

Abstract
Notwithstanding their genetic complexity, different cancers share a core group of perturbed pathways converging upon a few regulatory nodes that link the intracellular signaling network with the basic metabolic machinery. The clear implication of this view for cancer therapy is that instead of targeting individual genetic alterations one-by-one, the next generation of cancer therapeutics will target critical hubs in the cancer network. One such hub is the translation initiation complex eIF4F, which integrates several cancer-related pathways into a self-amplifying signaling system. When hyperactivated by apical oncogenic signals, the eIF4F-driven translational apparatus selectively switches the translational repertoire of a cell towards malignancy. This central integrative role of pathologically activated eIF4F has motivated the development of small molecule inhibitors to correct its function. A genome-wide, systems-level means to objectively evaluate the pharmacological response to therapeutics targeting eIF4F remains an unmet challenge.
Keywords: Cancer therapy, translation, eIF4F
Introduction
Biomedical scientists have recognized for three decades that components of the cap-dependent translation initiation machinery are more abundant and active in neoplastic cells than in their nonmalignant counterparts; an adjustment previously assumed to serve the increased metabolic needs of cancer. Here, we propose to review what is known about the eIF4F-driven translation initiation apparatus as an advanced example of a novel molecular target class for cancer drug discovery. Nearly all targets to date have been oncogenic variants of growth factors/growth factor receptors that generate sustained apical signals in proliferation and survival pathways. This new type of target is a component of the cellular machinery found downstream of such signals at hubs in the cancer-signaling network. Not only is eIF4F being actively explored as a target for cancer drug discovery by scientists in industry and academe; but it also illustrates a paradigm shift in targeted cancer therapy from a single molecule to a systems-based approach.
Bright and Dark Sides of Targeted Anticancer Therapy
During the past 5 decades, conventional cytotoxic anticancer therapy has greatly improved patient survival and quality of life; however, for the most common and lethal cancers we have reached the point of efficacy-limiting toxicity. As a result, it was with great optimism that the era of targeted anticancer therapy was launched, enabled by the mapping of signaling circuits that govern the decision of a cancer cell to proliferate or die (1). Many agents targeting cancer-related gene products are approved for clinical use, while others are in the midst of evaluation (2, 3). A major advantage of targeted cancer therapeutics is their low toxicity and selectivity for neoplastic targets conferring them with a high therapeutic index. Their target selectivity, however, can also be their undoing. Unlike testicular cancer or certain forms of leukemia in which tumorigenesis is predominantly driven by a single, targetable oncogene most solid tumors result from alterations of a large number of genes that create a complex network of collaborating, oncogenic circuits. Thus, the aberrant expression of a putative target does not by itself predict that a drug disabling that target will be effective. Thus, targeted agents can encounter de novo treatment resistance or facilitate the acquisition of resistance (4). This helps explain why available single-target agents might not be having the hoped for impact on long-term survival for most common malignancies.
What is the next step in anticancer therapy and rational drug discovery? One broadly accepted option is personalized cancer medicine - a combination of highly specific molecular-targeted drugs tailored to each patient’s tumor (2). This strategy follows the model of inborn errors of metabolism; conditions in which a critical rate limiting enzyme in a linear metabolic pathway mediates the illness (5). These assumptions are often not correct, since many naturally occurring tumors are genetically diverse and complex, comprised of cells harboring oncogenic signaling networks that are robust and flexible (6). In accord with this, clinical trials using drug combinations targeting several individual oncogenes have not lived up to expectation (7) as patients with the “correct” oncogenic lesions - with excellent initial responses - often relapse with tumors that with manifest drug resistance. A second possibility is a single multi-target agent, like tyrosine kinase inhibitors, that can hit key targets operating in several oncogenic pathways (3). Although attractive, major limitations of this approach are toxicity due to cross-reactivity with vital processes in normal cells and acquired drug resistance (3). A third option that we prefer is to develop agents targeting critical nodes and hubs in the cancer signaling network (8). These network elements reside downstream of the current growth factor/growth factor receptor/signal transduction targets; at loci where these pathways converge to usurp the basic cellular machinery and re-direct their functions toward oncogenesis.
The Paradigm: Targeting a Nexus of Cancer Pathways
The classical imperative for successful cancer therapy is to identify and attack those processes that are absolutely essential for malignancy yet dispensable for physiological function. To this end, a paramount question in cancer biology remains whether different malignancies share at least one lesion necessary for neoplastic function. The commonly accepted paradigm of oncogenesis as a multistep process implies that cell malignant conversion is mediated by the stochastic accumulation of genetic lesions that drives evolution of a cell population toward malignancy (9, 10). If true, then cancer would be an always-changing target that might be impossible to destroy. However, a series of observations indicating that neoplastic growth can be reversed by inactivating a pivotal oncogenic insult or targeting non-oncogenic pathways required for survival of cancer cells has revived the idea of an “Achilles Heel” in cancer. This idea has been conceptualized in the recent hypotheses of oncogene addiction (11) and synthetic lethality(12). These theories predict that tumorigenesis can be overturned by pharmacological inhibition of a single, or perhaps only a few, obligatory oncogenic alterations. Do all or many forms of cancer share a common Achilles Heel?
Most available targeted therapies operate at the apices of what have until recently been conceptualized as discrete oncogenic pathways. We now understand that cell surface signals and their transducers, whether physiological or oncogenic, form a branched network of interacting metabolic events that can take many routes to converge on key components of the cellular machinery. One innovative approach for the integrated analysis of the complex signaling network is a system biology-based strategy (13). It allows the efficient evaluation of functional interactions with among multiple cancer-related pathways and identification of critical convergence nodes and hubs in cancer circuits. Results of wide-ranging network modeling are consistent with the idea that proximal location of current therapeutic targets enables cells to develop or manifest drug resistance by switching to a collateral pathway that is still able to commandeer the core cellular apparatus controlling bioenergetics, viability, proliferation and gene expression (5). This provides a compelling biological rationale for mapping the cancer circuitry from its numerous and varied apical origins to key destinations in the network; and selecting those that are amenable to molecular targeted therapies (8, 14). The experimental data summarized here indicate that the distal arm of the PI3K/Akt/mTOR/translation axis functions as a regulatory hub in many critical cancer pathways and that the translation initiation machinery, eIF4F, represents a bona fide molecular target for anti-cancer drug discovery.
eIF4F Integrates and Amplifies Major Oncogenic Pathways
The eIF4F-mediated translational apparatus
Translation of mRNA into protein is the most energy expensive step in the flow of genetic information from the genome to the proteome. Not surprisingly, it is tightly regulated at several steps by a multilayered system of genome-wide and transcript-specific controls. Translational control provides a rapid, robust and highly specific mechanism by which a cell can respond to changes in its microenvironment (15, 16).
