

Abstract
Mutations in the cartilage oligomeric matrix protein gene (COMP) cause pseudoachondroplasia (PSACH). This dysplasia results from the intracellular retention of mutant COMP protein and premature death of growth-plate chondrocytes. Toward better understanding of these underlying mechanisms, we examined D469del-COMP activation of the unfolded protein response and cell death pathways in rat chondrosarcoma cells. Using an inducible expression system, we examined the effects of D469del-COMP retention after 4 days of mRNA expression and then 5 days without inducing agent. Retention of D469del-COMP stimulated Chop (Ddit3) and Gadd34 (Ppp1r15a) and triggered reactivation of protein translation that exacerbated intracellular retention. High levels of Nox4 and endoplasmic reticulum receptor stress-inducible Ero1β generated reactive oxygen species, causing oxidative stress. Increased expression of Gadd genes and presence of γH2AX indicated that DNA damage was occurring. The presence of cleaved apoptosis inducing factor (tAIF) and the absence of activated caspases indicated that retention of D469del-COMP triggers cell death in chondrocytes by necroptosis, a caspase-independent programmed necrosis. Loss of growth-plate chondrocytes by necroptosis was also found in our pseudoachondroplasia mouse model. These results suggest a model in which D469del-COMP expression induces persistent endoplasmic reticulum stress, oxidative stress, and DNA damage, thus priming chondrocytes for necroptosis. We define for the first time the precise mechanisms underlying D469del-COMP pathology in pseudoachondroplasia and suggest that oxidative stress and AIF may be promising therapeutic targets.
Cartilage oligomeric matrix protein (COMP) is a pentameric glycoprotein found in extracellular matrix (ECM) surrounding chondrocytes and other cell types.1–7 COMP (previously PSACH; also known as TSP5) belongs to the thrombospondin family, which are a group of proteins that mediate cell-cell and cell-matrix interactions.8 Multiple roles and functions have been suggested for COMP, including regulation of collagen fibril assembly, proliferation of chondrocytes, and interactions with other matrix proteins such as collagens type II and IX and matrilin-3 (MATN3).8–17 Two autosomal dominant skeletal dysplasias, pseudoachondroplasia and multiple epiphyseal dysplasia/epiphyseal dysplasia, multiple 1 (MED/EDM1), are caused by mutations in COMP.18,19 Pseudoachondroplasia is a short-limb type of dwarfism; MED/EDM1 is a similar but milder condition, in which stature is not dramatically reduced.20 Both conditions are associated with abnormal joint architecture, joint erosion, and early osteoarthritis.21 Pseudoachondroplasia and MED/EDM1 are defined at the cellular level by large endoplasmic reticulum (ER) cisternae filled with lamellar-appearing material.22 COMP localizes to these large ER cisternae, as do collagens type II and IX and MATN3, and prematurely forms an intracellular matrix network.3,23–27 This intracellular matrix ultimately leads to premature chondrocyte death and loss of growth-plate chondrocytes.24,28
Accumulation of misfolded proteins in the ER triggers the unfolded protein response (UPR), a mechanism that allows cells to cope with unfolded or misfolded proteins.29–31 The UPR is composed of both adaptive and apoptotic responses. The adaptive pathway functions to repress general translation, enhance protein folding and, degrade misfolded proteins. This is accomplished through a complex network of interacting proteins and transcription factors. For example, repression of general translation occurs by inactivation of eukaryotic initiation factor 2A (eIF2α) through PKR-like endoplasmic reticulum kinase (PERK; also known as eukaryotic translation initiation factor 2-alpha kinase 3, EIF2AK3).32,33 Protein folding requires the assistance of chaperone proteins. However, persistent misfolded protein should be targeted for degradation. When folding and degradation mechanisms fail, the apoptotic arm of the UPR persists through PERK and through DNA damage-inducible transcript 3 protein (DDIT3; hereafter referred to by the synonym CHOP), which ultimately leads to cell death.34 Persistent CHOP activation causes oxidative stress, which is also a contributor to cell death.35
Cell death occurs either as programmed cell death or as passive cell death.36–38 In programmed cell death, there are two pathways, apoptosis and necroptosis. In apoptosis, death occurs through the caspase-dependent pathway, in which chromatin is condensed and DNA is fragmented.38,39 In necroptosis, death occurs through apoptosis inducing factor (AIF), a mitochondrial oxidoreductase that is translocated to the nucleus, triggering chromatin condensation and large-scale DNA fragmentation.37,40,41 Passive cell death or necrosis is characterized by a loss of intracellular contents after swelling of the cell and rupture of the plasma membrane, often resulting from traumatic cellular events.36
Previously, in vivo and in vitro studies showed that expression of mutant COMP (T585M and D472Y) activated the apoptotic arm of the UPR, including PERK and phosphorylation of eIF2α.42,43 The presence of D472Y-COMP increased levels of P-eIF2α and apoptosis, compared with COS-7 cells expressing wild-type COMP (WT-COMP).43 In mice expressing T585M-COMP MED/EDM1 mutation, the UPR was activated, although mutant COMP retention was not detected in the ER of growth-plate chondrocytes.42 Despite the lack of COMP retention, expression of T585M-COMP up-regulated transcription of glucose-regulated protein 78 (Grp78; also known as Hspa5, Bip), calreticulin (Crt; also known as Calr, Crtc), and Chop, all of which are UPR genes. Additionally, T585M expression caused increased cleavage of caspase and reduction of B-cell lymphoma 2 (BCL-2) levels in chondrocytes.42 These findings suggest that the UPR pathway is activated by mutant COMP expression and that cell death occurs by caspase activation. However, expression of T585M-COMP does not model the pseudoachondroplasia chondrocyte pathology of intracellular retention and thus may not represent the true pseudoachondroplasia pathological mechanisms.
The present study was undertaken to define the UPR mechanisms involved when D469del-COMP, the common mutation causing pseudoachondroplasia, is expressed and retained in chondrocytes. Human D469del-COMP was expressed in rat chondrosarcoma (RCS) cells from an inducible adenoviral delivery system that we previously showed generates the pseudoachondroplasia chondrocyte pathology. This system allowed examination of the effect of retained D469del-COMP on the UPR. We show that D469del-COMP triggers the UPR but does not effectively activate the folding and degradation pathways. Furthermore, D469del-COMP expression induced persistent ER and oxidative stress, affecting mitochondrial metabolism and thereby priming chondrocytes for cell death through necroptosis.
Materials and Methods
Inducible Recombinant COMP Adenovirus Construct
An inducible recombinant COMP adenovirus construct (Figure 1A) was generated as described previously.44 This inducible system expresses either WT-COMP or D469del-COMP using the reverse transactivator (rtTA). D469del-COMP contains D469del, the common mutation responsible for 30% of pseudoachondroplasia cases.45 Both human WT-COMP and D469del-COMP have a FLAG-tag, and expression is driven by binding of doxycycline (DOX)-activated rtTA to the tetracycline responsive element (TRE) in the presence of DOX. Recombinant adenovirus was produced, isolated, and quantified using a Qbiogene AdEasy vector system kit (MP Biomedicals, Solon, OH). Amplification was performed by Vector BioLabs (Philadelphia, PA).
Figure 1.

Expression of DOX-inducible COMP in vitro. A: Schematic of the construct used to express WT-COMP and D469del-COMP. The ubiquitous cytomegalovirus promoter (pCMV) drives expression of rtTA, which in turn drives expression of the COMP gene in the presence of DOX. B: Western blots show WT-COMP and D469del-COMP expression. RCS cells were cultured in the presence of 1 μg/mL DOX for 2, 4, and 7 days. β-Actin was used as the loading control. C and D: Coimmunostaining of infected RCS cells with FLAG (green) and Golgi-specific 58K (red) at 4 days (C) or ER-specific CRT antibodies (red) at 7 days (D). DAPI (blue) indicates the nucleus. Scale bar = 5 μm.
