

Abstract
Malaria is a protozoal parasitic disease that is widespread in tropical and subtropical regions of Africa, Asia, and the Americas and causes more than 800,000 deaths per year. The continuing emergence of multi-drug-resistant Plasmodium falciparum drives the ongoing need for the development of new and effective antimalarial drugs. Our previous work has explored the preliminary structural optimization of 4(1H)-quinolone ester derivatives, a new series of antimalarials related to the endochins. Herein, we report the lead optimization of 4(1H)-quinolones with a focus on improving both antimalarial potency and bioavailability. These studies led to the development of orally efficacious antimalarials including quinolone analogue 20g, a promising candidate for further optimization.
Keywords: Malaria, Endochin, 4(1H)-quinolone, Orally Efficacious Antimalarial
INTRODUCTION
Malaria is a devastating disease that annually infects more than 240 million people and kills almost 1 million.1 Treatment and control of malaria has become increasingly difficult because of the spread of drug resistance to most antimalarial drugs including chloroquine, mefloquine, atovaquone/proguanil, primaquine, and sulphadoxine/pyrimethamine.2–5 Artemisinin-based combination therapy (ACT) has been mandated as the first-line treatment for malaria caused by P. falciparum by the WHO and led to a considerable decrease of the burden of this disease.1, 6 Nevertheless, ACT resistance appears to be emerging on the Cambodia-Thailand border, leading to concern about long term viability of this class of antimalarials.7, 8 Thus, there remains a need to discover and develop new and effective antimalarial agents.9–11 We have previously reported the discovery of the 4(1H)-quinolones, which are structurally related to a class of antimalarial compounds known as the endochins.12
Endochin (Figure 1, 1), discovered in the 1940s, possesses prophylactic and therapeutic activity in canaries infected with Plasmodium praecox or Plasmodium gallinaceum13, 14 but lacks activity in mammalian malarial models. The related 4(1H)-quinolone ester 2 (ICI 56,780), discovered in the 1960’s, exhibits blood schizonticidal activity against P. berghei in rodents as well as prophylactic and anti-relapse activity against P. Cynomolgi in monkeys.15,16 Compound 2 was abandoned after rapid acquisition of resistance was observed in a murine model. Dihydroacridinedione 3 (WR 243,251), discovered in the 1990’s, showed potent blood schizonticidal activity in P. falciparum-infected monkey model.17, 18 Mechanistic studies suggested that compound 3 targeted the parasite’s mitochondrial respiratory pathway and modest cross resistance was noted with atovaquone.19 The ketone hydrolysis product, a potential metabolite of compound 3, appeared to target the quinol oxidation site of the cytochrome bc1 complex.20 Recently, researchers in Riscoe and Manetsch groups reported several series of structurally related compounds displaying remarkable in vitro antimalarial activity.21–30 These data together with our prior work suggested that 4(1H)-quinolone derivatives might possess potential as antimalarial agents and warranted further development.
Figure 1.

Structures of Compounds 1 – 4
Our own series contained the same core quinolone pharmacophore as the previously studied series, but had several unique substituents including: a carboxylic ester functionality at the 3-postion, a meta substituted aromatic ring at the 2-position, and the absence of the long linear side chain at the 3-position. Herein, we report extensive structure-activity and structure-property profiling of 4(1H)-quinolones carried out to improve antimalarial potency and physicochemical properties as well as in vivo pharmacokinetics and efficacy studies of several 4(1H)-quinolone derivatives in murine models.
CHEMISTRY
All 4(1H)-quinolone compounds were synthesized using a previously reported general route (Scheme 1).12 Briefly, substituted anilines 5 were subjected to Gould-Jacobos cyclization with (2-ethoxymethylene) malonate to yield the corresponding quinolone intermediates 6. As described in our previous paper, simple bromination of 6 was not effective and a lengthier route was required.12 Then, the chloro, bromo intermediates 7 were prepared by sequential chlorination, oxidization, and bromination of quinolones 6. A variant aromatic group was introduced to the 2-position of quinolones 7 by Suzuki coupling reactions with the appropriate boronic acids. Subsequent hydrolysis afforded targeted quinolone compounds 8–20.
Scheme 1.

General route for synthesis of 4(1H)-quinolone derivatives 8–20
a Reagents and conditions: a) (2-ethoxymethylene)malonate, EtOH, 130 °C, 3 h; b) Ph2O, reflux 4–6 h; c) POCl3, 1,4-dioxane, 120 °C, 1 h; d) m-chloroperoxybenzoic acid, CHCl3, rt, 4 h; e) POBr3, CHCl3, 24 °C, 1h; f) appropriate boronic acid, Pd(PPh3)4, CsCO3, 1,4-dioxane/H2O, 75 °C, 3 h; g) AcOH/H2O (9:1), 120 °C, 1–2 h.
Quinolone derivatives with a methylthio substituent at the 7- position of the benzenoid ring could not be prepared using the general route. Instead, methylthio aniline 21 was reacted with benzoyl chloride 22 to afford amide 23. After the chlorination with phosphorus pentachloride, the resulting intermediate was treated with sodium diethyl malonate to give the phenylamino phenylmethylene malonate 24. Thermal ring closure afforded the desired 7-methylthioquinolone derivative 14 (Scheme 2).
Scheme 2.

Synthesis of 4(1H)-quinolone derivative 14
a Reagents and conditions: a) toluene, 24 °C, 3 h, 60%; b) PCl5, 1,4-dioxane, 110 °C, 1 h; c) sodium diethyl malonate, toluene, 110 °C, 6 h, 30% over 2 steps; d) Ph2O, 170 °C, 4 h, 28%.
Compound series 27 & 28 were prepared to examine the effect of groups at the 3-position on the antimalarial activity. As shown in Scheme 3, 3-ethyl carboxylic ester 25 was saponified to give carboxylic acid 26. The desired quinolone series 27 & 28 were then synthesized by coupling carboxylic acid 26 with alkyl alcohols amines in the presence of HBDU.
Scheme 3.

Synthesis of 4(1H)-quinolone derivative 27–28
a Reagents and conditions: a) KOH, H2O/EtOH (20/1), 75 °C, 40 h; b) DIPEA, HBTU, appropriate alkyl alcohol or alkyl amine, 40 °C, 2 h.
All compounds used in the studies described below were purified by reverse phase preparative HPLC to greater than 95% purity prior to further work. Identity was confirmed by both LC/MS and proton NMR analysis. Purity was confirmed by parallel LC/MS/ELSD/CLND with purity being assigned to the average purity determined by all spectral methods. Compounds were formulated as 10 mM stock solutions in DMSO with the concentrations being confirmed by CLND analysis.
RESULTS AND DISCUSSIONS
Structure-activity relationship studies
All compounds described in this work were evaluated for antimalarial activity against a panel of P. falciparum strains including K1 (chloroquine and pyrimethamine resistant), TM90-C2B (chloroquine, mefloquine, pyrimethamine, and atovaquone resistant), D10 (chloroquine sensitive), and D10_yDHOD (transfected D10 with yeast dihydroorate dehydrogenase, thereby DHODH inhibitor resistant) using a previously described assay.31 These strains were chosen based on the assumption that the cellular target for the quinolones was most likely either bc1 or DHODH. Each compound was tested in concentration-response experiments, using 10-point, 3-fold dilution schemes (spanning 15 µM to 0.7 nM), to define potency. Each experiment was performed in triplicate, and all experiments were independently replicated at least twice. Data are reported as average values based upon all replications of the experiments; EC50 values were derived by fitting pooled data from all replicate experiments; the standard deviations for these are reported in the Supporting Information. The data is summarized in a heat map and table in Supporting Information.
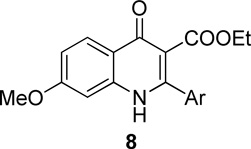
Our prior studies revealed that a meta-substituted aryl group at the 2-position appeared to be an essential feature of potent 4(1H)-quinolones. To investigate this trend more carefully, 7-methoxy-4(1H)-quinolones 8a-8aa, with various meta-substituted phenyl rings at the 2-position, were synthesized following a fusion of the Topliss and Hansch schemes, including systematic variation in hydrophobics, electrostatics, and sterics.32, 33 The antimalarial activity of 8a-8aa is summarized in Table 1. Quinolone 8a, the original lead, displayed an EC50 in the submicromolar range against both K1 (0.3 µM) and TM90-C2B (0.91 µM). Analog 8b, in which the (m,p-methylenedioxy)phenyl group at the 2-position were replaced by an unsubstituted phenyl ring, showed at least 2-fold reduced potency. Introduction of hydrophobic meta-substituents such as methyl 8c, vinyl 8d, and phenyl 8e gave equal or improved potency compared to 8a. Compounds 8f–n, bearing alkoxy groups, displayed great improvement of antimalarial potency against both K1 and TM90-C2B strains. In contrast to 8a, compound 8v bearing a N,N-dimethylamino substituent showed a reduced potency against K1 strain but an improved potency against TM90-C2B strain. Next, compounds 8p–r, containing meta-halo phenyls at the 2- position, were tested. Substitution with m-chloro or bromo gave a slightly improved potency, while the fluoro analog had a reduction in potency. Quinolones carrying a strongly electron-withdrawing group on 2-phenyl ring such as acetyl 8s, methylsulfone 8t, or nitro 8u, displayed weak antimalarial activity. Furthermore, compounds 8w–y containing an H-bond donor at the meta-position had poor or no antimalarial activity. Finally, compounds 8z and 8aa with a di-substituted phenyl ring, showed a similar potency in contrast to 8a. Overall, these studies show that hydrophobic electron-donating groups at the meta-position of the 2-phenyl are an essential feature for antimalarial potency. There is reasonable flexibility in the steric demand if this criteria is met.
