

Abstract
Background
Hepatoblastoma (HB) is an embryonal liver neoplasm of early childhood with a poor prognosis for patients with distant metastases and vascular invasion. We and others have previously shown that the overexpression of insulin-like growth factor 2 (IGF2), loss of imprinting at the IGF2/H19 locus, and amplification of pleomorphic adenoma gene 1 (PLAG1) are common features in HB, suggesting a critical role of the IGF axis in hepatoblastomagenesis. In this study, we investigated the role of the insulin-like growth factor binding protein 3 (IGFBP3), a known competitor of the IGF axis, in pediatric liver cancers.
Results
The IGFBP3 gene was highly expressed in normal pediatric livers but was heavily downregulated in four HB cell lines and the majority of HB primary tumors (26/36). Detailed methylation analysis of CpG sites in the IGFBP3 promoter region by bisulfite sequencing revealed a high degree of DNA methylation, which is causatively associated with the suppression of IGFBP3 in HB cell lines. Consequently, the treatment of HB cell lines with 5-aza-2'-deoxycytidine resulted in DNA demethylation and reactivation of the epigenetically silenced IGFBP3 expression. Interestingly, IGFBP3 promoter methylation predominantly occurred in metastatic HB with vascular invasion. Restoring IGFBP3 expression in HB cells resulted in reduced colony formation, migration, and invasion.
Conclusion
This study provides the first direct evidence that the reactivation of IGFBP3 decreases aggressive properties of pediatric liver cancer cells and that IGFBP3 promoter methylation might be used as an indicator for vessel-invasive tumor growth in HB patients.
Keywords: Hepatoblastoma, Epigenetics, Methylation, Invasion, IGF2
Background
Hepatoblastoma (HB) represents the most common primary liver tumor in childhood with an incidence of approximately one new case per million children less than 15 years of age [1]. Pathohistologically, HB resembles various stages of the developing liver, showing malignant epithelial cells with fetal and/or embryonal hepatic differentiation and foci of primitive blastemal cells. The mixed HB subtype also contains interspersed mesenchymal elements, such as immature fibrous tissue, spindle cells, and osteoid [1]. Although HB generally responds well to chemotherapy and the prognosis is usually good [2], the outcome of high-risk patients with metastatic tumors or invasion of large hepatic veins is fatal [3,4].
The type 1 insulin-like growth factor receptor and its ligands, IGF1 and IGF2, are upregulated in a variety of human cancers [5]. In pediatric tumors, such as rhabdomyosarcoma, nephroblastoma, and HB, the role of the IGF axis is particularly important [6]. We and others have shown that the fetal growth factor IGF2 is upregulated in almost all HB cases [7,8], even though the underlying molecular mechanism is still not understood. This upregulation could be explained in part by the observation that the loss of imprinting at the IGF2/H19 locus is evident in approximately 20% of all IGF2 overexpressing HB, thus leading to biallelic expression of the gene [9]. Moreover, the amplification and subsequent upregulation of the transcriptional IGF2 activator PLAG1 has been described in the majority of HB cases [10]. Collectively, these data suggest that several mechanisms could be responsible for the frequently observed upregulation of IGF2, which is characteristic for the molecular pathogenesis of HB.
The insulin-like growth factor binding protein 3 (IGFBP3) is a multifunctional protein predominantly produced by the liver, which mediates the growth suppression and induction of apoptosis by binding insulin-like growth factors [11]. Accordingly, IGFBP3 transgenic mice exhibit a significant reduction in both birth weight and litter size, with a reduction in some organ weights [12]. The stable transfection of IGFBP3 results in reduced growth rates of non-small cell lung cancer cells, both in vitro and in vivo, as xenotransplants in nude mice [13]. Moreover, the addition of recombinant IGFBP3 results in the massive induction of apoptosis, as shown in colon and prostate cancer [14,15]. Conversely, it has been postulated that the suppression of the putative tumor suppressor gene IGFBP3 could lead to elevated levels of insulin-like growth factors, thus promoting tumor growth. Because mutational inactivation has been precluded as being causative for IGFBP3 suppression [16], epigenetic inactivation by promoter methylation has recently been considered as an alternative mechanism [17,18]. It is a well-described phenomenon that the suppression of tumor suppressor genes could be facilitated by abnormal methylation of DNA at certain CpG islands that often lie in the promoter regions of these genes [19].
Because the activation of IGF signaling is characteristic for HB and IGFBP3 suppression contributes to the sustainment of IGF signaling, we wanted to determine the role of the IGFBP3 gene in the biology of pediatric liver cancers. We demonstrate that the downregulation of IGFBP3 expression is a common feature in HB, which is associated with CpG island promoter methylation in advanced, high-risk HB cases. In addition, we reveal that IGFBP3 is epigenetically silenced in HB cell lines and that the reintroduction of IGFBP3 leads to the inhibition of tumor cell migration and invasion. These findings indicate that the suppression of IGFBP3 displays an alternative mechanism for enhancing IGF signaling in the late stages of HB development.
Results
Downregulation of IGFBP3 is a common event in pediatric liver tumors
To define the IGF signaling status in our pediatric liver tumor collection, we initially investigated the endogenous expression of the ligand IGF2 and its positive regulator PLAG1. Real-time PCR analysis revealed that the mRNA level of IGF2 was markedly increased (> 3-fold induction compared to normal livers) in 23/36 (64%) of HB and 3/9 (33%) of hepatocellular carcinoma (HCC) cases (Figure 1A). Furthermore, we detected a strong upregulation of PLAG1 in 20/36 (56%) of HB and 1/9 (11%) of HCC tumors (Figure 1B). Interestingly, a high IGF2 expression correlated well with PLAG1 upregulation, predominantly in HB cases (Figure 1C).
Figure 1.

Expression analyses of pediatric liver cancers by real-time PCR. The relative IGF2 (A) PLAG1 (B), IGFBP3 (D), and TIA1 (E) mRNA expression levels of the 45 pediatric liver tumor cases and 7 normal liver samples were determined. The dots represent relative candidate gene expression in relation to the house-keeping gene TBP. Data from the HB and pediatric HCC cases are depicted as black and orange diamonds, respectively. Median expression values are given as red horizontal lines. Correlation analyses of pediatric liver cancers. The mRNA expression levels of IGF2 (C) and IGFBP3 (F) were correlated with PLAG1 and TIA1, respectively. Dots represent log2 values of the relative expression of candidate genes blotted versus each other in the 45 pediatric liver tumor samples (rho = Spearman's rank correlation coefficient).
