

Abstract
Macrophage differentiation and function are pivotal for cell survival from infection and involve the processing of microenvironmental signals that determine macrophage cell fate decisions to establish appropriate inflammatory balance. NADPH oxidase 2 (Nox2)–deficient chronic granulomatous disease (CGD) mice that lack the gp91phox (gp91phox−/−) catalytic subunit show high mortality rates compared with wild-type mice when challenged by infection with Listeria monocytogenes (Lm), whereas p47phox-deficient (p47phox−/−) CGD mice show survival rates that are similar to those of wild-type mice. We demonstrate that such survival results from a skewed macrophage differentiation program in p47phox−/− mice that favors the production of higher levels of alternatively activated macrophages (AAMacs) compared with levels of either wild-type or gp91phox−/− mice. Furthermore, the adoptive transfer of AAMacs from p47phox−/− mice can rescue gp91phox−/− mice during primary Lm infection. Key features of the protective function provided by p47phox−/− AAMacs against Lm infection are enhanced production of IL-1α and killing of Lm. Molecular analysis of this process indicates that p47phox−/− macrophages are hyperresponsive to IL-4 and show higher Stat6 phosphorylation levels and signaling coupled to downstream activation of AAMac transcripts in response to IL-4 stimulation. Notably, restoring p47phox protein expression levels reverts the p47phox-dependent AAMac phenotype. Our results indicate that p47phox is a previously unrecognized regulator for IL-4 signaling pathways that are important for macrophage cell fate choice.
Activated macrophages maintain inflammatory balance during tissue damage, stress, and immune reactions, and thereby preserve local homeostasis. Inflammation is a protective host response to injury caused by pathological and physiological insults such as microbial infection and tissue damage.1 It is a biphasic process; involving both vascular and cellular responses, and is triggered by irritants within the local tissue milieu. Macrophages are major innate immune cellular respondents to pathological and physiological environmental signals. Depending on the signal(s) received, macrophages differentiate along classical versus alternative activation pathways. Using this linear categorization, classically activated macrophages are typically defined immunologically as pro-inflammatory mediators, whereas the diverse population of alternatively activated macrophages (AAMac) are classified as anti-inflammatory.2 However, more recent classification of macrophages based on their homeostatic and regulatory roles such as host defense, wound healing, and immune regulation encompasses their homeostatic as well as immunological function.3,4 Various innate or adaptive signals initiate CD4+ T-cell activation and differentiation into polarized T helper 1 (TH1) and TH2 effectors that secrete cytokines such as interferon (IFN)–γ and interleukin (IL)–4, respectively. In turn, IFN-γ drives classical macrophage activation, whereas IL-4 drives alternative macrophage activation during adaptive immune responses.5
We recently reported that p47phox−/− mice,6 a model of the human immunodeficiency chronic granulomatous disease (CGD), develop AAMac-driven active chronic inflammation7 characterized by systemic exaggerated chitinase-like Ym1/Ym2 protein8 secretion and progressive crystalline macrophage pneumonia. p47phox is best recognized as an organizing adaptor protein for multicomponent NADPH oxidase (Nox)−2 holoenzyme–dependent superoxide generation.9,10 Mutations in the genes for p47phox and the membrane-associated catalytic gp91phox/Nox2 subunit comprise the major autosomal recessive and X-linked, respectively, forms of CGD.11–13 Phagocytes from p47phox−/−6 and gp91phox−/−14 mice are devoid of NADPH oxidase–dependent reactive oxygen species (ROS) production. In addition, similar to human CGD patients, p47phox−/− 6 and gp91phox−/−14 mice develop CGD-like lesions, including those of systemic infection, granulomatous inflammation, and active chronic hyperinflammatory diseases.13,15,16
Interestingly p47phox−/− mice spontaneously develop sterile progressive crystalline macrophage pneumonia in a pulmonary niche that also contains high levels of IL-12 and TH1, TH2, and TH17 cytokines.7 These findings reveal that the sterile spontaneous active chronic inflammation in the p47phox−/− mice is noncanonical, and thereby indicate that there is a more complex basis for the observed aberrant p47phox−/− AAMac-mediated inflammation that is likely to involve contributions from both macrophage intrinsic factors and the inflammatory niche in p47phox-deficient mice.
In this study, we explored the signaling pathways mediating p47phox−/− macrophage default to an AAMac phenotype during primary Lm infection. Here we show that AAMacs from p47phox−/− mice provide a protective function against Listeria monocytogenes (Lm) infection. A key finding is that STAT6 molecular signaling is upregulated and transcription of phenotypic AAMac transcripts is enhanced in p47phox−/− macrophages because the macrophages are hyperresponsive to IL-4 stimulation.
Materials and Methods
Mice
NADPH oxidase p47phox-deficient (p47phox−/−) mice have previously been described.6 The backcrossing of 14 generations with wild-type (WT) C57BL/6NTac generated congenic p47phox−/− mice on a C57BL/6NTac background. Both congenic p47phox−/− and WT control mice (C57BL/6NTac) were obtained from Taconic Farms (Hudson, NY).The p47phox−/− mice were housed in aseptic conditions and given water containing Bactrim (0.13 mg/mL trimethoprim and 0.67 mg/mL sulfamethoxazole; Actiatis MidAtlantic, Columbia, MD). gp91phox−/−/Nox2−/− B6.129S6-Cybbtm1Din/J mice14 were obtained from the Jackson Laboratory (Bar Harbor, ME). Nox1−/−17 mice were a generous gift from Dr. Chihiro Yabe-Nishimura (Kyoto Prefectural University of Medicine, Kyoto Japan). All mice were 6 to 8 weeks of age. This study (permit no. ASP LHD 11) was reviewed and approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (Public Health Service Assurance A4149-01). The study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health/National Institute of Allergy and Infectious Diseases.
Bacteria and Reagents
Recombinant Lm (rLM-OVA)18 strain 10403S expressing ovalbumin was a generous gift from Dr. Hao Shen (University of Pennsylvania, School of Medicine, Philadelphia, PA). Lm was subcultured on Difco Brain Heart Infusion Agar (BHI; BD, Sparks, MD). Polyethylene glycol SOD (PEG-SOD) was purchased from Sigma-Aldrich (St. Louis, MO), and diphenylene iodonium (DPI) was purchased from Calbiochem (EMD Chemicals, Inc., Gibbstown, NJ).