Table 1.
SAR of 7-Methoxy 4(1H)-quinolone derivatives 8a-aa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| CMPD | Ar (y) |
EC50 K1 (µM) |
EC50 C2B (µM) |
EC50 D10 (µM) |
EC50 D10_yD HOD (µM) |
EC50 D10_yD HOD+P G (µM) |
EC50 HEK293 (µM) |
|
| 8a | 0.30 | 0.91 | 0.06 | >15 | 0.07 | >22 | ||
| 8b | 0.82 | >15 | 0.60 | >15 | >15 | >22 | ||
| 8c | 0.30 | 0.06 | 0.01 | >15 | <0.001 | >22 | ||
| 8d | 0.22 | 0.18 | <0.001 | >15 | <0.001 | >22 | ||
| 8e | 0.07 | <0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8f | 0.13 | 0.21 | 0.04 | >15 | <0.001 | >22 | ||
| 8g | 0.07 | 0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8h | 0.02 | <0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8i | 0.16 | <0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8j | 0.03 | <0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8k | 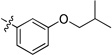 |
0.04 | <0.001 | 0.002 | >15 | <0.001 | >22 | |
| 8l | 0.08 | <0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8m | 0.03 | <0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8n | 0.11 | <0.001 | <0.001 | >15 | <0.001 | >22 | ||
| 8o | 0.22 | >15 | 0.01 | >15 | <0.001 | >22 | ||
| 8p | 0.55 | 7.7 | 0.27 | >15 | 2.1 | >22 | ||
| 8q | 0.18 | >15 | <0.001 | >15 | <0.001 | >22 | ||
| 8r | 0.13 | 1.8 | <0.001 | >15 | 0.41 | >22 | ||
| 8s | 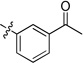 |
1.45 | <0.001 | 1.0 | >15 | >15 | >22 | |
| 8t | 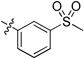 |
21 | 6.3 | >15 | >15 | >15 | >22 | |
| 8u | 0.87 | 2.3 | 0.27 | >15 | 0.41 | >22 | ||
| 8v | 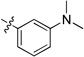 |
0.95 | 0.02 | 0.27 | >15 | 0.10 | >22 | |
| 8w | 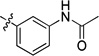 |
14 | 8.8 | >15 | >15 | >15 | >22 | |
| 8x | >15 | >15 | >15 | >15 | >15 | >22 | ||
| 8y | >15 | >15 | >15 | >15 | >15 | >22 | ||
| 8z | 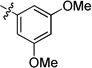 |
0.26 | 0.01 | 0.02 | >15 | <0.001 | >22 | |
| 8aa | 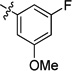 |
0.13 | 0.29 | <0.001 | >15 | <0.001 | >22 | |
| Atovaquone | 0.015 | >1.5a | 0.016 | >1.5 a | 0.002 | ndb | ||
the highest final concentration of atovaquone was 1.5 µM.
nd, not determined
The effects of hydrophobic, electronic, and steric factors were more formally examined using the methods of Hansch with regard to hydrophobicity (the relationship between pEC50 and Hansch constant π, Figure 2A) and electronic contributions (the relationship between pEC50 and Hammett constant σ, Figure 2B). We observed a significant linear correlation between pEC50 (K1) and hydrophobicity (slope = 0.7421, p = 3.6 × 10−6), while there was no linear correlation between antimalarial activity and electronic effects. While quinolones with more hydrophobic substituents clearly tended to be more potent as antimalarials, hydrophobicity of small molecules often reduces their permeability and aqueous solubility. Maintaining the balance between bioactivity and physicochemical properties is critical to finding drug-like small molecules. Thus, the subsequent studies were restricted to a small subset of the substitutents that had the best balance
Figure 2.

Quantitative analysis of structure activity relationships for quinolone derivatives 8a-aa in inhibition of P. falciparum K1 strain A) hydrophobicity; B) electronic effect.
Our original studies showed that analogs bearing a 7-methoxy group on the benzenoid ring generally had more antimalarial activity than compounds with a 5-methoxy substituent.12 This suggested that the substitution pattern of the benzenoid ring was another feature important for antimalarial potency. A series of compounds 9–20, varying substituents on the benzenoid ring while holding constant the meta-substituted phenyl ring at the 2-position was prepared and tested (Table 2). In this series of quinolone derivatives 9–14 the potency against the K1 strain of P. falciparum followed the trend of 7-MeO ~ 7-MeS > 7-OH ~ 7-iPrO > 7-Cl >> 7-CF3O > 7-CF3. This finding demonstrates that a small hydrophobic electron-donating group at the 7-position benefits the antimalarial potency of 4(1H)-quinolone analogues, while an electron-withdrawing group causes a significant decrease in potency.
Table 2.
SAR of 4(1H)-quinolone derivatives 9–14
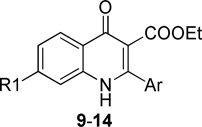 | ||||||||
|---|---|---|---|---|---|---|---|---|
| CMPD | R1 | Ar (y) |
EC50 K1 (µM) |
EC50 C2B (µM) |
EC50 D10 (µM) |
EC50 D10_yDH OD (µM) |
EC50 D10_yDH OD+PG (µM) |
EC50 HEK29 3 (µM) |
| 9a | -OiPr | 6.6 | >15 | >15 | >15 | >15 | >22 | |
| 9b | -OiPr | 0.081 | <0.001 | <0.001 | >15 | <0.001 | >22 | |
| 9c | -OiPr | 2.2 | 1.6 | 0.65 | >15 | 0.42 | >22 | |
| 10a | -OCF3 | >15 | >15 | >15 | >15 | >15 | >22 | |
| 10b | -OCF3 | 1.3 | >15 | >15 | >15 | >15 | >22 | |
| 10c | -OCF3 | >15 | 5.8 | 4.9 | >15 | 9.4 | >22 | |
| 11a | -CF3 | >15 | >15 | >15 | >15 | >15 | >22 | |
| 11b | -CF3 | 3.4 | >15 | >15 | >15 | >15 | >22 | |
| 11c | -CF3 | >15 | >15 | >15 | >15 | 3.3 | >22 | |
| 11d | -CF3 | >15 | >15 | >15 | >15 | >15 | >22 | |
| 12a | -OH | 0.12 | 0.01 | 0.06 | >15 | 0.02 | >22 | |
| 12b | -OH | 0.18 | 0.22 | 0.06 | >15 | 0.04 | >22 | |
| 13 | -Cl | 0.38 | <0.001 | 0.20 | >15 | <0.001 | >22 | |
| 14 | -SMe | 0.28 | 5.1 | 1.3 | >15 | 1.6 | >22 | |
| Atovaquone | 0.015 | >1.5a | 0.016 | >1.5 a | 0.002 | ndb | ||
the highest final concentration of atovaquone was 1.5 µM.
nd, not determined
This finding led to an examination of derivatives containing a fluoro, chloro, or methoxy group at the 7-position of the benzenoid ring 15–20 combined with a second substituent at the 5- or 6-position (Table 3). Analogues with 5,7-difluoro groups 15a–d and with 6,7-dichloro groups 18a–d showed submicromolar EC50 values against the K1 strain; however those di-halogen substituted quinolones were inactive against the multidrug resistant TM90-C2B strain. Compounds bearing 6,7-dimethoxy groups 16a–d exhibited a reduction in potency in comparison with their 7-methoxy counterparts. The simultaneous incorporation of 6-methoxy and 7-chloro substituents in analogs 17a–d, caused complete loss of antimalarial activity against both strains. Finally, compounds 19 and 20, containing the 6-halo-7-methoxy substitution pattern, possessed antimalarial potency ranging from submicromolar to nanomolar. For example, compounds 19d, 20b, and 20d containing a (meta-phenyl)phenyl or (meta-phenoxy) phenyl at the 2-position, showed EC50’s < 10 nM for both K1 and TM90-C2B strains. Although quinolone derivatives with a 2-(meta-halo)phenyl group, 19e–f and 20f–h, showed similar potency to the corresponding 7-methoxy quinolones 8p–r against the K1 strain, 19e–f and 20f–h displayed significant improvement of antimalarial activity against the TM90-C2B strain. Analogues 20j–l were designed to introduce a group that would force the 2-aryl group out-of-plane, which is one of strategies to improve solubility by disrupting crystal packing. These three compounds 20j–l displayed a slight decrease in activity against K1 strain compared to derivative 20b and 20d. The final two analogues 20m–n were prepared to quickly evaluate whether the presence of heterocyclic functional groups at the meta- position of 2-phenyl group would change the potency of quinolones. These analogues displayed reduced antimalarial activity.
Table 3.