Conceptually, the process of translation can be subdivided into four consecutive stages: initiation, elongation, termination and ribosome recycling (16). While control of mRNA translation occurs at each step, the primary target for regulation in eukaryotes is the process of initiation. At this stage, the 43S pre-initiation ribosome complex associates with the 5′ terminus of the mRNA through the translation initiation complex eIF4F and proceeds to scan its 5′UTR in the 3′ direction toward the initiation codon (16). eIF4F is a heterotrimer that binds the 5′-terminal cap structure m7GpppN (in which N is any nucleotide). It consists of the cap-binding protein eIF4E, the scaffolding protein eIF4G and the RNA helicase eIF4A (Supplementary Fig. S1). The rate-limited component of the eIF4F heterotrimer is the cap-binding protein eIF4E. Extracellular cues including peptide growth factors, extracellular matrix and direct cell-to-cell contacts positively regulate eIF4F through a network of signal transduction pathways. Integration of this signaling circuitry occurs at a translational control checkpoint governed by eIF4E and by mTORC1-mediated phosphorylation of members of the 4E-BP family of translational repressors(8) and 40S ribosomal protein kinases S6 (S6K) (17).
The primary regulation of eIF4F activity is exerted through the PI3K/Akt/mTORC1/S6K/4E-BP signaling axis, which is positively regulated by Ras and leads to hierarchical phosphorylation of the 4E-BPs. Ras can signal to mTORC1 by direct activation of PI3K or can operate through the canonical MAPK/ERK pathway (Fig. 1). When activated, Ras signals phosphorylation of eIF4E on Ser209 (18) via the MAPK-interacting protein kinases MNK1 and MNK2 (19). Myc can transcriptionally activate the eif4e gene through two functional Myc-binding sites (E-boxes) in the eIF4E promoter (20).
Figure 1.
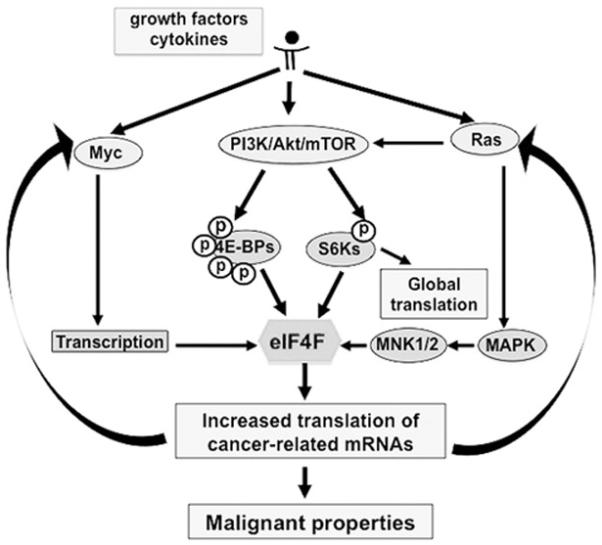
The eIF4F-signaling pathway. The distal arms of the major potentially oncogenic pathways converge to the translational machinery mediated by the initiation complex eIF4F, which functions as a major regulatory hub downstream of the receptor signaling circuit. When activated, eIF4F reprograms the cellular translational apparatus to amplify oncogenic signals and to regulate neoplastic capabilities in cancer cells.
The mRNAs most sensitive to the activity of eIF4F are those encoding proteins that regulate hallmark capabilities of cancer cells. Established examples of translationally regulated mRNAs include those for growth factors and cytokines (FGF-2, IGF II, PDGF, TGF-b, VEGF, IL-15, Wnt/b-catenin), protein kinases (Mos, Pim-1), transcription factors (Fos, Myc), polyamine biosynthetic enzymes (ornithine decarboxylase and ornithine aminotransferase), inhibitors of apoptosis (survivin, Bcl-2, Bcl-XL) and promoters of cell cycle transit (Ras, Cdk 4, cyclin D1) (21-23). Although some translationally activated proteins function as inhibitors of cell cycle progression and inducers of apoptosis (e.g. p27Kip, p53), the net effects of upregulated cap-dependent translation are increased proliferation and viability. These data indicate that the translation initiation apparatus can couple the major growth/survival circuits to a spectrum of trophic extracellular cues – making it a strategic downstream target of nearly any oncogenic mutation. Thus, eIF4F resides on the causal pathway to many cancers and functions as a critical regulatory hub where multiple oncogenic pathways converge or emerge.
Translational Addiction of Cancer
Cancer cells require constantly increased levels of eIF4F-mediated translation
Since eIF4F funnels and augments multiple oncogenic pathways, it is not surprising that cap-dependent translation is commonly deregulated in most - if not all - human malignancies(21). In the past decade, several causal links have emerged between deregulated translation and cancer, providing strong support for the idea that hyper-activated cap-dependent translation is a bone fide oncogenic pathway (8, 15, 24). Findings in mouse models of cancer demonstrate that germ line constitutive expression of eIF4E leads to multiple tumor formation (25) confirming earlier seminal studies(26) that overexpression of eIF4E is sufficient to transform rodent cells. In line with this, mouse germ line deficiency of two 4E-BP family members (4ebp1/2) leads to genome-wide translational activation of genes governing angiogenesis and cell proliferation; and sensitizes mice to lung tumorigenesis induced by the tobacco carcinogen 4-(methylnitrosamino)-I-(3-pyridyl)-1-butanone (NNK) (27). Moreover, normalizing deregulated translation in cancer cells by genetic (28) or pharmacological (29-31) interventions reverses the cell transformed phenotype and causes regression of xenograft tumors. Tumor cells of different origin commonly manifest increased levels of eIF4E phosphorylation (32). Interventions that increase eIF4E phosphorylation result in enhanced eIF4E-dependent nuclear export of growth-promoting mRNAs and contribute to cell transformation (33). Collectively, these data establish a direct causal link between dysfunction of the eIF4F-driven translational apparatus and acquisition of cancer-related capabilities; and support the notion that malignancies driven by different oncogenic insults are strictly dependent on the constant hyperactivity of the eIF4F-mediated translational apparatus.