Because IGFBP3 has been described to act as a negative regulator of the IGF axis by competitively binding IGFs [11], we were interested in whether the downregulation of this gene could also contribute to the activation of IGF signaling in HB. By using real-time PCR, we demonstrate that IGFBP3 mRNA levels are heavily decreased (> 3-fold reduction compared to normal livers) in 26/36 (72%) of HB cases (Figure 1D). As previously described for HCC in adults [17], we also detected a reduced IGFBP3 expression in 6/9 (67%) of pediatric HCC cases (Figure 1D) compared to normal childhood liver tissues. IGFBP3 has recently been described to be transcriptionally downregulated by binding T-cell-restricted intracellular antigen-1 (TIA1), which is also overexpressed in human HCC [20]. Correspondingly, TIA1 is also upregulated in the majority of HB cases (Figure 1E) and is inversely correlated with the expression of IGFBP3 (Figure 1F), although at a low level (rho = -0.3295) Altogether, these data suggest that the downregulation of IGFBP3 might significantly contribute to the activation of the IGF signaling cascade by sustaining the IGF2-induced stimulation in HB.
Promoter methylation causes IGFBP3 silencing in human HB cell lines
Promoter methylation has been described as a molecular mechanism to suppress the gene expression of negative regulators of tumor growth in a variety of cancers [19]. Because TIA1 upregulation does not completely explain the suppression of IGFBP3 in pediatric liver tumors (Figure 1F), we examined a CpG island located in the IGFBP3 promoter region for differential methylation in established HB cell lines, namely HUH6, HepT3, HepT1, and HepG2, and the non-hepatitis B virus-associated HCC cell line HUH7, as well as normal liver by means of bisulfite sequencing. We found that the entire IGFBP3 promoter region was heavily methylated in all four HB cell lines and heterogeneously methylated in HUH7, whereas the normal liver DNA was rarely methylated in this region (Figure 2A). Interestingly, promoter methylation was well correlated with very low IGFBP3 expression levels in HB cell lines and a detectable expression in HUH7 when compared to a normal liver, as revealed by real-time and RT-PCR (Figure 2B). Because promoter methylation has a strong impact on the transcriptional activity, we next wanted to determine whether treatment with the demethylating agent 5-Aza-dC could revert the methylation status of the IGFBP3 promoter region and re-establish IGFBP3 expression in these cell lines. After the 5-day 5-Aza-dC treatment and subsequent MSP analysis, we detected an increasing amount of demethylation in the IGFBP3 promoter, thereby qualifying MSP as an appropriate means to analyze DNA methylation (Figure 2C). Bisulfite sequencing of single clones of 5-Aza-dC-treated HepG2 and HUH6 cells revealed a decreased methylation rate of 12.2% and 12.0%, respectively (data not shown). Interestingly, 5-Aza-dC treatment significantly re-established IGFBP3 expression in all cell lines (Figure 2D), which was most prominent in the HepT1 and HepG2 cells. These data suggest that promoter hypermethylation is causatively associated with transcriptional silencing of the IGFBP3 gene in pediatric liver tumors.
Figure 2.

(A) Methylation status of the IGFBP3 promoter in HB cell lines. Bisulfite sequencing of the 5' region of IGFBP3 was performed in the indicated liver tumor cell lines and normal liver (NL) tissue. The open and filled circles represent unmethylated and methylated CpG sites, respectively. The location of each CpG site relative to the transcription start site (in base pairs) and the region used for MSP analysis, including the forward (F) and reverse (R) primer, is shown below. (B) mRNA level of IGFBP3 in HB cell lines. Expression of IGFBP3 was measured in normal liver tissue (NL) and in the liver tumor cell lines HepG2, HepT1, HepT3, HUH6, and HUH7 by real-time and RT-PCR. The values represent the relative gene expression in relation to the house-keeping gene TBP. (C) Demethylation of the IGFBP3 promoter region after 5-Aza-dC or trichostatin A treatment. Cells were incubated for 5 days with 5-Aza-dC and 24 hours with trichostatin A. Methylation status (U, unmethylated; M, methylated) of the IGFBP3 promoter region was determined using MSP. Representative images of the MSP experiments are given. (D) Restoration of IGFBP3 expression. The reactivation of IGFBP3 expression after 5 days of 5-Aza-dC treatment in HB cell lines, as revealed by real-time PCR. Statistically significant difference versus untreated cells: *P < 0.05 (unpaired Student's t-test).
The histone-deacetylase inhibitor trichostatin A has formerly been described to display strong effects on the transcriptional regulation of IGFBP3 [21]. Treatment of all five liver cancer cell lines with trichostatin A resulted in the strong demethylation (Figure 2C) and reexpression (data not shown) of IGFBP3, comparable to the effect communicated by 5-Aza-dC but in a much shorter period (24 h). Thus, it might be expected that both promoter methylation and histone-deacetylation may play important roles in the control of the IGFBP3 tumor suppressor in the liver.
IGFBP3 promoter methylation predominantly occurs in metastatic high-risk liver tumors with large vessel invasion
To assess whether IGFBP3 promoter methylation is clinically relevant, we performed a methylation analysis of our pediatric liver tumor collection using MSP. IGFBP3 methylation was detected in 9/36 (25%) of HB and 6/9 (66%) of pediatric HCC cases, whereas normal liver tissues had no bands for the methylated state (Figure 3A, B). However, there was no clear correlation between IGFBP3 promoter methylation and reduced IGFBP3 expression levels (data not shown). By analyzing clinicopathological features, such as gender, age at diagnosis, tumor differentiation, metastatic disease, outcome, multifocality, and vascular invasion (Table 1), we observed that IGFBP3 promoter methylation was significantly associated with metastases and invasion into large hepatic veins, two high-risk parameters for HB patients. Moreover, the overall survival of patients with IGFBP3 methylation was strongly reduced (Figure 3C). These data suggest that aberrant CpG island methylation of the IGFBP3 promoter region is a late event in the genesis of pediatric liver tumors and might predict the evolution of HB to a highly aggressive, metastatic, and vascular-invasive phenotype with worse outcomes.
Figure 3.