SAR of 4(1H)-quinolone derivatives 15–20
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CMPD | R1 | R2 | R3 | Ar (y) |
EC50 K1 (µM) |
EC50 C2B (µM) |
EC50 D10 (µM) |
EC50 D10_yD HOD (µM) |
EC50 D10_y DHOD +PG (µM) |
EC50 HEK 293 (µM) |
| 15a | -F | -H | -F | 0.04 | >15 | 0.02 | >15 | 0.01 | >22 | |
| 15b | -F | -H | -F | 0.06 | >15 | 0.06 | >15 | 0.05 | >22 | |
| 15c | -F | -H | -F | 0.01 | 1.2 | 0.01 | >15 | 0.01 | >22 | |
| 15d | -F | -H | -F | 0.36 | >15 | 0.33 | >15 | 0.58 | >22 | |
| 16a | -OMe | -OMe | -H | >15 | >15 | >15 | >15 | >15 | >22 | |
| 16b | -OMe | -OMe | -H | 0.38 | 0.03 | 0.35 | >15 | 0.08 | >22 | |
| 16c | -OMe | -OMe | -H | >15 | >15 | >15 | >15 | >15 | >22 | |
| 16d | -OMe | -OMe | -H | 1.5 | 0.47 | 1.4 | >15 | 0.50 | >22 | |
| 17a | -Cl | -OMe | -H | >15 | >15 | >15 | >15 | >15 | >22 | |
| 17b | -Cl | -OMe | -H | >15 | >15 | >15 | >15 | >15 | >22 | |
| 17c | -Cl | -OMe | -H | >15 | 1.5 | >15 | >15 | 6.9 | >22 | |
| 17d | -Cl | -OMe | -H | >15 | >15 | >15 | >15 | >15 | >22 | |
| 18a | -Cl | -Cl | -H | 0.10 | >15 | 0.14 | >15 | 0.16 | >22 | |
| 18b | -Cl | -Cl | -H | 0.23 | 0.02 | 0.17 | >15 | 0.04 | >22 | |
| 18c | -Cl | -Cl | -H | 0.54 | >15 | 0.66 | >15 | 0.34 | >22 | |
| 18d | -Cl | -Cl | -H | 0.05 | >15 | 0.05 | >15 | 0.05 | >22 | |
| 19a | -OMe | -Cl | -H | 0.03 | 0.05 | 0.03 | >15 | 0.035 | >22 | |
| 19b | -OMe | -Cl | -H | 0.02 | 0.01 | 0.02 | >15 | 0.004 | >22 | |
| 19c | -OMe | -Cl | -H | 0.06 | 0.12 | 0.05 | >15 | 0.07 | >22 | |
| 19d | -OMe | -Cl | -H | 0.01 | 0.01 | 0.01 | >15 | 0.002 | >22 | |
| 19e | -OMe | -Cl | -H | 0.17 | 0.11 | 0.13 | >15 | 0.13 | >22 | |
| 19f | -OMe | -Cl | -H | 0.18 | 0.12 | 0.11 | >15 | 0.11 | >22 | |
| 19g | -OMe | -Cl | -H | 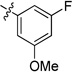 |
0.03 | 0.03 | 0.02 | >15 | 0.02 | >22 |
| 20a | -OMe | -F | -H | 0.21 | 0.55 | 0.12 | >15 | 0.12 | >22 | |
| 20b | -OMe | -F | -H | 0.01 | <0.001 | 0.01 | >15 | <0.001 | >22 | |
| 20c | -OMe | -F | -H | 0.09 | 0.17 | 0.06 | >15 | 0.05 | >22 | |
| 20d | -OMe | -F | -H | 0.01 | 0.01 | 0.01 | >15 | 0.01 | >22 | |
| 20e | -OMe | -F | -H | 0.13 | 0.10 | 0.09 | >15 | 0.09 | >22 | |
| 20f | -OMe | -F | -H | 0.95 | 1.9 | 0.87 | >15 | 1.1 | >22 | |
| 20g | -OMe | -F | -H | 0.08 | 0.20 | 0.05 | >15 | 0.07 | >22 | |
| 20h | -OMe | -F | -H | 0.12 | 0.15 | 0.10 | >15 | 0.11 | >22 | |
| 20i | -OMe | -F | -H | 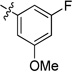 |
0.04 | 0.05 | 0.03 | >15 | 0.03 | >22 |
| 20j | -OMe | -F | -H | 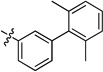 |
0.20 | 0.01 | 0.03 | >15 | 0.01 | >22 |
| 20k | -OMe | -F | -H | 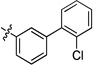 |
0.02 | <0.001 | 0.01 | >15 | <0.001 | >22 |
| 20l | -OMe | -F | -H | 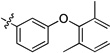 |
0.08 | 0.01 | 0.03 | >15 | 0.01 | >22 |
| 20m | -OMe | -F | -H | 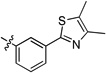 |
>15 | >15 | >15 | >15 | >15 | >22 |
| 20n | -OMe | -F | -H | 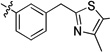 |
0.29 | 0.08 | 0.19 | >15 | 0.13 | >22 |
| Atovaquone | 0.015 | >1.5a | 0.016 | >1.5 a | 0.002 | ndb | ||||
the highest final concentration of atovaquone was 1.5 µM.
nd, not determined
The studies described above allowed the optimization of 4(1H) quinolones with respect to the aromatic ring at the 2-position and the benzenoid ring. The most promising quinolone analogues possessed the 6-halo-7-methoxy substitution pattern on the benzenoid ring along with a meta-substituted hydrophobic aromatic ring at the 2- position. While increasing hydrophobicity of the aromatic group at the 2-position enhanced antimalarial activity, it also reduced solubility.
Compound sets 27 and 28 were designed to test the possibility of maintaining antimalarial potency while improving solubility by introducing a solubilizing chain to the carbonyl functionality at the 3-position (Table 4). The length of the solubilizing chain varied between two and six carbons and incorporated morpholinyl, pyrrolidinyl, and N,N-dimethylamino functionalities. The antimalarial potency of this series followed the trend: morpholinylalkyl esters ~ pyrrolidinylalyl esters > N,N-dimethylamino alkyl esters. For analogues with morpholinylalkyl esters, the potency of compounds with a 3 carbon chain length 27b and 28b was higher than that with 2 carbon chain length 27a and 28a; however, the potency for compounds with N,N-dimethylamino alkyl esters 27c–e followed the trend of 2-carbon chain > 3-carbon chain > 6-carbon chain. Generally, compounds with carboxylic esters 28b–c were more potent than those with carboxylic amide groups 28d–e. Two compounds 28g–h containing glycol derivatives, exhibited low nanomolar antimalarial activity. Overall, all of these substitutions substantially reduced potency, without affording significant increases in solubility.
Table 4.
SAR of 4(1H)-quinolone derivatives 27–28
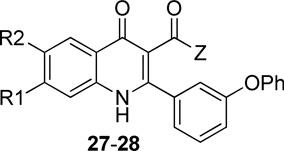 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| CMPD | R1 | R2 | Z | EC50 K1 (µM) |
EC50 C2B (µM) |
EC50 D10 (µM) |
EC50 D10_y DHO D (µM) |
EC50 D10_y DHO D+PG (µM) |
EC50 HEK2 93 (µM) |
| 27a | -OMe | -H | 0.53 | 0.11 | 0.22 | >15 | 0.07 | >22 | |
| 27b | -OMe | -H | 0.35 | 0.08 | 0.16 | >15 | 0.09 | >22 | |
| 27c | -OMe | -H | 0.64 | 0.45 | 0.34 | >15 | 0.24 | >22 | |
| 27d | -OMe | -H | 6.3 | >15 | >15 | 6.5 | 8.8 | >22 | |
| 27e | -OMe | -H | >15 | >15 | 1.1 | >15 | 0.66 | >22 | |
| 28a | -OMe | -H | 0.82 | 0.04 | 0.15 | >15 | 0.07 | >22 | |
| 28b | -OMe | -F | 0.16 | 0.02 | 0.07 | >15 | 0.04 | >22 | |
| 28c | -OMe | -F | 0.52 | 0.18 | 0.19 | >15 | 0.12 | >22 | |
| 28d | -OMe | -F | 7.3 | 0.10 | >15 | >15 | 0.34 | >22 | |
| 28e | -OMe | -F | 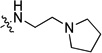 |
>15 | 1.8 | >15 | >15 | 8.5 | >22 |
| 28f | -OMe | -F | >15 | >15 | >15 | >15 | >15 | >22 | |
| 28g | -OMe | -F | 0.10 | 0.05 | 0.03 | >15 | 0.03 | >22 | |
| 28h | -OMe | -F | 0.42 | 0.11 | 0.22 | >15 | 0.16 | >22 | |
| Atovaquone | 0.015 | >1.5a | 0.016 | >1.5 a | 0.002 | ndb | |||
the highest final concentration of atovaquone was 1.5 µM.
nd, not determined.
Strain selectivity
All quinolone derivatives were screened against three P. falciparum strains known to be resistant to different antimalarial agents, including K1 (chloroquine resistant), TM90-C2B (multi-drug resistant, atovaquone resistant), and D10 (chloroquine sensitive). The resistance of K1 strain to chloroquine is conferred by mutation in pfcrt gene that codes for the chloroquine resistance transporter PfCRT.34, 35 The multi-drug-resistant strain TM90-C2B has a Tyr268Cys mutation in cytochrome bc1, within the quinol oxidization site, that produces the atovaquone resistance.19 Strain selectivity was used to evaluate the influence of these mechanisms on quinolone activity. The pEC50 values for K1 were plotted against that for D10 strain (Figure 3A), whereas the pEC50 values for multi-drug resistant strain TM90-C2B was plotted against pEC50 values for K1 (Figure 3B).
Figure 3.


Strain correlation plot. A) Plot of pEC50 for K1 vs. pEC50 for D10; B) Plot of pEC50 for TM90-C2B vs pEC50 forK1. Only compounds with precisely determined EC50 values for three strains have been considered in these two plots.
4(1H)-Quinolone compounds generally exhibited similar potencies against K1 and D10 strains with less than 3-fold difference, the historical minimal significant difference between EC50 values, although some outliers (compounds 8c, 8k, 8o, 8z, 14, and 20j) were observed. This implies that the mutation in the pfcrt gene does not influence the antimalarial potency of quinolone compounds.11 The majority of 4(1H)-quinolone compounds displayed minimal strain dependence between the TM90-C2B and K1 strains. However, a noticeable group of compounds showed more potency against TM90-C2B relative K1, while only one compound 14 exhibited better potency against K1. Among those outliers, compounds 20j, and 20l, in which functional groups were introduced to the meta-position of 2-phenyl ring to force it out-of-plane, showed >25-fold improvement in potency against TM90-C2B. Compounds with basic chain moiety at the 3-postion (including 27a, 27b, 28a, 28b, 28d) were another class of derivatives showing more activity against TM90-C2B. The results suggest that the mutation in the cytochrome bc1 of the TM90-C2B does not convey resistance to quinolones but that the compounds do interact with the quinol site of bc1.