Cell Culture and Adenovirus Infection
RCS cells were grown in monolayer with Dulbecco's modified Eagle's medium supplemented with 10% FBS. The cells were seeded in a 12-well plate at a density of 3 × 105 cells/well or in a 24-well plate at a density of 1.5 × 105 cells/well and infected with WT-COMP or D469del-COMP adenovirus in Dulbecco's modified Eagle's medium containing 2% of TET-free serum in the presence or absence of 1 μg/mL DOX. Multiplicity of infection values for recombinant adenovirus were selected to equalize for COMP expression levels in the infected cells: MOI = 40 for WT-COMP and MOI = 120 for D469del-COMP. Ad-TeTOn (rtTA) was used as adenovirus infection control (Vector BioLabs). For all adenovirus infections, the cells were cultured in 2% tetracycline-free FBS and Dulbecco's modified Eagle's medium in the presence or absence of 1 μg/mL DOX.
Histology and Immunohistochemistry
RCS cells were fixed in methanol at −20°C for 10 minutes either 4 or 7 days after infection. After three washes in PBS-Tween 20, cells were blocked in 10% normal donkey serum in PBS-Tween 20 for 1 hour at room temperature. The cells were incubated with the following primary antibodies: FLAG (green), 58K (red; Sigma-Aldrich, St. Louis, MO), calreticulin (red; CRT; Santa Cruz Biotechnology, Santa Cruz, CA) at 1:500 for 1 hour at 37°C. Secondary antibodies Alexa Fluor 488 (FLAG rabbit) and Alexa Fluor 594 (58K goat and CRT goat) were incubated at 1:800 dilutions for 1 hour at 37°C (Life Technologies; Invitrogen, Carlsbad, CA). Coverslips were mounted using ProLong Gold with DAPI (blue; Life Technologies). Images were captured on a BX51 inverted microscope (Olympus America, Center Valley, PA) attached to a Retiga 2000RV camera (QImaging, Surrey, BC, Canada) using SimplePCI6 software (Hamamatsu Photonics, Hamamatsu, Japan).
Immunoblot Analysis
Protein lysates were collected using radioimmunoprecipitation assay buffer. These lysates contained both cellular proteins and extracellular proteins found in the ECM surrounding RCS cells. Lysates were sonicated on ice three times for 15 seconds with a 45-second interval. Protein concentrations were determined using a Pierce BCA protein assay (Thermo Fisher Scientific, Rockford, IL) and samples were electrophoresed on gradient SDS-PAGE gels (4% to 12%) and transferred to polyvinylidene difluoride membranes. Western blots were incubated with antibodies against FLAG (dilution 1:5000; Sigma-Aldrich), eIF2α, phospho-eIF2α, caspase-3, phospho-Histone γH2AX (Ser139), AIF (at 1:1000 dilutions; Cell Signaling Technology, Inc), CHOP (GADD153), GADD34, GRP78, and GRP94 (at 1:500 dilutions; Santa Cruz Biotechnology). Secondary goat anti-rabbit IgG HRP was incubated at 1:7000 dilution (Life Technologies). β-actin-HRP antibody (Abcam, Cambridge, MA) was used at 1:200,000 dilution to detect the loading control, β-actin. Membranes were visualized using enhanced chemiluminescence (GE Healthcare, Piscataway, NJ).
mRNA Quantification
Total RNA was isolated from the RCS cells using TRIzol reagent (Life Technologies Corp) and treated with RNase-free DNase1 (Ambion; Applied Biosystems, Austin, TX). cDNA was made using an iScript kit (Bio-Rad Laboratories, Hercules, CA). Levels of UPR and Gapdh mRNAs were measured by quantitative PCR using a SYBR Green kit (Applied Biosystems). The primers used are listed in Table 1. Relative changes in mRNA levels were assessed using the comparative CT method. All of the measurements were normalized to the endogenous Gapdh mRNA.
Table 1.
Primers Used in Real-Time RT-PCR
| Gene⁎ | Primer sequences |
|---|---|
| hCOMP | Fwd-5′-GCAATGACACCATCCCAGAG-3′ |
| Rev-5′-CTTGTCATCGTCGTCCTTGTAGTC-3′ | |
| Chop | Fwd-5′-CCAGCAGAGGTCACAAGCAC-3′ |
| Rev-5′-CGCACTGACCACTCTGTTTC-3′ | |
| Grp78 | Fwd-5′-CCACCAGGATGCAGACATTG-3′ |
| Rev-5′-AGGGCCTCCACTTCCATAGA-3′ | |
| Xbp1(S)† | Fwd-5′-GTCTGCTGAGTCCGCAGCAGGT-3′ |
| Rev-5′-CAGACTCAGAATCTGAAGAGGC-3′ | |
| Edem1 | Fwd-5′-GCAATGAAGGAGAAGGAGAC-3′ |
| Rev-5′-CCATATGGCGTAGTAGAAGGC-3′ | |
| Gadd34 | Fwd-5′-AATCAGGACCCTGAGATTCCT-3′ |
| Rev-5′-CTGGTCCTGCCCAGACAG-3′ | |
| Ero1b | Fwd-5′-GACACCAAGACCCTTCTGCT-3′ |
| Rev-5′-CTTTAATGACTTGGCCCCC-3′ | |
| Gadd45a | Fwd-5′-CCGAAAGGATGGACACGGTG-3′ |
| Rev-5′-TTATCGGGGTCTACGTTGAGC-3′ | |
| Gapdh | Fwd-5′-AGTTCAACGGCACAGTCAAG-3′ |
| Rev-5′-TACTCAGCACCAGCATCACC-3′ |
Alternative gene names and symbols include the following. For Chop: DNA damage-inducible transcript 3 protein (Ddit3). For Gadd34: protein phosphatase 1, regulatory subunit 15A (Ppp1r15a). For Grp78: heat shock 70kDa protein 5 (glucose-regulated protein, 78kDa) (Hspa5).
Spliced isoform.
Reactive Oxygen Species Assay
Reactive oxygen species (ROS) levels were measured using an OxiSelect intracellular ROS assay kit (Cell Biolabs, San Diego, CA). Briefly, infected RCS cells were incubated with the cell-permeable fluorogenic probe 2′,7′-dichlorofluorescein-diacetate (DCFH-DA). After conversion of DCFH-DA into fluorescent DCF by ROS, the fluorescence was read with a plate reader. The concentration of DCF is proportional to the fluorescence intensity, which is used to measure the ROS levels.
Gene Expression PCR Array
Expression of genes involved in oxidative stress and antioxidant defense was analyzed using an RT2 Profiler PCR array, PARN-065E, from Qiagen SABiosciences (Frederick, MD). Three samples from the control (rtTA) and test (D469del-COMP) groups were prepared accordingly to the manufacturer's protocol. Samples containing 3.6 ng of cDNA were analyzed using a 4 × 96 plate format PCR array. Real-time PCR detection was performed on ABI 7900HT system using a two-step cycling program recommended by the manufacturer (Applied Biosystems). Software available through the SABiosciences website (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php) was used for data analysis.
Mitochondrial Membrane Potential Assay
Mitochondrial membrane potential in RCS cells was evaluated using a JC-1 mitochondrial membrane potential assay kit (Cayman Chemical, Ann Arbor, MI). Briefly, infected RCS cells were incubated with JC-1 staining solution according to manufacturer's protocol and were analyzed by fluorescent microscopy. Healthy mitochondria with J-aggregates (red) were visualized at 560/595 nm and unhealthy mitochondria with JC-1 monomers (green) were visualized at 485/535 nm.