Histopathology and Immunohistochemistry
Tissue sections (3 to 4 μm) from formalin-fixed paraffin-embedded tissues were stained with hematoxylin and eosin (H&E) and examined for routine histopathology. Similar unstained sections were used for immunofluorescence. For immunofluorescence, slides were deparaffinized in Xylene and rehydrated through graded concentrations of ethanol. A rabbit polyclonal antibody against YM1/YM2 proteins8 was incubated on the tissue sections at a dilution of 1:250 for 60 minutes at room temperature. The bound YM1/YM2 antibody was detected using a biotinylated anti-rabbit secondary antibody (Vector Labs, Burlingame, CA), followed by streptavidin conjugated with Alexa Fluor 594 (Invitrogen, Carlsbad, CA). For detecting murine macrophage mannose receptor (MMR), sections were incubated with a goat polyclonal antibody against MMR (R&D Systems, Inc., Minneapolis, MN) at a dilution of 1:100 for 60 minutes at room temperature. Bound antibody was detected using a biotinylated anti-goat secondary antibody (Vector Labs), followed by streptavidin conjugated with Alexa Fluor 594 (Invitrogen). Nuclei were stained with DAPI, and sections were mounted with ProLong Gold anti-fade mounting medium (Invitrogen).
Infection of Mice and Determination of Bacterial Load
Overnight rLM-OVA cultures were serially diluted in PBS to the desired dose and injected i.v. into the lateral tail vein or i.p. into the peritoneal cavity of mice as indicated. Inoculates were plated in sterile PBS on BHI agar (BD) to verify dose. Bactrim prophylaxis was discontinued 1 week before infection. Mice were infected with 5 × 104 CFU (0.1 LD50) by i. v. injection. Bacterial loads were determined by plating 10-fold serials dilutions of organ homogenates in sterile PBS on BHI agar (BD).
Macrophage Adoptive Transfer
Donor p47phox−/− mice received 5 × 104 CFU (0.1 LD50) Lm. in 1 mL of PBS by i.p. injection, or 1 mL of 3% w/v of thioglycolate 3 days before macrophage harvest. Ten million p47phox−/− inflammatory macrophages were i. v. injected in 10 gp91phox−/− recipient mice 1 day before Lm infection (as described above). A control group of 10 gp91phox−/− mice received 100 μL of PBS by i.v. injection.
Intracellular Stain and Flow-Cytometric Analysis
Spleens from C57BL/6 WT, p47phox−/− and gp91phox−/− mice infected by 0.1 LD50 rLM-OVA were aseptically removed and passed though a nylon mesh screen. Red blood cells were lysed with ACK lysing medium (Lonza, Walkersville, MD). Splenocytes (5 × 106 cells/mL) were resuspended in IMDM complete: IMDM (Gibco/Invitrogen Corp, Carlsbad, CA) containing 10% fetal bovine serum (Hyclone Laboratories, Logan, UT), 2.0 mmol/L l-glutamine (Hyclone), 50 μmol/L β-mercaptoethanol (Sigma Aldrich, St. Louis, MO), and 100 units/mL of penicillin and streptomycin 10 units/mL (Gibco/Invitrogen), and incubated with 10 μg/mL OVA257–264 or OVA323–339 peptides (synthesized by NIAID Research Technologies Branch- Peptide Synthesis and Analysis Unit, Bethesda, MD). A 1-μg/mL quantity of Golgistop (BD Pharmingen) was added for the final 2 hours of culture. After 5 hours, cells were stained with fluorochrome-labeled antibodies (anti-CD4, anti-CD8, and anti-IL-4; BD Pharmingen, San Diego, CA) using the Cytofix/Cytoperm Kit (BD Pharmingen) according to the manufacturer's instructions. Splenocytes were analyzed on a FACS Canto (BD Bioscience, San Jose, CA), and data were analyzed using FlowJo (Tree Star, Ashland, OR).
Macrophage Preparation and Culture
Peritoneal macrophages were harvested from thioglycolate (Sigma Aldrich, St. Louis, MO)–stimulated mice as previously described.7 Mice received 2 mL 3% w/v thioglycolate i.p. 3 days before macrophage harvest. Approximately 1 × 106 cells per sample were seeded into 24-well plates in volume of 1 mL. After a 3-hour incubation, the nonadherent cells were removed by washing with PBS. The adherent cells were incubated in DMEM (Gibco/Invitrogen) supplemented with 10% fetal bovine serum (ATCC, Manassas,VA), and stimulated as indicated.
Bone Marrow–Derived Macrophages
Briefly, bone marrow was flushed with PBS, resuspended in 1 mL of ACK lysing medium (Lonza), and incubated at room temperature for 5 minutes to eliminate red blood cells. The single-cell suspension was filtered though a 40-μm cell strainer (BD Falcon/BD Biosciences, San Jose, CA), re-suspended at a density of 3 × 106/mL in complete DMEM (Gibco/Invitrogen) supplemented with 10% fetal bovine serum (ATCC) and 10 ng/mL GM-CSF (R&D Systems), and then plated and incubated in a 5% CO2 incubator for 5 days. Nonadherent cells were decanted, and the remaining adherent cells were washed with fresh medium and stimulated as indicated.
Lm-Infected Macrophage Isolation and Culture
Mice received 5 × 104 CFU (0.1 LD50) Lm. in 1 mL of PBS by i.p. injection 7 days before macrophage harvest, as described above.
In Vitro Lm-Killing Assay
Bone marrow–derived macrophages were plated on a 24-well plate at 5 × 105 cells per well and cultured with complete DMEM alone or supplemented with IL-4 (1000 U/mL), heat-killed Lm (HKL; 1 × 105 CFU/mL), or lipopolysaccharide (100 ng/mL, InvivoGen, San Diego, CA) plus IFN-γ (50 U/mL, R&D Systems) for 24 hours. After washing with PBS, cells were inoculated with 5 × 105 CFU/mL of Lm in 1 mL of RPMI 1640 (Gibco/Invitrogen) plus 10% normal mouse serum without antibiotics for 30 minutes. The cells were then incubated with RPMI 1640 plus 10% FCS and 5 μg/mL Gentamicin (Sigma Aldrich) for 30 minutes to kill extracellular bacteria, washed with PBS, and incubated with RPMI 1640 plus 10% fetal calf serum for 5.5 hours (time 6 hours). After 6 hours, the cells were washed and lysed with 0.05% Triton X-100 (Sigma Aldrich). At the 30-minute time point, time 0 cells were washed and lysed with 1 mL of 0.05% Triton X-100 (Sigma Aldrich). The lysates were plated on BHI agar (BD) overnight, and Lm killing was determined by counting the number of CFU per well.