Simultaneously, the cytotoxicity of all quinolone compounds was investigated against four human cell lines HEK293, BJ, Raji, and HepG2 (The EC50 values on HEK293 are listed in Table 1–4. The EC50 values on BJ, Raji, and HepG2 are listed in Supporting Information). All quinolone compounds showed low cytotoxicity against all four human cell lines with EC50 values higher than 21.5 µM, the highest dose tested. This suggests there is no intrinsic cytotoxic behavior of the compounds that should lead to significant development difficulties.
Mitochondrial electron transport studies using D10 and D10_yDHOD
To further evaluate whether quinolone compounds targeted mitochondrial electron transport,36 all quinolone derivatives were tested against the transgenic strain D10_yDHOD, which over-expresses yeast dihydroorate dehydrogenase (DHODH). In the parasite electron transport chain, mitochondrial dehydrogenanses generate reduced coenzyme Q (CoQ), which then is re-oxidized by cytochrome bc1. Dihydroorotate dehydrogenase (DHODH) is one of the parasite mitochondrial dehydrogenanses, and is an essential enzyme for P. falciparum pyrimidine biosynthesis.37, 38 Therefore, the function of the cytochrome bc1 complex of parasites is linked to the pyrimidine biosynthesis pathway. The D10_yDHOD strain over-expresses transgenic yeast DHODH in the cytoplasm of the parasite, thus allowing pyrimidine synthesis. As a result, the pyrimidine biosynthesis pathway for D10_yDHOD is independent of cytochrome bc1 complex function. Compounds that exhibit reduced activity in D10_yDHOD in the comparison with D10, are likely to target either DHODH or cytochrome bc1 complex.39 When D10_yDHOD parasites are co-treated with bc1 inhibitors and proguanil together, the antimalarial activity of the bc1 inhibitor is restored because proguanil collapses mitochondrial membrane potential. Consequently, compounds displaying synergistic action with proguanil are likely to target the cytochrome bc1 complex of parasites whereas those that do not target DHODH. Furthermore, when compounds demonstrate synergistic action with proguanil and resistance to TM90-C2B, the cytochrome bc1 will most likely be the target, and the tyrosine residue at position 268 in wild-type cytochrome bc1 is within the binding area.
Most of screened quinolone derivatives demonstrated high potency against the D10 strain and significantly reduced potency against the D10_yDHOD strain. This finding suggests that DHODH and cytochrome bc1 complex of parasites were possible targets for these quinolone derivatives. Furthermore, antimalarial potencies against D10_yDHOD were restored when quinolone compounds were combined with proguanil suggesting that the quinolones most likely to target the mitochondrial cytochrome bc1 complex. The fact that some quinolones display both synergistic action with proguanil and retain activity in the TM90-C2B strain suggests that that the residue at position 268 of the cytochrome bc1 does not contribute the binding of quinolones to bc1.
Subsequently, enzyme inhibition assay were performed to test the molecular targets of the quinolones. Four quinolones with a wide range of EC50 values against the TM90-C2B strain (8p, 20a, 20d, and 20g) were selected for these studies. None showed inhibition of PfDHODH (IC50 > 30 µM, Supporting information). This result rules out PfDHODH as the target of quinolone compounds. Overall these data are very consistent with the quinolone series targeting mitochondrial electron flux, and potentially bc1, but doing so in ways that are subtly different from atovaquone.
Physicochemical properties, in vitro metabolism, and mouse systemic exposure profile
These SAR studies revealed several highly potent 4(1H)-quinolone derivatives (EC50 values < 10 nM). However, it is important to find the balance between in vitro bioactivity, physicochemical properties, and ADME properties. Therefore, all quinolone analogues were tested to assess aqueous solubility and membrane permeability. The complete data for physicochemical properties are listed in Supporting Information. The values for solubility and PAMPA permeability are reported as average values based upon three replications of each experiment. Generally the solubility ranged from poor to acceptable and the permeability for these compounds at pH 7.4 was in a reasonable range (> 100 × 106 cm−1 Pe) for all compounds. For the entire set of quinolone analogues, there were no clear associations between antimalarial activity and either solubility or permeability.
For selected compounds, melting point and the logP were also measured. The solubility correlated with both lipophilicity and melting point (Supporting information). Following the solubility equation of Yalkowsky and Banerjee,40 the solubility of quinolone derivatives will be predicted to decrease 10-fold when LogP value increases by 1 unit or melting point increases by 100 °C.
Furthermore, the physicochemical properties of analogue 28b were also evaluated at pH 4.0. Because of the ionizable basic chain moiety at the 3- position, compound 28b showed high solubility (78 µM) and low permeability (30 × 106 cm/s Pe) at pH 4.0 in comparison to low solubility (3.4 µM) and high permeability (880 × 106 cm/s Pe) at pH 7.4.
Based upon the best balance of in vitro antimalarial activity and physicochemical properties, 13 compounds were evaluated to assess their in vitro metabolic stability in human and mouse liver microsomes (Table 5). The aqueous solubility of these 13 quinolones at pH 7.4 varied from poorly soluble (< 10 µM) to soluble (> 50 µM). The most soluble compounds were 8f (74 µM), 8q (58 µM) and 20g (20 µM)
Table 5.
Physicochemical properties and in vitro intrinsic clearance in liver microsomes for selected 4(1H)-quinolones.
| MW | mp | solubility (µM) at pH 7.4 |
PAMPA permeability (cm−1) at pH7.4 |
CLint, in vitro (µL/min/mg) |
|||
|---|---|---|---|---|---|---|---|
| Human | Mouse | ||||||
| 8a | 367.35 | 234 | 14 | 100 | 12 | 12 | |
| 8e | 399.44 | 256 | 0.2 | 2900 | 5.0 | 5.7 | |
| 8f | 353.37 | 194 | 74 | 170 | 5.7 | 7.9 | |
| 8g | 367.40 | 208 | 8.7 | 260 | 5.7 | 12.0 | |
| 8h | 381.42 | 220 | 5.8 | 340 | 12 | 39 | |
| 8m | 415.44 | 197 | 4.3 | 750 | 0 | 35 | |
| 8q | 357.79 | 191 | 58 | 950 | 3.4 | 16 | |
| 20d | 433.43 | 231 | 4.3 | 560 | 0 | 16 | |
| 20g | 375.78 | 250 | 20 | 410 | 0.5 | 4.0 | |
| 20h | 420.23 | 258 | 7.3 | 670 | 1.2 | 2.7 | |
| 20i | 389.35 | 264 | 7.5 | 230 | 56 | 6.7 | |
| 28b | 532.56 | ndb | 3.4 (78)a |
880 (30)a |
11 | 24 | |
| 28h | 449.43 | ndb | 3.1 | 620 | 4.8 | 11 | |
the values were measured at pH 4.0.
nd, not determined.
Analogues 20g and 20h displayed most promising liver microsomal stability with CLint,in vitro values less than 4 µL/min/mg in both human and mouse microsomes, which suggested that these compounds should experience low rates of hepatic metabolism in vivo. Compounds 8a, 8e, 8f, 8g and 28h were also expected to be metabolically stable in human and mouse with CLint, in vitro values in the range of 4 – 12.5 mL/min/kg). Compounds 8h, 8m, 8q, 20d, 28b were more rapidly metabolized in mouse liver microsomes than in human (CLint in mouse was > 15 µL/min/mg; and CLint, in vitro value in mouse was at least two-fold higher than that in human), suggesting that hepatic metabolism could be a problematic clearance pathway for these compounds in the mouse efficacy model. In contrast, compound 20i exhibited a high intrinsic clearance value (50 µL/min/mg) in human microsomes, indicating that this compound would likely undergo significant in vivo hepatic metabolism in human. It is worth noting that predicted clearance did not correlate with solubility.
Based upon a combination of in vitro antimalarial potency and in vitro ADME performance, 9 compounds were evaluated for their systemic exposure in the mouse after a single oral dose (low dosage at 30 or 50 mg/kg, or high dosage at 200 mg/kg), with plasma concentration measured by LCMS and followed for 24 h after dosing (Table 6, and Figure 4). Because these compounds are still at the proof of concept stage, absolute bioavailabilities were not measured. Compounds 20g and 20h, both of which showed the highest hepatic metabolic stability in the liver microsome assay, displayed 38 µM and 31 µM maximum concentration in plasma (Cmax) at a 50 mg/kg dose, while the maximum concentration was 66 µM and 39 µM respectively at 200 mg/kg. These two compounds also showed significant AUC values at both dosages (> 125 µM·hr for 20g and > 70 µM·hr for 20h). For those two compounds, high oral doses (200 mg/kg) resulted in less than proportional increases in both AUCinf and Cmax. Less-than proportional increases in AUCinf were also recognized for compound 8e, 8f, 8g, 8h, 8i. Possible reasons for less-than proportional increases in AUCinf could be the impaired absorption caused by limited solubility or saturation of active membrane transporters at high doses.41 Compound 8a displayed a greater than proportional increase in AUCinf after oral administration at high doses, which may be due to decreased elimination.41 The high Cmax and AUC values after the oral administration of 20g and 20h indicated a significant systemic exposure for those two compounds relative to their potency. This profile suggests a high probability of in vivo activity (Figure 6) when administered by the oral route. The rest of seven compounds exhibited low Cmax and/or AUC values in mice, suggesting poor systemic exposure in vivo. Nevertheless, to fully explore PK/PD relationships for the series, all nine compounds were tested in vivo to evaluate efficacy in the murine P. berghei model.
