

Abstract
Post-translational modifications of amino acids can be used to generate novel cofactors capable of chemistries inaccessible to conventional amino acid side chains. The biosynthesis of these sites often requires one or more enzyme or protein accessory factors, the functions of which are quite diverse and often difficult to isolate in cases where multiple enzymes are involved. Herein is described the current knowledge of the biosynthesis of urease and nitrile hydratase metal centers, pyrroloquinoline quinone, hypusine, and tryptophan tryptophylquinone cofactors along with the most recent work elucidating the functions of individual accessory factors in these systems. These examples showcase the breadth and diversity of this continually expanding field.
Introduction
The role of post-translational modifications as regulatory or cell localization strategies has long been recognized. However, the observation that amino acids can be modified to generate cofactors with novel functions is relatively recent [1]. In some cases, the biosynthesis of these cofactors is autocatalytic, requiring only the proper protein fold and perhaps a second cofactor such as a heme or metal ion to initiate amino acid modification and complete cofactor formation. A well known example of such autocatalytic synthesis comes from the copper amine oxidases, where generation of the topaquinone (TPQ) cofactor from a Tyr residue requires only copper and oxygen [2]. In other cases, one or more accessory proteins are required for cofactor maturation. This review discusses some interesting examples of such systems and the recent advances in understanding the enzymes which generate these protein-derived cofactors.
Modified Amino Acid Ligands to Metal Cofactors: Urease and Nitrile Hydratase
Urease and nitrile hydratase are metalloproteins with post-translationally modified amino acid ligands to the metal(s) at the active site (Figure 1). In urease, a dinuclear Ni center is coordinated by a bridging carbamylated Lys residue [3]. Active site maturation in urease requires typically four accessory proteins (UreDEFG) as well as Ni2+, CO2 (derived from carbonate), and GTP. Despite intensive study over a number of years, the specific roles of these proteins in urease activation remain elusive. UreE is thought to function as a metallochaperone and UreG as a GTPase. GTP hydrolysis by UreG has been proposed to either cause a conformational change allowing access of Ni and CO2 to the urease active site, or to generate carboxyphosphate as a CO2 donor to the active site lysine residue. Less is known about the other accessory proteins. However, successful methods for the soluble expression of UreD (as a maltose binding protein fusion) [4] and a truncated UreF [5] were only recently achieved and may lead to significant advances in this field. The activation of Rubisco provides some interesting parallels to the urease system in that it also requires carbamylation of a lysine residue [6] in order to bind Mg2+ in the active site [7]. Here again an “activase” protein is involved, although it is not believed to participate directly in Lys carbamylation [8], which appears to occur spontaneously.
Figure 1.

Proposed pathways of cofactor biogenesis for Ni-urease, Co-nitrile hydratase, pyrroloquinoline quinone (PQQ), hypusine, and tryptophan tryptophylquinone (TTQ). Post-translational modifications made at each step are shown in red.
Low-molecular-mass nitrile hydratase (L-NHase) utilizes a mononuclear Co site coordinated by two oxidized Cys residues, one Cys-sulfenic acid (-SOH) and one Cys-sulfinic acid (-SO2H) [9]. Expression of the structural genes for the L-NHase α2β2 heterotetramer (nhlAB), in the absence of the downstream activator gene nhlE resulted in a protein with very little activity, low Co content, and no Cys-sulfinic acid (-SO2H) in the α-subunit [10]. Co-expression of nhlA with nhlE yields a trimeric complex (holo-αe2) which contains Co and modified Cys residues in the α-subunit. It was further demonstrated in vitro that NhlE was responsible for Co insertion and Cys oxidation [11]. The holo-αe2 complex is able to activate apo-α2β2 by a novel mechanism dubbed “self-subunit swapping” (Figure 2), where two holo-αe2 complexes exchange α-subunits with apo-α2β2 forming active holo-α2β2 and apo-αe2 [10,11]: this mechanism also holds true for the high-molecular-mass nitrile hydratase (H-NHase) [12]. The driving force for the exchange appears to be the formation of a salt bridge between two conserved Arg residues of the β-subunit and the negatively charged Cys-SO2− and Cys-SO− residues of the holo-α-subunit [10]. NhlE was consequently designated as the first self-subunit swapping chaperone as well as a metallochaperone. However, the sequence of NhlE has no known metal binding motifs and metal coordination by NhlE has never been shown directly. Thus, the question of how NhlE facilitates Co insertion and Cys oxidation in the α-subunit remains unclear.