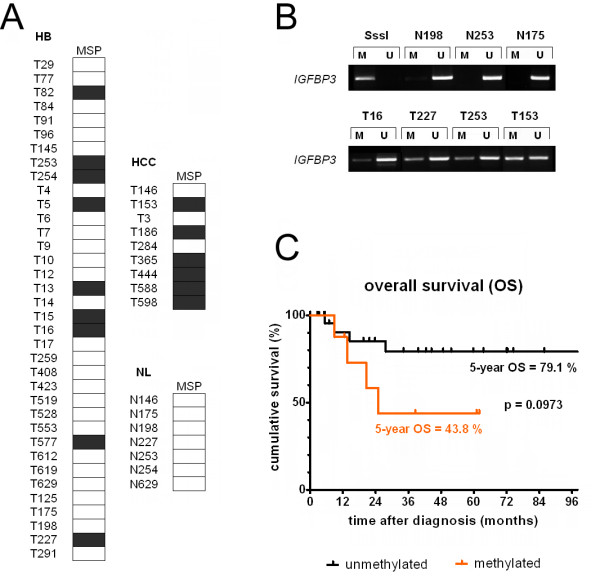
(A) Methylation status of the IGFBP3 promoter in pediatric liver cancers. Summary of the analyses of the IGFBP3 methylation status in the 36 hepatoblastomas (HB), 9 pediatric hepatocellular carcinomas (HCC), and 7 normal pediatric liver samples (NL), as determined by MSP. The black boxes represent the detection of methylated alleles, and the white boxes depict unmethylated alleles. (B) Methylation-specific PCR (MSP) of pediatric liver cancers. The promoter region of the IGFBP3 gene was analyzed for methylated CpG sites by MSP using bisulfite-treated genomic DNA from pediatric liver tumors (T) and normal liver tissue (N). Representative images of the MSP experiments are shown, revealing PCR products for unmethylated (U) and methylated alleles (M). In vitro methylated DNA (SssI) was used as a positive control for methylated alleles. (C) Overall survival (OS) stratified by methylation status. OS was calculated as the time from diagnosis to death from disease and is plotted for 8 HB patients with (orange line) and 25 without (black line) IGFBP3 promoter methylation over a period of 100 months. Statistical significance was calculated using Mantel-Cox test.
Table 1.
Associations of the clinical characteristics and IGFBP3 methylation status in 36 pediatric patients with hepatoblastoma
| IGFBP3 | |||||
|---|---|---|---|---|---|
| parameters | group | no. of tumors | methylated | unmethylated | P |
| gendera | m | 18 | 3 | 15 | 0.2482 |
| f | 18 | 6 | 12 | ||
| age at diagnosisb | < 2 | 21 | 4 | 17 | 0.3570 |
| > 2 | 12 | 4 | 8 | ||
| histological subtypec | f | 20 | 3 | 17 | 0.1213 |
| e | 16 | 6 | 10 | ||
| metastases | yes | 15 | 7 | 8 | 0.0047 |
| no | 19 | 1 | 18 | ||
| outcomed | NED | 25 | 4 | 21 | 0.0508 |
| DOD | 8 | 4 | 4 | ||
| multifocal growth | yes | 7 | 3 | 4 | 0.2662 |
| no | 19 | 4 | 15 | ||
| invasion of large hepatic veins | yes | 8 | 6 | 2 | 0.0001 |
| no | 25 | 2 | 23 | ||
am, male; f, female
bThe age of patients at diagnosis is given in months
cEpithelial component with predominantly fetal, f, or embryonal, e, differentiation
dNED, no evidence of disease; DOD, dead of disease
Restoring IGFPB3 has long-term effects on cell growth and apoptosis in HB
IGFBP3 is thought to mediate growth suppression and induce apoptosis by binding IGFs [11]. Thus, we determined whether the reintroduction of IGFBP3 into liver tumor cells could change the tumor's biological properties. Adding 1 μg/ml recombinant human IGFBP3 to tumor cell lines resulted in comparable growth rates over time (Figure 4A). In line with this, IGFBP3-substituted cells displayed no significant increase in apoptotic characteristics, such as elevated external appearance of phosphatidylserine (Figure 4B) or proteolytic cleavage of the PARP protein (Figure 4C). In order to see long-term effects, we used HepT1 cells stably transfected with an IGFBP3 expression plasmid that resulted in highly elevated IGFBP3 mRNA and protein levels (Figure 4D). Although stable transfectants displayed no reduction in growth within 96 h (Figure 4E), we found a significantly reduced clonogenic survival rate after 2 weeks, as evidenced by the lower number of colonies (Figure 4F). Furthermore, IGFBP3-transfected cells showed signs of apoptosis, such as cell shrinkage, membrane blebbing, and formation of apoptotic bodies, when compared to control-transfected cells (not shown) and an increase in the external appearance of phosphatidylserine (Figure 4G). Taken together, our results document that long-term reconstitution of IGFBP3 acts as a tumor suppressive factor in pediatric liver tumors.
Figure 4.

(A) Growth properties of IGFBP3 treated HB cells. The viability of tumor cells in the presence (IGFBP3) or absence (control) of 1 μg/ml recombinant human IGFBP3 was assessed at the time points indicated using MTT assays and optical density (OD) measurements. The values given represent the mean ratio of treated versus untreated cells from triplicate measurements ± SEM. (B) Annexin V staining. IGFBP3-treated cells were analyzed for phosphatidylserine membrane asymmetry using Cy5-conjugated annexin V and calcein staining. Representative plots of the flow-cytometric measurements depicting the percentages of apoptotic cells (annexin V and calcein positive) in the control (left panel) and recombinant human IGFBP3 treated cells (right panel) are shown. (C) Proteolytic cleavage of PARP. Activation of PARP by proteolytic cleavage was measured in tumor cells treated with or without recombinant human IGFBP3 by Western blot analysis using antibodies for cleaved PARP and β-actin as a loading control. (D) Stable IGFBP3 transfectants. HepT1 cells were transfected with an empty vector control (pIRES) or expression vector containing full-length IGFBP3 cDNA (pIGFBP3), selected with puromycin for 2 weeks and cloned by picking resistant colonies. Exogenous IGFBP3 expression was measured in transfected HepT1 cells using real-time PCR in relation to the house-keeping gene TBP as a calibrator (left) or Western blot analysis using antibodies for IGFBP3 and β-actin as a loading control (right). (E) Growth properties of IGFBP3 transfected HepT1 cells. The viability of stably transfected cells was assessed at the time points indicated using MTT assays and optical density (OD) measurements. The values given represent the mean of triplicate measurements ± SEM. (F) Colony formation assay. HepT1 cells were transfected with an empty vector control (pIRES) or IGFBP3 expression vector (pIGFBP3) and subsequently cultured in puromycin-containing media for 2 weeks. Colonies were stained with crystal violet, and representative assays were photographed (left) and counted (right). Statistically significant difference versus control: *P < 0.05 (unpaired Student's t-test). (G) Annexin V staining. IGFBP3 transfected cells were analyzed for phosphatidylserine membrane asymmetry using Cy5-conjugated annexin V and calcein staining. Representative plots of flow-cytometric measurements depicting 2.4% and 10.6% of apoptotic cells (annexin V and calcein positive) in the control (upper plot) and IGFBP3 transfectants (lower plot) are shown (left). The mean percentages of early apoptotic cells ± SEM of two independent annexin V experiments are given (right). Statistically significant difference versus vehicle: *P < 0.05 (unpaired Student's t-test).