Results
Inducible Expression of D469del-COMP in RCS Cells Generates the Pseudoachondroplasia Cellular Phenotype
To study the effect of human D469del-COMP in chondrocytes, we used a DOX-inducible adenoviral expression system to generate the pseudoachondroplasia chondrocyte phenotype in RCS cells (Figure 1A). RCS cells were used because they maintain the chondrocyte phenotype and produce ECM proteins in monolayer culture, whereas primary chondrocytes quickly dedifferentiate and cease to synthesize matrix proteins.46 We have previously established that D469del-COMP-transfected RCS cells mimic the pseudoachondroplasia chondrocyte trafficking defect.47 RCS cells were infected with either WT-COMP or D469del-COMP adenovirus, and COMP expression was induced by 1 μg/mL of DOX for 2, 4, and 7 days. WT-COMP and D469del-COMP were first detected at 4 days after infection and were present at 7 days (Figure 1B). The protein lysates contained both intra- and extracellular proteins. To evaluate the cellular trafficking of each recombinant COMP protein, coimmunostaining was performed with either 58K (Golgi-specific) or CRT (ER-specific) antibodies and a FLAG antibody that specifically recognizes recombinant COMP. WT-COMP colocalized with the 58K Golgi-specific marker (Figure 1C), indicating that the WT-COMP is processed through the ER to the Golgi and is being exported in the ECM. In contrast, D469del-COMP colocalized with CRT, indicating that the D469del-COMP is retained in the ER (Figure 1D). These results demonstrate that inducible D469del-COMP expression in RCS cells recapitulates the chondrocyte trafficking pathology seen in pseudoachondroplasia and in our mouse model of pseudoachondroplasia.18,19,22,25,27,48
Continuous Expression of COMP Induces Components of the UPR in RCS Cells
After confirming that this inducible system models the pseudoachondroplasia chondrocyte phenotype, we then examined which UPR pathways were triggered by WT-COMP or D469del-COMP after induction of expression for 2 and 4 days. The mRNA and protein levels of genes involved in the UPR pathways were assessed. After 2 days of expression, the presence of D469del-COMP induced higher levels of Grp78, X-box binding protein 1 spliced isoform [Xbp1(S)], Chop, and ER degradation-enhancing α-mannosidase-like 1 (Edem1) mRNAs, compared with cells expressing WT-COMP (Figure 2, A–D); however, protein levels of GRP78, GRP94, and protein disulfide isomerase (PDI), all folding chaperones, were similar in both WT-COMP and D469del-COMP expressing cells (Figure 2E). After 4 days of expression, D469del-COMP increased Chop and Edem1 mRNA (Figure 2, C and D). The presence of P-eIF2α by Western blot indicates that the UPR proapoptotic pathway is induced after 2 days of COMP expression (Figure 2F). CHOP protein was not detectable until after 4 days of expression (Figure 2F), even though D469del-COMP clearly increased Chop mRNA levels at both 2 and 4 days of expression (Figure 2D). These results show that folding (Grp78) and degradative [Edem1 and Xbp1(S)] pathways are induced at the mRNA level only. In contrast, induction of mRNA and protein levels of CHOP shows that the apoptotic arm is activated.
Figure 2.

D469del-COMP expression in RCS cells triggers UPR. A–D: mRNA levels of UPR genes Grp78 (A), Xbp1(S) (B), Edem1 (C), and Chop (D) were analyzed by qRT-PCR after expression of WT-COMP (white bars) and D469del-COMP (black bars) for 2 and 4 days. All qRT-PCR measurements were normalized to GAPDH. Assays were performed in triplicate; error bars indicate ± SD. **P < 0.01. E and F: Levels of UPR proteins from RCS cells expressing WT-COMP or D469del-COMP for 2 and 4 days were analyzed by Western blot: Grp78, Grp94, and PDI (E); CHOP, P-eIF2α, abd eIF2 α (F). β-Actin was used as the loading control.
Retained D469del-COMP Sustains the Apoptotic UPR Pathway in RCS Cells
After determining that 4 days of D469del-COMP expression was necessary for substantial intracellular retention and CHOP protein expression, we assessed the effect of retained D469del-COMP on the UPR pathway genes. WT-COMP or D469del-COMP expression was induced with DOX for 4 days (the expression period), and then DOX was removed and RCS cells were cultured for 5 days more (the retention period). To control for any effects of adenoviral infection, the TetOn adenovirus that expresses only reverse Tet transactivator (rtTA) was used. The absence of DOX turns off recombinant COMP synthesis, and during this period the effect of retained D469del-COMP was assessed. The levels of both WT-COMP and D469del-COMP mRNA dramatically decreased after removal of DOX (Figure 3A), indicating that removal of DOX effectively shuts down synthesis of COMP. High levels of intracellular D469del-COMP detected by Western blot after 5 days without DOX show that D469del-COMP was retained (Figure 3B). During the retention period, the levels of Grp78, Xbp1(S), Edem1, and Chop mRNA were significantly increased in cells with D469del-COMP, compared with controls (Figure 3, C–F). Moreover, levels of Xbp1(S) and Chop mRNAs increased approximately twofold from the expression to the retention period, even though there was no mRNA expression of D469del-COMP during the 5-day retention period (Figure 3, D and F). The levels of Grp78 and Edem1 remained unchanged during both expression and retention periods (Figure 3, C and E), indicating that retention of D469del-COMP does not effectively trigger the folding and degradation pathways of the UPR in RCS cells.
Figure 3.

Intracellular D469del-COMP maintains apoptotic UPR pathway in RCS cells. A and B: Levels of hCOMP mRNA and protein during the expression (dark bars) and retention periods (light bars) were assessed using qRT-PCR (A) and Western blot (B), in which human COMP expression was compared between negative controls, one with no virus (NV) and the other with rtTA virus (TA), and WT-COMP and D469del (MT)-COMP expressing viruses. β-Actin was used as the loading control for Western blots. C–F: mRNA levels of Grp78 (C), Xbp1(S) (D), Edem1 (E), and Chop (F) were analyzed by qRT-PCR during the WT-COMP and D469del-COMP expression and retention periods, normalized to GAPDH. Assays were performed in triplicate; error bars indicate ± SD. *P < 0.05; **P < 0.01.
Intracellular D469del-COMP Retention Increases Oxidative Stress
After determining that CHOP is critical to the D469del-COMP pseudoachondroplasia cellular pathology, we evaluated oxidative stress, which is known to be a consequence of sustained CHOP activation.35 Endoplasmic reticulum oxidoreductase 1beta (ERO1β) is an enzyme induced by ER stress and involved in the generation of ROS.49 Ero1β was significantly increased in the cells expressing D469del-COMP and continued to increase during the retention period (Figure 4A).
Figure 4.