Why do cancer cells switch to a sustained hyper-activated mode of eIF4F-mediated translation?
One simple explanation is that any actively propagating population of cells relies on increased global translation to meet their intrinsic metabolic needs. This, however, turns out not to be the case for cells with constitutively up-regulated eIF4F. In reality, cells harboring overactivated eIF4F display neoplastic phenotypic changes without significant changes in general translation. Instead, activated eIF4F dramatically stimulates recruitment of ribosomes to a specific subset of mRNA encoding proteins governing cancer-related functions (23, 34). Thus, oncogenic lesions promote sustained activation of eIF4F, which in turn redirects the translational repertoire of a cell to disable anti-cancer defenses and promote uncontrolled cell division. Once established, cancer cells are irrevocably addicted to this gene expression pattern.
Targeting the Signaling Pathways Leading to Pathological Activation of eIF4F
Targeting pathways leading to phosphorylation of 4E-BPs
In principle, normalizing deregulated eIF4F-mediated translation can be accomplished indirectly by interrupting upstream signals leading to eIF4F activation or by directly targeting the eIF4F complex (35-37). Phosphorylation of the eIF4F members and its antagonists is a common regulatory mechanism by which cells are signaled to modulate cap-dependent translation. Recent findings indicate that mTORC1-mediated phosphorylation of the 4E-BPs is required for Akt/mTOR-driven neoplastic growth and that how well a therapeutic targeting mTOR works, depends upon the degree to which it corrects the function of eIF4F (35). There is ample evidence supporting the role of the PI3K/AKT/mTOR pathway in cancer. This has motivated the development of a series of PI3K and mTORC1 inhibitors as cancer therapies; which in some trials have shown a promising anti-cancer signal (17, 38, 39). In addition, some agents widely used for other clinical indications, that also antagonize cancer, operate through the mTOR/eIF4F axis. For example, the anti-diabetic drug metformin inhibits tumor growth – an activity that is associated with metformin-induced inhibition of mTOR and reduction of cap-dependent translation (40). Likewise, the capacity of the antidepressant agent sertraline to antagonize breast cancer cell growth is linked to its ability to down regulate mTORC1-dependent phosphorylation of key translational regulators (41). Although the PI3K/Akt/mTOR axis has enjoyed attention as an attractive target for therapeutic interventions, several observations reveal significant limitations of targeting mTOR. One potential reason for limited efficacy of mTOR inhibitors as anticancer therapeutics is suppression of S6K mediated negative feedback loops, which leads to pro-oncogenic activation of PI3K/Akt(42) and MAPK/ERK (43) signaling. A second potential drawback to targeting the PI3K/AKT/mTOR axis as an upstream activator of eIF4F is that processes other than mTOR signaling can also activate eIF4F. Indeed, available data indicate that activation of eIF4F by phosphorylation of the 4E-BPs in cancer may be insensitive to rapamycin/rapalog treatments (44).
Targeting pathways leading to phosphorylation of eIF4E
Growth factors signal phosphorylation of eIF4F mostly through pathways separate from the PI3K/Akt/mTOR axis. These pathways have also been intensively examined for cancer drug discovery. For example, targeting the RAS pathway remains attractive. However, recent success treating melanoma with a small-molecule inhibitor of mutant BRAF kinase has been overshadowed by the rapid development of drug resistance due to activation of ERK and other downstream effectors of Ras(45). One such downstream effector of the Ras /MAPK/ERK axis is MNK kinase. To promote tumorigenesis, eIF4E must be phosphorylated by the MNK kinases (46). Using a high throughput screen, Graff and colleagues (47) identified an antifungal agent, cercosporamide, as a powerful and specific inhibitor of MNK kinase activity. At low micromolar doses, cercosporamide reduces eIF4F phosphorylation and inhibits tumorigenesis and metastases in mouse models of cancer. Thus, targeting eIF4E phosphorylation appears to be a promising new therapeutic strategy; with enthusiasm tempered at present by limited knowledge about the role of MNK kinase in phosphorylation of substrates other than eIF4E.
Collectively, these studies confirm earlier observations that concomitant inhibition of PI3K/Akt/mTOR and MAPK/ERK/MNK pathways can, under some circumstances, suppress eIF4F-mediated tumorigenesis(48). However, it is equally apparent that targeting proximal events upstream of eIF4F may result in undesired interruption of negative feedback signals and stimulation of collateral pro-neoplastic circuits. These findings support exploration of approaches to directly target eIF4F.
Strategies to Directly Target eIF4F
There are at least two readily apparent approaches for direct therapeutic targeting of eIF4F (Figs. 2 and 3, table 1): disrupting the integrity of eIF4F and antagonizing the eIF4E-mRNA cap interaction.
Figure 2.
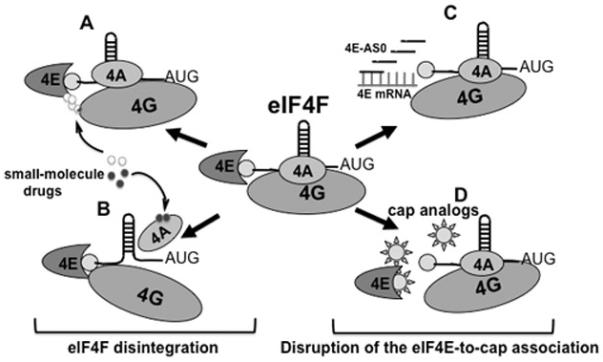
Disrupting assembly of a functional eIF4F heterotrimer. A and B, with deconstruction of eIF4F: A, disrupting association of eIF4E and eIF4G; B, disrupting association of eIF4A and eIF4G. C and D, targeting cap recognition: C, decreasing abundance of eIF4E; D, inhibiting the interaction between eIF4E and the mRNA cap. Abbreviations: 4E - eIF4E; 4A – eIF4A; 4G – eIF4G (See the text for details).
Figure 3.

Molecular structures of small-molecule inhibitors of eIF4F. A, compound antagonizing phosphorylation of eIF4E; B, agents preventing association of eIF4E and 7Me-GMP; C, compound disrupting association of eIF4E and eIF4Gs; D, agents inhibiting activity of eIF4A
Table 1.