Recombinant IGFBP3 slows the migratory and invasive capacity of liver tumor cells
As IGFBP3 has been described to suppress migration and invasion in several cancers [22,23], we desired to determine whether the restoration of IGFBP3 function has any impact on the migratory and invasive capacity of liver tumor cells. Using wound healing assays, we demonstrated that HepT1 cells stably transfected with IGFBP3 had a markedly slower cell migration into a cell-free wound within 48 h than their control transfected counterparts (Figure 5A). By choosing liver tumor cell lines with high migration rates, namely HepG2 and HUH7, migration assays using collagen-coated transwell inserts demonstrated a significantly decreased migration of tumor cells incubated with recombinant human IGFBP3 (Figure 5B). Moreover, tumor cells lost their invasiveness when recombinant human IGFBP3 was added to the culture medium, as evidenced by the transwell assays with Matrigel coated inserts (Figure 5C). Altogether, these data clearly indicate that restoring IGFBP3 function could dramatically diminish the migratory and invasive properties of liver tumor cells.
Figure 5.
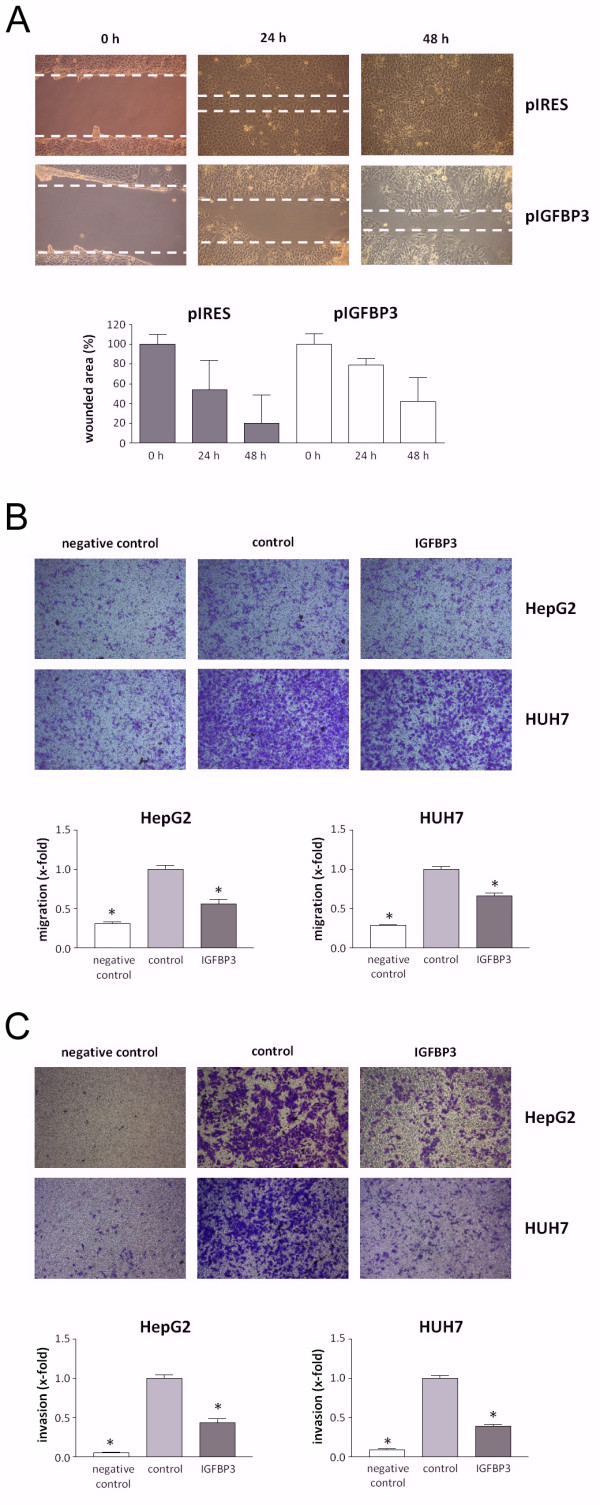
(A) Scratch assay. Representative images of HepT1 cells stably transfected with empty vector control (pIRES) or IGFBP3 migrating into a cell-free wound at 0, 24, and 48 h after wound infliction. Quantitative evaluation of wound closure shows the mean values of three independent experiments. (B) Cell migration and (C) invasion assays. Representative images of HepG2 and HUH7 cells migrating through collagen- (B) or Matrigel-coated (C) transwell plates (pore size, 8 μm) in the presence (IGFBP3) or absence (control) of 1 μg/ml recombinant human IGFBP3. 10% FCS and 50 ng/ml recombinant human HGF served as a chemoattractant in the lower compartment (IGFBP3 and control), whereas a negative control was run without a chemoattractant. The data are the mean ± SEM from three individual experiments. An asterisk indicates P < 0.05.
Discussion
Binding of the IGF2 ligand and the subsequent activation of the IGF1 receptor is known to confer a survival advantage for a wide range of cell types [24]. Consequently, constitutive activation of the IGF axis is a common feature of tumor cells, especially those of early childhood cancers [6,25]. The prevailing mechanism for IGF pathway activation in HB has been allocated to the overexpression of IGF2, which is a result of genetic and epigenetic alterations at the PLAG1 and IGF2/H19 locus [7,8,10,26] and causes activation of the downstream serine/threonine kinase and survival factor AKT [27]. The present study adds an alternative activation mechanism, namely the augmentation of the IGF/IGF1R interaction through downregulation of the IGF2 competitor IGFBP3. We provide evidence that low IGFBP3 expression is a common phenomenon in HB that may contribute to the activation of the IGF axis at the physiological level by the loss of ligand sequestration. Furthermore, the loss of IGFBP3 expression could be attributed to the methylation of the IGFBP3 promoter in at least some primary HB cases, with a predominant occurrence of this epigenetic alteration in metastatic and vascular-invasive high-risk tumors. Our data support the hypothesis that IGFBP3 silencing may contribute to enhanced IGF2/IGF1R signaling and thus the survival and progression of transformed liver cells at a late stage of the disease, which may eventually have considerable clinical implications.