Table 6.
Summary of mouse exposure data following a single oral dose for selected 4(1H)-quinolones.
| dose (mg/kg) |
Cmax (µM) |
AUCinf (µM·hr) |
|
|---|---|---|---|
| 8a | 30 | 1.5 | 6.1 |
| 200 | 5.0 | 65 | |
| 8e | 30 | 0.23 | 1.7 |
| 200 | 0.43 | 4.3 | |
| 8f | 30 | 8.0 | 8.0 |
| 200 | 5.8 | 7.8 | |
| 8g | 30 | 1.9 | 5.1 |
| 200 | 2.4 | 10 | |
| 8h | 30 | 0.30 | 1.6 |
| 200 | 1.8 | 7.7 | |
| 8m | 30 | 0.06 | 0.15 |
| 200 | 0.40 | 1.0 | |
| 20g | 50 | 38 | 130 |
| 200 | 66 | 280 | |
| 20h | 50 | 31 | 71 |
| 200 | 39 | 160 | |
| 20i | 50 | 0.71 | 9.3 |
| 200 | 1.6 | 19 |
Figure 6.

Plasma concentration vs time profiles in mouse for 20g, 20h, and 20i after a single oral dose of 50 mg/kg and 200 mg/kg respectively. The EC90 value of each compound on 3D7 strain is presented as dashed lines.
In vivo antimalarial activity
The in vivo efficacies of the nine quinolone compounds were determined using a Thompson test, measuring parasite clearance and the survival of mice after oral administration of the test compound on days 3 to 5 post-infection. Parasitemia was measured on day 6 and survival followed for 30 days or until patent infection required sacrifice of the animals. Compounds were considered active when the survival time of the treated mice increased 2-fold relative to mice treated with vehicle. For all experiments, amodiaquine was used as a positive control.
Only compounds 20g and 20h suppressed parasite growth (Figure 7); the other seven compounds (8a, 8e, 8f, 8g, 8h, 8m and 20i) had no in vivo efficacy. In the control experiments, mice treated with 30 mg/kg of Amodiaquine started to die on day 15; on day 17, 60% of mice were alive; on day 23 all mice in this group were dead. Compound 20g showed 57% suppression of parasitemia at 30 mg/kg dosage (po) on day 6 post-infection. However, it did not significantly inhibit parasite growth at 10 mg/kg. All of mice treated with 30 mg/kg of 20g were alive at day 8, 60% of mice still survived on day 16, and the last mouse died on day 22. Thus, compound 20g has suppression activity comparable to amodiaquine but is not curative. For compound 20h, mice treated with either 10 or 30 mg/kg exhibited ~ 25% suppression of parasitemia on day 6. Mice treated with 10 mg/kg of 20h were alive on day 8, 20% survived to day 9, there was no mice survived after day 14. Increasing the dosage of 20h did not improve the survival time of mice. The minimal increase in survival time was possibly due the toxicity of 20h, which was apparent after repeat dosing. This toxicology, which was not detected by cellular models, is most likely related to ATP depletion after inhibition of the mammalian mitochondrial respiratory chain.42
Figure 7.

In vivo efficacy of compounds 20g and 20h in the murine P. berghei model. The zero time point represents the day of infection; once daily oral dosing was performed at 72 h, 96 h and 120 h post-infection. Five mice were dosed per group. A) Percentage of inhibition of 20g and 20h on day 6 of post infection. B) Survival curve for infected mice treated with 20g; C) Survival curve for infected mice treated with 20h.
Of all the analogs tested, compound 20g displayed the best balance between potency (EC50 of 83 nM), physicochemical properties (solubility: 20.3 µM, PAMPA permeability : 407 × 106 cm−1), and in vitro mouse liver microsome stability (predicted in vivo mouse CL′inf, : 9.4 mL/min/kg). These findings were mirrored in vivo with compound 20g showing the best pharmacokinetic profile and the best efficacy. Overall its performance was equivalent to that of the control drug amodiaquine. Overall this profile suggests that close analogs to 20g show promise for further development as antimalarials.
CONCLUSION
This paper describes studies aimed at selecting an early lead compound from the 4(1H)-quinolone ester series. The efforts were focused on improving antimalarial potency and physicochemical properties, with chemistry focused on exploring meta-substituted phenyl at the 2-position, the substituents on the benzenoid ring, and the solubilizing groups at the 3-position. The best antimalarial activity was observed if 1) a hydrophobic group is presented at the meta position of 2-phenyl ring, 2) 6-halo and 7-methoxy groups were substituted on the benzenoid ring, 3) ethyl ester group was maintained at the 3-position.
Mechanism of action was explored using a series of strain sensitivity studies with genetically characterized strains of P. falciparum and biochemical assays. The 4(1H)-quinolone esters appear to act on mitochondrial electron flux, most likely targeting the cytochrome bc1 complex. Toxicity seen in vivo with repeat dosing of some compounds in the series is most likely related to inhibition of the mammalian cytochrome bc1 complex. Selectivity between the parasite and mammalian activities will be a critical parameter in further lead optimization.
Two analogues, 20g and 20h, were identified with good liver microsome stability and oral systemic exposure in mice. Subsequent murine efficacy testing revealed that 20g had the suppression activity at a 30 mg/kg dosage, equivalent to amodiaquine. The in vivo potency of these compounds places them about 10-fold weaker than endochin based series being developed by the Riscoe and Manetsch groups, but with the possibility of significant improvements in oral bioavailability in optimized analogs due to the lower molecular weight of this series. Overall, 4(1H)-quinolone esters show promise as antimalarials but would likely would benefit from additional optimization to improve exposure and potency in vivo.
EXPERIMENTAL SECTION
General methods
3,4-Methylenedioxphenyl boronic acid was purchased from Boron Molecular, and all other chemical reagents were from Acros, Aldrich, or Combi-Blocks. All materials were obtained from commercial suppliers and used without further purification. Thin layer chromatography was performed using a silica gel 60 F254 plate from EMD. Purification of compounds was done by normal phase column chromatography (Biotage SP1). 1H NMR, 13C NMR, and 2D NOE spectra were recorded on a Bruker 400 MHz. Chemical shifts were expressed in ppm relative to tetramethyl silane, which was used as an internal standard. Purity was estimated using high-performance liquid chromatography/mass spectrometry (Alliance HT, Micromass ZQ 4000, and RP-C18 Xterra column, 5 µm, 6 × 50 mm [Waters]) or ultra-performance liquid chromatography/mass spectrometry (Acquity PDA detector, Acquity SQ detector, and Acquity UPLC BEH-C18 column, 1.7 µm, 2.1 × 50 mm [Waters]). FTIR spectrums were recorded on a Thermo Nicolet IR 100 FTIR spectrometer. Compounds prepared in our laboratory were generally 90–98% pure. Efficacy data were obtained only on compounds that were at least 95% pure.
General procedure for synthesis of 8–13, 15–20
Quinolone derivatives 8–20 were synthesized as previous described,12 except 7-methylthio quinolone derivatives 14. Briefly, the intermediate 4(1H)-quinolone 6 were prepared from appropriate aniline 5 (15 mL) and 2-(ethoxymethylene)malonate (16.5 mmol), followed by Gould-Jacobs cyclization. Then, to the solution of 6 (6.5 mmol) in 6 mL 1,4-dioxane was added POCl3 (7.8 mmol). The mixture was stirred at 120 °C for 1 hour. After cooling to room temperature, the reaction mixture was poured into ice water and then neutralized by aqueous K2CO3. The resulting 4-chloro quinolone derivatives were extracted by CH2Cl2, which were used for next step without purification. 4-Chloro quinolone derivatives were then treated with m-chloroperbenzoic acid (7.8 mmol) in 30 mL of CHCl3 at room temperature for 4 hours to afford N-oxide 4-chloro-quinoline intermediate which then reacted with POBr3 (7.2 mmol) in 30 mL CHCl3 at room temperature for 1 hour. The crude mixture was quenched by adding ice water and then neutralized by aqueous K2CO3. 2-Bromo-4-chloro-quinoline derivatives 7 were then extracted by CH2Cl2 and purified by flash chromatography.
The flask was charged with 2-bromo-4-chloro quinolone intermediate 7 (0.3 mmol), appropriate boronic acid (0.31 mmol), Pd(PPh3)4 (8.6 mg, 0.0075 mmol) in 2 mL of 1,4-dioxane. The flask was degassed three times. To the mixture was added the solution of CsCO3 (0.195 g, 0.6 mmol) in 0.6 mL of H2O. The flask was degassed again three times. The reaction mixture was stirred at 75 °C for 3 h. After being cooled to rt, the bottom aqueous layer was removed, and the organic layer was concentrated and purified by flash column chromatography to produce the intermediate compound ethyl 2-aryl-4-chloroquinoline-3-carboxylate.
The solution of ethyl 2-aryl-4-chloroquinoline-3-carboxylate intermediate (0.2 mmol) in 1 mL of AcOH/H2O (9:1) was refluxed at 120 °C for 1 h. The reaction mixture was concentrated and neutralized by adding 5 drops of NH4OH. Purification was performed by reverse-phase HPLC to produce the desired compounds 8–13, 15–20. (Waters Xterra preparative C18 column, MeOH/H2O, 10mM NH4HCO3). For characterization of 8–13, 15–20 see Supporting Information.
