

Abstract
Coxsackieviruses are significant human pathogens causing myocarditis, meningitis, and encephalitis. We previously demonstrated the ability of coxsackievirus B3 (CVB3) to persist within the neonatal central nervous system (CNS) and to target neural stem cells. Given that CVB3 is a cytolytic virus and may therefore damage target cells, we characterized the potential reduction in neurogenesis within the developing brain and the subsequent developmental defects that occurred after the loss of these essential neural stem cells. Neonatal mice were inoculated with a recombinant CVB3 expressing eGFP (eGFP-CVB3), and alterations in neurogenesis and brain development were evaluated over time. We observed a reduction in proliferating cells in CNS neurogenic regions simultaneously with the presence of nestin+ cells undergoing apoptosis. The size of the brain appeared smaller by histology, and a permanent decrease in brain wet weight was observed after eGFP-CVB3 infection. We also observed an inverse relationship between the amount of virus material and brain wet weight up to day 30 postinfection. In addition, signs of astrogliosis and a compaction of the cortical layers were observed at 90 days postinfection. Intriguingly, partial brain wet weight recovery was observed in mice treated with the antiviral drug ribavirin during the persistent stage of infection. Hence, long-term neurological sequelae might be expected after neonatal enteroviral infections, yet antiviral treatment initiated long after the end of acute infection might limit virus-mediated neuropathology.
Coxsackieviruses (CV) are a group of small, positive, single-stranded RNA viruses that commonly cause human disease, including meningo-encephalitis1,2 and myocarditis.3 Neonatal CV infections are particularly damaging,4,5 and infection of newborn infants may eventually lead to aseptic meningitis3 and severe myocarditis.6 Of note, antibodies against CV and other enteroviruses can be detected in roughly 75% of the population.7 However, the long-term consequences of an early CV infection in surviving individuals are largely unknown. Correlations between prior infection and CNS disorders have been examined in children with viral infections at infancy who were thought to have fully recovered. One study found that 5 years after an initial viral infection, these children had lower intellectual levels as compared with those in control groups.8 These epidemiological studies demonstrate the necessity of an animal model in evaluating the lasting effects of an early CV infection on the developing CNS.
The B serotype of CV is characterized by spastic paralysis in suckling mice.9 Six coxsackievirus B serotypes have been identified, and each is associated with acute disease in humans. In particular, coxsackievirus B3 (CVB3) has been shown to be associated with infection of the heart, central nervous system (CNS), and pancreas. The determinants of CVB3 tropism in these organs remain poorly understood. Our laboratory has previously shown that CVB3 preferentially infects nestin+, proliferating neural stem cells,10 and myeloid cells entering the CNS in response to early infection.11 Also, we have recently demonstrated the ability of CVB3 to efficiently replicate and induce cytopathic effects within neurospheres grown in culture, which comprise neural stem cells and their progeny at different stages of development12,13 The main target sites of early CVB3 infection in the neonatal brain included regions of neurogenesis.10,14 Neural stem cells can be found in two principal regions: the subventricular zone (SVZ), which lines the lateral ventricles,15,16 and the dentate gyrus within the hippocampus.17 Given that CVB3 efficiently infects both of these regions, we hypothesized that CNS development and might be greatly affected in the surviving host. Indeed, mice infected with Theiler's murine encephalomyelitis virus, a murine picornavirus, experience disruption in their spatial memory resulting from major damage to the pyramidal neurons of the hippocampus.18
Although CVB3 is known to be cytolytic, persistence of viral material may be observed within the heart19 and CNS20 by mechanisms that remain unclear. Persistence of CVB3 in target organs might involve virus attenuation following the accumulation of genomic deletions within 5′ untranslated regions that may drastically slow virus replication.21–23 However, it remains uncertain how an attenuated CVB3 might evade the host innate and adaptive immune response for extended periods. CVB3 has been previously shown to attach to target cells through two main receptors: decay-accelerating factor (DAF),24,25 and murine coxsackievirus and adenovirus receptor (mCAR),26 which has been found to be highly expressed in the developing brain.27 The high levels of mCAR expressed in neonatal organs might play a role in enhanced susceptibility to CVB3 infection and disease. However, high levels of cell proliferation (including neural stem cells) expected during development may also increase the number of potential target cells for CVB3 infection.28 Potential CVB3-induced damage to neural stem cells and immature neurons may point to an underlying mechanism of neurological disease. In this regard, CV and related enteroviruses have been linked to neurological disorders such as acute disseminated encephalomyelitis,29 schizophrenia,30 amyotrophic lateral sclerosis,31,32 and, more recently, demyelination33 or white matter damage.34
Therefore, we inspected the long-term effects of CVB3 infection on the central nervous system in our neonatal murine model of infection. More specifically, we evaluated the ability of CVB3 to induce apoptosis within neural progenitor and stem cells (NPSCs). We also inspected potential reductions in brain wet weight values within mice surviving a prior CVB3 infection. Finally, we assessed the degree of astrogliosis and cortical layer alterations after CVB3 infection. Our results suggest that a prior CVB3 infection during the neonatal period may lead to lasting neurological disease in the surviving host.
Materials and Methods
Ethics Considerations
This study was performed in strict accordance with the requirements pertaining to animal subjects protections within the Public Health Service Policy and United States Department of Agriculture Animal Welfare Regulations. All experimental procedures with mice were approved by the San Diego State University Institutional Animal Care and Use Committee (Animal Protocol Form #10-05-013F), and all efforts were made to minimize suffering.
Mice and Viral Inoculations
BALB/c mice were obtained from Harlan Sprague Dawley (Harlan Laboratories, San Diego, CA). Breeding mice ranged between 6 and 8 weeks of age, and breeding pairs were monitored daily to ensure that pups were identified within 24 hours of birth. The procedure for intracranial inoculation of pups has been described previously.14 Briefly, 1 or 3 days after the recorded date of birth, pups were inoculated i.c. with 25 μl Dulbecco's modified Eagle's medium (DMEM; Cellgro, Manassas, VA) (mock-infected control pups), or with either 106 or 107 pfu of eGFP-CVB3. At various time points after infection, pups were euthanized by placing them in a bell jar with isoflurane followed by immediate decapitation. Alternatively, adult mice were euthanized by isoflurane followed by cervical dislocation. Brains were harvested, and brain wet weight values were recorded immediately. One half of each brain was fixed by immersion in 10% formaldehyde followed by paraffin embedding; the other half of the brain was placed into a tube and placed on dry ice. These samples were eventually used to determine viral load by plaque assay and real-time reverse transcription (RT)–PCR.
Isolation and Production of Recombinant Coxsackievirus
The generation of a recombinant coxsackievirus expressing eGFP has been described previously.28 All virus stocks were grown on HeLa RW cells maintained in DMEM supplemented with 10% fetal bovine serum. Virus titrations were performed as described previously.28
Virus Titration
Half of the brain saved for viral load determination was weighed and homogenized with 1 mL of DMEM for 45 seconds using the Mini-BeadBeater (BioSpec Products, Bartlesville, OK). Viral titers were measured using a standard CVB3 plaque assay, as published previously28 Briefly, HeLa RW cells were plated at 3.00 × 105 cells per well in a six-well plate and were grown until 70% confluent (1 to 2 days). Tenfold serial dilutions were made, and incubated with cells for 1 hour and then overlaid with 2X complete DMEM mixed with 1.2% agar. After 40 to 48 hours of incubation at 37°C and 5% CO2, the cells were fixed with 2 mL per well of methanol/acetic acid (3:1 v/v) for 10 minutes. The fixative and plugs were then removed, and plaques were stained with 1 mL per well of 0.25% Crystal Violet overnight. The plates were then rinsed in water (three times), and plaques were counted.