One interesting finding of the current study is that promoter hypermethylation is one possible mechanism for IGFBP3 silencing in HB. We unequivocally demonstrated that DNA is heavily methylated throughout the entire IGFBP3 promoter region of all four HB cell lines under investigation, which conveys a strong suppression of IGFBP3 transcription. These repressive modifications could be removed by the addition of the demethylating agent 5-Aza-dC to the cycling cells, thereby re-establishing IGFBP3 expression. Aberrant DNA methylation has been shown to play an important role in the silencing of IGFBP3 expression in several human cancers, including gastric, colorectal, breast [18], ovarian [28], and renal cancer [29], as well as HCC in adults [17]. However, because DNA methylation only explains the downregulation of IGFBP3 in a subset of primary HB cases, molecular mechanisms other than DNA methylation might also be responsible for the low IGFBP3 expression levels found in the majority of primary HB tumors. Degradation of IGFBP3 by cathepsin D, a specific protease of IGFBP3, has been envisaged as an alternative suppression mechanism of IGFBP3, at least at the protein level [17]. Upregulation of the regulatory protein TIA1 that binds to the AU-rich region of the 3'-UTR of IGFBP3 has recently been described to be associated with downregulation of IGFBP3 in primary HCC [20]. As we have detected an inverse correlation of TIA1 and IGFBP3, it could be assumed that this suppressive mechanism could act in pediatric liver tumors. In addition, histone deacetylation may also play an important role in the suppression of IGFBP3, as shown in this and other studies [21]. Nevertheless, technical restrictions, such as heterogeneity of tumor samples, which comprise the stromal components and the adjacent normal liver tissue in low proportions, might have contributed to an underestimation of HB cases with a methylated IGFBP3 promoter in our study. Noteworthy, a discrepancy between high methylation rates in tumor cell lines and relative low rates in primary tumors is a common phenomenon [30-32]. It has been suggested that a large proportion of CpG hypermethylation found in cancer cell lines reflects an intrinsic property of mammalian cells grown in culture rather than a dependency on the cell of origin. Furthermore, the accumulation of epigenetic changes during the prolonged culture of human embryonal stem cell lines and their derivatives has been described [33]. Alternatively, it might be speculated that subclones within primary cancers with aberrant CpG island methylation may be preferentially selected during cell passage and/or that cancers with high levels of aberrant CpG methylation could be more likely to become established as cell lines. Nevertheless, our functional data clearly show that IGFBP3 silencing is not just a cell culture artifact, but instead, it plays an important role in driving adverse growth characteristics of liver cancer cells originating from advanced stages of liver tumor development.
In addition to its mechanistic role in gene silencing, IGFBP3 promoter methylation might also have clinical implications as a biomarker. It has been reported that IGFBP3 is frequently methylated and significantly associated with a poor prognosis in early-stage non-small-cell lung [34,35], ovarian [28], and prostate cancer [36]. In contrast to these studies, in which hypermethylation of the IGFBP3 promoter is a common and early event during tumorigenesis, we found only 9/36 of HB tumor cases to be methylated, seven of which were high-risk metastatic tumors, indicating a late event in the development of HB. Moreover, as IGFBP3 promoter methylation was significantly associated with vascular invasion in HB and occurred more frequently in pediatric HCC, the detection of this epigenetic alteration might be used as an attractive biomarker for stratifying patients for risk-adapted therapy. Congruent with our assumption, high promoter hypermethylation frequencies of tumor suppressor genes, including IGFBP3, already serve as an indicator for a distinct subclass of advanced HCC in adults with a poor prognosis [37]. This relationship, in turn, suggests that demethylating drugs, which have already been under clinical evaluation [38], might be a novel therapeutic option to treat high-risk liver tumor patients. However, further studies in a large cohort of HB patients are warranted.
Our finding that IGFBP3 restoration results in reduced tumor cell migration and invasion, while leaving growth and apoptosis merely unaffected, also underscores the assumption that IGFBP3 acts at more advanced stages of liver tumor development in children. Furthermore, IGFBP3 has been shown to suppress migration and invasion in adult HCC [22] and melanoma [23]. Interestingly, low IGFBP3 levels have been found to correlate with higher portal invasion and worse prognosis in HCC [39]. Altogether, these data suggest that IGFBP3 downregulation likely has a major role in the vascular invasive and metastatic growth properties of pediatric liver tumors.
Conclusions
In summary, our study clearly documents the following regarding IGFBP3: i) it is downregulated in a high proportion of pediatric liver tumors; ii) it is epigenetically silenced in a subset of HB, indicating that additional repressive mechanisms must exist for this gene; iii) promoter methylation is a late event and predominantly occurs in progressed metastatic and vessel-invasive HB, which may be of clinical significance for HB patients by proposing adapted therapies; and iv) it prevents the migration and invasiveness of HB. Thus, it is intriguing to speculate that restoring IGFBP3 expression and/or use of demethylating drugs could contribute to new therapeutic strategies for HB, especially with the existence of additional epigenetically silenced genes in this tumor type, such as HHIP, RASSF1, SOCS1, APC and CASP8 [40].
Methods
Subjects and tumor cell lines
A total of 45 liver tumor specimens were obtained from pediatric patients undergoing surgical resection in our clinic. Normal liver matching was available from seven patients (N146, N175, N198, N227, N253, N254, and N629). Written informed consent was obtained from each patient, and the study protocol was approved by the Committee of Ethics of the Ludwig-Maximilians-University of Munich. We used the HB cell lines HUH6 (Japanese Collection of Research Bioresources, JCRB, Osaka, Japan), HepT1 [41], HepT3 [7], and HepG2 [42], as well as the hepatocellular carcinoma (HCC) cell line HUH7 (JCRB). All cell lines were maintained as the suppliers recommended.
Real-time reverse transcription-PCR (RT-PCR)
The total RNA was extracted from macroscopically dissected frozen tumor tissue (at least 80% tumor cells), frozen normal liver tissue and HB cell lines, depleted from residual DNA, and reverse transcribed as previously described [43]. PCR amplifications were carried out with 40 ng of cDNA, 500 nM forward and reverse primers and iTaq SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) on a Mastercycler Realplex2 cycler (Eppendorf, Hamburg, Germany) with 40 cycles consisting of a 15 sec denaturation at 95°C, primer annealing for 15 sec at 55°C, and extension for 30 sec at 72°C. We used the following primer pairs (5'- > 3' orientation): IGF2, CCTCCGACCGTGCTTCC, GGTGGACTGCTTCCAGGTGT; PLAG1, ACAAGTGCATACAACAAGACTGCA, CAGGAGAATGAGTAGCCATGTGC; IGFBP3, GTCCAAGCGGGAGACAGAATAT, CCTGGGACTCAGCACATTGA; TIA1, TTAGCCAGATTGGACCTTGTAAAAA, CGATGCTCATGAAACTCCACA; TBP, GCCCGAAACGCCGAATAT, CCGTGGTTCGTGGCTCTCT. Amplification of the house-keeping gene TATA-Box-binding-Protein (TBP) was performed to standardize the amount of sample RNA according to a previous study [44]. PCR efficiencies for all assays were determined, with slopes ranging from -3.34 to -3.69. The relative quantization of gene expression was performed using the ΔΔct method as previously described [45].
Methylation analyses
Genomic DNA from frozen tumor and normal liver samples (see above) and cell lines was extracted with phenol and chloroform, precipitated with ethanol and dissolved in TE buffer following standard procedures. Genomic DNA (10 μg) from a healthy person was methylated in vitro using 40 U CpG methyltransferase (SssI), S-adenosylmethionine, and NEBuffer2 (New England Biolabs, Frankfurt, Germany) at 37°C for 4 h, precipitated with ethanol, dissolved in TE buffer, and used as a positive control for methylated alleles. Genomic DNA (2 μg) was bisulfite-treated using the EpiTect® Bisulfite Kit (Qiagen, Hilden, Germany) and amplified using primers not specific for methylation status (IGFBP3-BS-F, GGTGTTGAGTTGGTTAGGAGT; IGFBP3-BS-R, AAACAACACCAACAAAATCAA). We cloned the PCR products into the pCR2.1 TOPO vector (Invitrogen, Karlsruhe, Germany) and sequenced six independent clones per sample (MWG Biotech, Ebersberg, Germany).