Ethyl 2-(3-chlorophenyl)-6-fluoro-7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate 20g
Compound 20g was synthesized following the general procedure described in Scheme 1. A solution of 4-fluoro-3-methoxyaniline (4.0 g, 28.3 mmol) and diethyl 2-(ethoxymethylene) malonate (6.4 g, 29.7 mmol) in ethanol (15 mL) was stirred at 120 °C for 2 h. The reaction mixture was cooled to room temperature then concentrated in vacuuo and the crude intermediate utilized without further purification. The crude anilinomethylenemalonate was dissolved in diphenyl ether (10mL) and heated to reflux for 1 h. The reaction mixture was allowed to cool to room temperature and diethyl ether was added giving a precipitate that was isolated by filtration, washed with diethyl ether, and dried in vacuuo to give compound 6 (6.2 g, 81% yield in 2 steps). To a solution of 6 (1.2 g, 6.5 mmol) in 1,4-dioxane (6 mL) was added POCl3 (0.50 mL, 7.8 mmol). The mixture was heated to 120 °C and stirred for 1 hour. After cooling to room temperature, the reaction mixture was poured into ice water and then neutralized using aqueous K2CO3. The resulting 4-chloro quinolone were extracted into CH2Cl2, dried, and treated with m-chloroperbenzoic acid (1.3 g, 7.8 mmol) in 30 mL of CHCl3 at room temperature for 4 hours. The resulting N-oxide 4-chloro-quinoline was then allowed to react with POBr3 (2.0 g, 7.2 mmol) in 30 mL CHCl3 at room temperature for 1 hour. The crude mixture was quenched by adding ice water and the resulting solution neutralized with aqueous K2CO3. The 2-Bromo-4-chloro-quinoline isolated by extraction into CH2Cl2, dried with magnesium sulfate, and dried in vacuuo. The solid product was purified by flash chromatography (Biotage SP1) eluting with 10–40 % of ethyl acetate/hexane to obtain the desired product 7 (1.72 g, 73% yield in 3 steps).
A flask was charged with ethyl 2-bromo-4-chloro-6-fluoro-7-methoxyquinoline-3-carboxylate 7 (1.08 g, 0.3 mmol), 3-chlorophenylboronic acid (0.48 g, 0.31 mmol), and Pd(PPh3)4 (8.6 mg, 0.0075 mmol) in 1,4-dioxane (2 mL) and degassed. To the mixture was added the solution of CsCO3 (0.195 g, 0.6 mmol) in H2O (0.6 mL). The flask was degassed again. The reaction mixture was heated to 75 °C and stirred for 3 h. After being cooled to room temperature, the aqueous layer was removed, and the organic layer was concentrated and purified by flash chromatography (Biotage SP1) to give ethyl 4-chloro-2-(3-chlorophenyl)-6-fluoro-7-methoxyquinoline-3-carboxylate. A solution of ethyl 4-chloro-2-(3-chlorophenyl)-6-fluoro-7-methoxyquinoline-3-carboxylate (0.788 g, 0.2 mmol) in AcOH/H2O (9:1, 1 mL) was heated to reflux and stirred at 120 °C for 1 h. After cooling to room temperature, the reaction mixture neutralized by adding 5 drops of NH4OH and concentrated. The crude product was isolated directly by reverse-phase HPLC (XbridgeTM C18 5um OBD, 30 × 50 mm, mobile phase: Water with 0.1% Formic Acis : Acetonitrile with 0.1% Formica Acid, Gradient (0~90% AcCN), Flow Rate: 4.0 mL/min) to afford the desired compound 20g (326 mg, 29% yield over 2 steps). 1H NMR (400 MHz, DMSO) δ 11.99 (s, 1H), 7.72 (d, J = 11.6, 1H), 7.30 (d, J = 7.4, 1H), 7.08 (ddd, J = 9.8, 9.1, 1.7, 3H), 6.14 (s, 2H), 4.02 (q, J = 7.1, 2H), 3.93 (s, 3H), 1.02 (t, J = 7.1, 3H). 13C NMR (101 MHz, DMSO) δ 172.39, 166.48, 151.38, 151.24, 150.58, 148.79, 148.58, 148.14, 147.35, 137.71, 122.52, 118.17, 114.64, 109.85, 109.67, 108.42, 101.75, 60.20, 56.20, 13.80. MS (ESI) cald C20H16FNO6 for [M+H]+, 386.10, found 386.24
General procedure for synthesis of 14
Quinolone derivative 14 were synthesized as previous described with minor mortification.43 To the solution of benzoyl chloride 22 (6.70 mmol) was added the solution of 3-methylthio Aniline 21 (8.04 mmol) in 10 mL toluene drop wise. After the reaction mixture was stirred at room temperature for 3 hours, solvents were evaporated to afford the crude amide product 23, which was purified by flash column chromatography. Then, the amide 23 (4 mmol) in 10 mL of 1,4-dioxan was treated with PCl5 (4 mmol). After refluxing at 110 ° for 1 hour, the solvent of the reaction mixture was evaporated under vacuum. The residues were re-dissolved in 5 mL of toluene and dropped into the solution of refresh prepared sodium diethyl malonate in 10 mL of toluene. The mixture was then refluxed at 110° for 6 hours. The crude product 24 was purified by flash column chromatography. The compound 14 was prepared by the cyclization of 24 (0.7 mmol) in 1 mL of Ph2O under 170 °C for 4 hours. The reverse-phase HPLC was used to purify the desired compound 14. For characterization of 14 see Supporting Information.
General procedure for synthesis of 27–28
To the solution of KOH (12 mmol) in 10 mL of water and 0.5 mL of EtOH was added quinolone ester 25. After stirring at 75°C for 40 hours, the reaction mixture was cooled to room temperature and neutralized by adding 2N HCl until pH reached to 7. The white solids were collected by filtration and washed by water twice. This crude product 26 was used for the next step without purification. Then, to the mixture of acid 26 (0.05 mmol) in 0.5 mL of DMF was added DPIEA (0.075 mmol) and HBTU (0.05 mmol). The mixture was stirred at rt for 30 min. After stirring at room temperature for 30 min, appropriate alkyl alcohol or alkyl amine (0.150 mmol) was added into the reaction mixture. The mixture continued to stir at 40 °C for 2 hours. After the solvent was evaporated, the crude product 27–28 was purified by reverse-phase HPLC. For characterization of 27–28 see Supporting Information.
Anti-malarial Potency Assays
Two P. falciparum strains, CQ-S 3D7 and CQ-R K1, were used in this study and were provided by the MR4 Unit of the American Type Culture Collection (Manassas, VA).
Asynchronous parasites were maintained in culture based on the method of Trager. Parasites were grown in the presence of fresh group O-positive erythrocytes (Lifeblood, Memphis, TN) in Petri dishes at a hematocrit of 4–6% in RPMI-based medium. It consisted of RPMI 1640 supplemented with 0.5% AlbuMAX II, 25 mM HEPES, 25 mM NaHCO3 (pH 7.3), 100 µg/mL hypoxanthine, and 5 µg/mL gentamicin. Cultures were incubated at 37 °C in a gas mixture of 90% N2, 5% O2, and 5% CO2. For EC50 determinations, 20 µl of RPMI 1640 with 5 µg/mL gentamicin were dispensed per well in an assay plate (384-well microtiter plate, clear-bottom, tissue-treated). Next, 40 nL of compound, previously serial diluted in a separate 384-well white polypropylene plate, were dispensed in the assay plate, and then 20 µL of a synchronized culture suspension (1% rings, 10% hematocrit) were added per well to make a final hematocrit and parasitemia of 5% and 1%, respectively. Assay plates were incubated for 72 h, and the parasitemia was determined by a method previously described. Briefly, 10 µL of 10X Sybr Green I, 0.5% v/v Triton, and 0.5 mg/mL saponin solution in RPMI were added per well. Assay plates were shaken for 30 s, incubated in the dark for 4 h, and then read with the Envision spectrofluorometer at Ex/Em 485 nm/535 nm. EC50s were calculated using proprietary software developed in house (RISE) in the Pipeline Pilot environment that pools data from all replicates of the experiment and fits a consensus model.
Solubility Assays
Solubility assays were carried out on a Biomek FX lab automation workstation (Beckman Coulter, Inc., Fullerton, CA) using µSOL Evolution software (pION Inc., Woburn, MA) as follows: 10 µL of compound stock was added to 190 µL of 1-propanol to make a reference stock plate. Next, 5 µL of this reference stock plate was mixed with 70 µL of 1-propanol and 75 µL of PBS (pH 7.4 and 4, respectively) to make the reference plate, and the UV spectrum (250–500 nm) of the reference plate was read. Then, 6 µL of 10 mM test compound stock was added to 600 µL of PBS in a 96-well storage plate and mixed. The storage plate was sealed and incubated at rt for 18 h. The suspension was then filtered through a 96-well filter plate (pION Inc., Woburn, MA). Next, 75 µL of filtrate was mixed with 75 µL of 1-propanol to make the sample plate, and the UV spectrum of the sample plate was read. Calculations were done using µSOL Evolution software based on the area under the curve (AUC) of the UV spectrum of the sample plate and the reference plate. All compounds were tested in triplicate.