Virus Production and Transduction
VSV-G–pseudotyped lentiviral–human p47phox and e-green fluorescent protein (eGFP) were generated by transient co-transfection of the specific transfer vector plasmid with the three packaging plasmids (pMDLg/pRRE, the gag-pol plasmid; pRSV-Rev, a Rev-expressing plasmid; and pMD.G, a VSV-G envelope–expressing plasmid) into 293T cells (ATCC) as described previously.19 Lentiviral vector supernatant was filtered, concentrated by ultracentrifugation (43,000 × g for 3 hours), and aliquots were stored at −70°C. For transient expression, 20 μL of the concentrated virus was added to the cell culture media, followed by a 30-minute centrifugation (2500 rpm) for 3 consecutive days to enhance the transduction. Transduction was confirmed by microscopic and flow-cytometric detection of eGFP expression using FACS (BD Bioscience).
Quantitative Real-Time PCR
Total RNA was extracted by using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Quantitative real-time PCR (qPCR) was performed with the 7500 Real Time PCR System (Applied Biosystems, Foster City, CA) and performed with 1 ng of RNA samples and specific Gene Expression Assay (Applied Biosystems) using the TaqMan RNA-to-CT 1-Step Kit (Applied Biosystems). The Gene Expression Assays for YM1, Arg1, GAPDH, PKG1, and RPS29 were purchased from Applied Biosystems. The relative change in gene expression was calculated by the ΔΔ Ct method (Applied Biosystems) from triplicate determinations using RPS29 as a reference.
Immunoblotting
Cell lysates were prepared in M-PER Buffer (Pierce Biotechnology, Rockford, IL) supplemented with Halt Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail (Pierce Biotechnology). The protein concentrations were determined by the Bradford assay. Proteins were resolved by NuPage 4 to 12% Bis-Tris Gel (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes by iBlot Gel Transfer (Invitrogen). Immunoblotting analyses were performed by SNAPid system (Millipore, Billerica, MA). Immunocomplexes were reacted with Western Blotting Reaction Reagent (GE Healthcare, Piscataway, NJ). Proteins were then visualized by exposed to HyBlot CL film (Denville Scientifics, Metuchen, NJ). Scanned images were analyzed by ImageJ20 for the relative protein quantification, anti–β-actin monoclonal antibody (Sigma), anti-p47phox polyclonal antibody (Millipore, Billerica, MA), anti–ECF-L monoclonal antibody (R&D Systems), anti-Arg1 monoclonal antibody (BD), anti-phosphorylated Stat6 (pY641) polyclonal antibody (BD) and anti-Stat6 polyclonal antibody (BD) were used as primary antibodies. The secondary antibodies, ECL Mouse IgG, HRP-Linked Whole antibody (from sheep), ECL Rabbit IgG, HRP-Linked Whole antibody (from donkey) and ECL Rat IgG, HRP-Linked Whole antibody (from goat) were purchased from GE Healthcare.
Statistical Analysis
Multigroup test analysis using repeated-measures analysis of variance with the Tukey, Bonferroni, or Newman–Keuls post test using a value of α = 0.05 for the 99.9% confidence interval was performed for the real-time PCR and the severity score analyses. Multigroup test analysis using repeated-measures analysis of variance with the Tukey, Bonferroni or Newman–Keuls post test, and two-way analysis of variance with a Bonferroni post test using a value of α = 0.001 for the 99.9% confidence interval was performed for the in vitro killing assays. Differences between gp91phox−/− recipient mice in the survival analysis were determined by Mantel–Cox log-rank test and the log-rank test for trends (Prism 5; GraphPad Software, San Diego, CA).
Results
p47phox−/− Macrophages Discriminate Classical Versus Alternative Activation Signals
We previously demonstrated that thioglycolate-elicited inflammatory macrophages from p47phox−/− mice discriminate classical versus alternative macrophage activation signals in vitro, and that the inflammatory macrophages from the peritoneal cavities of p47phox−/− mice also secrete Ym1 protein ex vivo.7 Using IL-4 to simulate alternative activation or IFN-γ in combination with heat-killed Lm (HKL/γ) or LPS (LPS/γ) to simulate classical activation, we observed that, similar to inflammatory macrophages, bone marrow–derived p47phox−/− macrophages (BMMacs) also discriminate classical versus alternative macrophage activation signals in vitro (Figure 1). Using real-time PCR quantification, we found that whereas IL-4 stimulated p47phox−/− BMMacs express more Ym1 than WT or NADPH oxidase 2 catalytic subunit–deficient gp91phox−/− BMMacs, Ym1 gene expression was not detected in WT, p47phox−/− or gp91phox−/− BMMacs treated with classical pathway stimuli (Figure 1A). We also found that, in response to IL-4 as well as classical pathway stimuli, transcription for the classical activation marker iNos was similar in BMMacs from each genotype (Figure 1A).
Figure 1.
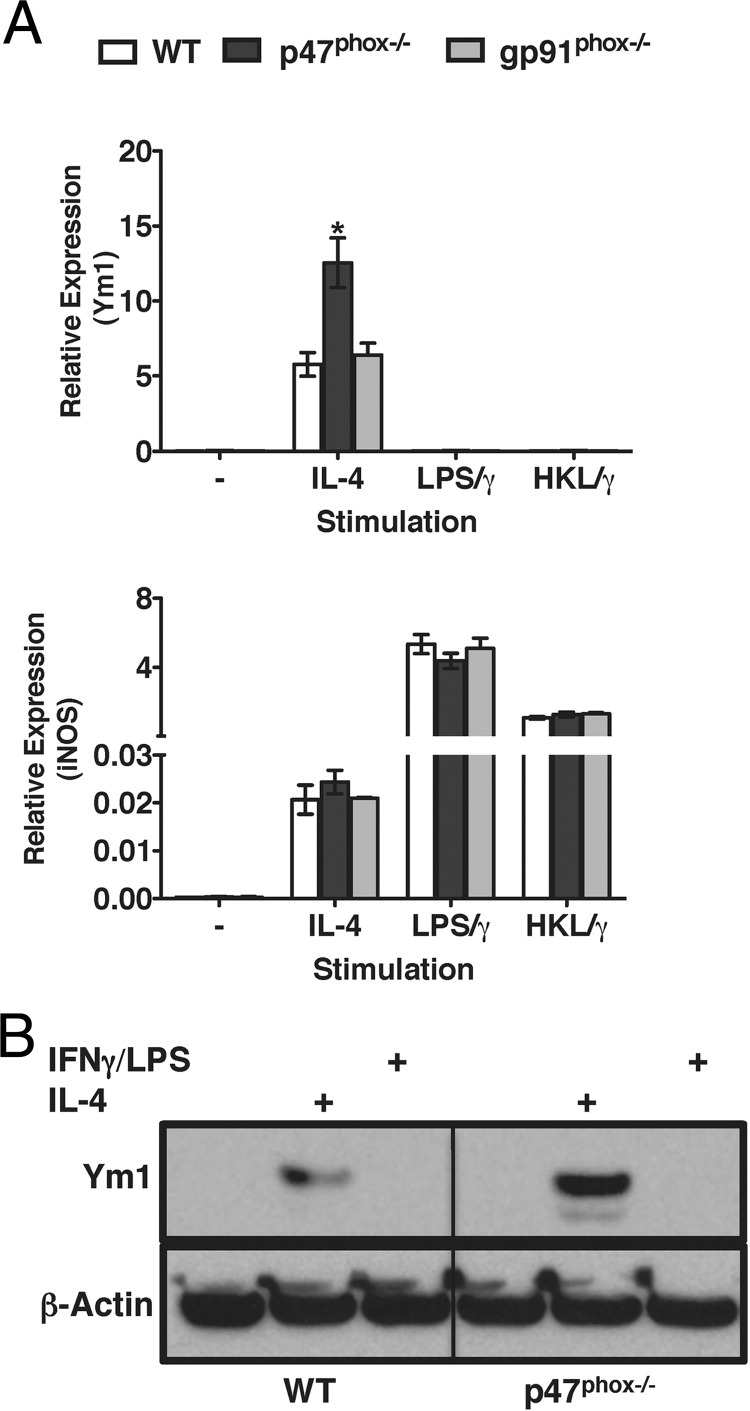
p47phox−/− macrophages discriminate activation signal in vitro. Bone marrow–derived macrophages were stimulated overnight with 1000 units/mL IL-4, heat-killed Lm (HKL/γ or LPS/γ). A: Real-time PCR quantification of Ym1 and iNos. Values are relative to expression of the gene encoding RPS29. Data represent three independent experiments including three to four of each genotype per experiment. *P < 0.05. B: Total cell lysates from BMMacs stimulated overnight with IL-4 or LPS/γ were blotted with anti-Ym1 or anti–β-actin. Results are representative of two independent experiments including three of each genotype per experiment.