In addition, the methylation status of the promoter region of IGFBP3 (from -314 to -147 bp upstream of transcriptional start site) gene was analyzed by methylation-specific-PCR (MSP) using the following primer sets 5'- > 3' orientation): methylated (IGFBP3-M-F, TGATTCGGGTTTCGGGCGTGC; IGFBP3-M-R, GCCGACCGCTATATAAAAACCG) and unmethylated (IGFBP3-U-F, GGTGATTTGGGTTTTGGGTGTGTGTAT; IGFBP3-U-R, AAACACACCAACCACTATATAAAAACCAAA). MSP primer design and PCR conditions were performed according to [43]. For DNA demethylation experiments, we used 0.5 μM 5-aza-2'-deoxycytidine (5-Aza-dC; Sigma-Aldrich, Seelze, Germany) for HUH6 and HepT3 cells and 1.25 μM for HepT1, HepG2 and HUH7 cells; 5-the Aza-dC was applied for 5 days and changed daily. Alternatively, Trichostatin A (Sigma-Aldrich) was applied for 24 h in a concentration of 0.1 μM (HUH6 and HepT3) and 0.25 μM (HepT1, HepG2 and HUH7).
Stable transfection
HepT1 cells (5 × 105 cells/6-well plate) were transfected with 1 μg DNA of the pIRES-IGFBP3 expression vector containing full-length IGFBP3 cDNA [46] or the empty vector control using the FuGene 6 transfection reagent (Roche Diagnostics, Mannheim, Germany). After 24 h of transfection, the cells were changed to media containing 1 μg/ml puromycin (Sigma-Aldrich). After 2 weeks of selection, puromycin-resistant colonies were selected and cultured as stable transfected HepT1 clones. Western blot analysis was performed using rabbit polyclonal anti-IGFBP3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit anti-human β-actin (Cell Signaling, Technology, Danvers, MA, USA) antibodies, as previously described [43].
Cell viability assay
For the proliferation assay, 5 × 103 cells were seeded into 96-well plates, and the viability was assessed at the time points indicated using the Cell Proliferation Kit I (Roche Diagnostics) according to the manufacue's protocol. The optical density was measured at a wavelength of 595 nm after the addition of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) labeling reagent on the GENios microplate reader (Tecan, Männedorf, Switzerland).
Colony formation assay
HepT1 cells (5 × 105 cells/well) were transfected in a 6-well plate format with 1 μg of the pIRES-IGFBP3 expression vector or control vector using the FuGene 6 transfection reagent (Roche Diagnostics). They were subsequently cultured in selection media containing 1 μg/ml puromycin (Sigma-Aldrich) for 2 weeks. Colonies were fixed with 100% methanol, stained with 0.1% crystal violet and counted.
Apoptosis analyses
For annexin V-based apoptosis analysis, cells were trypsinized, washed with PBS, and suspended in 500 μl of calcium-containing binding buffer. Cy5-conjugated annexin V (1:100; BioVision, Mountain View, CA, USA) and 5 μM calcein (Invitrogen) were added to the cell suspension. Early apoptotic cells (annexin V and calcein positive) were detected using cell fluorescence assays with an Agilent 2100 Bioanalyzer. The cleavage of poly(ADP-ribose) polymerase was detected as previously described [47] using antibodies for human poly(ADP-ribose) polymerase and human β-actin (both from Cell Signaling Technology, Danvers, MA, USA).
Cell migration assay
HB cells were seeded into 24-well plates and grown to confluency. A wound of approximately 1 mm was inflicted to cell monolayers with a pipette-tip. The wells were washed twice with PBS to remove detached cells and incubated at 37°C with medium in the presence or absence of 1 μg/ml recombinant human IGFBP3 (R&D systems, Wiesbaden, Germany) for 72 h. Images were taken at 0, 24, and 48 h after scratching, and the wound widths were measured and quantified.
Transwell assays
Transwell permeable supports (8 μm pore polycarbonate inserts; Corning Incorporated, Corning, NY, USA) were coated either with collagen or 10% Matrigel® (BD Biosciences, Heidelberg, Germany) in DMEM and subsequently added to 24-wells containing DMEM (negative control) or DMEM/10% FCS/50 ng/ml recombinant human HGF (NatuTec, Frankfurt, Germany) as a chemoattractant. Cells (1 × 105) were seeded in DMEM in the inside compartments and allowed to migrate for 16 h (HUH7) or 72 h (HepG2) in the presence or absence of 1 μg/ml recombinant human IGFBP3 (R&D systems). Afterwards, the inserts were stained with crystal violet solution. Cells from the upper side of the insert were removed by using cotton swabs. Cells attached to the bottom side of the insert were photographed using a Zeiss Axiovert 25 microscope and a Canon 450D camera. For each sample, eight pictures were taken, and the number of cells was calculated by using ImageJ (National Institute of Health, Bethesda, MD, USA) and the Particle Counter plugin. Data for three independent experiments were analyzed using GraphPad Prism Version 3.0 (GraphPad Software, La Jolla, CA, USA).
Statistical analysis
The data are presented either as dot plots or bar graphs, indicating the mean ± SEM. The statistical analyses and Kaplan Meier calculations were performed with GraphPad Prism Version 3.0 using Student's unpaired t-test, Mantel-Cox test, Mann-Whitney-U test, Spearman's rank correlation, one-way ANOVA and Dunnett's test. P < 0.05 was considered to be significant.
Abbreviations
HCC: Hepatocellular carcinoma; HB: Hepatoblastoma; IGF2: Insulin-like growth factor 2; IGFBP3: Insulin-like growth factor binding protein 3; MSP: Methylation-specific-PCR; PLAG1: Pleomorphic adenoma gene 1; RT-PCR: reverse transcription-PCR; TBP: TATA-Box-binding-Protein; TIA1: T-cell-restricted intracellular antigen-1; 5-Aza-dC: 5-aza-2'-deoxycytidine.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
IR carried out the processing of clinical samples, expression measurements, migration assays, 5-Aza-dC treatments, and the methylation-specific PCR, and she participated in the analysis and interpretation of the data. ME performed the Western blot and apoptosis assays. SJ conducted the bisulfite-sequencing study and interpreted the methylation data. JL performed the invasion and migration assays. BH collected the clinical samples and carried out the statistical analyses of the clinical data. JMH performed the pathological diagnosis and staging of tumor specimens. AV participated in the design of the study and helped in the analysis and interpretation of the data. DvS participated in the design of the study and helped to draft the manuscript. RK designed the study, participated in the analysis and interpretation of data, and drafted the manuscript. All of the authors read and approved the final version of this manuscript.