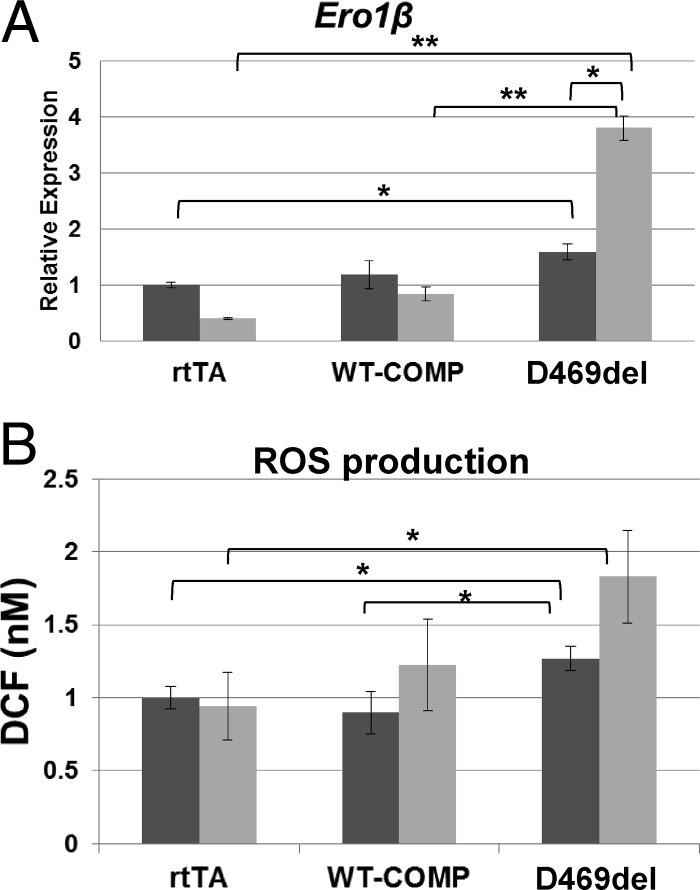
Intracellular D469del-COMP induces generation of ROS in RCS cells. A: mRNA levels of Ero1β were analyzed by qRT-PCR after the WT-COMP and D469del-COMP expression (dark bars) and retention (light bars) periods. Measurements were normalized to GAPDH. Assays were performed in triplicate; error bars indicate ± SD. *P < 0.05; **P < 0.01. B: Intracellular levels of ROS were measured in RCS cells after the WT-COMP and D469del-COMP expression and retention periods using a fluorogenic probe, DCFH-DA. The DCF fluorescence was detected using a standard fluorescence plate reader and compared with a standard curve. *P < 0.05.
To determine whether D469del-COMP retention alters the redox state of chondrocytes, we measured the level of intracellular ROS using DCF, which measures the conversion of the DCFH-DA probe by ROS.50,51 The level of ROS production was significantly higher in the cells infected with D469del-COMP, compared with WT-COMP or rtTA, during both the expression and the retention periods (Figure 4B). After determining that ROS stress plays a role in the D469del-COMP pseudoachondroplasia chondrocyte pathology, we evaluated the expression of 84 genes involved in oxidative stress and antioxidant defense using a PCR array (RT2 Profiler PARN-065E; Qiagen). The genes showing a twofold or greater differential expression of antioxidant and oxidative stress genes during the retention period are listed in Table 2.
Table 2.
Genes Involved in Oxidative Stress That Are Regulated by Retention of MT-COMP in RCS Cells Compared with Control rtTA
| Rat gene symbol | Gene name | Fold change | P value |
|---|---|---|---|
| Antioxidants | |||
| Apc | Adenomatous polyposis coli | 11.32 | 0.0018 |
| Gsr | Glutathione reductase | 2.35 | 0.0065 |
| Gstk1 | Glutathione-S-transferase kappa1 | 4.42 | 0.032 |
| Prdx3 | Peroxiredoxin 3 | 6.15 | 0.0057 |
| Prdx5 | Peroxiredoxin 5 | 3.48 | 0.001 |
| Prdx6 | Peroxiredoxin 6 | 3.53 | 0.009 |
| Ptgs1 | Prostaglandin-endoperoxide synthase 1 | 2.08 | 0.01 |
| Ptgs2 | Prostaglandin-endoperoxide synthase 2 | 16.56 | 0.037 |
| Tmod1 | Tropomodulin 1 | 12.29 | 0.0031 |
| Txnrd1 | Thioredoxin reductase 1 | 5.36 | 0.0001 |
| Txnrd2 | Thioredoxin reductase 2 | 8.36 | 0.001 |
| Genes involved in reactive oxygen species metabolism | |||
| Genes involved in superoxide metabolism | |||
| Nox4 | NADPH oxidase 4 | 107.55 | 0.0030 |
| Oxidative stress responsive genes | |||
| Als2 | Amyotrophic lateral sclerosis (juvenile) homolog human | 4.53 | 0.0005 |
| Apoe | Apolipoprotein E | 3.73 | 0.0001 |
| Duox2 | Dual oxidase 2 | −2.77 | 0.037 |
| Ercc2 | Excision repair cross-complementing rodent repair deficiency, complementation group 2 | 5.09 | 0.0235 |
| Nqo1 | NAD(P)H dehydrogenase, quinone 1 | 4.25 | <0.0001 |
| Park7 | Parkinson disease (autosomal recessive, early onset) 7 | 2.34 | 0.0145 |
| Prnp | Prion protein | 10.58 | 0.0006 |
| Psmb5 | Proteasome (prosome, macropain) subunit, beta type 5 | 2.26 | 0.0001 |
| Txnip | Thioredoxin-interacting protein | 29.89 | 0.0016 |
| Oxygen transporter genes | |||
| Cygb | Cytoglobin | 4.85 | 0.0031 |
| Dnm2 | Dynamin 2 | 4.17 | 0.0402 |
| Fancc | Fanconi anemia, complementation group C | 3.92 | 0.0046 |
| Hba-a2 | Hemoglobin alpha, adult chain 2 | 8.85 | 0.0002 |
| Ift172 | Intraflagellar transport 172 homolog (Chlamydomonas) | 3.73 | 0.0025 |
| Slc38a1 | Solute carrier family 38, member 1 | 19.22 | 0.0076 |
| Slc38a4 | Solute carrier family 38, member 4 | 98.46 | 0.0028 |
Out of 84 genes present in the rat oxidative stress and antioxidant defense PCR array (PARN-065E RT2 Profiler PCR array, Qiagen SABiosciences, Frederick, MD), only genes that presented a twofold or greater change with significance at P < 0.05 are shown.
Intracellular D469del-COMP up-regulated the expression of many antioxidant genes 2- to 16-fold. In contrast, thioredoxin interacting protein (TXNIP) was induced 30-fold by D469del-COMP retention. TXNIP is known to inhibit a major antioxidant, thioredoxin (TRX).52 Most importantly, retention of D469del-COMP increased NADPH oxidase 4 (NOX4) >100-fold. NOX4 is an enzyme involved in ROS generation during the UPR. In addition, dual oxidase 2 (Duox2), an oxidative stress response gene, was down-regulated approximately threefold. Two solute carrier family 38 genes, Slc38a1 and Slc38a4, both involved in transport of neutral amino acids, were up-regulated 19- and 98-fold, respectively, in the retention period. Neutral amino acids play a role in the biosynthesis of reduced glutathione (GSH), a major intracellular antioxidant.53 Taken together, these results indicate that D469del-COMP retention generates oxidative stress in RCS cells by substantially increasing the expression of enzymes involved in ROS production (Ero1β and Nox4) and by limiting antioxidant activity through Txnip.
Intracellular D469del-COMP Induces DNA Damage
To determine whether the oxidative stress generated by D469del-COMP retention causes DNA damage, we assessed the mRNA levels of DNA damage marker genes, including growth arrest and DNA damage 34 (Gadd34), Gadd45A, and assessed the protein level of Ser139-phosphorylated histone H2AX (γH2AX). Expression of Gadd34 was significantly increased during the D469del-COMP expression period (Figure 5A). Gadd34 is directly activated by CHOP.35 Gadd45A gene expression was higher in cells expressing and retaining D469del-COMP (Figure 5B). Similarly, levels of γH2AX were increased during D469del-COMP the expression and retention periods (54% and 43%, respectively), compared with WT-COMP control (6% and 27%, respectively; Figure 5C). These findings indicate that retention of D469del-COMP causes DNA damage in RCS cells.
Figure 5.