Figure 2.
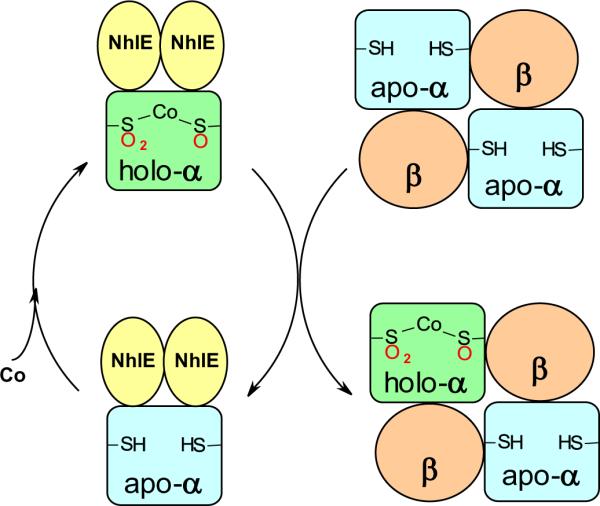
Proposed mechanism of nitrile hydratase active site maturation by “self-subunit swapping”.
The Pyrroloquinoline Quinone Cofactor
Pyrroloquinoline quinone (PQQ) biosynthesis can be viewed as an unusual case of posttranslational modification that generates a stand-alone cofactor primarily utilized by bacterial alcohol and sugar dehydrogenases. The cofactor is synthesized from Tyr and Glu residues within a peptide encoded by pqqA (Figure 1). In Klebsiella pneumonia five additional genes (pqqB–F) are required beyond pqqA [13]. Of these gene products, PqqB is homologous to β-lactamases, and a crystal structure is available (PDB ID: 3JXP): it is thought to be a PQQ carrier, as some PQQ synthesis occurs in its absence [13]. The protein contains a conserved zinc-finger motif, but has undergone no in vitro functional studies at this time, and so the role of this and other conserved residues waits to be elucidated. The protein PqqF is thought to be a protease that may be involved in excising a cross-linked Tyr-Glu intermediate from the PqqA peptide.
Earlier work primarily focused on PqqC; the final enzyme in the pathway [14]. PqqC is a cofactorless oxidase and catalyzes the final ring closure and an 8-electron oxidation to generate PQQ. Using a combination of kinetics, site-directed mutagenesis and crystal structures, current studies have broadly begun to define the roles of individual amino acids, and an observed open and closed conformation [15]. Three equivalents of O2 are required for a single turnover producing two equivalents of H2O2, indicating that one equivalent of H2O2 produced by the enzyme is used as an electron acceptor during PQQ synthesis [14]. The activation of O2 in the absence of a metal or cofactor is of particular interest. Diatomic electron density was observed bound by a group of conserved amino acids [16] at a distance of 3.4 Å from a carbon of a trapped quinoid or quinol reaction intermediate [15].