Contributor Information
Ivonne Regel, Email: ivonne.regel@lrz.tum.de.
Melanie Eichenmüller, Email: melanie.eichenmueller@med.uni-muenchen.de.
Saskia Joppien, Email: saskiajoppien@web.de.
Johanna Liebl, Email: johanna.liebl@cup.uni-muenchen.de.
Beate Häberle, Email: beate.haeberle@med.uni-muenchen.de.
Josef Müller-Höcker, Email: josef.mueller-hoecker@med.uni-muenchen.de.
Angelika Vollmar, Email: angelika.vollmar@cup.uni-muenchen.de.
Dietrich von Schweinitz, Email: dietrich.schweinitz@med.uni-muenchen.de.
Roland Kappler, Email: roland.kappler@med.uni-muenchen.de.
Acknowledgements
We are grateful to Nicole Stadler and Fatemeh Promoli for their technical assistance and to Stefan Zahler for critically reading the manuscript. We thank Dr. Michael A. Tainsky (Wayne State University School of Medicine, Detroit) for kindly providing the pIRES-IGFBP3 expression vector. This work was supported by grants from the Deutsche Forschungsgemeinschaft (KA2274/3-1), the Bettina Bräu foundation, Munich, and the Projekt Omnibus foundation, Munich (to R.K.).
References
- Weinberg AG, Finegold MJ. Primary hepatic tumors of childhood. Hum Pathol. 1983;14:512–537. doi: 10.1016/S0046-8177(83)80005-7. [DOI] [PubMed] [Google Scholar]
- Perilongo G, Shafford E, Plaschkes J. SIOPEL trials using preoperative chemotherapy in hepatoblastoma. Lancet Oncol. 2000;1:94–100. doi: 10.1016/S1470-2045(00)00018-8. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Rydzynski J, Von Schweinitz D, Bode U, Hecker H, Weinel P, Burger D, Harms D, Erttmann R, Oldhafer K, Mildenberger H. Pretreatment prognostic factors and treatment results in children with hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer. 2002;95:172–182. doi: 10.1002/cncr.10632. [DOI] [PubMed] [Google Scholar]
- Haberle B, Bode U, von Schweinitz D. Differentiated treatment protocols for high- and standard-risk hepatoblastoma-an interim report of the German Liver Tumor Study HB99. Klin Padiatr. 2003;215:159–165. doi: 10.1055/s-2003-39375. [DOI] [PubMed] [Google Scholar]
- Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- Scotting PJ, Walker DA, Perilongo G. Childhood solid tumours: a developmental disorder. Nat Rev Cancer. 2005;5:481–488. doi: 10.1038/nrc1633. [DOI] [PubMed] [Google Scholar]
- Hartmann W, Waha A, Koch A, Goodyer CG, Albrecht S, von Schweinitz D, Pietsch T. p57(KIP2) is not mutated in hepatoblastoma but shows increased transcriptional activity in a comparative analysis of the three imprinted genes p57(KIP2), IGF2, and H19. Am J Pathol. 2000;157:1393–1403. doi: 10.1016/S0002-9440(10)64652-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Adam G, Cui H, Sandstedt B, Ohlsson R, Ekstrom TJ. Expression, promoter usage and parental imprinting status of insulin-like growth factor II (IGF2) in human hepatoblastoma: uncoupling of IGF2 and H19 imprinting. Oncogene. 1995;11:221–229. [PubMed] [Google Scholar]
- Honda S, Arai Y, Haruta M, Sasaki F, Ohira M, Yamaoka H, Horie H, Nakagawara A, Hiyama E, Todo S, Kaneko Y. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer. 2008;99:1891–1899. doi: 10.1038/sj.bjc.6604754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatkova A, Rouillard JM, Hartmann W, Lamb BJ, Kuick R, Eckart M, von Schweinitz D, Koch A, Fonatsch C, Pietsch T. et al. Amplification and overexpression of the IGF2 regulator PLAG1 in hepatoblastoma. Genes Chromosomes Cancer. 2004;39:126–137. doi: 10.1002/gcc.10307. [DOI] [PubMed] [Google Scholar]
- Clemmons DR. Insulin-like growth factor binding proteins and their role in controlling IGF actions. Cytokine Growth Factor Rev. 1997;8:45–62. doi: 10.1016/S1359-6101(96)00053-6. [DOI] [PubMed] [Google Scholar]
- Modric T, Silha JV, Shi Z, Gui Y, Suwanichkul A, Durham SK, Powell DR, Murphy LJ. Phenotypic manifestations of insulin-like growth factor-binding protein-3 overexpression in transgenic mice. Endocrinology. 2001;142:1958–1967. doi: 10.1210/en.142.5.1958. [DOI] [PubMed] [Google Scholar]
- Hochscheid R, Jaques G, Wegmann B. Transfection of human insulin-like growth factor-binding protein 3 gene inhibits cell growth and tumorigenicity: a cell culture model for lung cancer. J Endocrinol. 2000;166:553–563. doi: 10.1677/joe.0.1660553. [DOI] [PubMed] [Google Scholar]
- Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem. 1997;272:12181–12188. doi: 10.1074/jbc.272.18.12181. [DOI] [PubMed] [Google Scholar]
- Williams AC, Collard TJ, Perks CM, Newcomb P, Moorghen M, Holly JM, Paraskeva C. Increased p53-dependent apoptosis by the insulin-like growth factor binding protein IGFBP-3 in human colonic adenoma-derived cells. Cancer Res. 2000;60:22–27. [PubMed] [Google Scholar]
- Zou T, Fleisher AS, Kong D, Yin J, Souza RF, Wang S, Smolinski KN, Abraham JM, Meltzer SJ. Sequence alterations of insulin-like growth factor binding protein 3 in neoplastic and normal gastrointestinal tissues. Cancer Res. 1998;58:4802–4804. [PubMed] [Google Scholar]
- Hanafusa T, Yumoto Y, Nouso K, Nakatsukasa H, Onishi T, Fujikawa T, Taniyama M, Nakamura S, Uemura M, Takuma Y. et al. Reduced expression of insulin-like growth factor binding protein-3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2002;176:149–158. doi: 10.1016/S0304-3835(01)00736-4. [DOI] [PubMed] [Google Scholar]
- Tomii K, Tsukuda K, Toyooka S, Dote H, Hanafusa T, Asano H, Naitou M, Doihara H, Kisimoto T, Katayama H. et al. Aberrant promoter methylation of insulin-like growth factor binding protein-3 gene in human cancers. Int J Cancer. 2007;120:566–573. doi: 10.1002/ijc.22341. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- Subramaniam K, Ooi LL, Hui KM. Transcriptional down-regulation of IGFBP-3 in human hepatocellular carcinoma cells is mediated by the binding of TIA-1 to its AT-rich element in the 3'-untranslated region. Cancer Lett. 2010;297:259–268. doi: 10.1016/j.canlet.2010.05.019. [DOI] [PubMed] [Google Scholar]