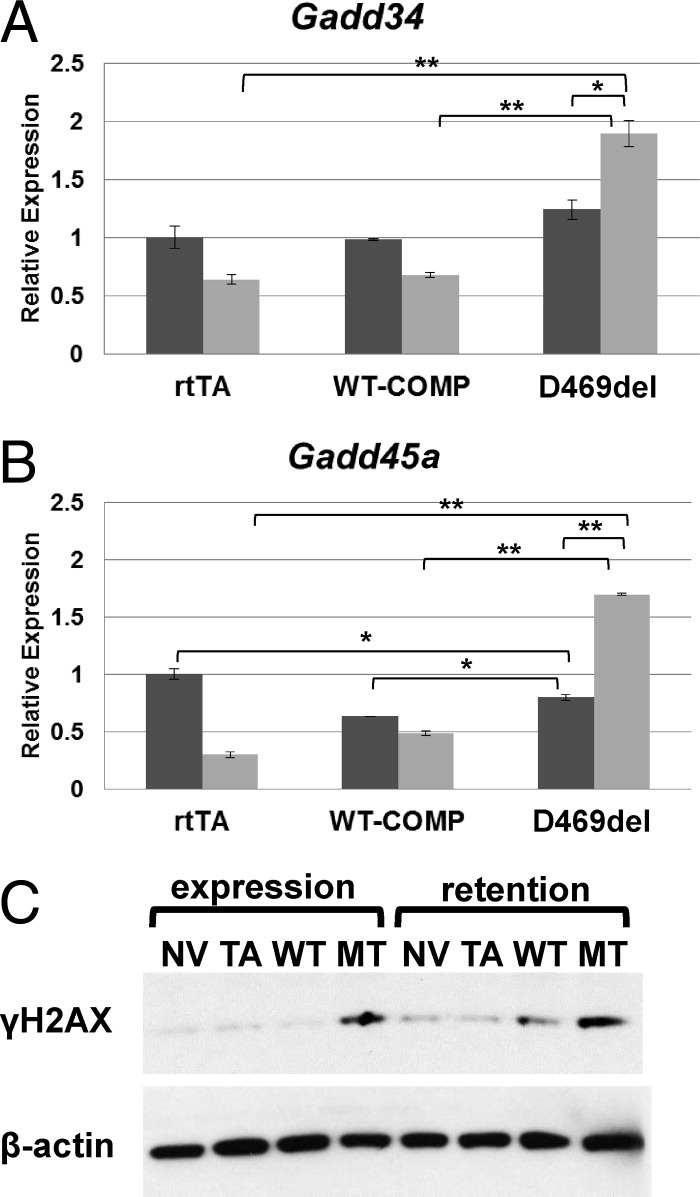
Intracellular D469del-COMP induces DNA damage in RCS cells. mRNA levels of Gadd34 (A) and Gadd45a (B) were analyzed by qRT-PCR after the WT-COMP and D469del-COMP expression (dark bars) and retention (light bars) periods. Measurements were normalized to GAPDH. Assays were performed in triplicate; error bars indicate ± SD. *P < 0.05; **P < 0.01. C: Levels of γH2AX in RCS cells after the WT-COMP and D469del-COMP expression and retention periods were analyzed by Western blot. γH2AX expression was compared between negative controls, one with no virus (NV) and the other with rtTA virus (TA), and WT-COMP and D469del (MT)-COMP expressing viruses. β-Actin was used as the loading control.
Intracellular D469del-COMP Induces Cell Death through Necroptosis
To determine which type of cell death is elicited by retained D469del-COMP, we evaluated cleavage of caspases 3, 8, 9, and 12, mitochondrial integrity, and AIF (necroptosis specific). Expression and retention of D469del-COMP did not trigger activation of caspase 3 (Figure 6A). Similar results were obtained for caspases 8, 9, and 12 (data not shown). We also assessed the levels of survivin, a member of the inhibitor of apoptosis (IAP) family that inhibits caspase cleavage.54,55 During the retention period, D469del-COMP expressing cells showed a higher level of survivin than the controls, which may explain the lack of caspase cleavage (Figure 6B).
Figure 6.

Intracellular D469del-COMP induces necroptosis in RCS cells. Protein levels of cleaved caspase 3 (A) and survivin (B) were analyzed by Western blot after the WT-COMP and D469del-COMP expression and retention periods. Expression was compared between negative controls, one with no virus (NV) and the other with rtTA virus (TA), and WT-COMP and D469del (MT)-COMP expressing viruses. C: Mitochondrial membrane potential was measured by detecting color changes in fluorescence of the dye JC-1. RCS cells were cultured in the presence of 1 μg/mL DOX for expression (EXP) and retention (RET) periods. JC-1 changes color from green to red as it forms J-aggregates in healthy mitochondria with elevated membrane potential; it remains green in the monomeric form in mitochondria with low membrane potential. The loss of red fluorescence and increased green mitochondria are indicators of low mitochondrial membrane potential and cell death. D: Protein levels of AIF and cleaved AIF (asterisk) were analyzed by Western blot after expression and retention periods. β-Actin was used as the loading control.
Mitochondrial membrane potential is often compromised during cell death and therefore, we investigated whether mitochondria are involved in D469del-COMP-induced cell death. Mitochondrial transmembrane potential (Δψm) was measured by detecting color changes in fluorescent dye JC-1 (5,5′,6,6′-tetrachloro-1, 1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide). The JC-1 monomer changes from green to red as it forms J-aggregates in healthy mitochondria with high membrane potential.56 In contrast, unhealthy mitochondria have a low membrane potential and form few J-aggregates. D469del-COMP retention compromises mitochondrial membrane potential, because no J-aggregates were detected during the retention period (green) (Figure 6C). In contrast, the control cells had a mixture of healthy and unhealthy mitochondria (red and green; Figure 6C).
In a proapoptotic environment, AIF is cleaved, released from the mitochondria to the cytosol, and translocated to the nucleus, where it induces caspase-independent cell death (ie, programmed necrosis, or necroptosis). We next examined whether AIF is involved in the cell death of chondrocytes during the D469del-COMP retention period. More cleaved AIF was detected (12%) in cells retaining D469del-COMP, compared with controls (2.6%; Figure 6D). Moreover, the amount of full-length AIF with expression of WT-COMP and D469del-COMP was equivalent when normalized to β-actin. These results demonstrate that D469del-COMP retention in chondrocytes negatively affects mitochondria function and triggers cell death through necroptosis.
Discussion
Pseudoachondroplasia is a well-characterized, severe, and debilitating dwarfism caused by mutations in COMP.18,19 It has long been known that the pseudoachondroplasia chondrocyte ER becomes massively engorged, but the pathological mechanism or mechanisms have been difficult to unravel.22 Recent in vivo and in vitro studies have shown that different COMP mutations activate the UPR and that this activation is not dependent on whether mutant COMP is retained.42,43 However, none of these studies followed the UPR beyond activation, and thus there is little information about the actual processes that lead to chondrocyte death and short-limb dwarfism. To better understand the process by which mutant COMP expression and retention mediates chondrocyte toxicity, we expressed the common D469del-COMP mutation in a RCS pseudoachondroplasia model system. Although both the adaptive (protein folding and degradation) and apoptotic arms of the UPR are activated, only the apoptotic arm is functional (Figure 7). Moreover, we show that necroptosis, a specific type of apoptosis, causes cell death through downstream CHOP signaling and oxidative stress. These novel results now define the mechanisms underlying this ER chondrocyte storage disorder. Importantly, this mechanism has been confirmed in our transgenic D469del-COMP (D469del) mouse that exhibits the phenotypic and chondrocyte pathology of pseudoachondroplasia.48
Figure 7.