The enzyme PqqD has engendered the most interest as it has no homology to any characterized protein, and its crystal structure was recently determined (PDB ID: 3G2B) [17]. Initially the focus was on its assumed interaction with PqqC, as it is found as a pqqCD fusion in some organisms [18]. However, recently, an interaction has been determined with PqqE [19], a radical S-adenosyl-L-methionine (SAM) enzyme containing two [4Fe4S]2+ clusters [20]. PqqE is highly sensitive to O2, with catalytic SAM cleavage to generate the 5'-deoxyadenosine radical only occurring in PqqE that has been expressed and purified under anaerobic conditions [20]. The addition of PqqD to anaerobic PqqE in the presence of SAM leads to an increase in α-helical content compared to the individual proteins, and changes in the electronic environment of the [4Fe4S]2+ clusters indicating a specific interaction between the two proteins [19]. It has been proposed that PqqE may be the first enzyme in the pathway catalyzing C-C bond formation, but thus far no reactivity with PqqA has been reported. Perhaps one of the most intriguing mysteries is the incongruity that PqqD binds to both a strictly anaerobic PqqE and an O2-dependent PqqC.
The Hypusine Residue of eIF5A
Eukaryotic translational initiation factor 5A (eIF5A) is conserved among eukaryotes and is involved in cell proliferation. Although initially described as a translation initiation factor, eIF5A is now considered to be primarily involved in translation elongation [21]. However, the transcripts which rely upon it for efficient translation and its exact role in the cell cycle have remained elusive. It has been established that eIF5A contains a unique and essential post-translational modification of a specific lysine residue to form hypusine in two enzymatic steps (Figure 1) [22, 23]. The first step involves the formation of deoxyhypusine from spermidine and the eIF5A precursor lysine and is catalyzed by deoxyhypusine synthase (DHS). The second step is catalyzed by deoxyhypusine hydroxylase (DOHH) and involves hydroxylation of the deoxyhypusine intermediate to give hypusine. DHS has been structurally characterized and the mechanism of deoxyhypusine formation is well understood. In contrast, little is known about DOHH. Homology modeling and mutational analysis suggest a structure with two arms separated by a flexible linker, with each arm composed of 4 consecutive α-hairpin repeats known as Huntingtin Elongation factor 3 protein phosphatase 2A TOR1 (HEAT) motifs: a motif consisting of two α-helices separated by a nonhelical region. Conserved His and Glu residues on each arm coordinate a bridging diiron center [22]. Consistent with this, native gel electrophoresis and SAXS show a more compact conformation for Fe-bound versus apo-DOHH [24]. Although many of the bacterial diiron monoxygenases also utilize a His/Glu coordinated diiron center [25], DOHH appears to be structurally and sequentially unrelated to these enzymes.
Recombinant expression and purification of human DOHH yields a blue solution. Extensive spectroscopic characterization including EPR, Mössbauer, resonance Raman, and X-ray absorption spectroscopy confirmed the identity of the blue species as a (μ-1,2-peroxo)diiron(III) complex (Figure 3) [26]. The complex can be chemically reduced by dithionite and regenerated by exposure to air. Most importantly, incubation of the peroxo species with deoxyhypusine-containing eIF5A leads to nearly stoichiometric formation of mature, hypusine-containing eIF5A.
Figure 3.
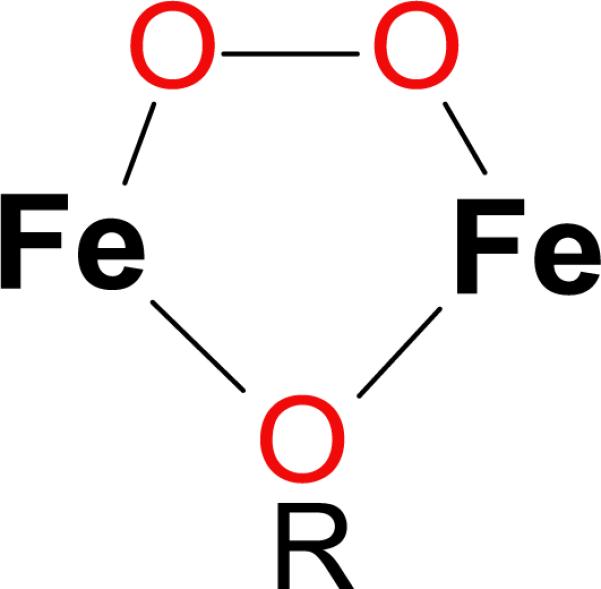
Proposed structure of the diiron-peroxo species of deoxyhypusine hydroxylase. The R reflects that the single atom bridging ligand is not expected to be an oxo bridge, but its exact identity is unknown.