Permeability Assays
A parallel artificial membrane permeability assay (PAMPA) was conducted on a Biomek FX lab automation workstation (Beckman Coulter, Inc., Fullerton, CA) with PAMPA evolution 96 command software (pION Inc., Woburn, MA) as follows: 3 µL of 10 µM test compound stock was mixed with 600 µL of system solution buffer, pH 7.4 or 4 (pION Inc., Woburn, MA) to make diluted test compound. Then 150 µL of diluted test compound was transferred to a UV plate (pION Inc., Woburn, MA), and the UV spectrum was read as the reference plate. The membrane on a preloaded PAMPA sandwich (pION Inc., Woburn, MA) was painted with 4 µL of GIT lipid (pION Inc., Woburn, MA). The acceptor chamber was then filled with 200 µL of acceptor solution buffer (pION Inc., Woburn, MA), and the donor chamber was filled with 180 µL of diluted test compound. The PAMPA sandwich was assembled, placed on the Gut-Box controlled environment chamber and stirred for 30 min. The aqueous boundary layer was set to 40 µm for stirring. The UV spectrum (250–500 nm) of the donor and the acceptor were read. The permeability coefficient was calculated using PAMPA Evolution 96 Command software (pION Inc., Woburn, MA) based on the AUC of the reference plate, the donor plate, and the acceptor plate. All compounds were tested in triplicate.
Cytotoxicity screens
BJ, HEK293, HepG2, and Raji cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured according to recommendations. Cell culture media were purchased from ATCC. Cells were routinely tested for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza). Exponentially growing cells were plated in Corning 384 well white custom assay plates, and incubated overnight at 37° C in a humidified, 5% CO2 incubator. DMSO inhibitor stock solutions were added the following day to a top final concentration of 25 µM, 0.25% DMSO and then diluted 1/3 for a total of ten testing concentrations. Cytotoxicity was determined following a 72-hour incubation using Promega Cell Titer Glo Reagent according to the manufacturer’s recommendation. Luminescence was measured on an Envision plate reader (Perkin Elmer).
PfDHOD inhibition assays
Compounds were screened for P. falciparum dihydroorotate dehydrogenase (PfDHOD) inhibitory activity using a previous described protocol.44 Briefly, Assay buffer solution was: 100 mM HEPES pH 8.0, 150 mM NaCl, 10% Glycerol, 0.05% Triton X-100, 20 µM CoQ0, 200 µM L-dihydroorotate, 120 µM 2,6-dichloroindophenol. The assay was started by the addition of a 5 µL of stock solution of PfDHOD enzyme (10 nM final in assay plate) and monitored at 600 nm for 5–10 min at 20 °C. The initial rates were used to determine the reaction velocity in the absence and presence of inhibitors. Compounds were added over a range of 0.05–100 µM using a 3-fold dilution series.
In vitro liver microsome assays
1.582 mL of mouse liver microsomes (20 mg/mL, female CD-1 mice, ~75 pooled, Fisher Scientific, #NC9567486) were mixed with 0.127 mL of 0.5M EDTA solution and 48.3 mL potassium phosphate buffer (0.1M, pH 7.4, 37°C) to make 50 mL of mouse liver microsome solution. Human liver microsomal solution was made with human liver microsomes (50 pooled mix gender, Fisher Scientific # 50-722-516) using the same procedure. 1 volume of 10 mM DMSO compound stock was mixed with 4 volume of acetonitrile to make 2 mM diluted compound stock in DMSO and acetonitrile. 37.83 µL diluted compound stock was added to 3 mL liver microsomal solution and vortexed to make microsomal solution with compound. 1 mL of liver microsomal solution with compound is added to each well of a master storage plate (pION Inc., MA, #110323). All compounds were tested in triplicate. Mouse and human liver microsomes were tested side by side on the same plate. 175 µL of each well was dispensed from the master plate into 5 storage plates. For the 0 hour time point, 450 µL pre-cooled (4 °C) internal standard (10 µM warfarin in methanol) was added to the first plate before the reaction starts. 5.25 mL of microsome assay solution A (Fisher Scientific, #NC9255727) was combined with 1.05 mL of solution B (Fisher Scientific, #NC9016235) in 14.7 mL of potassium phosphate buffer (0.1 M, pH 7.4). 45 µL of this A+B solution was added to each well of all the 96-well storage plates and mixed briefly. The plates were sealed, and all plates except the 0-hr plate were incubated at 37 °C, shaken at a speed of 60 rpm. 0.5 h, 1 h, 2 h and 4 h time points were taken. At each time point, 450 µL pre-cooled internal standard was added to the plate to quench the reaction. The quenched plate was then centrifuged (model 5810R, Eppendorf, Westbury, NY) at 4000 rpm for 20 minutes. 150 µL supernatant was transferred to a 96-well plate and analyzed by UPLC-MS (Waters Inc., Milford, MA). The compounds and internal standard were detected by SIR. The log peak area ratio (compound peak area / internal standard peak area) was plotted vs. time (h) and the slope was determined to calculate the elimination rate constant [k = (−2.303) * slope]. The half-life (h) was calculated as t (1/2) = 0.693 / k. Intrinsic clearance was calculated as CLint, in vitro= (0.693 / (t1/2)) * (1/microsomal concentration in the reaction solution), where microsomal concentration in the reaction solution is 0.5 mg/mL.
Pharmacokinetic studies in female CD1 mice
All pharmacokinetic studies were performed in accordance with SRI International’s animal care policies in an AAALAC and OLAW accredited facility. The procedure for the pharmacokinetics studies followed the previously described method.45 Briefly, the plasma pharmacokinetics of selected quinolone derivative were determined in female CD1 mice after administration of a single dose (30, 50, or 200 mg/kg, n=3) by oral gavage. Blood was collected from three mice per test article and at 15, 30, and 60 min and 2, 4, 6, 8, 10, 24 h after dose administration.
In vivo antimalarial efficacy studies in female ICR mice
For all experiments, Swiss outbreed (ICR) female mice (15–20 g) were purchased form Harlan, (N. America). All animals were housed in cages in an animal facility with alternative light and dark cycles in pellet food and tap water ad libitum. P. berghei (NK-65) were maintained by serial passaging in infected blood in female mice until the experiment was initiated. All animal protocols were approved by University of South Florida IACUC and experiments were conducted in accordance to animal care policies.
The oral activities of selected nine quinolone compounds were tested against P. berghei (NK-65) infected ICR by modified Thompson’s test.46 Briefly, the animals on day 0 were intraperitoneally inoculated with 1×106 infected red blood cells diluted in 0.1 ml plasma obtained from uninfected donor mice. Positivity of infection and validity of the test model was checked by making thin blood films from tail vein puncture of each individual mouse on Day 3 before dosing. The animals were treated with single oral dose of either the test compound or a reference drug AMDQ with a concentration ranging from 10–100 mg/kg on days 3, 4 and 5 post infections. All drugs were prepared in 0.5% hydroxyethylcellulose (HEC) dissolved in water and control mice received only the vehicle. Survival of the mice was monitored and recorded daily. The trend of parasitemia was monitored by preparing thin blood films on Day 3 and 6 post inoculation, then at weekly intervals (days 13, 20 and 27) through day 30. Percent parasitemia was determined by observing at least a total of 1000 cells in the methanol fixed and Giemsa stained blood smears from individual mice microscopically (magnification, X1,000).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Taosheng Chen and Jimmy Cui from the St Jude High Throughput Screening Core for their support in antimalarial and cytotoxicity screening. We acknowledge Michael Riscoe, Isaac Forquer, and Rolf Winter of Oregon Health Sciences University; Roman Manetsch of the University of South Florida; Akhil Vaidya of Drexel University; and Jeremy Burrows of the Medicines for Malaria Venture for many discussions concerning structure activity relationships and mechanism of this and related series that informed this work. We acknowledge Akhil Vaidya for providing the yDHODH strain of P. falciparum. This work was supported by the National Institutes of Health, (NIAID AI075517 and NIAID AI075594), the American Lebanese Syrian Associated Charities (ALSAC), and St. Jude Children’s Research Hospital. MAP holds the Carolyn R. Bacon Professorship in Medical Science and Education and acknowledges support of the Welch Foundation (I-1257).