Agents targeting eIF4F
| Primary target | Compound | Compound class | Tested cancer | Study | Reference |
|---|---|---|---|---|---|
| eIF4E phosphorylation |
Cercosporamide | Small organic | Xenograft melanoma |
Preclinical | 47 |
|
eIF4E synthesis |
4E-ASO LY2275796 |
Oligonucleotide |
Multiple human cancers |
Phase I |
30,54 |
| eIF4E-cap interaction |
4Ei-1 | Small organic | Breast carcinoma cell lines |
Laboratory | 60,61,62 |
| 4EGI-1 | Small organic | Multiple human cancer cell lines |
Laboratory | 31 | |
| eIF4E-eIF4G interaction |
[DLys6]GnRH- | Oligopeptide | Xenograft ovarian | Laboratory | 32 |
| 4EBP1-WT 4E1RCat |
Small organic | Mouse lymphoma | Laboratory | 50 | |
| eIF4A activity | Pateamine A | Small organic | Xenograft melanoma |
Laboratory | 51, 52 |
| Silvestrol | Small organic | Mouse lymphoma | Laboratory | 29,50,53 |
Disrupting the integrity of eIF4F
To pharmacologically mimic the anti-eIF4F effect of the 4E-BPs, a research group led by Gerhard Wagner identified a compound designated 4EGI-1 in a high throughput screen (31). At micromolar doses in vitro, 4EGI-1 attenuated neoplastic properties including expression of the c-Myc and Bcl-XL oncoproteins, apoptosis resistance and clonogenic expansion. Although 4EGI-1 did suppress cell growth, cancer cells revealed only a 2-fold higher susceptibility to 4EGI-1 than their non-transformed counterparts. This relatively modest therapeutic index, together with more recent data demonstrating that 4EGI-1 promotes cell death through a mechanism that is independent of the eIF4F-driven translational apparatus(49), dampens enthusiasm for this agent.
Nonetheless, targeting the eIF4E-to-eIF4G interaction remains an attractive option. One elegant approach for disrupting eIF4E/eIF4G is based on a 19-residue fragment of 4E-BP1 containing the Y(X)4LΦ motif. Fusion with an analog of gonadotropin-releasing hormone enables it to use a hormone receptor that is widely expressed on ovarian, breast and other endocrine epithelial cancers to enter its cellular target (32). This compound suppresses tumor growth in a mouse xenograft model of ovarian cancer without significant general toxicity. More recently, Pelletier and colleagues identified a non-toxic small-molecule inhibitor of the eIF4E-to-eIF4G association designated 4E1RCat with highly promising biochemical, and therapeutic characteristics (50). This compound blocks association of eIF4E with eIF4G by interaction with the eIF4E binding motif on the dorsal surface of eIF4E; and reverses chemoresistance in a Myc-dependent lymphoma murine model.
The marine sponge natural product pateamine A (PatA) stabilizes the closed confirmation of eIF4A preventing its association with mRNA (51). Active at nigh nanomolar concentrations, PatA suppresses cap-dependent and IRES-driven translation and induces apoptosis in cancer cells (52). Another natural product, the plant cyclopenta[b]benzofuran flavagline silvestrol depletes the eIF4F complex of eIF4A and inhibits cancer cell growth by inducing mitochondrion-dependent apoptosis (29). It suppresses the growth of lung, prostate and breast cancer cells in vitro (53) inhibits hematopoietic malignancies in vivo (29) and synergizes with conventional chemotherapy (29, 50). These effects of silvestrol are associated with a preferential reduction in the translation of cancer-related mRNAs with highly structured 5′ UTRs (50). Although promising in preliminary testing, both PatA and silvestrol antagonize both cap-dependent and IRES-driven translation – a feature that might lead to undesired effects.
Antagonizing the eIF4E-mRNA cap interaction
The only constituent of eIF4F able to recognize the cap structure is eIF4E. Two approaches to targeting eIF4E are being actively investigated: decreasing eIF4E abundance and antagonizing association of eIF4E with the 5′ mRNA cap. Graff and colleagues from the Lilly Research Laboratory successfully applied an ASO-based strategy to down regulate eIF4E and suppress tumorigenesis (30). They were able to circumvent technical problems typical for ASOs in vivo by using a second-generation, nuclease resistant ASO modified to penetrate the cell membrane, designated 4E-ASO. Nanomolar concentrations of 4E-ASO normalize eIF4E levels in cancer cells; and doses in the 5 to 25 mg/kg range suppress tumorigenicity in xenograft models of breast cancer without any signs of toxicity. A phase I/II clinical trial of 4E-ASO (LY2275796 ClinicalTrials.gov Identifier: NCT00903708) has recently been completed (54). The data show that 4E-ASO is well tolerated up to 1,000 mg. Because up to 80% reduction in eIF4E mRNA did not result in tumor response, the author suggested the next step would be to examine the agent as a chemosensitizer for combinatorial chemotherapy.
Antagonizing the eIF4E-to-cap interaction has appeal because an effective compound should in principle selectively target the most eIF4E sensitive transcripts at the lowest doses. One approach to this is the guanosine ribonucleoside analog ribavirin, which is well established as therapy for hepatitis C and respiratory syncytial virus infections. Ribavirin inhibits growth of breast cancer cells (55) and cooperates with established therapeutics in primary acute myeloid leukemia (AML). It has shown promising results in preliminary clinical testing in AML patients and is currently under investigation in phase I-II clinical trials for AML and metastatic breast cancer. Ribavirin was originally thought to suppress neoplastic growth by competing with the cap structure for binding to eIF4E. However, two independent research groups found that ribavirin is unable to function as a cap-analog in either cell-free or cell-based assay systems (56, 57). These data indicate that the anti-cancer activity of ribavirin may not be directly related to its ability to repress of cap-dependent translation.