DOHH is the first example of a eukaryotic hydroxylase capable of oxygen activation at a non-heme diiron site. Of particular interest is the remarkable stability of the peroxo species of DOHH, which persists for days whereas other diiron(III)-peroxo complexes decay with half-lives on the order of milliseconds to minutes [27]. EXAFS results suggest that the source of this enhanced stability may be a single atom bridging ligand between the Fe atoms [26]. Substrate addition to peroxo-DOHH enhances its decay rate and results in substrate hydroxylation, suggesting that substrate binding causes a conformational change at the active site, facilitating O-O bond cleavage and substrate oxidation. Tantalizingly, the slow rate of activity observed in vitro may imply that there are other accessory factors required to maximize the reaction rate.
Tryptophan Tryptophylquinone
Tryptophan tryptophylquinone (TTQ) is an in situ cofactor generated from two tryptophans within the polypeptide (Figure 1) [1,28]. It is found in the active site of the bacterial enzymes methylamine dehydrogenase (MADH) and aromatic amine dehydrogenase. The synthesis of TTQ has been studied in the Paracoccus denitrificans MADH system, and involves the βTrp57 and βTrp108 of the α2β2 MADH tetramer. Four gene products are required for maturation of MADH, and thus far only the final enzyme responsible for TTQ biosynthesis, MauG, has been studied. MauG is a di-heme c enzyme that catalyzes a 6-electron oxidation to complete formation of TTQ in an O2 + reducing equivalents or H2O2-dependent reaction [1,28]. The MADH substrate for MauG (preMADH) has a partially assembled cofactor (preTTQ) consisting of a monohydroxylated βTrp57 at C7 of the indole ring. The enzyme responsible for the addition of this initial −OH group is currently unknown. The reaction consists of three MauG-dependent 2-electron events: (1) cross-link formation between βTrp57 and βTrp108, (2) hydroxylation of βTrp57 C6 to form quinol, and (3) oxidation of quinol to quinone. The order of these events remains to be determined. MauG catalysis is highly unusual for a number of reasons: (1) it contains a c-type heme that can bind and activate O2, (2) the two hemes act in concert as a single redox cofactor, and (3) the catalytic MauG oxidant is a unique bis-Fe(IV) species consisting of ferryl heme (Fe(IV)=O) and the second heme as six-coordinate Fe(IV) [29,30].
The crystal structure of MauG in complex with preMADH (PDB ID: 3L4M), and the finding that the crystals were catalytically active, has been the cornerstone of recent work (Figure 4) [31]. This structure demonstrated that MauG did not need to dissociate between the three 2-electron events, and that no significant structural rearrangements were required. The fold of preMADH was essentially that of the mature enzyme, and the βTrp108 and monohydroxylated βTrp57 of preTTQ were at the same site as mature TTQ. Neither residue was in direct contact with either heme of MauG: the closest atom of preTTQ being 15.5 and 37.5 Å from each of the porphyrin macrocycles. Therefore, oxidation of preTTQ to TTQ involves long-range electron transfer. The kinetics of the first 2-electron cycle [32] and the MauG-dependent oxidation of dithionite-reduced MADH (quinol to quinone) [33] have recently been reported. One of the kinetic surprises was that the reaction was random order: the potent bis-Fe(IV) species could form in the absence of preMADH [29,32] and is extremely long-lived for one that has the potential to cause significant non-specific oxidative damage (decay to diferric MauG being 0.0002 s−1) [32]. Indeed, cycles of formation and decay of the bis-Fe(IV) species in the absence of preMADH leads to suicide inactivation [34]. The site of H2O2 and O2 binding was shown to be the heme furthest from preMADH [31,35,36]. The heme closest to preMADH has an unusual His/Tyr axial ligation required for bis-Fe(IV) formation [37]. MauG Trp199 at the interface with preMADH was shown to be critical for TTQ synthesis, although formation of the bis-Fe(IV) was unimpaired [38]. W199F and W199K MauG mutants did not support initiation of TTQ biosynthesis, implicating a MauG-dependent hopping mechanism that requires formation of a Trp199 radical.