ABBREVIATIONS USED
- P. falciparum
Plasmodium falciparum
- ACT
Artemisinin-based combination therapy
- WHO
the World Health Organization
- HPLC
high-performance liquid chromatography
- rt
room temperature
- POCl3
Phosphorus oxychloride
- POBr3
Phosphorus oxybromide
- Pd(PPh3)4
Tetrakis (triphenylphosphine) Palladium
- PCl5
Phosphorus pentachloride
- DIPEA
diisopropyl ethylamine
- HBDU
2-(1H-Benzotriazole-1-yl)-1,1,3,3-Tetramethyluronium hexafluorophosphate
- DHODH
dihydroorotate dehydrogenase
- yDHODH
yeast dihydroorotate dehydrogenase
- pfDHODH
Plasmodium falciparum dihydroorotate dehydrogenase
- rt
room temperature
- SAR
structure-activity relationship
- ADME
absorption, distribution, metabolism and excretion
- PAMPA
parallel artificial membrane permeability assay
- Cint
intrinsic clearance
- AUC
area under curve
Footnotes
SUPPORTING INFORMATION
Synthetic procedures and tabulated spectral data for quinolone derivatives. Complete antimalarial data, permeability and solubility data for all listed compounds. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 2004;22:245–252. doi: 10.1016/j.tibtech.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Wellems TE, Plowe CV. Chloroquine-resistant malaria. J. Infect. Dis. 2001;184:770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]
- 3.Hyde JE. Drug-resistant malaria. Trends in Parasitology. 2005;21:494–498. doi: 10.1016/j.pt.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessl JJ, Meshnick SR, Trumpower BL. Modeling the molecular basis of atovaquone resistance in parasites and pathogenic fungi. Trends in Parasitology. 2007;23:494–501. doi: 10.1016/j.pt.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Sidhu ABS, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nature Reviews Microbiology. 2009;7:864–874. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wongsrichanalai C, Meshnick SR. Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border. Emerg. Infect. Dis. 2008;14:716–719. doi: 10.3201/eid1405.071601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, DM NL, Socheat D, White NJ. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature. 2002;415:686–693. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- 10.Gamo F-J, Sanz LM, Vidal J, Cozar Cd, Alvarez E, Lavandera J-L, Vanderwall DE, Green DVS, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 11.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jiménez-Díaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, Mazouni FE, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, DeRisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Voorhis WCV, Avery VM, Guy RK. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Guiguemde WA, Sigal M, Zhu F, Connelly MC, Nwaka S, Guy RK. Synthesis and structure–activity relationships of antimalarial 4-oxo-3-carboxyl quinolones. Biorg. Med. Chem. 2010;18:2756–2766. doi: 10.1016/j.bmc.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephen JML, Tonkin IM, Walker J. Tetrahydroacridones and Related Compounds as Antimalarials. J. Chem. Soc. 1947:1034–1039. doi: 10.1039/jr9470001034. [DOI] [PubMed] [Google Scholar]
- 14.Coatey GR, Cooper WC. Symposium on Exoerythrocytic Forms of Malaria Parasites III: The Chemotherapy of Malaria in Relation to Our Knowledge of Exoerythrocytic Forms. J. Parasitol. 1948;34:275–289. [PubMed] [Google Scholar]
- 15.Ryley JF, Peters W. Antimalarial activity of some quinolone esters. Ann. Trop. Med. Parasitol. 1970;64:209–222. doi: 10.1080/00034983.1970.11686683. [DOI] [PubMed] [Google Scholar]
- 16.Puri SK, Dutta GP. Quinoline Esters as Potential Antimalarial-Drugs - Effecto on Relapses of Plasmodium-Cynomolgi Infections in Monkeys. Trans. R. Soc. Trop. Med. Hyg. 1990;84:759–760. doi: 10.1016/0035-9203(90)90066-n. [DOI] [PubMed] [Google Scholar]
- 17.Kesten SJ, Degnan MJ, Hung J, McNamara DJ, Ortwine DF, Uhlendorf SE, Werbel LM. Antimalarial drugs. 64. Synthesis and antimalarial properties of 1-imino derivatives of 7-chloro-3-substituted-3,4-dihydro-1,9(2H,10H)-acridinediones and related structures. J. Med. Chem. 1992;35:3429–3447. doi: 10.1021/jm00097a001. [DOI] [PubMed] [Google Scholar]
- 18.Berman J, Brown L, Miller R, Andersen SL, McGreevy P, Schuster BG, Ellis W, Ager A, Rossan R. Antimalarial Activity of WR 243251, a Dihydroacridinedione. Antimicrob. Agents Chemother. 1994;38:1753–1756. doi: 10.1128/aac.38.8.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suswam E, Kyle D, Lang-Unnasch N. Plasmodium falciparum: The Effects of Atovaquone Resistance on Respiration. Exp. Parasitol. 2001;98:180–187. doi: 10.1006/expr.2001.4639. [DOI] [PubMed] [Google Scholar]
- 20.Biagini GA, Fisher N, Berry N, Stocks PA, Brigitte Meunier, Williams DP, Bonar-Law R, Bray PG, Owen A, O’Neill PM, Ward SA. Acridinediones: Selective and Potent Inhibitors of the Malaria Parasite Mitochondrial bc1 Complex. Mol. Pharmacol. 2008;73 doi: 10.1124/mol.108.045120. [DOI] [PubMed] [Google Scholar]
- 21.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK. Antimalarial quinolones: Synthesis, potency, and mechanistic studies. Exp. Parasitol. 2008;118:487–497. doi: 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cross RM, Monastyrskyi A, Mutka TS, Burrows JN, Kyle DE, Manetsch R. Endochin Optimization: Structure-Activity and Structure-Property Relationship Studies of 3-Substituted 2-Methyl-4(1H)-quinolones with Antimalarial Activity. J. Med. Chem. 2010;53:7076–7094. doi: 10.1021/jm1007903. [DOI] [PubMed] [Google Scholar]
- 23.Cross RM, Maignan JR, Mutka TS, Luong L, Sargent J, Kyle DE, Manetsch R. Optimization of 1,2,3,4-tetrahydroacridin-9(10H)-ones as antimalarials utilizing structure-activity and structure-property relationships. J. Med. Chem. 2011;54:4399–4426. doi: 10.1021/jm200015a. [DOI] [PubMed] [Google Scholar]
- 24.Cross RM, Manetsch R. Divergent route to access structurally diverse 4-quinolones via mono or sequential cross-couplings. J. Org. Chem. 2010;75:8654–8657. doi: 10.1021/jo1014504. [DOI] [PubMed] [Google Scholar]
- 25.Cross RM, Monastyrskyi A, Mutka TS, Burrows JN, Kyle DE, Manetsch R. Endochin optimization: structure-activity and structure-property relationship studies of 3-substituted 2-methyl-4(1H)-quinolones with antimalarial activity. J. Med. Chem. 2010;53:7076–7094. doi: 10.1021/jm1007903. [DOI] [PubMed] [Google Scholar]
- 26.Kelly JX, Smilkstein MJ, Brun R, Wittlin S, Cooper RA, Lane KD, Janowsky A, Johnson RA, Dodean RA, Winter R, Hinrichs DJ, Riscoe MK. Discovery of dual function acridones as a new antimalarial chemotype. Nature. 2009;459:270–273. doi: 10.1038/nature07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly JX, Smilkstein MJ, Cooper RA, Lane KD, Johnson RA, Janowsky A, Dodean RA, Hinrichs DJ, Winter R, Riscoe M. Design, synthesis, and evaluation of 10-N-substituted acridones as novel chemosensitizers in Plasmodium falciparum. Antimicrob. Agents Chemother. 2007;51:4133–4140. doi: 10.1128/AAC.00669-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winter R, Kelly JX, Smilkstein MJ, Hinrichs D, Koop DR, Riscoe MK. Optimization of endochin-like quinolones for antimalarial activity. Exp. Parasitol. 2011;127:545–551. doi: 10.1016/j.exppara.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Bagby GC, Rathbun RK, Levin JI, Hinrichs D, Riscoe MK. Evaluation and lead optimization of anti-malarial acridones. Exp. Parasitol. 2006;114:47–56. doi: 10.1016/j.exppara.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 30.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK. Antimalarial quinolones: synthesis, potency, and mechanistic studies. Exp. Parasitol. 2008;118:487–497. doi: 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madrid PB, Wilson NT, DeRisi JL, Guy RK. Parallel Synthesis and Antimalarial Screening of a 4-Aminoquinoline Library. J. Comb. Chem. 2004;6:437–442. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansch C, Leo A, Taft RW. A survey of hammett substituent constants and resonance and field parameters. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 33.Hansch C, Leo A, Hoekman D. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants. Wahsington DC: An American Chemical Society Publication; 1995. [Google Scholar]
- 34.Sidhu ABS, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium faciparum malaria parasites conferred by pfCRT mutations. Science. 2002;298 doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DJ, Fidock DA, Mungthin M, Lakshmanan V, Sidhu ABS, Bray PG, Ward SA. Evidence for a central role for PfCRT in conferring Plasmodium falciparum resistance to diverse antimalarial agents. Mol. Cell. 2004;15 doi: 10.1016/j.molcel.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 37.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan M-S, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaidya AB, Mather MW. A post-genomic view of the mitochondrion in malaria parasites. Curr. Top. Microbiol. Immunol. 2005;295:233–250. doi: 10.1007/3-540-29088-5_9. [DOI] [PubMed] [Google Scholar]
- 39.Ke H, Morrisey JM, Ganesan SM, Painter HJ, Mather MW, Vaidya AB. Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain function. Eukaryotic Cell. 2011;10:1053–1061. doi: 10.1128/EC.05049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yalkowsky S, Banerjee S. Aqueous solubility: Methods of estimation for organic compounds. NY: Marcel Dekker: New York: 1992. [Google Scholar]
- 41.Lin JH. Dose-dependent pharmacokinetics: experimental observations and theoretical considerations. Biopharm. Drug Dispos. 1994;15:1–31. doi: 10.1002/bdd.2510150102. [DOI] [PubMed] [Google Scholar]
- 42.Scatena R, Bottoni P, Botta G, Martorana GE, Giardina B. The role of mitochondria in pharmacotoxicology: a reevaluation of an old, newly emerging topic. American journal of physiology. Cell physiology. 2007;293:C12–C21. doi: 10.1152/ajpcell.00314.2006. [DOI] [PubMed] [Google Scholar]
- 43.Lai Y-Y, Huang L-J, Lee K-H, Xiao Z, Bastow KF, Yamori T, Kuo S-C. Synthesis and biological relationships of 3',6-substituted 2-phenyl-4-quinolone-3-carboxylic acid derivatives as antimitotic agents. Biorg. Med. Chem. 2005;13:265–275. doi: 10.1016/j.bmc.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 44.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. Triazolopyrimidine-based dihydrooratate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J. Med. Chem. 2008;51:3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ray S, Madrid PB, Catz P, LeValley SE, Furniss MJ, Rausch LL, Guy RK, DeRisi JL, Iyer LV, Green CE, Mirsalis JC. Development of a New Generation of 4-Aminoquinoline Antimalarial Compounds Using Predictive Pharmacokinetic and Toxicology Models. J. Med. Chem. 2010;53:3685–3695. doi: 10.1021/jm100057h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miroshnikova OV, Hudson TH, Gerena L, Kyle DE, Lin AJ. Synthesis and Antimalarial Activity of New Isotebuquine Analogues. J. Med. Chem. 2007;50:889–896. doi: 10.1021/jm061232x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.