Viral RNA Extraction and Quantitative Real-Time RT-PCR
Viral RNA was isolated from brain tissue using TRIzol reagent (Gibco-BRL, Rockville, MD), as described by the manufacturer. The quantitative real-time RT-PCR procedure has been described previously.20 Briefly, reverse transcription reaction mixtures included 1 μg of total RNA and were performed using SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA) following the procedure described by the manufacturer. Separate RT reaction mixtures containing 1 μl of RNaseOUT (Invitrogen Inc.) and reverse primer or forward primer were used to quantify the number of positive- and negative-sense viral genomes. PCR amplification was done using Platinum quantitative PCR SuperMix-UDG ready-to-use cocktail (Invitrogen), which contained all of the components needed except the primers and probe, as described by the manufacturer. Quantitative analysis of viral RNA was performed using an iQ5 Real Time PCR Detection System in 96-well optical reaction plates heated to 50°C for 2 minutes to digest dUTP-containing contaminants, followed by 95°C for 2 minutes to deactivate uracil N-glycosylase and to activate Platinum TaqDNA polymerase. A total of 40 cycles of denaturation at 95°C for 15 seconds and annealing and extension at 60°C for 30 seconds were performed. All samples were evaluated in triplicate amplification reactions. The procedure to normalize the amount of RNA in each samples has been described previously.20 CVB3 RNA generated via our infectious viral clone (pH3 plasmid) was used to generate the standard curve. The standard curve was based on threshold cycle (CT) values, and CT values from unknown samples were compared with the standard curve to determine viral RNA copy numbers.
Histochemical and Immunofluorescence Staining
Paraffin-embedded brain sections (3- to 4 μm thick) were stained by hematoxylin and eosin (H&E) or by Nissl stain. For immunofluorescence microscopy, paraffin-embedded sections were deparaffinized with three washes of xylene, three washes with ethanol (100%, 95%, and 70%), and followed by washes in PBS and water. The detection of apoptosis, neuronal class III β-tubulin, nestin, Ki-67, GFAP, and GFP required high-temperature antigen unmasking in low-pH citrate buffer (Vector Labs, Burlingame, CA). Immunofluorescence staining procedures have been described previously.14 Apoptosis was detected using the ApopTag Red In Situ Apoptosis Detection Kit (Chemicon/Millipore, Billerica, MA) as described by the manufacturer. However, when the kit was used in combination with an antibody against nestin (Covance Research Products, Cumberland, VA), the proteinase K step was removed from the protocol. Neuronal class III β-tubulin was detected using a rabbit primary antibody (Covance Research Products), diluted at 1:100. Nestin was detected using a rabbit primary antibody (nestin tail; Covance Research Products) diluted at 1:100. Active caspase-3 was detected using a rabbit primary antibody (Cell Signaling Technology) diluted at 1:100. Ki-67 was detected using a mouse primary antibody (Vector Labs) diluted at 1:100. The Mouse-On-Mouse Kit (Vector Labs) was used for mouse primary antibodies as instructed by the manufacturer. GFP was detected using a rabbit primary antibody (Molecular Probes, Carlsbad, CA) diluted at 1:100. The secondary antibody used was a biotinylated goat anti-rabbit IgG [heavy and light chains (H+L)] (Vector Laboratories, Burlingame, CA) diluted to 1:100 in 2% normal goat serum and incubated on sections for 30 minutes. All sections were then washed twice with PBS and incubated for 30 minutes with a streptavidin-green or streptavidin-red complex (Molecular Probes/Invitrogen, Carlsbad, CA) diluted at 1:500 in 2% normal goat serum. Detection of nuclei was done using Vectashield mounting medium containing DAPI (Vector Laboratories). Sections were observed by fluorescence microscopy (Zeiss Axio Observer D1 with an attached Zeiss MRc camera; Zeiss, Oberkochen, Germany) for the indicated cellular marker (green or red), and DAPI (blue). Green, red, and blue channel images were merged using AxioVision software (Zeiss).
Brain Wet Weight Values
Wet weight measurements were taken at time of harvest by placing the whole brain on a scale and recording the weight. Wet weights values were analyzed using GraphPad Prism 3.0 software, and the data were displayed graphically. For the average wet weights graph, the infected wet weights for each time point were compared with the average wet weights of the mock-infected samples. Each infected brain wet weight value was divided by the average mock weight for each time point, and the data were analyzed using GraphPad Prism 3.0. Each time point brain wet-weight value was compared with the respective viral load quantities (viral titers or viral copies) and graphed using Microsoft Excel scatter plots. Data were analyzed by adding a trend line to decipher the relationship between the two variables.
Ribavirin Treatment
Day 30 and day 90 postinfection (PI) mice received ribavirin (100 mg/kg body weight) diluted in DMEM via subcutaneous injections in the neck. Ribavirin treatment was given every 3 days, starting at day 14 (for day 30 PI; total, 16 days) or day 60 (for Day 90 PI; total, 30 days). Mice were euthanized, brains were harvested, and the brain wet weight values were immediately determined.
Quantification of ApopTag Signal in Subventricular Zone
SVZ sections of infected and mock-infected animals (three animals per treatment and time point) were used for ApopTag image collection using a Zeiss Axio Observer D1 inverted microscope. Quantification of apoptotic cells was performed, and values were entered into GraphPad Prism 3.0 software, analyzed, and displayed graphically. Each apoptotic cell was then examined to determine co-localization with nestin staining. The total amount of apoptotic cells in sections from both infected and mock-infected mice were graphed using Microsoft Excel. Values represent the amount of nestin+ or nestin− apoptotic cells.
Results
Lethal Inoculation of CVB3 in Neonatal Mice Causes Delay in Early CNS Development
Previously, we generated a neonatal murine model of infection using a recombinant coxsackievirus B3 expressing eGFP (eGFP-CVB3) to study virus tropism and pathogenesis.10,14 Lesions were observed in the olfactory bulb, cortex, and hippocampal regions of the mouse brain after acute CVB3 infection.14 The degree of pathology and the number of lesions were dependent on the level of initial viral inoculum. Notably, these lesions were observed for up to 9 months postinfection.20 To determine early CNS developmental changes after CVB3 infection, 1-day-old mice were mock-infected or infected i.c. with a high inoculum (2 × 106 pfu) of eGFP-CVB3. Mice were sacrificed on days 1, 2, 3, and 5 PI, and the brains were evaluated by H&E staining (Figure 1). The exact time of pup birth was determined within a 24-hour window. Subsequently, many pups infected with the high viral inoculum did not survive longer than 4 days PI. Some pups survived longer than 4 days PI (∼20% of mice inoculated with 2 × 106 pfu i.c.); we propose that animals surviving longer than 4 days PI may have been slightly older pups inoculated at or after ∼16 hours postbirth.
Figure 1.