Model of UPR and apoptosis induction by intracellular D469del-COMP retention. Intracellular D469del-COMP retention triggers the UPR. Several adaptive mechanisms of the UPR pathway (decreased translation, refolding, and ER-associated degradation) are unable to successfully restore normal ER function. Activation of the proapoptotic CHOP UPR pathway results in expression of GADD34, which in turn restarts protein translation, thereby exacerbating D469del-COMP accumulation. Overexpression of CHOP-dependent ERO1β and NOX4 leads to increased ROS production and subsequently to oxidative stress. Oxidative stress causes DNA damage (elevated levels of Gadd45A mRNA and protein γH2AX) and decreased mitochondrial membrane potential (ΔΨm). Ultimately, tAIF is released from the impaired mitochondria and translocates to the nucleus, where it associates with γH2AX to initiate necroptosis.
Necroptosis is a type of cell death that has features of both apoptosis (programmed cell death) and necrosis (passive cell death).57 Mitochondrial dysfunction and oxidative stress play important roles in necroptosis.40,57 Mild oxidative stress causes apoptosis through the activation of caspase cleavage.58 In contrast, excessive or prolonged oxidative stress suppresses cleavage of caspases and mediates cell death through caspase-independent mechanisms.59 Here, we show that D469del-COMP retention lowers mitochondrial transmembrane potential and induces oxidative stress through Ero1β and Nox4 (Figures 4 and 6C). Additionally, no caspase 3 activation or cleavage was detected (Figure 6A), and similarly none for caspases 8, 9, and 12 (data not shown), indicating that the caspase-dependent apoptosis is not activated by D469del-COMP. Other studies have shown that high levels of ROS inhibit activation of caspases, and this is likely occurring in RCS cells that express and retain D469del-COMP.58,59 Additionally, two necroptosis executioners have been identified: mitochondrial protein AIF and receptor interacting protein (RIP-1).40,41,60 Necroptosis is initiated by the association of cleaved AIF with γH2AX, a marker of DNA double-strand breaks. Subsequently, the endonuclease CypA is recruited to join the γH2AX/AIF complex and initiates chromatin condensation, DNA fragmentation, and finally cell death. We show that cleaved AIF and γH2AX are both up-regulated by retained D469del-COMP (Figure 6D). Taken together, these findings indicate that the mechanisms underlying necroptosis, rather than apoptosis, are occurring in the RCS cells expressing and retaining D469del-COMP.
The generation of ROS and ER stress are closely linked.49 ROS production during UPR stress involves several enzymes, including ERO1β, PDI, and NOX4.49 ERO1β maintains an oxidative environment necessary for correct protein folding in the ER.61 D469del-COMP retention increased expression of CHOP and Ero1β (CHOP-dependent; Figure 4A), which is consistent with previous descriptions of the CHOP-induced UPR pathway.35 Nox4 expression was also markedly increased in response to retained D469del-COMP (Table 2). Increases in Ero1β and Nox4 are known to generate ROS, which could further stimulate incorrect disulfide bonding, leading to additional protein misfolding and impairment of chaperone function.61 In addition, Txnip, an important inhibitor of the antioxidant thioredoxin was strongly induced after D469del-COMP retention, most likely exacerbating oxidative stress. Furthermore, our results showed that DNA damage occurred in cells retaining D469del-COMP, as indicated by up-regulation of Gadd45A expression and the presence of γH2AX. Extensive DNA damage is a well-recognized mediator of cell death, and D469del-COMP-induced DNA damage is most likely due to oxidative stress. Overall, our data suggest a model in which persistent presence of misfolded proteins in the ER results in excessive oxidation, causing damage to DNA and ultimately cell death through necroptosis (Figure 7).
Retention of D469del-COMP indicates that UPR clearance mechanisms are ineffective, and here we show that, even though the adaptive UPR responses are activated, downstream processes do not proceed. D469del-COMP induced more protein-folding capacity and ER-associated degradation, through increased synthesis of Xbp1(S), Grp78, and Edem1 (Figures 2 and 3). However, despite increased mRNA levels, D469del-COMP retention did not stimulate an increase in the Grp78 protein needed for protein folding (Figure 2). Edem1 is an ER enzyme involved in retrotranslocation of misfolded proteins to the cytoplasm and targeting the proteins for proteasomal degradation. D469del-COMP was not cleared from the ER, even though the level of Edem1 mRNA was increased after D469del-COMP expression in RCS cells, indicating that the UPR clearance mechanisms are ineffective.
A calcium scaffold is necessary for correct COMP folding in the ER.62 The D469del mutation causes the loss of calcium-binding, which negatively affects the ability of COMP to undergo protein folding,11,63,64 and so D469del-COMP is likely resistant to refolding efforts. Repetitive folding attempts by chaperones would therefore be unsuccessful, which is likely another cause of excessive ROS production.35,65 Additionally, we have previously shown that retained D469del-COMP prematurely associates with other ECM proteins in the ER and assembles a matrix network in chondrocytes both in vivo and in vitro.26,27 The matrix assembly likely traps D469del-COMP, limiting retrotranslocation from the ER to the proteasome.26,27 Taken together, these and previous results suggest that D469del-COMP cannot be delivered to the proteasome, because the size of the retained material renders Edem1 ineffective.
Thus far, we have shown that UPR mechanisms to clear retained D469del-COMP from RCS cells are ineffective, thus driving UPR toward the CHOP-dependent proapoptotic pathway. Several other studies have evaluated the UPR in cells expressing different COMP mutations. Expression of mutant D472Y COMP in COS-7 cells increased i) the level of P-eIF2α, ii) the number of TUNEL-positive cells, and iii) DNA fragmentation.43 In the mutant T585M COMP knock-in mouse, increases in P-eIF2α protein and Chop mRNA levels generated mild rER stress, despite little accumulation.42 Increases in caspase 12 cleavage and TUNEL-positive cells with a decrease in BCl-2 expression were observed.42 In both studies, however, D469del-COMP was not retained, which may limit the level of ER stress and thereby limit ROS production. In those models, mild oxidative stress may lead to apoptotic cell death.58,59 In our RCS model, which has extensive D469del-COMP retention similar to that of pseudoachondroplasia, high levels of ROS cause cell death through necroptosis. Moreover, we show in our D469del-COMP mouse, which recapitulates the pseudoachondroplasia chondrocyte phenotype, that oxidative stress and cleaved AIF are up-regulated, indicating that necroptosis is occurring in growth-plate chondrocytes.40,41
The role of oxidative stress in D469del-COMP chondrocyte pathology has not previously been appreciated. The present novel findings suggest that antioxidants and drugs that reduce ROS production may be beneficial for pseudoachondroplasia and MED/EDM1. These easily accessible therapies, which have a long history of safe and effective use, provide a new avenue to address the underlying chondrocyte pathology. This class of therapeutics has been shown to improve protein secretion and cellular function with other ER-stress-related disorders.66 Specifically, a mixture of dietary antioxidants and anti-inflammatories improved cognitive function in an Alzheimer disease mouse model.67 Additionally, a combination of vitamin C and E has been shown to decrease inflammation and oxidative stress and to improve insulin sensitivity in type 2 diabetes.68–70 Investigations of exciting new oxidative stress therapies suggest that limiting ROS production by use of NOX inhibitors may be more effective than antioxidant therapy.71 At present, only symptomatic interventions are available for pseudoachondroplasia and MED/EDM1. Oxidative stress therapy is a novel approach to prevent premature chondrocyte loss and dwarfism.
Footnotes
Supported by NIH grant 1RO1AR057117 (J.T.H), a Shriners Hospital for Children grant (J.T.H.), and the Leah Lewis Foundation.