Rational drug design has guided the synthesis of nucleotide derivatives designed to displace capped mRNAs from the eIF4F complex. Two groups (58, 59) have developed libraries of 7-MeGMP analogues with favorable drug-like properties. Using a multi-step test system, we identified a new class of cell permeable inhibitors of the eIF4E-cap interaction and have extensively evaluated one member of this class, 7-benzyl guanosine monophosphate (7-BnGMP), the compound with the highest affinity for eIF4E (60). While effective in mammalian cell free test systems and in zebra fish embryos, 7-BnGMP displayed low cell membrane permeability. One approach to circumventing this obstacle is to develop a membrane permeable pro-drug (pro-nucleotide), which can be bioactivated within the cell. Phosphoramidation produces a class of water-soluble stable nucleotide derivatives with high membrane permeability (61). Within a cancer cell, nucleoside phosphoramidates can be efficiently converted into their corresponding 5′-monophosphate nucleotides by a family of enzymes termed human histidine triad nucleotide binding protein (hHINT) (62). When injected in zebra fish embryos, phosphoramidated 7-BnGMP (designated 4Ei-1) was converted from a pro-drug into functionally active 7-BnGMP (60). Pro-drug processing was accompanied by efficient down regulation of eIF4F-mediated translation and prevention of phenotypic alterations induced by over expressed eIF4E without causing developmental abnormalities. More recent findings show that 4Ei-1 enters human breast carcinoma cells where it is converted into 7-BnGMP which in turn down regulates cap-dependent translation. When applied together with the cytostatic drug 5-Fluorouracil (5-FU), 4Ei-1 enhances the cytostatic/cytotoxic effect of 5FU against some breast carcinoma cell lines (63).
Overall, these results show that targeting aberrantly up-regulated eIF4F-mediated translation reduces tumor growth in most types of malignancies and sensitizes tumor cells to low doses of genotoxic chemotherapy. Agents causing up to a 60% reduction of cap-dependent translation are well tolerated during embryogenesis and do not disrupt physiological processes in adult vertebrate organisms. Thus, selective eIF4F inhibitors can serve as useful tools for probing the role of eIF4F-mediated translation in critical cancer-related processes such as angiogenesis, metastasis and the epithelial-mesenchymal transition. In addition anti-eIF4F interventions are ready to be optimized in light of our current understanding of the eIF4F-mediated step in the posttranscriptional control of cancer.
Molecular Biomarkers of Successful Anti-Translational Therapy
A means to objectively evaluate the pharmacological response to therapeutics targeting the translation initiation machinery remains an unmet challenge. Traditionally, studies investigating the genome-wide control of gene expression in cancer have focused on transcriptional gene expression profiling. The OMICS era has been witness to an avalanche of reports aiming to identify reliable genome-wide prognostic markers that predict response to anti-cancer therapy (64). Gene expression-based drug discovery has the potential to identify compounds that convert malignant cells, as defined by their transcriptional gene-expression signature, to a less malignant and/or chemosensitive state (65). However, there are several drawbacks to transcriptional gene-expression profiling as a biological marker for cellular responses to anti-eIF4F agents (66). First, many transcripts present in a cell are translationally silent. Second, the effect of translational modulators on recruitment of ribosomes to available cellular mRNAs is much greater than alterations in mRNA abundance. Third, and most importantly for anti-eIF4F drug discovery, genome-wide analysis of polyribosome bound mRNAs reveal that translational modulators affect the cellular phenotype by differentially altering the translational efficiency of physiologically important transcripts that are preferentially selected by the modified translational apparatus from among the pool of available mRNAs (see previous section for details). For these reasons, a “translational signature” based on groups of genes that are actively recruited on or off ribosomes in response to drug treatments may better reflect the efficacy of anti-eIF4F agents (15, 66). This strategy accounts for both the transcriptional and translational effects of therapeutic interventions (67); and provides clues to pharmacological mechanism that can be definitively tested by examining causal connections between changes in gene expression and ablation of specific cancer-related functions.
Why Won’t Inhibition of eIF4F Kill All Cells – Not Just Cancer?
The concept of targeted anti-eIF4F therapy has emerged over the past decade; and presently it is regarded as a promising new approach for attacking cancer. Initially, however, there was little enthusiasm for translational inhibitors as a strategy for cancer therapy. This skepticism was based on the concern that suppressors of protein synthesis would not only kill cancer, but also would harm normal cells. At some tipping point, suppression of cap-dependent translation will indeed be toxic to all cells, not just cancer. Fortunately that tipping point for normal cells appears to differ dramatically from that for cancer. Hints that this might be true were evident from observations indicating that translation initiation is only reduced, but not shut down in normal cells even after removal of the cap-structure from transcripts (68). This helps explain why non-transformed cells tolerate a broad range of eIF4F activity. In contrast, malignant cells require constitutively hyperactivated eIF4F-driven translation to maintain their malignant state. Therefore, the rational approach to eIF4F-targeted therapy is not complete inhibition of cap-dependent translation but rather reversal of eIF4F hyperactivation in cancer cells with proper dosing. Accepting this, what is the theoretical and experimentally determined therapeutic margin between normal cells and cancer? Experimental data show that cancer cells manifest about a 2-fold increase in cap-dependent translation; and that suppressing eIF4F-mediated activity 50% by genetic interventions is sufficient to promote apoptotic death in cancer cells while leaving non-transformed cells unharmed (69). In vivo, eIF4E ASO suppresses eIF4E-mediated translation by 60%. This intervention inhibits tumor growth in mice without any deleterious effects (30, 70). Detailed analysis of protein expression in these mice documents a selective decrease in the abundance of oncogenic proteins with minimal effect on physiological protein expression.
Why does eIF4F inhibition target cancer cells while sparing normal cell? Both in vitro and in vivo data presented here indicate that decreasing eIF4F-mediated translation in nonmalignant cells, including chemosensitive cells in the bone marrow and gastrointestinal epithelium, does not noticeably affect cell viability. In sharp contrast, normalizing cap-dependent translation in cancer cells results in apoptotic death. These findings show that when eIF4F switches from its inducible mode in nonmalignant cells to a constitutively up-regulated mode in cancer cells, it functions to defeat the apoptotic tumor surveillance pathway. Thus, cancer cells are extremely vulnerable to reversing aberrant activity of eIF4F because up-regulated eIF4F is an essential component of the strategy employed by cancer cells to disable the innate oncogene-triggered anticancer defense program.