Figure 4.

Position of hemes and residues relevant to TTQ synthesis in the crystal structure of MauG-preMADH. MauG is shown in pink cartoon, the α and β subunits of preMADH are shown in blue and green, respectively. β-preMADH Trp57 and Trp108 and MauG Trp199, Trp93, hemes and heme ligands are drawn in stick form. Figure produced using Pymol (http://www.pymol.org).
Concluding Remarks
The number and diversity of cofactors derived from post-translational modification of protein residues is considerable and continually expanding. Here we have touched upon a few examples that showcase recent advances in our understanding of their biosynthesis. As the field matures, new cofactors and systems will surely come to light, highlighting new ways in which nature has expanded the chemistry available to the 20 conventional amino acids.
Highlights
Maturation of metal centers with modified amino acid ligands; urease and nitrile hydratase
Biosynthesis of pyrroloquinoline quinone from a peptide precursor
Spectroscopic characterization of an enzyme required for hypusine synthesis
Long range electron transfer is required for tryptophan tryptophylquinone biosynthesis
Acknowledgements
This work was supported by National Institutes of Health grants GM-66569 (CMW) and GM-97779 (ETY). Computing was provided by the Minnesota Supercomputing Institute for Advanced Computational Research.
ABBREVIATIONS
- TPQ
topaquinone
- Rubisco
Ribulose-1,5-bisphosphate carboxylase oxygenase
- L-NHase
low-molecular-mass nitrile hydratase
- H-NHase
high-molecular-mass nitrile hydratase
- PQQ
pyrroloquinoline quinone
- SAM
S-adenosyl methionine
- eIF5A
eukaryotic translation initiation factor 5A
- DHS
deoxyhypusine synthase
- DOHH
deoxyhypusine hydroxylase
- HEAT
(Huntingtin Elongation factor 3 protein phosphatase 2A TOR1) a motif consisting of two α-helices separated by a non-helical region
- SAXS
small angle x-ray scattering
- EPR
electron paramagnetic resonance
- EXAFS
extended x-ray absorption fine structure
- TTQ
tryptophan tryptophyl quinone
- MADH
methylamine dehydrogenase
- preMADH
biosynthetic precursor protein of MADH with incompletely synthesized TTQ (preTTQ)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ••1.Davidson VL. Generation of protein-derived redox cofactors by posttranslational modification. Molecular BioSystems. 2011;7:29–37. doi: 10.1039/c005311b. [DOI] [PubMed] [Google Scholar]; This recent review is a great starting point for those wanting a general survey of the field, rather than the current review which focuses on the most recent developments.
- 2.Dubois JL, Klinman JP. Mechanism of post-translational quinone formation in copper amine oxidases and its relationship to the catalytic turnover. Arch Biochem Biophys. 2005;433:255–265. doi: 10.1016/j.abb.2004.08.036. [DOI] [PubMed] [Google Scholar]
- •3.Kaluarachchi H, Chan Chung KC, Zamble DB. Microbial nickel proteins. Nat Prod Rep. 2010;27:681–694. doi: 10.1039/b906688h. [DOI] [PubMed] [Google Scholar]; This is an excellent review of bacterial nickel proteins including a concise description of the individual accessory factors required for urease activation.