References
- 1.Riessen R., Fenchel M., Chen H., Axel D.I., Karsch K.R., Lawler J. Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:47–54. doi: 10.1161/01.atv.21.1.47. [DOI] [PubMed] [Google Scholar]
- 2.Recklies A.D., Baillargeon L., White C. Regulation of cartilage oligomeric matrix protein synthesis in human synovial cells and articular chondrocytes. Arthritis Rheum. 1998;41:997–1006. doi: 10.1002/1529-0131(199806)41:6<997::AID-ART6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 3.Hecht J.T., Deere M., Putnam E., Cole W., Vertel B., Chen H., Lawler J. Characterization of cartilage oligomeric matrix protein (COMP) in human normal and pseudoachondroplasia musculoskeletal tissues. Matrix Biol. 1998;17:269–278. doi: 10.1016/s0945-053x(98)90080-4. [DOI] [PubMed] [Google Scholar]
- 4.DiCesare P., Hauser N., Lehman D., Pasumarti S., Paulsson M. Cartilage oligomeric matrix protein (COMP) is an abundant component of tendon. FEBS Lett. 1994;354:237–240. doi: 10.1016/0014-5793(94)01134-6. [DOI] [PubMed] [Google Scholar]
- 5.Müller G., Michel A., Altenburg E. COMP (cartilage oligomeric matrix protein) is synthesized in ligament, tendon, meniscus, and articular cartilage. Connect Tissue Res. 1998;39:233–244. doi: 10.3109/03008209809021499. [DOI] [PubMed] [Google Scholar]
- 6.Fife R.S., Brandt K.D. Identification of a high-molecular-weight (greater than 400 000) protein in hyaline cartilage. Biochim Biophys Acta. 1984;802:506–514. doi: 10.1016/0304-4165(84)90370-2. [DOI] [PubMed] [Google Scholar]
- 7.Hedbom E., Antonsson P., Hjerpe A., Aeschlimann D., Paulsson M., Rosa-Pimentel E., Sommarin Y., Wendel M., Oldberg A., Heinegård D. Cartilage matrix proteins: An acidic oligomeric protein (COMP) detected only in cartilage. J Biol Chem. 1992;267:6132–6136. [PubMed] [Google Scholar]
- 8.Adams J.C. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 9.Holden P., Meadows R.S., Chapman K.L., Grant M.E., Kadler K.E., Briggs M.D. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem. 2001;276:6046–6055. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- 10.Mann H.H., Ozbek S., Engel J., Paulsson M., Wagener R. Interactions between the cartilage oligomeric matrix protein and matrilins: Implications for matrix assembly and the pathogenesis of chondrodysplasias. J Biol Chem. 2004;279:25294–25298. doi: 10.1074/jbc.M403778200. [DOI] [PubMed] [Google Scholar]
- 11.Thur J., Rosenberg K., Nitsche D.P., Pihlajamaa T., Ala-Kokko L., Heinegård D., Paulsson M., Maurer P. Mutations in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen I, II, and IX. J Biol Chem. 2001;276:6083–6092. doi: 10.1074/jbc.M009512200. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg K., Olsson H., Mörgelin M., Heinegård D. Cartilage oligomeric matrix protein shows high affinity zinc-dependent interaction with triple helical collagen. J Biol Chem. 1998;273:20397–20403. doi: 10.1074/jbc.273.32.20397. [DOI] [PubMed] [Google Scholar]
- 13.Kipnes J., Carlberg A.L., Loredo G.A., Lawler J., Tuan R.S., Hall D.J. Effect of cartilage oligomeric matrix protein on mesenchymal chondrogenesis in vitro. Osteoarthritis Cartilage. 2003;11:442–454. doi: 10.1016/s1063-4584(03)00055-4. [DOI] [PubMed] [Google Scholar]
- 14.Chen F.H., Thomas A.O., Hecht J.T., Goldring M.B., Lawler J. Cartilage oligomeric matrix protein/thrombospondin 5 supports chondrocyte attachment through interaction with integrins. J Biol Chem. 2005;280:32655–32661. doi: 10.1074/jbc.M504778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawler J. The functions of thrombospondin-1 and -2. Curr Opin Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 16.Xu K., Zhang Y., Ilalov K., Carlson C.S., Feng J.Q., Di Cesare P.E., Liu C.J. Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J Biol Chem. 2007;282:11347–11355. doi: 10.1074/jbc.M608744200. [DOI] [PubMed] [Google Scholar]
- 17.Halász K., Kassner A., Mörgelin M., Heinegård D. COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282:31166–31173. doi: 10.1074/jbc.M705735200. [DOI] [PubMed] [Google Scholar]
- 18.Briggs M.D., Hoffman S.M.G., King L.M., Olsen A.S., Mohrenweiser H., Leroy J.G., Mortier G.R., Rimoin D.L., Lachman R.S., Gaines E.S., Cekleniak J.A., Knowlton R.G., Cohn D.H. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995;10:330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 19.Hecht J.T., Nelson L.D., Crowder E., Wang Y., Elder F.F., Harrison W.R., Francomano C.A., Prange C.K., Lennon G.G., Deere M., Lawler J. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet. 1995;10:325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 20.Unger S.L., Briggs M.D., Holden P., Zabel B., Ala-Kokko L., Paassilta P., Lohiniva J., Rimoin D.L., Lachman R.S., Cohn D.H. Multiple epiphyseal dysplasia: radiographic abnormalities correlated with genotype. Pediatr Radiol. 2001;31:10–18. doi: 10.1007/s002470000362. [DOI] [PubMed] [Google Scholar]
- 21.McKeand J., Rotta J., Hecht J.T. Natural history study of pseudoachondroplasia. Am J Med Genet. 1996;63:406–410. doi: 10.1002/(SICI)1096-8628(19960517)63:2<406::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 22.Cooper R.R., Ponseti I.V., Maynard J.A. Pseudoachondroplastic dwarfism: A rough-surfaced endoplasmic reticulum storage disorder. J Bone Joint Surg Am. 1973;55:475–484. [PubMed] [Google Scholar]
- 23.Hecht J.T., Hayes E., Haynes R., Cole W.G. COMP mutations, chondrocyte function and cartilage matrix. Matrix Biol. 2005;23:525–533. doi: 10.1016/j.matbio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Hecht J.T., Makitie O., Hayes E., Haynes R., Susic M., Montufar-Solis D., Duke P.J., Cole W.G. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J Orthop Res. 2004;22:759–767. doi: 10.1016/j.orthres.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Hecht J.T., Montufar-Solis D., Decker G., Lawler J., Daniels K., Duke P.J. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol. 1998;17:625–633. doi: 10.1016/s0945-053x(98)90113-5. [DOI] [PubMed] [Google Scholar]
- 26.Merritt T.M., Alcorn J.L., Haynes R., Hecht J.T. Expression of mutant cartilage oligomeric matrix protein in human chondrocytes induces the pseudoachondroplasia phenotype. J Orthop Res. 2006;24:700–707. doi: 10.1002/jor.20100. [DOI] [PubMed] [Google Scholar]
- 27.Merritt T.M., Bick R., Poindexter B.J., Alcorn J.L., Hecht J.T. Unique matrix structure in the rough endoplasmic reticulum cisternae of pseudoachondroplasia chondrocytes. Am J Pathol. 2007;170:293–300. doi: 10.2353/ajpath.2007.060530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duke J., Montufar-Solis D., Underwood S., Lalani Z., Hecht J.T. Apoptosis staining in cultured pseudoachondroplasia chondrocytes. Apoptosis. 2003;8:191–197. doi: 10.1023/a:1022926811397. [DOI] [PubMed] [Google Scholar]
- 29.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 30.Malhotra J.D., Kaufman R.J. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schröder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harding H.P., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [Erratum appeared in Nature 1999;398:90] [DOI] [PubMed] [Google Scholar]
- 33.Harding H.P., Zhang Y., Bertolotti A., Zeng H., Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 34.Kim R., Emi M., Tanabe K., Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 35.Marciniak S.J., Yun C.Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H.P., Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Festjens N., Vanden Berghe T., Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta. 2006;1757:1371–1387. doi: 10.1016/j.bbabio.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 37.Golstein P., Aubry L., Levraud J.P. Cell-death alternative model organisms: why and which? Nat Rev Mol Cell Biol. 2003;4:798–807. doi: 10.1038/nrm1224. [DOI] [PubMed] [Google Scholar]