Lethal inoculum of eGFP-CVB3 during the neonatal period hinders CNS development. One-day-old pups were infected with eGFP-CVB3 (2 × 106 pfu i.c.) or were mock infected. Brains were harvested and fixed in 10% neutral buffered formalin at the indicated time points PI. Paraffin sections were stained by H&E or immunostained for the cellular proliferation antigen (Ki-67, red) and viral protein expression (GFP, green) and observed by fluorescence microscopy. A: Two color channels were merged into a single image (original magnification, ×5) and composites of overlapping fields were assembled to illustrate a complete transverse section of the neonatal CNS for two representative mice. Normal CNS development was observed in mock-infected pups. As would be expected for regions of neurogenesis in the CNS, a large number of Ki-67+ cells were observed in the SVZ near the lateral ventricles and in the cerebellum. In pups infected with eGFP-CVB3 and harvested 5 days later, the overall size of the brain was significantly smaller and the ventricles were severely distended. Furthermore, CNS lesions were seen in the hippocampus and cortex regions, in parallel with high levels of viral protein expression. The cerebellum (only partly visible) had little to no detectable viral protein levels and displayed similar numbers of proliferating (Ki-67+) cells compared with mock-infected control mice. Higher magnification for day 5 fluorescent images revealed that the number of proliferating (Ki-67+) cells was substantially reduced in the SVZ (white arrow) near the lateral ventricles, as compared with mock-infected control mice. B: High levels of Ki-67+ signal were observed in the SVZ of mock-infected mice. C: In contrast, the level of Ki-67+ signal was sharply reduced within infected mice. D: Quantification of Ki-67+ signal in the SVZ showed substantially reduced levels of staining in the SVZ of infected mice (*P = 0.0327). Quantification of Ki-67+ cells was done in triplicate (three mice per group and average of three fields per mouse) using ImageJ software as described previously.10
In those animals lasting 5 days PI, clear histological differences were observed in the infected brain, and the overall size of the brain and olfactory bulbs appeared considerably smaller than mock-infected control mice (Figure 1A). Furthermore, high levels of viral protein expression were observed in regions of the cortex and hippocampus (Figure 1A), as determined previously.14 Taken together with our previously published results,10 these results suggested that CVB3 may affect normal CNS development by targeting neural progenitor cells and their downstream cell lineage products, immature and mature neurons. We hypothesized that a reduction in the number of proliferating NPSCs by apoptosis after CVB3 infection may be responsible for the observed delay in CNS development. Consequently, the number of proliferating (Ki-67+) cells in the neurogenic regions of the developing CNS of infected mice appeared to be reduced in number, as compared with those in mock-infected control mice (Figure 1A) at a time when H&E staining revealed severe developmental deficiencies in the CNS.
Loss of Proliferating Cells in the SVZ After Lethal CVB3 Infection
We quantified the relative level of Ki-67+ proliferating cells in the SVZ on day 5 PI in 1-day-old neonatal mice infected with a high inoculum of eGFP-CVB3 (2 × 106 pfu) and mock-infected control mice, as determined by immunofluorescence microscopy. As shown in Figure 1, B and C, a clear reduction in the number of proliferating cells in the SVZ was observed within infected mice, suggesting alterations in normal levels of neurogenesis after CVB3 infection. The reduction of proliferating cells at day 4 PI in infected animals (three mice per treatment) was quantified by analysis with ImageJ software (Figure 1D) and shown to be statistically significant (P = 0.0327), as compared with values in mock-infected mice.
Lethal CVB3 Infection Induces Apoptosis in Neurogenic Regions of Developing CNS
Our previous studies identified CVB3-infected neurons undergoing caspase-3–mediated apoptosis in the neonatal hippocampus and entorhinal cortex at day 3 and day 5 PI.14 The observed reduction of proliferating (Ki-67+) cells within neurogenic areas of the CNS, in combination with delayed CNS development, suggested to us that neural stem cells also may be depleted in the SVZ after CVB3 infection. Therefore, we inspected the level of apoptosis within neurogenic regions of the CNS at early (day 1 and day 2 PI) and later (day 5 PI) time points after infection with eGFP-CVB3 to determine whether the delay in CNS development may be due to the programed cell death of infected NPSCs within the SVZ.
Intriguingly, apoptotic cells (ApopTag staining, red) were observed within the rostral migratory stream (RMS, outlined by neuronal class III β-tubulin expression shown in green) as soon as day 1 PI (Figure 2A). These apoptotic cells were identified as neuroblasts by their expression of neuronal class III β-tubulin. In contrast, mock-infected neonatal mice showed little to no apoptotic activity within the RMS (Figure 2B). Also, numerous cells undergoing apoptosis were observed within the SVZ 2 days PI (Figure 2C). Regions of apoptosis overlapped directly with areas of infection (green), as determined by viral protein expression. Higher magnification revealed apoptotic cells adjacent to infected cells near the ependymal cell layer (Figure 2D). By day 5 PI, high levels of viral protein expression and apoptosis were observed in the hippocampus and cortex regions of the neonatal CNS (Figure 2E), similar to what was described previously after infection with eGFP-CVB3.14 Higher magnification of the hippocampus revealed ApopTag staining within the dentate gyrus and CA1-3 regions of the hippocampus (Figure 2F). Although strong viral protein expression levels were evident in the dentate gyrus, reduced levels were detectable in other regions of the hippocampus, perhaps reflecting the reduced numbers of viable cells in Ammon's horn at later time points. In contrast, little or no apoptosis was observed in the hippocampus or SVZ of mock-infected control animals (Figure 2G).
Figure 2.

High levels of apoptosis in neurogenic regions observed in both nestin+ and nestin- cells following a lethal inoculation with eGFP-CVB3. Pups 1 or 3 days old were infected with eGFP-CVB3 (2 × 106 – 1 × 107 pfu i.c.) or mock infected. Brain sections from pups harvested up to day 5 PI were examined for apoptotic activity by ApopTag staining. A: An increase in the level of apoptotic cells (red) was seen in the RMS of infected pups as compared with mock-infected mice. These apoptotic cells were identified as neuroblasts which migrate through the RMS by staining for neuronal β-tubulin (green). B: Little to no apoptotic cells were identified in mock-infected control mice. C: Clear apoptotic activity (red) was also observed in the SVZ as early as day 2 PI. The white dotted line represents the boundary between the ependymal cell layer and the lateral ventricle. D: Higher magnification revealed apoptotic cells located adjacent or within infected (GFP+) cells. E: By day 5 PI, high levels of apoptosis were observed near the lateral ventricles, the hippocampus, and regions of the cortex. These regions also supported high levels of viral protein expression. F: Higher magnification of the hippocampus demonstrated apoptosis in the dentate gyrus and in the CA3 region of Ammon's horn. G: In contrast, mock-infected control pups showed little or no apoptotic activity in the CNS. H–J: Using nestin (green) as a marker we identified neural stem and progenitor cells undergoing apoptosis within the SVZ. Immunofluorescence microscopy identified nestin+ cells in the SVZ undergoing apoptosis (red) at 24 (H), 36 (I), and 48 hours PI (J), (white arrows), respectively. K: More than 50% of cells undergoing apoptosis were identified as neural stem and progenitor cells by nestin staining. Also, the total number of apoptotic cells was significantly higher in infected mice, as compared with mock-infected mice (*P = 0.0042). For quantification, three mice were used per group and time point.
Nestin+ and Nestin− Cells in Neurogenic Regions Undergo Apoptosis After Lethal CVB3 Infection
Although cells undergoing apoptosis were observed within neurogenic regions, we determined whether these apoptotic bodies represented NPSCs or other glial cells. Therefore, we quantified the total number of apoptotic nestin+ and nestin− cells in the SVZ after CVB3 infection. By fluorescence microscopy, many nestin+ cells (green) were observed in the SVZ undergoing apoptosis (red) at 24, 36, and 48 hours PI (Figure 2, H, I, and J, respectively). The total number of apoptotic nestin+ and nestin− cells was quantified in CVB3-infected and mock-infected mice (Figure 2K). Greater numbers of apoptotic cells were observed in the SVZ of infected mice at all three time points, as compared with mock-infected control animals (P = 0.0042). Also, a greater number of nestin+ cells undergoing apoptosis was observed, as compared with nestin− cells within infected mice at all three time points after infection, although these values were not statistically significant. We hypothesize that nestin− cells undergoing apoptosis may represent downstream neural precursor cells or unidentified cells present in the SVZ.
