2011;55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- 56.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery andsignaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima M, Iwata JI, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 58.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011;21:113–119. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012:8. doi: 10.4161/auto.8.1.16618. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 62.Periyasamy-Thandavan S, Jiang M, Schoenlein P, Dong Z. Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297:F244–256. doi: 10.1152/ajprenal.00033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chien CT, Shyue SK, Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation. 2007;84:1183–1190. doi: 10.1097/01.tp.0000287334.38933.e3. [DOI] [PubMed] [Google Scholar]
- 64.Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, Uchiyama Y, Takahara S, Imai E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun. 2008;368:100–106. doi: 10.1016/j.bbrc.2008.01.059. [DOI] [PubMed] [Google Scholar]
- 65.Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15:1875–1886. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- 66.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 67.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 69.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Motzer RJ, Bander NH, Nanus DM. Renal cell carcinoma. N Engl J Med. 1996;335:865–875. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- 72.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 73.Kaelin WG., Jr Treatment of kidney cancer: insights provided by the VHL tumor-suppressor protein. Cancer. 2009;115:2262–2272. doi: 10.1002/cncr.24232. [DOI] [PubMed] [Google Scholar]
- 74.Di Sole F. Adenosine and renal tubular function. Curr Opin Nephrol Hypertens. 2008;17:399–407. doi: 10.1097/MNH.0b013e32830321e1. [DOI] [PubMed] [Google Scholar]; Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Novoa JM, Rodriguez-Pena AB, Ortiz A, Martinez-Salgado C, Lopez Hernandez FJ. Etiopathology of chronic tubular, glomerular and renovascular nephropathies: clinical implications. J Transl Med. 2011;9:13. doi: 10.1186/1479-5876-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–913. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Isaka Y, Kimura T, Takabatake Y. The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells. Autophagy. 2011;7:1085–1087. doi: 10.4161/auto.7.9.16465. [DOI] [PubMed] [Google Scholar]
- 78.Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181–1192. doi: 10.2353/ajpath.2010.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Periyasamy-Thandavan S, et al. Autophagy is cytoprotective during cysplatin induced injury of renal proximal tubular cells. Kidney Int. 2008;74:631–640. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 80.Jiang M, Wang CY, Huang S, Yang T, Dong Z. Cisplatin-induced apoptosis in p53-deficient renal cells via the intrinsic mitochondrial pathway. Am J Physiol Renal Physiol. 2009;296:F983–993. doi: 10.1152/ajprenal.90579.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, Yoshimori T, Isaka Y, Rakugi H. Autophagy guards against Cisplatin-induced acute kidney injury. Am J Pathol. 2012;180:517–25. doi: 10.1016/j.ajpath.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 82.Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de Bruïne AP. VHL and HIF signalling in renal cell carcinogenesis. J Pathol. 2010;221:125–138. doi: 10.1002/path.2689. [DOI] [PubMed] [Google Scholar]
- 83.Morselli E, Galluzzi L, Kepp O, Mariño G, Michaud M, Vitale I, Maiuri MC, Kroemer G. Oncosuppressive functions of autophagy. Antioxid Redox Signal. 2011;14:2251–2269. doi: 10.1089/ars.2010.3478. [DOI] [PubMed] [Google Scholar]
- 84.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 85.Jefferson JA, Nelson PJ, Najafian B, Shankland SJ. Podocyte disorders: Core Curriculum 2011. Am J Kidney Dis. 2011;58:666–677. doi: 10.1053/j.ajkd.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 87.Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hanna Z, Kay DG, Rebai N, Guimond A, Jothy S, Jolicoeur P. Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell. 1998;95:163–175. doi: 10.1016/s0092-8674(00)81748-1. [DOI] [PubMed] [Google Scholar]
- 89.Zhong J, Zuo Y, Ma J, Fogo AB, Jolicoeur P, Ichikawa I, Matsusaka T. Expression of HIV-1 genes in podocytes alone can lead to the full spectrum of HIV-1-associated nephropathy. Kidney Int. 2005;68:1048–1060. doi: 10.1111/j.1523-1755.2005.00497.x. [DOI] [PubMed] [Google Scholar]
- 90.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Quaggin SE, George AL., Jr Apolipoprotein L1 and the genetic basis for racial disparity in chronic kidney disease. J Am Soc Nephrol. 2011;22:1955–1958. doi: 10.1681/ASN.2011090932. [DOI] [PubMed] [Google Scholar]
- 92.Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- 93.Yamamoto T, Ryan RO. Role of leucine zipper motif in apoE3 N-terminal domain lipid binding activity. Biochim Biophys Acta. 2006;1761:1100–1106. doi: 10.1016/j.bbalip.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 94.Vanhollebeke B, Nielsen MJ, Watanabe Y, Truc P, Vanhamme L, Nakajima K, Moestrup SK, Pays E. Distinct roles of haptoglobin-related protein and apolipoprotein L-I in trypanolysis by human serum. Proc Natl Acad Sci U S A. 2007 Mar 6;104(10):4118–23. doi: 10.1073/pnas.0609902104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Paturiaux-Hanocq F, Andris F, Lins L, Pays E. C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog. 2009;5:e1000685. doi: 10.1371/journal.ppat.1000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harrington JM, Howell S, Hajduk SL. Membrane permeabilization by trypanosome lytic factor, a cytolytic human high density lipoprotein. J Biol Chem. 2009;284:13505–13512. doi: 10.1074/jbc.M900151200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Molina-Portela MP, Samanovic M, Raper J. Distinct roles of apolipoprotein components within the trypanosome lytic factor complex revealed in a novel transgenic mouse model. J Exp Med. 2008;205:1721–1728. doi: 10.1084/jem.20071463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vanhollebeke B, De Muylder G, Nielsen MJ, Pays A, Tebabi P, Dieu M, Raes M, Moestrup SK, Pays E. A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science. 2008 May 2;320(5876):677–81. doi: 10.1126/science.1156296. [DOI] [PubMed] [Google Scholar]
- 99.Lugli EB, Pouliot M, Portela Mdel P, Loomis MR, Raper J. Characterization of primate trypanosome lytic factors. Mol Biochem Parasitol. 2004;138:9–20. doi: 10.1016/j.molbiopara.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 100.Wheeler RJ. The trypanolytic factor-mechanism, impacts and applications. Trends Parasitol. 2010;26:457–464. doi: 10.1016/j.pt.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 101.Vanhollebeke B, Truc P, Poelvoorde P, Pays A, Joshi PP, Katti R, Jannin JG, Pays E. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N Engl J Med. 2006;355:2752–2756. doi: 10.1056/NEJMoa063265. [DOI] [PubMed] [Google Scholar]
- 102.Freedman BI, Murea M. Target Organ Damage in African American Hypertension: Role of APOL1. Curr Hypertens Rep. 2012;14:21–28. doi: 10.1007/s11906-011-0237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Medapalli RK, He JC, Klotman PE. HIV-associated nephropathy: pathogenesis. Curr Opin Nephrol Hypertens. 2011;20:306–311. doi: 10.1097/MNH.0b013e328345359a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Papeta N, Sterken R, Kiryluk K, Kalyesubula R, Gharavi AG. The molecular pathogenesis of HIV-1 associated nephropathy: recent advances. J Mol Med (Berl) 2011;89:429–436. doi: 10.1007/s00109-010-0719-x. [DOI] [PubMed] [Google Scholar]