Potential Loss of Anti-Oncogenic Feedback Loops Downstream of Deregulated eIF4F
One theoretical limitation of single-agent eIF4F inhibitors is activation of pro-oncogenic feedback/feedforward loops. Genome-wide analysis of transcripts recruited to ribosomes in response to eIF4F activation shows that coordinated activation of mRNAs encoding positive regulators of cell proliferation and inhibitors of apoptosis is accompanied by recruitment of ribosomes to transcripts encoding potential tumor suppressors (22). Decreasing eIF4F activity, therefore, might have the unintended consequence of attenuating homeostatic repression of some pro-survival and cell growth pathways. The role of signaling feedback loops in anti-eIF4F therapy remains a subject for future investigations by methodologies that examine ribosome recruitment genome-wide.
Clinical Implementations: Testing Therapies that Normalize Aberrant Translation
An important question that will arise in clinical trials is whether anti-translational interventions should be tested as single-agents or as one component of a multidrug regimen. While the ability of eIF4F inhibitors as monotherapy to suppress tumor growth in preclinical models is impressive, available information about mechanism indicates that the combination of eIF4F antagonists with already established treatment protocols might be the most promising option. This prediction is based on data indicating that formation of eIF4F determines cancer cell responsiveness to drugs targeting the EGF and HER2 receptors (71). As a result cells harboring hyperactivated eIF4F become vulnerable to apoptotic stimuli and to interruption of growth factor signaling. Indeed, available data show that inhibitors of cap-dependent translation exhibit synergy when combined with an inhibitor of the innate apoptosis antagonist BCL-2 (72) or with classical genotoxic therapies (50) such as gemcitabine (73) or 5-FU (63). Since the constitutive activity of eIF4F rescues cells from drug-induced apoptosis and triggers the PI3K/Akt/mTOR, Ras/Raf/MAPK and other classical oncogenic kinase cascades, an anti-eIF4F intervention should in principle sensitize cancers to conventional cytotoxic agents and to a wide range of targeted therapies. Data showing that suppression of eIF4F-mediated translation by the MNK kinase inhibitor CGP57380 overcomes leukemia cell resistance to the small-molecule tyrosine kinase inhibitor imatinib (74) supports this view. It remains to be seen whether such impressive anti-cancer activity shown in vitro and in preclinical in vivo models will be replicated in clinical trials.
Conclusion and Future Challenges
Despite improvements in cancer therapies, many cancers remain incurable. Although cultured primary human cells require only five or six discrete genetic alterations before acquiring tumorigenicity (75) genetic analysis of naturally occurring tumors reveals a high level of complexity. Despite this complexity, different cancers and even cancers of the same type manifest common features at the protein pathway level sharing a core group of about a dozen perturbed pathways that converge to several crucial regulatory hubs linking the intracellular signaling network with the basic metabolic machinery. This supports the view that cancer is a protein-network disease. The clear implication of this view for cancer therapy is that instead of targeting individual genetic alterations one-by-one, the next generation of cancer therapeutics will target regulatory hubs in the cancer protein-network.
Available evidence indicates that one such hub is eIF4F, which integrates multiple cancer-related pathways into a self-amplifying oncogenic system whose integrity and sustained activity are essential for tumorigenicity. The upstream mTORC1 node combines nutrient inputs with signals from mitogens, stressors, survival factors and energy. Its major effector eIF4F integrates this input with other pro-oncogenic inputs from the RAS/MAPK/ERK and MYC pathways to reprogram a cell towards cancer by selectively shifting which transcripts are recruited to ribosomes. This central integrative role of pathologically activated eIF4F in the oncogenic circuitry motivated the development of small molecule inhibitors of eIF4F as potential cancer therapies.
Hyperactivated eIF4F does not increase the translation of all mRNA uniformly; rather it selectively switches the translational repertoire of a cell towards malignancy. This occurs because the genes that are most dependent on the activity of eIF4F are those encoding regulators of cell proliferation, viability and adaptation; although the precise genes involved are different in each type of malignancy and can differ between drug sensitive and resistant subtypes of a given malignancy. In addition, different types of cancer will depend to a different degree on hyperactivated eIF4F for each of its neoplastic properties. As a result, therapies reversing aberrant eIF4F function will provide critical data linking changes in the genome-wide translational profile to therapeutic effect. In principle, such a genome-wide systems level profile could serve as a biomarker of therapeutic success or failure, greatly facilitating subsequent clinical trials. It could also provide mechanistic guidance to the selection of companion therapies that depend on the elimination of an eIF4F-dependent neoplastic function for their own efficacy. As new eIF4F inhibitors that act through different pharmacological mechanisms are developed, it is hoped that each will provide important new insights into the translational control of cancer; unveiling new vulnerabilities that can be exploited therapeutically.
Supplementary Material
{kind=link}
Acknowledgment
We thank members of the Bitterman’s and Polunovsky’s research group for the productive discussions and Svetlana Avdulov and Daniel Rossi for excellent technical assistance
Grant Support
Vitaly Polunovsky is supported by the NIH/NCI 3R01 CA-11338-05S1 and University of Minnesota Masonic Cancer Center Bridge Grant NIH/NHLBI. Peter Bitterman is supported by the NIH/NHLBI 1R01 HL08924
Financial Support: The NIH/NCI 3R01 CA-11338-05S1 and University of Minnesota Masonic Cancer Center Bridge Grant for V. Polunovsky and NIH/NHLBI 1R01 HL089249 for P. Bitterman
Footnotes
Disclosure of Potential Conflict Interests: No potential conflict interests were disclosed.