Establishment of Viral Persistence in the Neonatal Mouse CNS After Inoculation with Nonlethal Dose of CVB3
Our results clearly demonstrate that neonatal mice inoculated with a lethal dose of eGFP-CVB3 showed reduced levels of neurogenesis and profound CNS developmental delays at early time points (up to day 5 PI). However, we speculated whether CVB3 infection may lead to lasting consequences in the CNS within surviving mice. Therefore, we inspected the later stages of infection, ongoing neuropathology, and long-term consequences on CNS development in neonatal mice inoculated with a nonlethal dose of virus determined by a survival curve in a previous study.20 Three-day-old mice inoculated with eGFP-CVB3 (2 × 107 pfu i.c.) survived acute infection and established a persistent infection in the CNS (Figure 3).20 High viral titers were observed up to day 5 PI (Figure 3A). By day 10 PI and beyond, infectious virus could no longer be detected by conventional plaque assay. However, viral RNA was observed by real-time RT-PCR up to day 90 PI, although RNA abundance decreased substantially over time (Figure 3B).
Figure 3.

Viral titers over time in 3-day-old mice after a nonlethal inoculation with eGFP-CVB3. Pups 3 days old were infected with an amount of viral inoculum whereby mice survived infection (eGFP-CVB3 at 1 × 107 pfu i.c.). Brains were harvested and virus titers in brain homogenates were determined by plaque assay. A: Early time points (days 1, 2, and 5) showed high viral titers for multiple animals. At later time points (days 10, 30, and 90), no detectable infectious virus was observed. B: In contrast, viral RNA was detected by real-time RT-PCR up to day 90 PI (three mice per time point) demonstrating the presence of viral genomic material for extended periods.
High Levels of Apoptosis in SVZ of Mice Surviving CVB3 Infection
We quantified the level of apoptosis in the SVZ of mice inoculated with a sublethal dose of eGFP-CVB3. Infected and mock-infected mice were stained for apoptotic cells in the SVZ and observed by fluorescence microscopy over time (Figure 4). For each mouse (three mice per group), three representative brain sections were quantified by fluorescence microscopy to determine the amount of apoptotic cells within the SVZ. As shown for mice inoculated with a lethal dose of eGFP-CVB3, mice surviving infection exhibited relatively high levels of apoptosis within the SVZ for up to day 10 PI, as compared with mock-infected control mice (Figure 4A). By day 30 PI and beyond, few apoptotic cells could be seen in the SVZ of both infected and mock-infected mice. These results suggest that acute CVB3 infection rapidly depleted target cells within the SVZ. Eventually, the level of virus-mediated apoptosis waned in parallel with a drop in viral titers and/or a reduction in target cell populations as the animals aged.
Figure 4.

Surviving mice infected with eGFP-CVB3 showed higher levels of apoptosis in the SVZ up to 10 days PI, as compared with mock-infected mice. Three day-old pups were infected with eGFP-CVB3 (1 × 107 pfu i.c.) or were mock infected. A: Levels of apoptosis were quantified in the SVZ for both infected and mock-infected mice by ApopTag staining. Higher levels of apoptosis were observed specifically within infected mice during acute time points (days 1, 2, and 5 PI). By day 90 PI, levels of ApopTag staining were below detection limits for both infected and mock-infected mice. B and D: Active caspase-3 (red) was detected by immunofluorescence microscopy within the hippocampus and in neurogenic regions at 36 hours PI. High levels of active caspase-3 were observed near sites of infection in the RMS (B) and in the CA2 region of the hippocampus (D; green). C and E: Higher magnification of (B) and (D), respectively, showed colocalization of active caspase-3 with cells expressing high levels of viral protein (white arrows). Single-channel images for active caspase-3 and viral protein demonstrated the overlapping red and green signal. Nuclei/DNA were stained with DAPI (blue).
Also, infected cells undergoing apoptosis were identified in the hippocampus and in neurogenic regions after infection (Figure 4, B and D). Immunofluorescence microscopy showed the induction of active caspase-3 (red) in the RMS, the SVZ (data not shown), and within infected regions of the CA2 region of the hippocampus. Higher magnification demonstrated colocalization of infected cells with those expressing active-caspase 3 in both the RMS and the hippocampus (Figure 4, C and E, respectively) suggesting eGFP-CVB3 could directly induce apoptosis in neurogenic regions. Single channel images confirmed the overlap of active caspase-3 and viral protein (Figure 4, C and E). These results fit well with our recent findings showing eGFP-CVB3-induced cytopathicity in NPSCs grown in culture.12 Although high levels of active caspase-3 were also observed in nearby cells having no detectable levels of viral protein, these cells might represent infected cells that no longer express detectable levels of viral protein because of the compromised status of the cell, or cleavage of viral proteins by proteases induced during apoptosis. Alternatively, eGFP-CVB3 may indirectly induce apoptosis of nearby cells, as shown for Theiler's murine encephalomyelitis virus.35
Reduced Brain Wet Weights in Mice Surviving CVB3 Infection
We hypothesized that any reduction of NPSCs in mice surviving infection might alter normal brain development and size. Therefore, we quantified brain wet weights in both infected and mock-infected control mice during acute infection and in persistently infected mice. As seen in Figure 5, a significant decrease in brain wet weight values of infected mice was observed at days 1, 5, 10, 30, and 90 PI, as compared with mock-infected control mice. Also, an inverse relationship was observed between brain wet weight values and viral titers (as measured by plaque assay) within infected mice - the higher the viral load the lower the brain wet weight. However, the trend was no longer evident at day 90 PI, possibly because of the relatively low viral copy levels detected at this later time point.
Figure 5.

Surviving mice infected with eGFP-CVB3 exhibited reduced brain wet weights at early and late time points, as compared with mock-infected mice. Pups 3 days old were infected with eGFP-CVB3 (1 × 107 pfu i.c.) or mock infected and harvested up to day 90 PI. At time of harvest, brain wet weights for infected or mock-infected mice were determined. Infected mice showed a statistically significant reduction in brain wet weights on day 1 PI (*P = 0.0468), day 2 PI (†P = 0.7162), day 5 PI (‡P = 0.0339), day 10 PI (§P = 0.037), day 30 PI (¶P < 0.05), and day 90 PI (‖P < 0.03). Also, an inverse relationship trend was observed between the brain wet weights of infected mice and viral RNA load up to day 30 PI (as determined by real time RT-PCR): the lower the brain wet weight, the higher the viral RNA load. Student's t-test was used to compare brain wet weights for infected and mock-infected animals. The antiviral drug ribavirin was injected i.p. into infected mice at late time points to determine whether viral replication during persistence contributed to the observed reduced brain wet weights. Different treatment regimens were used for day 30 (administered 2 weeks before harvest; treatment every 3 days) and day 90 (administered 30 days before harvest; treatment every 3 days). Although no significant change in brain wet weights was observed for day 30 ribavirin-treated infected mice, analysis of variance analysis with Newman–Keuls method revealed a significant increase (*P < 0.03) in brain wet weight for day 90 ribavirin-treated infected mice, as compared with untreated infected animals.
Administration of Antiviral Drugs During Persistent Stage of CVB3 Infection Leads to Partial Brain Wet Weight Recovery
Previously, we detected the presence of attenuated CVB3 within the CNS of persistently infected mice that continued to replicate on passage in HeLa cells.36 We hypothesized that an antiviral drug such as ribavirin given during the persistent phase of infection in vivo might inhibit the low levels of ongoing viral replication in the CNS. We predicted that any reduction in viral load during persistent infection might lead to brain wet weight recovery. Ribavirin targets replicating RNA viruses by increasing the amount of nucleotide mutations to an unsustainable high rate whereby the virus is unable to retain genomic stability and in driven into “lethal mutagenesis.”37
Published reports have described the ability of ribavirin to inhibit coxsackievirus replication in culture or during early infection in vivo.38 Also, ribavirin has been previously shown to enter through the blood–brain barrier at levels sufficient to inhibit some neurotropic viral infections, including enterovirus infections. Therefore, we injected ribavirin i.p. into persistently infected mice in an effort to observe brain wet weight recovery. For day 30 PI persistently infected mice, ribavirin was injected at day 14 and every third day thereafter until harvest (total of 16 days treatment). For day 90 PI persistently infected mice, ribavirin was injected at day 60 and every third day thereafter until harvest (total of 30 days treatment). A 16-day treatment of ribavirin failed to show brain wet weight recovery in day 30 PI mice (Figure 5). In contrast, day 90 PI mice treated with ribavirin for 30 days showed a statistically significant brain wet weight recovery (Figure 5; P < 0.03). These results suggest that ribavirin treatment during the persistent phase of CVB3 infection may be beneficial in limiting CNS pathology. Ribavirin is a nucleoside analog, and would be expected to only be effective during active viral replication. Its apparent efficacy at day 90 PI, but not at day 30 PI, suggests that CVB replication may fluctuate over the course of a persistent infection. A comparison of brain wet weight normalized to average mock-infected brain wet weight for all animals analyzed at each time point illustrated the highly significant reduction in brain size of mice infected with eGFP-CVB3 across all time points analyzed (Figure 6; P < 0.0001).