- Gray SG, Kytola S, Lui WO, Larsson C, Ekstrom TJ. Modulating IGFBP-3 expression by trichostatin A: potential therapeutic role in the treatment of hepatocellular carcinoma. Int J Mol Med. 2000;5:33–41. doi: 10.3892/ijmm.5.1.33. [DOI] [PubMed] [Google Scholar]
- Luo SM, Tan WM, Deng WX, Zhuang SM, Luo JW. Expression of albumin, IGF-1, IGFBP-3 in tumor tissues and adjacent non-tumor tissues of hepatocellular carcinoma patients with cirrhosis. World J Gastroenterol. 2005;11:4272–4276. doi: 10.3748/wjg.v11.i27.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AA, Majid S, Nosrati M, de Semir D, Federman S, Kashani-Sabet M. Functional modulation of IGF-binding protein-3 expression in melanoma. J Invest Dermatol. 2010;130:2071–2079. doi: 10.1038/jid.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- Gray SG, Eriksson T, Ekstrom C, Holm S, von Schweinitz D, Kogner P, Sandstedt B, Pietsch T, Ekstrom TJ. Altered expression of members of the IGF-axis in hepatoblastomas. Br J Cancer. 2000;82:1561–1567. doi: 10.1054/bjoc.1999.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann W, Kuchler J, Koch A, Friedrichs N, Waha A, Endl E, Czerwitzki J, Metzger D, Steiner S, Wurst P. et al. Activation of phosphatidylinositol-3'-kinase/AKT signaling is essential in hepatoblastoma survival. Clin Cancer Res. 2009;15:4538–4545. doi: 10.1158/1078-0432.CCR-08-2878. [DOI] [PubMed] [Google Scholar]
- Wiley A, Katsaros D, Fracchioli S, Yu H. Methylation of the insulin-like growth factor binding protein-3 gene and prognosis of epithelial ovarian cancer. Int J Gynecol Cancer. 2006;16:210–218. doi: 10.1111/j.1525-1438.2006.00299.x. [DOI] [PubMed] [Google Scholar]
- de Ibanez Caceres I, Dulaimi E, Hoffman AM, Al-Saleem T, Uzzo RG, Cairns P. Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006;66:5021–5028. doi: 10.1158/0008-5472.CAN-05-3365. [DOI] [PubMed] [Google Scholar]
- Antequera F, Boyes J, Bird A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 1990;62:503–514. doi: 10.1016/0092-8674(90)90015-7. [DOI] [PubMed] [Google Scholar]
- Paz MF, Fraga MF, Avila S, Guo M, Pollan M, Herman JG, Esteller M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003;63:1114–1121. [PubMed] [Google Scholar]
- Smiraglia DJ, Rush LJ, Fruhwald MC, Dai Z, Held WA, Costello JF, Lang JC, Eng C, Li B, Wright FA. et al. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum Mol Genet. 2001;10:1413–1419. doi: 10.1093/hmg/10.13.1413. [DOI] [PubMed] [Google Scholar]
- Shen Y, Chow J, Wang Z, Fan G. Abnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cells. Hum Mol Genet. 2006;15:2623–2635. doi: 10.1093/hmg/ddl188. [DOI] [PubMed] [Google Scholar]
- Chang YS, Wang L, Liu D, Mao L, Hong WK, Khuri FR, Lee HY. Correlation between insulin-like growth factor-binding protein-3 promoter methylation and prognosis of patients with stage I non-small cell lung cancer. Clin Cancer Res. 2002;8:3669–3675. [PubMed] [Google Scholar]
- Chang YS, Wang L, Suh YA, Mao L, Karpen SJ, Khuri FR, Hong WK, Lee HY. Mechanisms underlying lack of insulin-like growth factor-binding protein-3 expression in non-small-cell lung cancer. Oncogene. 2004;23:6569–6580. doi: 10.1038/sj.onc.1207882. [DOI] [PubMed] [Google Scholar]
- Perry AS, Loftus B, Moroose R, Lynch TH, Hollywood D, Watson RW, Woodson K, Lawler M. In silico mining identifies IGFBP3 as a novel target of methylation in prostate cancer. Br J Cancer. 2007;96:1587–1594. doi: 10.1038/sj.bjc.6603767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA, Schroeder I, Factor VM, Thorgeirsson SS. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117:2713–2722. doi: 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJ, Anzalone L, Pezzani L, Di Giacomo AM, Fonsatti E. et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol. 2007;212:330–344. doi: 10.1002/jcp.21066. [DOI] [PubMed] [Google Scholar]
- Aishima S, Basaki Y, Oda Y, Kuroda Y, Nishihara Y, Taguchi K, Taketomi A, Maehara Y, Hosoi F, Maruyama Y. et al. High expression of insulin-like growth factor binding protein-3 is correlated with lower portal invasion and better prognosis in human hepatocellular carcinoma. Cancer Sci. 2006;97:1182–1190. doi: 10.1111/j.1349-7006.2006.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappler R, von Schweinitz D. In: Pediatric Liver Tumors. Zimmermann A, Perilongo G, editor. Heidelberg: Springer; 2011. Molecular aspects of hepatoblastoma; pp. 27–42. [Google Scholar]
- Pietsch T, Fonatsch C, Albrecht S, Maschek H, Wolf HK, von Schweinitz D. Characterization of the continuous cell line HepT1 derived from a human hepatoblastoma. Lab Invest. 1996;74:809–818. [PubMed] [Google Scholar]
- Lopez-Terrada D, Cheung SW, Finegold MJ, Knowles BB. Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol. 2009;40:1512–1515. doi: 10.1016/j.humpath.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Eichenmuller M, Gruner I, Hagl B, Haberle B, Muller-Hocker J, von Schweinitz D, Kappler R. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology. 2009;49:482–490. doi: 10.1002/hep.22649. [DOI] [PubMed] [Google Scholar]
- Specht K, Kremer M, Muller U, Dirnhofer S, Rosemann M, Hofler H, Quintanilla-Martinez L, Fend F. Identification of cyclin D1 mRNA overexpression in B-cell neoplasias by real-time reverse transcription-PCR of microdissected paraffin sections. Clin Cancer Res. 2002;8:2902–2911. [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman AL, Rosati R, Li Q, Tainsky MA. Epigenetic and functional analysis of IGFBP3 and IGFBPrP1 in cellular immortalization. Biochem Biophys Res Commun. 2007;357:785–791. doi: 10.1016/j.bbrc.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Eichenmuller M, von Schweinitz D, Kappler R. Betulinic acid treatment promotes apoptosis in hepatoblastoma cells. Int J Oncol. 2009;35:873–879. doi: 10.3892/ijo_00000402. [DOI] [PubMed] [Google Scholar]