Protein analysis revealed that neither resting WT nor p47phox−/− BMMacs express Ym1 protein in the absence of in vitro stimulation. However, similar to inflammatory p47phox−/− macrophages, IL-4–stimulated p47phox−/− BMMacs express more Ym1 protein than similarly treated WT BMMacs (Figure 1B). These findings indicated that although p47phox−/− macrophages discriminated classical versus alternative macrophage activation signals, IL-4 stimulation induced a more robust AAMac phenotype in p47phox−/− macrophages.
p47phox Protein Is Essential for Attenuating IL-4–Dependent Alternative Macrophage Activation
In macrophages, IL-4 induced STAT6 activation, which initiated transcription of specific alternative macrophage activation genes such as Ym1, arginase 1 (Arg1), and FIZZ1.5 To further discriminate a role for p47phox protein in regulating macrophage cell fate decisions, we next assessed IL-4–induced STAT6 activation, and transcriptional changes in STAT6-regulated AAMac target genes in inflammatory macrophages. Relative to either WT or gp91phox−/− macrophages, STAT6 phosphorylation was significantly enhanced in p47phox−/− macrophages that were treated with 10 and 50 units of IL-4 (Figure 2A). In contrast, there was no difference in STAT6 phosphorylation in WT, p47phox−/−, or gp91phox−/− macrophages that were stimulated with a saturating dose of 5000 units of IL-4. Consistent with the finding that Ym1 transcripts are upregulated in IL-4–stimulated p47phox−/− BMMacs relative to similarly treated WT macrophages (Figure 1), transcripts encoding Ym1, Arg1, and FIZZ1 were also upregulated in IL-4–stimulated p47phox−/− inflammatory macrophages (Figure 2B). Interestingly, IL-4–induced transcripts for Ym1, Arg1, and FIZZ1 were similar in WT and gp91phox−/− inflammatory macrophages (Figure 2B). Furthermore, unlike WT or gp91phox−/− macrophages, transcription of FIZZ1 was significantly upregulated in response to 10 units of IL-4 stimulation, and transcripts for FIZZ1, Ym1, and Arg1 were significantly upregulated in response to 50 units of IL-4 stimulation in p47phox−/− macrophages (Figure 2B). These data clearly demonstrate that, unlike WT or gp91phox−/− macrophages, p47phox−/− macrophages are hyperresponsive to IL-4 stimulation, which indicates that the skewed AAMac phenotype is cell intrinsic and due to the loss of p47phox protein expression.
Figure 2.

p47phox protein attenuates IL-4 signaling in macrophages independent of the gp91phox catalytic subunit. A: Immunoblot analysis of STAT6 phosphorylation in inflammatory macrophages from each genotype stimulated with IL-4 for 20 minutes as indicated. Results of one representative experiment of three independent experiments are shown. B: Real-time PCR analysis of Arg1, Ym, and FIZZ1 mRNA from inflammatory macrophages from each genotype stimulated with IL-4 for 18 hours as indicated. Values are relative to expression of the gene encoding RPS29. Data represent three independent experiments including three of each genotype per experiment. *P < 0.05.
Notably, IL-4 did not trigger STAT6 hyperphosphorylation or enhance transcription of target AAMac genes in gp91phox−/− macrophages in vitro (Figure 2). We also found that Ym1 protein expression was similar in IL-4–stimulated macrophages from WT mice, and Nox catalytic subunit gp91phox and Nox117-deficient mice (see Supplemental Figure S1A at http://ajp.amjpathol.org). Furthermore, scavenging endogenous superoxide, using the membrane-permeable ROS scavenger polyethylene glycol–superoxide dismutase (PEG-SOD), which converts superoxide into hydrogen peroxide (see Supplemental Figure S1B at http://ajp.amjpathol.org), and the selective Nox inhibitor diphenylene iodonium (DPI;9 see Supplemental Figure S1C at http://ajp.amjpathol.org) did not enhance IL-4–induced STAT6 activation in inflammatory WT macrophages. These findings further support the essential role for p47phox protein rather that gp91phox/Nox2 enzymatic activity for regulating IL-4–dependent alternative macrophage activation.
p47phox−/−–Dependent AAMac Phenotype Is Rescued by Restoring p47phox Expression
To assess the significance of the p47phox protein for modulating IL-4–dependent alterative macrophage activation, we used a retroviral vector that expresses human p47phox to restore p47phox protein expression in p47phox−/− murine macrophages. Compared with p47phox−/− macrophages transfected with a control vector that expresses green fluorescent protein, when p47phox protein expression was re-established in IL-4–stimulated p47phox−/− macrophages, STAT6 phosphorylation was attenuated (Figure 3A), and Ym1 and Arg1 protein expression were significantly reduced (Figure 3B). Moreover, STAT6 activation was also attenuated (Figure 3A), and Ym1 and Arg1 protein expression were reduced (Figure 3B) in WT macrophages where p47phox is overexpressed. These data support that p47phox protein is necessary and sufficient to revert the WT macrophage phenotype.