Figure 6.
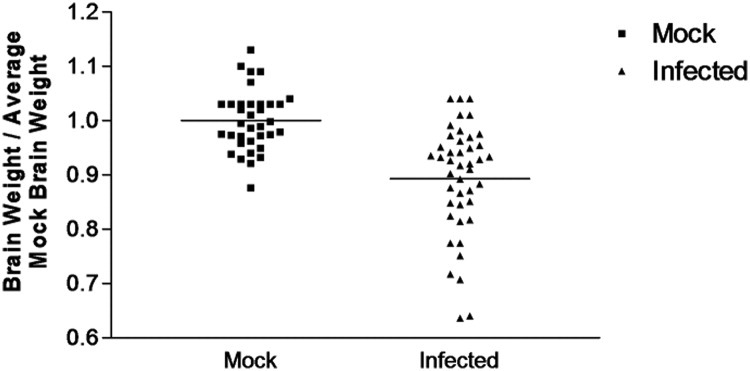
Surviving mice infected with eGFP-CVB3 from all time points showed reduced brain wet weights, as compared with averaged brain weights of mock-infected mice. Pups 3 days old were infected with eGFP-CVB3 (as described in Figure 5) or mock-infected. Brain wet weights from infected and mock-infected mice were normalized to average mock-infected brain wet weights for each time point. The normalized brain wet weight results for animals from all time points (days 1, 2, 5, 10, 30, and 90 PI) are graphically displayed. A highly significant difference was observed between infected and mock-infected brain wet weights when mice from all time points were compared (P < 0.0001; Student's t-test).
Astrogliosis and Neurological Abnormalities in Mice Surviving CVB3 Infection
The brains of day 30 and day 90 PI mice were inspected for the presence of lasting neuropathology that may be attributed to a reduction in neurogenesis after eGFP-CVB3 infection. We previously observed CNS lesions and microgliosis in persistently infected mice. By immunofluorescence microscopy, high levels of GFAP staining in the hippocampus of persistently infected mice as compared with mock-infected control mice indicated the presence of astrogliosis in persistently infected mice at day 30 (Figure 7, A and B) and day 90 PI (Figure 7, D and E). Astrogliosis represents an increase in the proliferation of astrocytes after the loss of nearby neurons. The quantification of GFAP signal per section clearly demonstrated an increase in astrogliosis after eGFP-CVB3 infection in surviving mice (Figure 7, C and F). We hypothesize that the observed astrogliosis may represent either residual viral replication during the persistent phase of infection, or alternatively may reflect earlier viral insult to the surrounding neurons which remained at later time points. CNS lesions within the CA3 region of Ammon's horn and dentate gyrus of the hippocampus were also observed by Nissl staining at day 90 PI (Figure 7, G and H). In addition, we also observed a reduction in the size of the brain cortex after eGFP-CVB3 infection at day 90 PI by Nissl staining (Figure 7, I and J). Higher magnification of Figure 7, I and J, revealed a compaction of the cortical layers and reduced germinal cells in the SVZ layer of infected mice, as compared with mock-infected control mice (Figure 7, K and L).
Figure 7.

Astrogliosis and neurological abnormalities in mice surviving CVB3 infection. Pups 3 days old were infected with eGFP-CVB3 (1 × 107 pfu i.c.) or mock-infected. Brains from days 30 and 90 PI were inspected for signs of lasting CNS defects and neuropathology., Astrogliosis in the hippocampus was quantified by GFAP immunofluorescence in both mock-infected (A) and infected (B) mice. Relatively higher levels of GFAP expression were observed within infected mice at 30 days PI, as compared with mock-infected control mice. C: Higher GFAP levels seen for infected mice 30 days PI were quantified by ImageJ analysis and the results were shown to be statistically significant (P = 0.0503). Compared with mock-infected mice (D), relatively higher levels of GFAP remained within the hippocampus of infected mice (E) as long as 90 days PI. F: GFAP levels in the hippocampus remained higher at 90 day PI, as quantified by ImageJ analysis (P = 0.0407). Compared with mock-infected mice (G), the overall size of the hippocampus within infected mice (H) appeared smaller 90 days PI by Nissl staining. Also, some infected mice exhibited lesions in the CA3 region of Ammon's horn and in the dentate gyrus (green arrows). Compared with mock-infected mice (I), the cortex of infected mice (J) also appeared to be smaller in width by Nissl staining. Compared with mock-infected mice (K), closer inspection of the cortex revealed the overall compaction of the cortical layers and reduced germinal cells in the SVZ layer of infected mice (L).
Our model of lasting CVB3-mediated neuropathology is described in Figure 8. We propose that type B, C, and A stem cells are the main early targets of this highly cytolytic virus, and may undergo early apoptosis after infection. CVB3-mediated apoptosis of infected nestin+ NPSCs and their differentiated counterparts may lead to a rapid decline in neurogenesis and may eventually cause developmental defects reflected in our study by reduced brain wet weight values and alterations in cortex size. Finally, ribavirin may act on persistently infected stem cells and/or neurons, and a reduction in viral replication may lead to partial brain wet weight recovery.
Figure 8.

Model for coxsackievirus-induced depletion of progenitor cells and neurons in the CNS. The SVZ of noninfected mice (left column) contains three populations of neural stem and progenitor cells (NPSCs; type B, C, and A progenitor cells). Type B cells give rise to type C cells, which eventually produce migratory neuroblasts (type A cells). NPSCs migrate through the rostral migratory stream (RMS) or radial glia (RG) and differentiate into mature neurons in the olfactory bulb or cortex. In an infected mouse (center column), CVB3 infects neural NPSCs. Some infected NPSCs undergo apoptosis depleting the resident population of cells. Other infected NPSCs survive initial infection and migrate and differentiate into neurons within the CNS. The infected migratory cells assist in the dissemination of virus. Many infected neurons eventually undergo apoptosis. Apoptosis of NPSCs and neurons following CVB3 infection may lead to a reduction in neurons and neurodevelopmental defects. In an infected mouse treated with ribavirin (right column), ongoing sporadic viral replication during persistence may be reduced. After ribavirin treatment, progenitor cells and neurons may be protected from ongoing infection and apoptosis, thereby leading to partial brain wet-weight recovery in the mouse.