References
- 1.Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–7. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 2.Collins I, Workman P. New approaches to molecular cancer therapeutics. Nat Chem Biol. 2006;2:689–700. doi: 10.1038/nchembio840. [DOI] [PubMed] [Google Scholar]
- 3.Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009;8:709–23. doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]
- 4.Raguz S, Yague E. Resistance to chemotherapy: New treatments and novel insights into an old problem. Br J Cancer. 2008;99:387–91. doi: 10.1038/sj.bjc.6604510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 6.Stratton MR. Exploring the genomes of cancer cells: Progress and promise. Science. 2011;331:1553–8. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 7.Arteaga CL, O’Neill A, Moulder SL, Pins M, Sparano JA, Sledge GW, et al. A phase I-II study of combined blockade of the ErbB receptor network with trastuzumab and gefitinib in patients with HER2 (ErbB2)-overexpressing metastatic breast cancer. Clin Cancer Res. 2008;14:6277–83. doi: 10.1158/1078-0432.CCR-08-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polunovsky VA, Bitterman PB. The cap-dependent translation apparatus integrates and amplifies cancer pathways. RNA Biol. 2006;3:10–7. doi: 10.4161/rna.3.1.2718. [DOI] [PubMed] [Google Scholar]
- 9.Foulds L. The experimental study of tumor progression: A review. Cancer Res. 1954;14:327–39. [PubMed] [Google Scholar]
- 10.Renan MJ. How many mutations are required for tumorigenesis? implications from human cancer data. Mol Carcinog. 1993;7:139–46. doi: 10.1002/mc.2940070303. [DOI] [PubMed] [Google Scholar]
- 11.Weinstein IB. Cancer. addiction to oncogenes--the achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 12.Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145:30–8. doi: 10.1016/j.cell.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 13.Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH. Proof of concept: Network and systems biology approaches aid in the discovery of potent anticancer drug combinations. Mol Cancer Ther. 2010;9:3137–44. doi: 10.1158/1535-7163.MCT-10-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bitterman P, Polunovsky VA. Translational control of cancer: Imlpications for targeted therapy. In: Polunovsky VA, Houghton P, editors. mTOR Pathway and mTOR Inhibitors in Cancer Therapy. Humana Press; Springer: 2010. pp. 237–55. [Google Scholar]
- 15.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–66. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 16.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zoncu R, Efeyan A, Sabatini DM. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi B, Cai AL, Keiper BD, Minich WB, Mendez R, Beach CM, et al. Phosphorylation of eukaryotic protein synthesis initiation factor 4E at ser-209. J Biol Chem. 1995;270:14597–603. doi: 10.1074/jbc.270.24.14597. [DOI] [PubMed] [Google Scholar]
- 19.Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol. 1999;19:1871–80. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruggero D. The role of myc-induced protein synthesis in cancer. Cancer Res. 2009;69:8839–43. doi: 10.1158/0008-5472.CAN-09-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 22.Larsson O, Li S, Issaenko OA, Avdulov S, Peterson M, Smith K, et al. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814–24. doi: 10.1158/0008-5472.CAN-07-0752. [DOI] [PubMed] [Google Scholar]
- 23.Mamane Y, Petroulakis E, Martineau Y, Sato TA, Larsson O, Rajasekhar VK, et al. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS One. 2007;2:e242. doi: 10.1371/journal.pone.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh AC, Ruggero D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin Cancer Res. 2010;16:4914–20. doi: 10.1158/1078-0432.CCR-10-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 26.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–7. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- 27.Kim YY, Von Weymarn L, Larsson O, Fan D, Underwood JM, Peterson MS, et al. Eukaryotic initiation factor 4E binding protein family of proteins: Sentinels at a translational control checkpoint in lung tumor defense. Cancer Res. 2009;69:8455–62. doi: 10.1158/0008-5472.CAN-09-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell. 2004;5:553–63. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 29.Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SM, Wendel HG, et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest. 2008;118:2651–60. doi: 10.1172/JCI34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007;117:2638–48. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–67. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 32.Fan S, Ramalingam SS, Kauh J, Xu Z, Khuri FR, Sun SY. Phosphorylated eukaryotic translation initiation factor 4 (eIF4E) is elevated in human cancer tissues. Cancer Biol Ther. 2009;8:1463–9. doi: 10.4161/cbt.8.15.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Topisirovic I, Ruiz-Gutierrez M, Borden KL. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res. 2004;64:8639–42. doi: 10.1158/0008-5472.CAN-04-2677. [DOI] [PubMed] [Google Scholar]
- 34.Koromilas AE, Lazaris-Karatzas A, Sonenberg N. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. Embo J. 1992;11:4153–8. doi: 10.1002/j.1460-2075.1992.tb05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–61. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mavrakis KJ, Wendel HG. Translational control and cancer therapy. Cell Cycle. 2008;7:2791–4. doi: 10.4161/cc.7.18.6683. [DOI] [PubMed] [Google Scholar]
- 37.Pelletier J, Graff JR. mTOR Pathway and mTOR Inhibitors in Cancer Therapy. Humana Press; Springer: 2010. Downstream from mTOR: Theraputic approachs to targeting the eIF4E translation initiation complex; pp. 257–285. [Google Scholar]
- 38.Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 39.van der Heijden MS, Bernards R. Inhibition of the PI3K pathway: Hope we can believe in? Clin Cancer Res. 2010;16:3094–9. doi: 10.1158/1078-0432.CCR-09-3004. [DOI] [PubMed] [Google Scholar]
- 40.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–12. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 41.Lin CJ, Robert F, Sukarieh R, Michnick S, Pelletier J. The antidepressant sertraline inhibits translation initiation by curtailing mammalian target of rapamycin signaling. Cancer Res. 2010;70:3199–208. doi: 10.1158/0008-5472.CAN-09-4072. [DOI] [PubMed] [Google Scholar]
- 42.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–20. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Downward J. Targeting RAF: Trials and tribulations. Nat Med. 2011;17:286–8. doi: 10.1038/nm0311-286. [DOI] [PubMed] [Google Scholar]