Figure 3.

Restoring p47phox protein attenuates signaling downstream of the IL-4R. Immunoblot analysis of p47phox−/− bone marrow–derived macrophages (BMMacs) expressing human p47phox (hNCF1). BMMacs were transduced by viral infection with hNCF1 or control vector (rGFP). A: Immunoblot analysis of IL-4–stimulated STAT6 phosphorylation. Quantification relative to total STAT6 is indicated. B: Immunoblot analysis of IL-4 stimulated Ym1 and ArgI expression. Results are representative of three independent experiments including three of each genotype per experiment.
Enhanced Lm Killing by IL-4–Primed p47phox−/− Macrophages Is Mediated by IL-1α
To investigate p47phox−/− macrophage responsiveness in vivo, we also assessed macrophage differentiation in p47phox−/− and catalytic subunit gp91phox−/− CGD mice in the context of Lm infection.21 Lm, is an intracellular bacterium that induces a robust innate immune response including triggering macrophage NADPH oxidase and nitric oxide–dependent microbial killing; as well as CD4+ TH1 and CD8+ T lymphocyte IFN-γ–mediated adaptive immunity.22 Thus, Lm infection typically triggers classical macrophage activation and therefore is not expected to induce an AAMac phenotype. We inoculated Lm i.p. into WT, gp91phox−/−, and p47phox−/− mice and found that the Lm bacterial burdens were significantly lower in p47phox−/− peritoneal exudate cells than gp91phox−/− peritoneal exudate cells 3 days postinfection (Figure 4A), which suggests that Lm elicited p47phox−/− macrophages have enhanced Lm killing. To further investigate the mechanism for the lower Lm bacterial load in p47phox−/− Lm elicited p47phox−/− macrophages, we next performed in vitro Lm-killing assays using BMMacs primed with IFN-γ and heat-killed Lm (HKL) to simulate classical macrophage activation, or BMMacs prime with IL-4 to simulate alternative macrophage activation. As expected, we found that the classical pathway induction improved Lm killing in both WT and p47phox−/− macrophages. However, we also found that there was significantly more killing of Lm in the IL-4–primed p47phox−/− macrophages but not in IL-4–primed WT macrophages (Figure 4B).
Figure 4.

Enhanced Lm killing by p47phox−/− AAMacs. Mice were infected with 5 × 104 CFU of Lm by i.p. injection. Tissues were harvested 3 days postinfection. A:Lm peritoneal bacterial burdens 3 days postinfection. Data are mean (± SEM) for six individual mice from two separate experiments with three of each genotype. B:In vitro Lm killing by WT and p47phox−/− BMMacs primed overnight as indicated, before infection with 5 × 105 CFU Lm. Data are mean (± SEM) of seven independent experiments with pooled cells from three of each genotype/experiment. **P < 0.001. C:In vitro Lm killing by p47phox−/− BMMacs primed overnight as indicated, before infection with 5 × 105 CFU Lm. Data are mean (± SEM) of three independent experiments with pooled cells from three p47phox−/− mice/experiment. **P < 0.001.
We previously reported that although IL-4 induces primarily anti-inflammatory cytokine secretion in WT and p47phox−/− macrophages, both classically and alternatively activated p47phox−/− macrophages hypersecrete IL-1α,7 which enhances resistance against Lm infection.23 Although the mechanism for the enhanced IL-1α secretion is not yet defined, we speculated that this pro-inflammatory cytokine secretion might be the basis for the enhanced Lm killing in IL-4–primed p47phox−/− macrophages. In agreement with this hypothesis, we found that the in vitro killing by IL-4–treated p47phox−/− macrophages that were also treated with anti–IL-1α antibody before Lm infection was similar to that in the untreated p47phox−/− macrophage population (Figure 4C). Thus, blocking the IL-1α activity abolishes the enhanced in vitro Lm killing demonstrated by IL-4–biased p47phox−/− macrophages, thereby supporting that by secreting IL-1α the p47phox−/− macrophages do kill intracellular Lm better than unprimed p47phox−/− macrophages.
p47phox−/− Mice Survive Primary Lm Infection and Show Less Severe Tissue Pathology
We next investigated whether p47phox−/− AAMacs would convey protection against Lm in vivo. Similar to what we recently reported,7 p47phox−/− and gp91phox−/− mice developed progressive crystalline macrophage pneumonia 3 days after Lm infection (Figure 5). This pneumonia was more severe in p47phox−/− mice when compared with gp91phox−/− mice (Figure 5), and was characterized by intracellular and extracellular accumulation of YM1/YM2-positive crystals (Figure 6A) and intracellular macrophage mannose receptor (Figure 6B), and by increased numbers of alveolar macrophages and multinucleated giant cells. The spleens of gp91phox−/− and p47phox−/− mice were markedly enlarged when compared with Lm-infected WT mice, and the white pulp was expanded by infiltrates of macrophages admixed with variable numbers of neutrophils (Figure 5). Splenic inflammation and disruption of the micro-architecture was more extensive and severe in Lm-infected gp91phox−/− mice relative to Lm-infected WT and p47phox−/− mice 3 days postinfection. In addition, the spleens of Lm-infected gp91phox−/− mice contained multiple areas of parenchymal necrosis, which were not evident in either Lm-infected WT or p47phox−/− mice. Although spleens from Lm-infected p47phox−/− mice were abnormal, livers from these animals were generally unremarkable and contained small scattered foci of inflammation (Figure 5), comparable to what was seen in livers of Lm-infected WT mice. In contrast to the mild changes observed in the livers of Lm-infected p47phox−/− and WT mice, livers from Lm-infected gp91phox−/− showed multiple, often extensive areas of inflammation and necrosis. In gp91phox−/− mice, this severe inflammation and necrosis in both the spleen and liver at 3 days after Lm infection was associated with reduced survival (75% versus 100% in p47phox−/− mice).
Figure 5.

H&E-stained tissue sections from gp91phox−/− (A–C), p47phox−/− (D–F), and WT (G–I) mice showing differences in severity of pathology in multiple tissue types. Changes in the liver and spleen of gp91phox−/− mice at 3 days post-Lm infection were more severe than in those of p47phox−/− and WT mice and show evidence of tissue necrosis (original magnification, ×200). Lungs of gp91phox−/− and p47phox−/− contained infiltrates of macrophages and multinucleated giant cells and crystalline pneumonia (original magnification, ×400). Results are representative of five each genotype examined.
Figure 6.