Discussion
Little is known regarding the long-term consequences of a neurotropic CVB3 infection on the CNS of the developing host. We previously demonstrated that CVB3 infects NPSCs in the SVZ and RMS of the CNS.10,14 The SVZ is located near the lateral ventricles of the brain and comprises NPSCs that migrate through the RMS and radially differentiate into functional olfactory neurons.39–41 Also, CVB3 preferentially replicated within proliferating cells,28 which might explain its preference for targeting NPSCs.10,42 Susceptibility to CVB3 is highly dependent on the age of the host, perhaps because of the greater number of proliferating cells in the developing host, which might serve as highly susceptible target cells.14 Despite infection, NPSCs continued to migrate and differentiate into neurons,14 although many infected neurons eventually underwent caspase-3–induced apoptosis. We hypothesized that potential reduction of neurogenesis because of stem cell death after CVB3 infection might lead to developmental defects and CNS dysfunction.18
Here, we show that a lethal inoculation of eGFP-CVB3 also induced apoptosis within nestin+ NPSCs in the SVZ and decreased proliferation in neurogenic regions of the neonatal CNS. Expression of eGFP by our recombinant CVB3 has been very helpful in characterizing neural progenitor cell tropism of the virus and tracking virus dissemination using our neonatal murine model of infection. Previous studies have used eGFP expressed under the promoter of neural stem and progenitor cell-specific genes such as nestin, GFAP, or Sox2 to successfully label and track neural progenitor cells with no apparent impact on neurogenesis or CNS development.43–45
Neuroblasts in the RMS were directly observed undergoing apoptosis after infection, which might account for the reduction in the size of olfactory bulbs, as compared with mock-infected mice. More than 50% of apoptotic cells at 24, 36, and 48 hours PI were found to express nestin, a marker for NPSCs. The remaining cells undergoing apoptosis have not yet been identified. However, because of their location within the SVZ in the neonatal CNS, these cells might represent downstream progenitor cells lacking detectable levels of nestin expression. Considerable CNS damage was observed in lethally inoculated mice; as early as day 5 postinfection (PI) a reduction in the size of the brain was seen simultaneously with high levels of virus protein expression in the SVZ, hippocampus, and cortex.
One-day-old mice infected with CVB3 exhibited high lethality; hence, these studies did not allow long-term analysis of CNS defects to be conducted. Therefore, we used 3-day-old mice to extend the lifespan of infected mice and determine long term consequences of infection on the developing CNS. By ApopTag staining, we determined that a sublethal inoculum of CVB3 also induced high levels of apoptosis in the SVZ, as compared with mock-infected control mice. We hypothesized that CVB3 directly harmed NPSCs by inducing apoptosis. However, technical limitations associated with ApopTag staining limited our ability to directly identify infected cells undergoing apoptosis. Therefore we used active caspase-3 staining, an early marker of programed cell death, to directly show apoptosis of infected cells within neurogenic regions of the CNS. These results parallel our recent publication demonstrating cytopathic effects and cell death by trypan staining in NPSCs infected in culture.12
Nonetheless, we cannot rule out the possibility that CVB3 may indirectly cause apoptosis in some cells by the induction of the innate immune response and/or infiltrating immune cells.35 For example, we have recently described the influx of Iba1+ macrophages/microglia that engulfed infected myeloid cells undergoing apoptosis within the choroid plexus at early time points after infection.11 These cells were also found in the SVZ following infection at a time point when nestin+ and nestin− cells were observed undergoing apoptosis. Therefore Iba1+ macrophages/microglia, or similar immune cells responding to infection, may play a role in NPSC apoptosis or the subsequent removal of dying cells by the process of phagocytosis.
As might be expected, a reduction in brain wet weight values were observed during acute infection (days 1, 2, and 5 PI) and during persistent infection (day 10, 30, and 90 PI) at a time when viral RNA could be detected in the absence of infectious virus. These results suggested CNS developmental defects after infection, most likely because of targeting of NPSCs. The amount of viral material (infectious virus or viral RNA) was analyzed with respect to brain wet weight values for each mouse to shed light on the association between the amount of virus and brain size. An inverse relationship between virus and brain wet weight values was clearly observed and appeared to grow stronger over time. However by day 90 PI, the inverse relationship was no longer seen, perhaps because of the relatively low viral load observed at this time point as determined by real-time RT-PCR.
Ribavirin has been successfully used to combat enterovirus infections. For example in one study examining enterovirus 71 infection in mice, the authors observed a reduction in mortality of mice after the administration of ribavirin because of a decrease in viral replication.24 Ribavirin can successfully cross the blood–brain barrier,46 suggesting that this antiviral drug might be effective in limiting CVB3 infection within the CNS. Antiviral drugs, such as ribavirin typically target the replication cycle of viruses. Ribavirin is thought to induce “lethal mutagenesis” in viral populations47 by increasing the viral mutation rate over a threshold of tolerable mutations. Therefore, we assessed the ability of ribavirin to improve brain wet-weight values in infected mice. However, rather than treat infected mice during acute infection (a point in time when an antiviral drug would most likely reduce viral titers), we delayed ribavirin treatment until the persistent stage of infection. We hypothesize that any improvement in brain wet weight values in mice treated at later time points (day 14 or day 60) might indicate ongoing virus replication during the persistent stage of infection. Infected mice treated with ribavirin at day 60 PI (every 3 days) and harvested at day 90 PI (antiviral treatment for a total of 30 days) showed a significant recovery of brain wet weight values, as compared with untreated, infected mice. Nevertheless, the brain wet weight values of ribavirin-treated mice remained below the values for mock-infected mice. These results suggest that antiviral drugs given during the persistent stage of infection may be beneficial in improving brain recovery. In addition, these results indirectly suggest that CVB3 may continue to replicate in the CNS at late time points (from day 60 to 90 PI), despite the absence of infectious virus as measured by plaque assay. In contrast, brain wet weight values of infected mice treated with ribavirin at day 14 PI (every 3 days) and harvested at day 30 PI (antiviral treatment for a total of 16 days) failed to improve. We suggest that the 16 day ribavirin treatment was insufficient to contribute to brain wet weight recovery in these mice.
Previous studies have suggested causal links between virus infection and behavioral disorders in humans.8 Some reports have suggested an association of coxsackievirus infection with neurological disorders, such as schizophrenia.30 Based on our existing data, we suggest that acute CVB3 infection may cause a significant amount of damage to neurogenic regions of the CNS, and the brain may be unable to fully recover. Other viruses have also been shown to target neural stem cells and alter neurogenesis including human cytomegalovirus,48,49 Japanese encephalitis virus50–54 and human immunodeficiency virus.55 Our results suggest that CNS injury and pathology after CVB3 infection may involve a direct effect of virus infection on NPSCs. Nonetheless, the immune response against viral infection may also play a critical role as the mechanism of CVB3 neuropathology in our neonatal murine model. For example, acute disseminated encephalomyelitis following an enteroviral infection29 might be considered a postinfectious condition with an underlying immunopathological response. Treatment with steroids and other anti-inflammatory and immunosuppressive therapies have shown a beneficial effect in patients with acute disseminated encephalomyelitis.56
Also, we have previously described viral persistence and chronic immunopathology in mice surviving CVB3 infection during the neonatal period. Most likely, our observed developmental defects and reduced brain wet-weight values after CVB3 infection might reflect a combination of direct infection of neural stem cells with resultant cytopathicity, along with an ongoing and deleterious immunopathological response against viral infection. Our recently published manuscript12 indicates that acute CVB3 infection may alter neural progenitor cell differentiation by increasing the percentage of cells expressing neuronal class III β-tubulin after their differentiation in the presence of fetal bovine seum. However, more studies are necessary to determine which lineage may show the greatest impact after CVB3 infection in the host. For example, a total reduction across all cell lineages may occur after infection despite a percentage increase in neuronal, as compared with glial cell lineages.
A more comprehensive assessment of the geographic distribution of viral infection in the CNS over multiple time points has been evaluated previously.10,14,20 Nevertheless, our previous studies and the current study directly demonstrate the susceptibility of neural stem and progenitor cells in the subventricular zone to CVB3 infection. Also, infection of the dentate gyrus suggests that neural stem cells in the hippocampus may be susceptible to infection. In contrast, the cerebellum and cerebellar progenitor cells may be less susceptible to infection. Hence, the susceptibility of neural progenitor cells may not be completely uniform across the brain. We have recently isolated neural progenitor cells from all three neurogenic regions of the CNS to determine whether CVB3 prefers a subpopulation of neural progenitor cells. These studies may help to determine whether particular regions of the CNS may be impacted after infection. Even so, the results described in the manuscript revealed a compaction of the cortical layers and reduced germinal cells in the SVZ layer of infected mice, as compared with mock-infected control mice (Figure 7), suggesting depletion of neural progenitor cells in the subventricular zone.