- 46.Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–7. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011;71:1849–57. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- 48.She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan S, Li Y, Yue P, Khuri FR, Sun SY. The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia. 2010;12:346–56. doi: 10.1593/neo.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011;108:1046–51. doi: 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, et al. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell. 2005;20:709–22. doi: 10.1016/j.molcel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 52.Korneeva NL. Translational dysregulation by pateamine A. Chem Biol. 2007;14:5–7. doi: 10.1016/j.chem-biol.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim S, Hwang BY, Su BN, Chai H, Mi Q, Kinghorn AD, et al. Silvestrol, a potential anticancer rocaglate derivative from aglaia foveolata, induces apoptosis in LNCaP cells through the mitochondrial/apoptosome pathway without activation of executioner caspase-3 or -7. Anticancer Res. 2007;27:2175–83. [PMC free article] [PubMed] [Google Scholar]
- 54.Hong DS, Kurzrock R, Oh Y, Wheler JJ, Naing A, Brail L, et al. A phase 1 dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clin Cancer Res. 2011;17:6582–91. doi: 10.1158/1078-0432.CCR-11-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pettersson F, Yau C, Dobocan MC, Culjkovic-Kraljacic B, Retrouvay H, Puckett R, et al. Ribavirin treatment effects on breast cancers overexpressing eIF4E, a biomarker with prognostic specificity for luminal B-type breast cancer. Clin Cancer Res. 2011;17:2874–84. doi: 10.1158/1078-0432.CCR-10-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Westman B, Beeren L, Grudzien E, Stepinski J, Worch R, Zuberek J, et al. The antiviral drug ribavirin does not mimic the 7-methylguanosine moiety of the mRNA cap structure in vitro. Rna. 2005;11:1505–13. doi: 10.1261/rna.2132505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan Y, Svitkin Y, Lee JM, Bisaillon M, Pelletier J. Ribavirin is not a functional mimic of the 7-methyl guanosine mRNA cap. Rna. 2005;11:1238–44. doi: 10.1261/rna.2930805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh P, Park C, Peterson MS, Bitterman PB, Polunovsky VA, Wagner CR. Synthesis and evaluation of potential inhibitors of eIF4E cap binding to 7-methyl GTP. Bioorg Med Chem Lett. 2005;15:2177–80. doi: 10.1016/j.bmcl.2005.01.080. [DOI] [PubMed] [Google Scholar]
- 59.Grudzien E, Stepinski J, Jankowska-Anyszka M, Stolarski R, Darzynkiewicz E, Rhoads RE. Novel cap analogs for in vitro synthesis of mRNAs with high translational efficiency. Rna. 2004;10:1479–87. doi: 10.1261/rna.7380904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghosh B, Benyumov AO, Ghosh P, Jia Y, Avdulov S, Dahlberg PS, et al. Nontoxic chemical interdiction of the epithelial-to-mesenchymal transition by targeting cap-dependent translation. ACS Chem Biol. 2009;4:367–77. doi: 10.1021/cb9000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagner CR, Chang SL, Griesgraber GW, Song H, McIntee EJ, Zimmerman CL. Antiviral nucleoside drug delivery via amino acid phosphoramidates. Nucleosides Nucleotides. 1999;18:913–9. doi: 10.1080/15257779908041599. [DOI] [PubMed] [Google Scholar]
- 62.Chou TF, Baraniak J, Kaczmarek R, Zhou X, Cheng J, Ghosh B, et al. Phosphoramidate pronucleotides: A comparison of the phosphoramidase substrate specificity of human and escherichia coli histidine triad nucleotide binding proteins. Mol Pharm. 2007;4:208–17. doi: 10.1021/mp060070y. [DOI] [PubMed] [Google Scholar]
- 63.Jia Y, Chiu TL, Amin EA, Polunovsky V, Bitterman PB, Wagner CR. Design, synthesis and evaluation of analogs of initiation factor 4E (eIF4E) cap-binding antagonist Bn7-GMP. Eur J Med Chem. 2010;45:1304–13. doi: 10.1016/j.ejmech.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Borok Z, Whitsett JA, Bitterman PB, Thannickal VJ, Kotton DN, Reynolds SD, et al. Proc Am Thorac Soc; Cell plasticity in lung injury and repair: Report from an NHLBI workshop; april 19-20, 2010; 2011. pp. 215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rajasekhar VK, Holland EC. Postgenomic global analysis of translational control induced by oncogenic signaling. Oncogene. 2004;23:3248–64. doi: 10.1038/sj.onc.1207546. [DOI] [PubMed] [Google Scholar]
- 66.Larsson O, Bitterman PV. Genome-wide analysis of translational control. In: mTOR Pathway and mTOR Inhibitors in Cancer Therapy. Humana Press; Springer: 2010. pp. 217–36. [Google Scholar]
- 67.Provenzani A, Fronza R, Loreni F, Pascale A, Amadio M, Quattrone A. Global alterations in mRNA polysomal recruitment in a cell model of colorectal cancer progression to metastasis. Carcinogenesis. 2006:27–1323. doi: 10.1093/carcin/bgi377. [DOI] [PubMed] [Google Scholar]
- 68.Gunnery S, Maivali U, Mathews MB. Translation of an uncapped mRNA involves scanning. J Biol Chem. 1997;272:21642–6. doi: 10.1074/jbc.272.34.21642. [DOI] [PubMed] [Google Scholar]
- 69.Li BD, Gruner JS, Abreo F, Johnson LW, Yu H, Nawas S, et al. Prospective study of eukaryotic initiation factor 4E protein elevation and breast cancer outcome. Ann Surg. 2002;235:732–8. doi: 10.1097/00000658-200205000-00016. discussion 738-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soni A, Akcakanat A, Singh G, Luyimbazi D, Zheng Y, Kim D, et al. eIF4E knockdown decreases breast cancer cell growth without activating akt signaling. Mol Cancer Ther. 2008;7:1782–8. doi: 10.1158/1535-7163.MCT-07-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zindy P, Berge Y, Allal B, Filleron T, Pierredon S, Cammas A, et al. Formation of the eIF4F translation-initiation complex determines sensitivity to anticancer drugs targeting the EGFR and HER2 receptors. Cancer Res. 2011;71:4068–73. doi: 10.1158/0008-5472.CAN-11-0420. [DOI] [PubMed] [Google Scholar]
- 72.Cencic R, Carrier M, Trnkus A, A PJ, Jr, Minden M, Pelletier J. Synergistic effect of inhibiting translation initiation in combination with cytotoxic agents in acute myelogenous leukemia cells. Leuk Res. 2010;34:535–41. doi: 10.1016/j.leukres.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobson BA, Alter MD, Kratzke MG, Frizelle SP, Zhang Y, Peterson MS, et al. Repression of cap-dependent translation attenuates the transformed phenotype in non-small cell lung cancer both in vitro and in vivo. Cancer Res. 2006;66:4256–62. doi: 10.1158/0008-5472.CAN-05-2879. [DOI] [PubMed] [Google Scholar]
- 74.Zhang M, Fu W, Prabhu S, Moore JC, Ko J, Kim JW, et al. Inhibition of polysome assembly enhances imatinib activity against chronic myelogenous leukemia and overcomes imatinib resistance. Mol Cell Biol. 2008;28:6496–509. doi: 10.1128/MCB.00477-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao JJ, Roberts TM, Hahn WC. Functional genetics and experimental models of human cancer. Trends Mol Med. 2004;10:344–50. doi: 10.1016/j.molmed.2004.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.