Immunofluorescence for YM1/YM2 (A) and MMR protein (B) in the lung of a p47phox−/− mouse 3 days post Lm infection. YM1/YM2–positive crystals are present within the cytoplasm of macrophages and multinucleated cells (original magnification, ×400). Results are representative of five each genotype examined. Splenocyte cultures from Lm-infected mice were incubated for 5 hours with 1 μmol/L OVA323–339 and/or OVA257–264 peptides for CD4 and CD8 T lymphocyte stimulation, respectively. Monensin was added for the final 2 hours of culture, and the percentage of CD4+ or CD8+ lymphocytes expressing IL-4 was determined by flow cytometry (C). Data are mean (± SEM) of six individual mice from two separate experiments with 3 of each genotype/experiment. *P = 0.043.
Our in vitro investigations reveal that p47phox−/− macrophages are hyperresponsive to IL-4 stimulation (Figure 2). Therefore, to determine whether the rapid and pronounced AAMac accumulation in Lm-infected p47phox−/− tissues was due to aberrant cytokine secretion, we used flow cytometry to assess IL-4 protein expression in T cells from Lm-infected mice. As shown in Figure 6C, the percentage of IL-4–positive CD4+ lymphocytes in the spleens of Lm-infected p47phox−/− mice was marginally higher than similarly treated WT T lymphocytes, at 9.5% and 5.9% respectively (P = 0.043). Furthermore, the difference in the percentage of IL-4–positive CD8+ lymphocytes in the Lm-infected spleens of WT and p47phox−/− as well as WT and gp91phox−/− mice was not statistically significant (P = 0.057 and P = 0.075, respectively). Thus, these findings indicate that the exaggerated AAMac phenotype in the Lm-infected p47phox−/− mice was not driven solely by excessive IL-4 secretion.
Adoptive Transfer of AAMacs from p47phox−/− Mice into Lm-Infected gp91phox−/− Mice Reduces Tissue Pathology and Enhances Survival
The finding that p47phox−/− macrophages differentiate into an AAMac phenotype spontaneously in sterile inflammatory lesions7 and during acute Lm infection suggest that selective differentiation along the alternative activation pathway may be advantageous in p47phox−/− mice. To discern whether this is the case in the context of in vivo Lm infection, we adoptively transferred Lm or thioglycolate elicited inflammatory p47phox−/− macrophages (see Materials and Methods) into gp91phox−/− mice before Lm infection. All of the recipients of p47phox−/− macrophages survived primary Lm infection, compared with 60% of gp91phox−/− mice that were sham injected with PBS before Lm infection (Figure 7A). Moreover, gp91phox−/− recipients of p47phox−/− AAMacs showed a reduction in the extent and severity of inflammation and necrosis in both the spleen and liver (Figure 6B). Collectively these observations suggest that AAMacs in Lm-infected p47phox−/− mice function to moderate Lm-induced disease pathogenesis, particularly with respect to hepatic and splenic necrosis, thereby enhancing survival in these mice.
Figure 7.
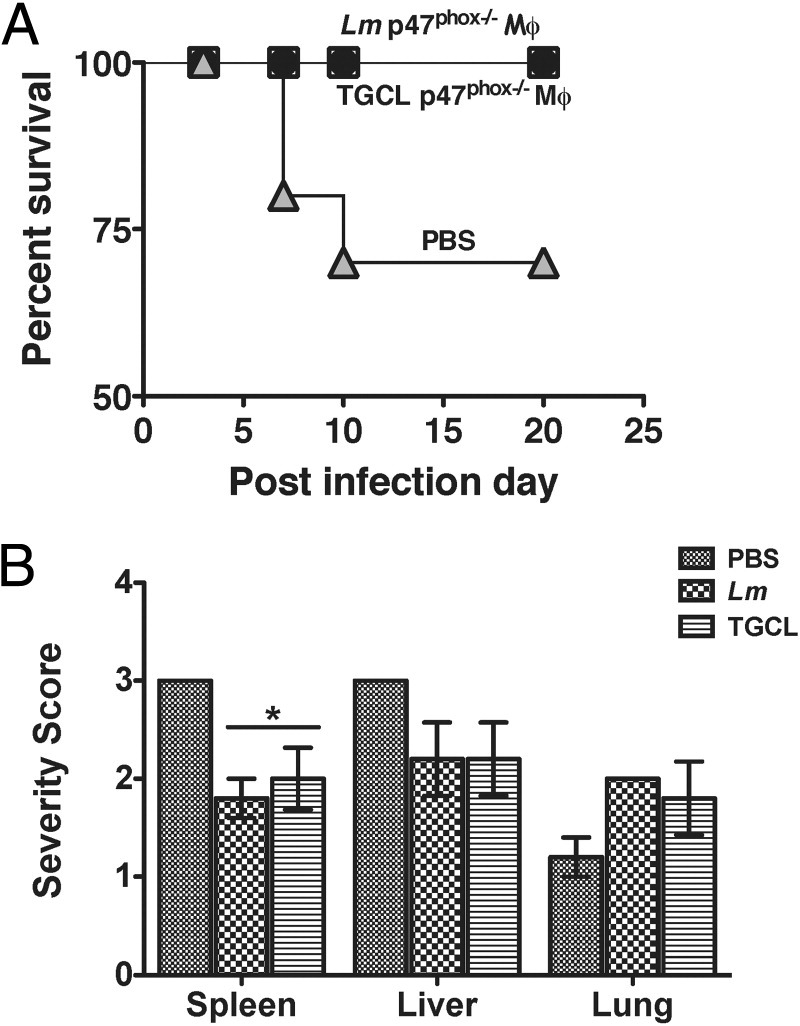
Enhanced survival of gp91phox−/− mouse recipients of p47phox−/− AAMacs. A: gp91phox−/− Mouse survival after adoptive transfer of 10 × 106Lm or thioglycolate (TGCL)) elicited p47phox−/− inflammatory macrophages before Lm infection. PBS indicates survival of gp91phox−/− control recipients that received PBS by i.v. injection. n = 10 per group. P = 0.048. B: Tissue severity scores for gp91phox−/− recipients of Lm or TGCL elicited p47phox−/− inflammatory macrophages. n = 5 per group. *P < 0.05.