The presence of persistent viral RNA that continue to replicate at a low or undetectable level might contribute to lasting CNS damage after CVB3 infection. Of note, we anticipate substantial possible learning disabilities after neonatal enterovirus infection based on these studies. One of the main targets of CVB3 appears to be the hippocampus, a region responsible for learning and memory. Hence, any hippocampal damage occurring after infection may impact learning and memory tasks in surviving individuals.18 Our ongoing studies inspecting murine behavior suggest memory alterations in C57 BL/6 mice surviving an early CVB3 infection (manuscript in preparation). The observed damage in the CNS of surviving mice after CVB3 infection necessitates further investigations to clarify possible long-term repercussions in those individuals diagnosed with neurotropic enterovirus infections during infancy, and a need for designing antiviral drugs and vaccines for controlling enterovirus infections in the human host.
Footnotes
Supported by an NIH R01 award NS054108 (R.F.), NIH R01 awards AI042314 and HL093177 (J.L.W.), an NIH Research Supplement to Promote Diversity in Health-Related Research Award 3R01NS054108-01A2S1 (R.F. and S.M.R.), and a National Institutes of Mental Health (NIMH) Minority Research Infrastructure Support Program (M-RISP) R24 Faculty Fellow Award MH065515 (R.F.). J.M.T.-G. is a recipient of an Achievement Rewards for College Scientists Foundation Scholarship. S.M.R. is a recipient of the Rees-Stealy Research Foundation and the San Diego State University Heart Institute Fellowship. S.M. is a recipient of the SDSU Minority Biomedical Research Support Program (NIGMS, NIH grant 2R25GM058906-09A2).
References
- 1.Berger J.R., Chumley W., Pittman T., Given C., Nuovo G. Persistent Coxsackie B encephalitis: report of a case and review of the literature. J Neurovirol. 2006;12:511–516. doi: 10.1080/13550280601090546. [DOI] [PubMed] [Google Scholar]
- 2.Kamei S., Hersch S.M., Kurata T., Takei Y. Coxsackie B antigen in the central nervous system of a patient with fatal acute encephalitis: immunohistochemical studies of formalin-fixed paraffin-embedded tissue. Acta Neuropathol (Berl) 1990;80:216–221. doi: 10.1007/BF00308928. [DOI] [PubMed] [Google Scholar]
- 3.Whitton J.L., Cornell C.T., Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol. 2005;3:765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]
- 4.Romero J.R. Pediatric group B coxsackievirus infections. Curr Top Microbiol Immunol. 2008;323:223–239. doi: 10.1007/978-3-540-75546-3_10. [DOI] [PubMed] [Google Scholar]
- 5.Wikswo M.E., Khetsuriani N., Fowlkes A.L., Zheng X., Penaranda S., Verma N., Shulman S.T., Sircar K., Robinson C.C., Schmidt T., Schnurr D., Oberste M.S. Increased activity of Coxsackievirus B1 strains associated with severe disease among young infants in the United States, 2007–2008. Clin Infect Dis. 2009;49:e44–e51. doi: 10.1086/605090. [DOI] [PubMed] [Google Scholar]
- 6.Verma N.A., Zheng X.T., Harris M.U., Cadichon S.B., Melin-Aldana H., Khetsuriani N., Oberste M.S., Shulman S.T. Outbreak of life-threatening coxsackievirus B1 myocarditis in neonates. Clin Infect Dis. 2009;49:759–763. doi: 10.1086/605089. [DOI] [PubMed] [Google Scholar]
- 7.Sawyer M.H. Enterovirus infections: diagnosis and treatment. Semin Pediatr Infect Dis. 2002;13:40–47. doi: 10.1053/spid.2002.29756. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain R.N., Christie P.N., Holt K.S., Huntley R.M., Pollard R., Roche M.C. A study of school children who had identified virus infections of the central nervous system during infancy. Child Care Health Dev. 1983;9:29–47. doi: 10.1111/j.1365-2214.1983.tb00301.x. [DOI] [PubMed] [Google Scholar]
- 9.Hyypia T., Kallajoki M., Maaronen M., Stanway G., Kandolf R., Auvinen P., Kalimo H. Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res. 1993;27:71–78. doi: 10.1016/0168-1702(93)90113-2. [DOI] [PubMed] [Google Scholar]
- 10.Feuer R., Pagarigan R.R., Harkins S., Liu F., Hunziker I.P., Whitton J.L. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J Neurosci. 2005;25:2434–2444. doi: 10.1523/JNEUROSCI.4517-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabor-Godwin J.M., Ruller C.M., Bagalso N., An N., Pagarigan R.R., Harkins S., Gilbert P.E., Kiosses W.B., Gude N.A., Cornell C.T., Doran K.S., Sussman M.A., Whitton J.L., Feuer R. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J Neurosci. 2010;30:8676–8691. doi: 10.1523/JNEUROSCI.1860-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsueng G., Tabor-Godwin J.M., Gopal A., Ruller C.M., Deline S., An N., Frausto R.F., Milner R., Crocker S.J., Whitton J.L., Feuer R. Coxsackievirus preferentially replicates and induces cytopathic effects in undifferentiated neural progenitor cells. J Virol. 2011;85:5718–5732. doi: 10.1128/JVI.02261-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhoades R.E., Tabor-Godwin J.M., Tsueng G., Feuer R. Enterovirus infections of the central nervous system. Virology. 2011;411:288–305. doi: 10.1016/j.virol.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feuer R., Mena I., Pagarigan R.R., Harkins S., Hassett D.E., Whitton J.L. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol. 2003;163:1379–1393. doi: 10.1016/S0002-9440(10)63496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alvarez-Buylla A., Garcia-Verdugo J.M., Tramontin A.D. A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci. 2001;2:287–293. doi: 10.1038/35067582. [DOI] [PubMed] [Google Scholar]
- 16.Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- 17.Gage F.H. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 18.Buenz E.J., Rodriguez M., Howe C.L. Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol Dis. 2006;24:266–273. doi: 10.1016/j.nbd.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Kandolf R., Sauter M., Aepinus C., Schnorr J.J., Selinka H.C., Klingel K. Mechanisms and consequences of enterovirus persistence in cardiac myocytes and cells of the immune system. Virus Res. 1999;62:149–158. doi: 10.1016/s0168-1702(99)00041-6. [DOI] [PubMed] [Google Scholar]
- 20.Feuer R., Ruller C.M., An N., Tabor-Godwin J.M., Rhoades R.E., Maciejewski S., Pagarigan R.R., Cornell C.T., Crocker S.J., Kiosses W.B., Pham-Mitchell N., Campbell I.L., Whitton J.L. Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J Virol. 2009;83:9356–9369. doi: 10.1128/JVI.02382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim K.S., Tracy S., Tapprich W., Bailey J., Lee C.K., Kim K., Barry W.H., Chapman N.M. 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol. 2005;79:7024–7041. doi: 10.1128/JVI.79.11.7024-7041.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim K.S., Chapman N.M., Tracy S. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol. 2008;82:2033–2037. doi: 10.1128/JVI.01774-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman N.M., Kim K.S., Drescher K.M., Oka K., Tracy S. 5′ Terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology. 2008;375:480–491. doi: 10.1016/j.virol.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu K.H., Lonberg-Holm K., Alstein B., Crowell R.L. A monoclonal antibody specific for the cellular receptor for the group B coxsackieviruses. J Virol. 1988;62:1647–1652. doi: 10.1128/jvi.62.5.1647-1652.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergelson J.M., Mohanty J.G., Crowell R.L., St John N.F., Lublin D.M., Finberg R.W. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55) J Virol. 1995;69:1903–1906. doi: 10.1128/jvi.69.3.1903-1906.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergelson J.M. Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host Microbe. 2009;5:517–521. doi: 10.1016/j.chom.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Xu R., Crowell R.L. Expression and distribution of the receptors for coxsackievirus B3 during fetal development of the Balb/c mouse and of their brain cells in culture. Virus Res. 1996;46:157–170. doi: 10.1016/S0168-1702(96)01398-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feuer R., Mena I., Pagarigan R., Slifka M.K., Whitton J.L. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol. 2002;76:4430–4440. doi: 10.1128/JVI.76.9.4430-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saitoh A., Sawyer M.H., Leake J.A. Acute disseminated encephalomyelitis associated with enteroviral infection. Pediatr Infect Dis J. 2004;23:1174–1175. [PubMed] [Google Scholar]