- 38.Hengartner M.O. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 39.Bassik M.C., Scorrano L., Oakes S.A., Pozzan T., Korsmeyer S.J. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baritaud M., Boujrad H., Lorenzo H.K., Krantic S., Susin S.A. Histone H2AX: the missing link in AIF-mediated caspase-independent programmed necrosis. Cell Cycle. 2010;9:3166–3173. doi: 10.4161/cc.9.16.12887. [DOI] [PubMed] [Google Scholar]
- 41.Artus C., Boujrad H., Bouharrour A., Brunelle M.N., Hoos S., Yuste V.J., Lenormand P., Rousselle J.C., Namane A., England P., Lorenzo H.K., Susin S.A. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29:1585–1599. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piróg-Garcia K.A., Meadows R.S., Knowles L., Heinegård D., Thornton D.J., Kadler K.E., Boot-Handford R.P., Briggs M.D. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum Mol Genet. 2007;16:2072–2088. doi: 10.1093/hmg/ddm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hashimoto Y., Tomiyama T., Yamano Y., Mori H. Mutation (D472Y) in the type 3 repeat domain of cartilage oligomeric matrix protein affects its early vesicle trafficking in endoplasmic reticulum and induces apoptosis. Am J Pathol. 2003;163:101–110. doi: 10.1016/S0002-9440(10)63634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Posey K.L., Liu P., Wang H.R., Veerisetty A.C., Alcorn J.L., Hecht J.T. RNAi reduces expression and intracellular retention of mutant cartilage oligomeric matrix protein. PLoS One. 2010;5:e10302. doi: 10.1371/journal.pone.0010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Unger S., Hecht J.T. Pseudoachondroplasia and multiple epiphyseal dysplasia: new etiologic developments. Am J Med Genet. 2001;106:244–250. [PubMed] [Google Scholar]
- 46.King K.B., Kimura J.H. The establishment and characterization of an immortal cell line with a stable chondrocytic phenotype. J Cell Biochem. 2003;89:992–1004. doi: 10.1002/jcb.10571. [DOI] [PubMed] [Google Scholar]
- 47.Chen T.L., Stevens J.W., Cole W.G., Hecht J.T., Vertel B.M. Cell-type specific trafficking of expressed mutant COMP in a cell culture model for PSACH. Matrix Biol. 2004;23:433–444. doi: 10.1016/j.matbio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Posey K.L., Veerisetty A.C., Liu P., Wang H.R., Poindexter B.J., Bick R., Alcorn J.L., Hecht J.T. An inducible cartilage oligomeric matrix protein mouse model recapitulates human pseudoachondroplasia phenotype. Am J Pathol. 2009;175:1555–1563. doi: 10.2353/ajpath.2009.090184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santos C.X., Tanaka L.Y., Wosniak J., Laurindo F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11:2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 50.Brandt R., Keston A.S. Synthesis of diacetyldichlorofluorescin: a stable reagent for fluorometric analysis. Anal Biochem. 1965;11:6–9. doi: 10.1016/0003-2697(65)90035-7. [DOI] [PubMed] [Google Scholar]
- 51.Keston A.S., Brandt R. The fluorometric analysis of ultramicro quantities of hydrogen peroxide. Anal Biochem. 1965;11:1–5. doi: 10.1016/0003-2697(65)90034-5. [DOI] [PubMed] [Google Scholar]
- 52.Schulze P.C., De Keulenaer G.W., Yoshioka J., Kassik K.A., Lee R.T. Vitamin D3-upregulated protein-1 (VDUP-1) regulates redox-dependent vascular smooth muscle cell proliferation through interaction with thioredoxin. Circ Res. 2002;91:689–695. doi: 10.1161/01.res.0000037982.55074.f6. [DOI] [PubMed] [Google Scholar]
- 53.Mackenzie B., Erickson J.D. Sodium-coupled neutral amino acid (system N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004;447:784–795. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- 54.Shin S., Sung B.J., Cho Y.S., Kim H.J., Ha N.C., Hwang J.I., Chung C.W., Jung Y.K., Oh B.H. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry. 2001;40:1117–1123. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 55.Tamm I., Wang Y., Sausville E., Scudiero D.A., Vigna N., Oltersdorf T., Reed J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58:5315–5320. [PubMed] [Google Scholar]
- 56.Salvioli S., Ardizzoni A., Franceschi C., Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997;411:77–82. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- 57.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 58.Hampton M.B., Orrenius S. Redox regulation of apoptotic cell death. Biofactors. 1998;8:1–5. doi: 10.1002/biof.5520080101. [DOI] [PubMed] [Google Scholar]
- 59.Hampton M.B., Fadeel B., Orrenius S. Redox regulation of the caspases during apoptosis. Ann N Y Acad Sci. 1998;854:328–335. doi: 10.1111/j.1749-6632.1998.tb09913.x. [DOI] [PubMed] [Google Scholar]
- 60.Delavallée L., Cabon L., Galán-Malo P., Lorenzo H.K., Susin S.A. AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics. IUBMB Life. 2011;63:221–232. doi: 10.1002/iub.432. [DOI] [PubMed] [Google Scholar]
- 61.Sevier C.S., Kaiser C.A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta. 2008;1783:549–556. doi: 10.1016/j.bbamcr.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 62.Kvansakul M., Adams J.C., Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004;23:1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen H., Deere M., Hecht J.T., Lawler J. Cartilage oligomeric matrix protein is a calcium-binding protein, and a mutation in its type 3 repeats causes conformational changes. J Biol Chem. 2000;275:26538–26544. doi: 10.1074/jbc.M909780199. [DOI] [PubMed] [Google Scholar]
- 64.Hou J., Putkey J.A., Hecht J.T. Delta 469 mutation in the type 3 repeat calcium binding domain of cartilage oligomeric matrix protein (COMP) disrupts calcium binding. Cell Calcium. 2000;27:309–314. [Google Scholar]
- 65.McCullough K.D., Martindale J.L., Klotz L.O., Aw T.Y., Holbrook N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malhotra J.D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S.W., Kaufman R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parachikova A., Green K.N., Hendrix C., LaFerla F.M. Formulation of a medical food cocktail for Alzheimer's disease: beneficial effects on cognition and neuropathology in a mouse model of the disease. PLoS One. 2010;5:e14015. doi: 10.1371/journal.pone.0014015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ristow M., Zarse K., Oberbach A., Klöting N., Birringer M., Kiehntopf M., Stumvoll M., Kahn C.R., Blüher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garcia-Bailo B., El-Sohemy A., Haddad P.S., Arora P., Benzaied F., Karmali M., Badawi A. Vitamins D, C, and E in the prevention of type 2 diabetes mellitus: modulation of inflammation and oxidative stress. Biologics. 2011;5:7–19. doi: 10.2147/BTT.S14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Devaraj S., Leonard S., Traber M.G., Jialal I. Gamma-tocopherol supplementation alone and in combination with alpha-tocopherol alters biomarkers of oxidative stress and inflammation in subjects with metabolic syndrome. Free Radic Biol Med. 2008;44:1203–1208. doi: 10.1016/j.freeradbiomed.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Drummond G.R., Selemidis S., Griendling K.K., Sobey C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]