Discussion
A physiological aim of inflammation is to restore tissue homeostasis.1 However, cancer, autoimmunity, and active inflammatory disease in immunodeficiency each pose distinct tissue insults that drive chronic inflammation that is not restorative. Furthermore, persistent insults can alter the function of inflammatory mediators and effectors, which include macrophages and T cells. During cell-mediated immune responses, TH1-derived IFN-γ and pathogen recognition receptor activation prime macrophages to become classically activated. These macrophages produce reactive oxygen and nitrogen intermediates to facilitate their microbicidal function, secrete pro-inflammatory cytokines, and can be tumoricidal.3,5 In contrast, TH2-driven pathogenesis and TH2-derived IL-4 and/or IL-13 adaptive immune responses, and local tissue injury, induce alternative macrophage activation. These macrophages promote wound healing and inhibit pro-inflammatory cytokine production.3,5 We also show, for what we believe is the first time, that p47phox−/− and gp91phox−/− CGD mice develop distinct acute, innate cellular responses and granulomatous inflammation in response to Lm infection. Although both Lm-infected CGD mice develop granulomatous inflammation, the extent and severity of pathology observed differ between the two strains. We show that during acute primary Lm infection, gp91phox−/− mice develop an aggressive granulomatous influx with necrotizing lesions in the liver and spleen, whereas Lm-infected p47phox−/− mice develop less severe granulomatous inflammation in lung and spleen, without evidence of necrosis.
Abscess and granuloma formation are characteristic CGD defense reactions to infection with pathogens that are typically eliminated by ROS. In addition, both lesions cause local tissue injury. However, our investigations reveal that the CGD knockout mouse inflammatory response to acute sublethal Lm infection is different. Although the acute granulomatous lesions in the p47phox−/− mice accumulate large numbers of Ym1+ AAMacs, the gp91phox−/− mouse lesions are more typical of those seen in IFN-γ– and tumor necrosis factor–α–driven suppurative granulomas that are a hallmark of chronic Lm infection.24–26 The finding of such dramatically disparate acute tissue reactions in the CGD mice strongly suggests that there is a cell-intrinsic basis for the observed enhanced AAMac differentiation in p47phox−/− macrophages. Indeed, we discerned that unlike WT or gp91phox−/− macrophages, p47phox−/− macrophages are hyperresponsive to IL-4 stimulation. Relative to both WT and gp91phox−/− macrophages, STAT6 activation and STAT6 specific transcripts were significantly upregulated in p47phox−/− macrophages at lower doses of IL-4. Moreover, restoring p47phox protein function corrected the aberrant IL-4 driven p47phox−/− AAMac phenotype, which suggests that p47phox protein regulates IL-4 signaling upstream of STAT6 activation.
Potential regulatory mechanisms for p47phox protein upstream of STAT6 are not entirely clear. A previous report showed an association between p47phox and the IL-4R α in Epstein–Barr virus–transformed human B cells.27 Interestingly, preliminary investigations revealed that p47phox co-immunoprecipitates with the IL-4R-α in Raw264 monocyte-macrophage cells. Therefore, future studies will be extended to primary macrophages once high-affinity antibodies are available for comprehensive analyses of IL-4–induced protein–protein interactions that involve the p47phox protein.
CGD is a multifaceted clinical disease and a unique model of host immune defense. The genetically engineered murine models of the most common genetic variants of CGD6,14 each recapitulate that the phagocyte respiratory burst is critical for combating microbial infection. Furthermore, these models allow complex analyses of innate and adaptive immune responses that are not possible in human CGD patients. In addition, the murine model can also be used to assess whether p47phox, the essential adaptor protein for Nox2-dependent ROS activity, functions independently of the catalytic gp91phox/Nox2 subunit, and to investigate clinical consequences of p47phox protein deficiency. Although it has been reported that IL-4 induced Nox1 and Nox5-dependent ROS,28 our investigations showed that STAT6 activation and target transcripts are similar in IL-4–stimulated WT and gp91phox−/− macrophages. In addition, neither gp91phox−/− nor Nox1-deficient macrophages hyperproduced signature AAMac proteins in response to IL-4 stimulation, as observed in p47phox−/− macrophages. Collectively, these findings indicate that p47phox works independent of the gp91phox/Nox2 catalytic subunit to attenuate IL-4–stimulated macrophage activation. Consistent with our observations, recent investigations have revealed that, independent of its role in generating Nox2-dependent ROS, p47phox is an important protein for integrating signaling downstream of TLR9 in murine dendritic cells.29
Granulomas are organized microenvironments that provide physical barriers to contain pathogens that escape from macrophages that are incompetent because of protein dysfunction or microbial manipulation. Although granulomas are controlled inflammatory sites, they are also dynamic structures composed of active effector cells that also cause tissue injury. An early, innate response to tissue injury is mast cell– and basophil-derived IL-4 production,3,30 which could drive the AAMac differentiation observed in both CGD mice during acute Lm infection, albeit by different mechanisms. The AAMac phenotype in Lm-infected gp91phox−/− mice could be explained by tissue IL-4 production in response to extensive tissue damage and necrosis. In contrast, although there is a relative lack of tissue necrosis in Lm-infected p47phox−/− mice, consistent with the CGD phenotype, these mice develop abscesses that cause tissue injury and can thereby trigger local IL-4 production. Therefore, the exaggerated AAMac phenotype in Lm-infected p47phox−/− mice could be explained by the p47phox−/− macrophage predisposition to hyperrespond to IL-4 produced in the damaged tissues.
Our original report of the p47phox−/− mice showed that they could develop severe, spontaneous systemic infection with granulomatous inflammation. Furthermore, we also reported that the tissues from the spontaneous lesion in the p47phox−/− mice contained variable amounts of granuloma, abscess, and infection.6 Subsequently, we demonstrated that these mice developed granulomatous inflammation without infection.7 In this report, using a controlled acute infection model, we now show that with low-level bacterial stimulation, during the acute phase of Lm infection, the p47phox−/− mice develop granulomatous inflammation with AAMacs. In addition, our current findings suggest that these AAMacs also convey a relative protection. In conclusion, using histological analysis as well as ex vivo molecular and cellular investigations, we report a novel mechanism by which p47phox−/− mice develop extensive, systemic alternative macrophage activation. These findings allow broader speculation about the basis for the nonrestorative chronic inflammation that is observed in CGD patients, and may explain why clinical disease in X-linked CGD patients can be more severe than in autosomal recessive CGD patients.16
Acknowledgments
We thank Kevin Gardner for helpful discussions and critique of this manuscript. We also thank Sharon Wahl, Harry Malech, and Warren Strober for careful review and critique of this manuscript.
Footnotes
Supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH and in part by the National Institute on Minority Health and Health Disparities, NIH.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.11.019.
Supplementary data
p47phox Protein attenuates IL-4 signaling in macrophages independent of the gp91phox catalytic subunit. A: Immunoblot analysis of Ym1 expression in inflammatory macrophages from each genotype stimulated overnight with IL-4 as indicated. Immunoblot analysis of STAT6 phosphorylation in inflammatory macrophages from WT mice that were treated with (B) PEG-SOD or (C) DPI as indicated before IL-4 stimulation for 20 minutes.