- 30.Rantakallio P., Jones P., Moring J., Von W.L. Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int J Epidemiol. 1997;26:837–843. doi: 10.1093/ije/26.4.837. [DOI] [PubMed] [Google Scholar]
- 31.Woodall C.J., Riding M.H., Graham D.I., Clements G.B. Sequences specific for enterovirus detected in spinal cord from patients with motor neurone disease. BMJ. 1994;308:1541–1543. doi: 10.1136/bmj.308.6943.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodall C.J., Graham D.I. Evidence for neuronal localisation of enteroviral sequences in motor neurone disease/amyotrophic lateral sclerosis by in situ hybridization. Eur J Histochem. 2004;48:129–134. doi: 10.4081/877. [DOI] [PubMed] [Google Scholar]
- 33.Berger J.R., Fee D.B., Nelson P., Nuovo G. Coxsackie B meningoencephalitis in a patient with acquired immunodeficiency syndrome and a multiple sclerosis-like illness. J Neurovirol. 2009;15:282–287. doi: 10.1080/13550280902913263. [DOI] [PubMed] [Google Scholar]
- 34.Verboon-Maciolek M.A., Groenendaal F., Cowan F., Govaert P., van Loon A.M., de Vries L.S. White matter damage in neonatal enterovirus meningoencephalitis. Neurology. 2006;66:1267–1269. doi: 10.1212/01.wnl.0000208429.69676.23. [DOI] [PubMed] [Google Scholar]
- 35.Buenz E.J., Sauer B.M., Lafrance-Corey R.G., Deb C., Denic A., German C.L., Howe C.L. Apoptosis of hippocampal pyramidal neurons is virus independent in a mouse model of acute neurovirulent picornavirus infection. Am J Pathol. 2009;175:668–684. doi: 10.2353/ajpath.2009.081126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doetsch F. A niche for adult neural stem cells. Curr Opin Genet Dev. 2003;13:543–550. doi: 10.1016/j.gde.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Crotty S., Andino R. Implications of high RNA virus mutation rates: lethal mutagenesis and the antiviral drug ribavirin. Microbes Infect. 2002;4:1301–1307. doi: 10.1016/s1286-4579(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 38.Kishimoto C., Crumpacker C.S., Abelmann W.H. Ribavirin treatment of murine coxsackievirus B3 myocarditis with analyses of lymphocyte subsets. J Am Coll Cardiol. 1988;12:1334–1341. doi: 10.1016/0735-1097(88)92618-6. [DOI] [PubMed] [Google Scholar]
- 39.Pencea V., Bingaman K.D., Freedman L.J., Luskin M.B. Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol. 2001;172:1–16. doi: 10.1006/exnr.2001.7768. [DOI] [PubMed] [Google Scholar]
- 40.Doetsch F., Caille I., Lim D.A., Garcia-Verdugo J.M., varez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 41.Alvarez-Buylla A., Garcia-Verdugo J.M. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feuer R., Whitton J.L. Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr Top Microbiol Immunol. 2008;323:149–173. doi: 10.1007/978-3-540-75546-3_7. [DOI] [PubMed] [Google Scholar]
- 43.Mignone J.L., Kukekov V., Chiang A.S., Steindler D., Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol. 2004;469:311–324. doi: 10.1002/cne.10964. [DOI] [PubMed] [Google Scholar]
- 44.Kim Y., Comte I., Szabo G., Hockberger P., Szele F.G. Adult mouse subventricular zone stem and progenitor cells are sessile and epidermal growth factor receptor negatively regulates neuroblast migration. PLoS One. 2009;4:e8122. doi: 10.1371/journal.pone.0008122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barraud P., Thompson L., Kirik D., Bjorklund A., Parmar M. Isolation and characterization of neural precursor cells from the Sox1-GFP reporter mouse. Eur J Neurosci. 2005;22:1555–1569. doi: 10.1111/j.1460-9568.2005.04352.x. [DOI] [PubMed] [Google Scholar]
- 46.Connor E., Morrison S., Lane J., Oleske J., Sonke R.L., Connor J. Safety, tolerance, and pharmacokinetics of systemic ribavirin in children with human immunodeficiency virus infection. Antimicrob Agents Chemother. 1993;37:532–539. doi: 10.1128/aac.37.3.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Outbreaks of aseptic meningitis associated with echoviruses 9 and 30 and preliminary surveillance reports on enterovirus activity–United States, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:761–764. [PubMed] [Google Scholar]
- 48.Luo M.H., Schwartz P.H., Fortunato E.A. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol. 2008;82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheeran M.C., Hu S., Ni H.T., Sheng W., Palmquist J.M., Peterson P.K., Lokensgard J.R. Neural precursor cell susceptibility to human cytomegalovirus diverges along glial or neuronal differentiation pathways. J Neurosci Res. 2005;82:839–850. doi: 10.1002/jnr.20682. [DOI] [PubMed] [Google Scholar]
- 50.Das S., Basu A. Japanese encephalitis virus infects neural progenitor cells and decreases their proliferation. J Neurochem. 2008;106:1624–1636. doi: 10.1111/j.1471-4159.2008.05511.x. [DOI] [PubMed] [Google Scholar]
- 51.Das S., Chakraborty S., Basu A. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J Neurochem. 2010;115(2):537–549. doi: 10.1111/j.1471-4159.2010.06951.x. [DOI] [PubMed] [Google Scholar]
- 52.Luo M.H., Hannemann H., Kulkarni A.S., Schwartz P.H., O'Dowd J.M., Fortunato E.A. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol. 2010;84:3528–3541. doi: 10.1128/JVI.02161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Odeberg J., Wolmer N., Falci S., Westgren M., Sundtrom E., Seiger A., Soderberg-Naucler C. Late human cytomegalovirus (HCMV) proteins inhibit differentiation of human neural precursor cells into astrocytes. J Neurosci Res. 2007;85:583–593. doi: 10.1002/jnr.21144. [DOI] [PubMed] [Google Scholar]
- 54.Odeberg J., Wolmer N., Falci S., Westgren M., Seiger A., Soderberg-Naucler C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol. 2006;80:8929–8939. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okamoto S., Kang Y.J., Brechtel C.W., Siviglia E., Russo R., Clemente A., Harrop A., McKercher S., Kaul M., Lipton S.A. HIV/gp120 decreases adult neural progenitor cell proliferation via checkpoint kinase-mediated cell-cycle withdrawal and G1 arrest. Cell Stem Cell. 2007;1:230–236. doi: 10.1016/j.stem.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 56.Shahar E., Andraus J., Savitzki D., Pilar G., Zelnik N. Outcome of severe encephalomyelitis in children: effect of high-dose methylprednisolone and immunoglobulins. J Child Neurol. 2002;17:810–814. doi: 10.1177/08830738020170111001. [DOI] [PubMed] [Google Scholar]