References
- 1.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 2.Gordon S., Taylor P.R. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 3.Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stout R.D., Watkins S.K., Suttles J. Functional plasticity of macrophages: in situ reprogramming of tumor-associated macrophages. J Leukoc Biol. 2009;86:1105–1109. doi: 10.1189/jlb.0209073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon S., Martinez F.O. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Jackson S.H., Gallin J.I., Holland S.M. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Q., Cheng L.I., Yi L., Zhu N., Wood A., Changpriroa C.M., Ward J.M., Jackson S.H. p47phox Deficiency induces macrophage dysfunction resulting in progressive crystalline macrophage pneumonia. Am J Pathol. 2009;174:153–163. doi: 10.2353/ajpath.2009.080555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward J.M., Yoon M., Anver M.R., Haines D.C., Kudo G., Gonzalez F.J., Kimura S. Hyalinosis and Ym1/Ym2 gene expression in the stomach and respiratory tract of 129S4/SvJae and wild-type and CYP1A2-null B6, 129 mice. Am J Pathol. 2001;158:323–332. doi: 10.1016/S0002-9440(10)63972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 10.El-Benna J., Dang P.M., Gougerot-Pocidalo M.A., Marie J.C., Braut-Boucher F. p47phox, the Phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. 2009;41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark R.A., Malech H.L., Gallin J.I., Nunoi H., Volpp B.D., Pearson D.W., Nauseef W.M., Curnutte J.T. Genetic variants of chronic granulomatous disease: prevalence of deficiencies of two cytosolic components of the NADPH oxidase system. N Engl J Med. 1989;321:647–652. doi: 10.1056/NEJM198909073211005. [DOI] [PubMed] [Google Scholar]
- 12.Roos D. The genetic basis of chronic granulomatous disease. Immunol Rev. 1994;138:121–157. doi: 10.1111/j.1600-065x.1994.tb00850.x. [DOI] [PubMed] [Google Scholar]
- 13.Stasia M.J., Li X.J. Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol. 2008;30:209–235. doi: 10.1007/s00281-008-0121-8. [DOI] [PubMed] [Google Scholar]
- 14.Pollock J.D., Williams D.A., Gifford M.A., Li L.L., Du X., Fisherman J., Orkin S.H., Doerschuk C.M., Dinauer M.C. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 15.Schappi M.G., Jaquet V., Belli D.C., Krause K.H. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol. 2008;30:255–271. doi: 10.1007/s00281-008-0119-2. [DOI] [PubMed] [Google Scholar]
- 16.Rosenzweig S.D. Inflammatory manifestations in chronic granulomatous disease (CGD) J Clin Immunol. 2008;28(Suppl 1):S67–S72. doi: 10.1007/s10875-007-9160-5. [DOI] [PubMed] [Google Scholar]
- 17.Matsuno K., Yamada H., Iwata K., Jin D., Katsuyama M., Matsuki M., Takai S., Yamanishi K., Miyazaki M., Matsubara H., Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 18.Foulds K.E., Rotte M.J., Seder R.A. IL-10 is required for optimal CD8 T cell memory following Listeria monocytogenes infection. J Immunol. 2006;177:2565–2574. doi: 10.4049/jimmunol.177.4.2565. [DOI] [PubMed] [Google Scholar]
- 19.Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasband W.S. U. S. National Institutes of Health; Bethesda, MD: 1997–2011. ImageJ. [Google Scholar]
- 21.Mackaness G.B. Cellular resistance to infection. J Exp Med. 1962;116:381–406. doi: 10.1084/jem.116.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pamer E.G. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 23.Rogers H.W., Sheehan K.C., Brunt L.M., Dower S.K., Unanue E.R., Schreiber R.D. Interleukin 1 participates in the development of anti-Listeria responses in normal and SCID mice. Proc Natl Acad Sci USA. 1992;89:1011–1015. doi: 10.1073/pnas.89.3.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popov A., Abdullah Z., Wickenhauser C., Saric T., Driesen J., Hanisch F.G., Domann E., Raven E.L., Dehus O., Hermann C., Eggle D., Debey S., Chakraborty T., Kronke M., Utermohlen O., Schultze J.L. Indoleamine 2,3-dioxygenase-expressing dendritic cells form suppurative granulomas following Listeria monocytogenes infection. J Clin Invest. 2006;116:3160–3170. doi: 10.1172/JCI28996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vazquez-Boland J.A., Kuhn M., Berche P., Chakraborty T., Dominguez-Bernal G., Goebel W., Gonzalez-Zorn B., Wehland J., Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mielke M.E., Peters C., Hahn H. Cytokines in the induction and expression of T-cell-mediated granuloma formation and protection in the murine model of listeriosis. Immunol Rev. 1997;158:79–93. doi: 10.1111/j.1600-065x.1997.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 27.Izuhara K., Arinobu Y., Sumimoto H., Nunoi H., Takeya R., Higuchi K., Takeshige K., Hamasaki N., Harada N. Association of the interleukin-4 receptor alpha chain with p47phox, an activator of the phagocyte NADPH oxidase in B cells. Mol Immunol. 1999;36:45–52. doi: 10.1016/s0161-5890(98)00111-4. [DOI] [PubMed] [Google Scholar]
- 28.Sharma P., Chakraborty R., Wang L., Min B., Tremblay M.L., Kawahara T., Lambeth J.D., Haque S.J. Redox regulation of interleukin-4 signaling. Immunity. 2008;29:551–564. doi: 10.1016/j.immuni.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richter C., Juan M.H., Will J., Brandes R.P., Kalinke U., Akira S., Pfeilschifter J.M., Hultqvist M., Holmdahl R., Radeke H.H. Ncf1 provides a reactive oxygen species-independent negative feedback regulation of TLR9-induced IL-12p70 in murine dendritic cells. J Immunol. 2009;182:4183–4191. doi: 10.4049/jimmunol.0800795. [DOI] [PubMed] [Google Scholar]
- 30.Loke P., Gallagher I., Nair M.G., Zang X., Brombacher F., Mohrs M., Allison J.P., Allen J.E. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J Immunol. 2007;179:3926–3936. doi: 10.4049/jimmunol.179.6.3926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
p47phox Protein attenuates IL-4 signaling in macrophages independent of the gp91phox catalytic subunit. A: Immunoblot analysis of Ym1 expression in inflammatory macrophages from each genotype stimulated overnight with IL-4 as indicated. Immunoblot analysis of STAT6 phosphorylation in inflammatory macrophages from WT mice that were treated with (B) PEG-SOD or (C) DPI as indicated before IL-4 stimulation for 20 minutes.