- 4.Carter EL, Hausinger RP. Characterization of the Klebsiella aerogenes urease accessory protein UreD in fusion with the maltose binding protein. J Bacteriol. 2010;192:2294–2304. doi: 10.1128/JB.01426-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lam R, Romanov V, Johns K, Battaile KP, Wu-Brown J, Guthrie JL, Hausinger RP, Pai EF, Chirgadze NY. Crystal structure of a truncated urease accessory protein UreF from Helicobacter pylori. Proteins. 2010;78:2839–2848. doi: 10.1002/prot.22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lorimer GH, Miziorko HM. Carbamate formation on the epsilon-amino group of a lysyl residue as the basis for the activation of ribulosebisphosphate carboxylase by CO2 and Mg2+ Biochemistry. 1980;19:5321–5328. doi: 10.1021/bi00564a027. [DOI] [PubMed] [Google Scholar]
- 7.Andersson I, Knight S, Schneider G, Lindqvist Y, Lundqvist T, Branden C-I, Lorimer GH. Crystal structure of the active site of ribulose-bisphosphate carboxylase. Nature. 1989;337:229–234. [Google Scholar]
- 8.Portis AR., Jr. Rubisco activase. Biochim Biophys Acta. 1990;1015:15–28. doi: 10.1016/0005-2728(90)90211-l. [DOI] [PubMed] [Google Scholar]
- 9.Prasad S, Bhalla TC. Nitrile hydratases (NHases): at the interface of academia and industry. Biotechnol Adv. 2010;28:725–741. doi: 10.1016/j.biotechadv.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Z, Hashimoto Y, Shiraki K, Kobayashi M. Discovery of posttranslational maturation by self-subunit swapping. Proc Natl Acad Sci U S A. 2008;105:14849–14854. doi: 10.1073/pnas.0803428105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •11.Zhou Z, Hashimoto Y, Kobayashi M. Self-subunit swapping chaperone needed for the maturation of multimeric metalloenzyme nitrile hydratase by a subunit exchange mechanism also carries out the oxidation of the metal ligand cysteine residues and insertion of cobalt. J Biol Chem. 2009;284:14930–14938. doi: 10.1074/jbc.M808464200. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that NhlE is responsible for Co insertion and Cys oxidation of the alpha subunit of L-NHase, demonstrating a metallochaperone and redox function for this protein as well as the unusual subunit swapping mechanism.
- 12.Zhou Z, Hashimoto Y, Cui T, Washizawa Y, Mino H, Kobayashi M. Unique biogenesis of high-molecular mass multimeric metalloenzyme nitrile hydratase: intermediates and a proposed mechanism for self-subunit swapping maturation. Biochemistry. 2010;49:9638–9648. doi: 10.1021/bi100651v. [DOI] [PubMed] [Google Scholar]
- 13.Puehringer S, Metlitzky M, Schwarzenbacher R. The pyrroloquinoline quinone biosynthesis pathway revisited: a structural approach. BMC Biochem. 2008;9:8. doi: 10.1186/1471-2091-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magnusson OT, Toyama H, Saeki M, Rojas A, Reed JC, Liddington RC, Klinman JP, Schwarzenbacher R. Quinone biogenesis: Structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc Natl Acad Sci U S A. 2004;101:7913–7918. doi: 10.1073/pnas.0402640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puehringer S, RoseFigura J, Metlitzky M, Toyama H, Klinman JP, Schwarzenbacher R. Structural studies of mutant forms of the PQQ-forming enzyme PqqC in the presence of product and substrate. Proteins. 2010;78:2554–2562. doi: 10.1002/prot.22769. [DOI] [PubMed] [Google Scholar]
- 16.RoseFigura JM, Puehringer S, Schwarzenbacher R, Toyama H, Klinman JP. Characterization of a protein-generated O2 binding pocket in PqqC, a cofactorless oxidase catalyzing the final step in PQQ production. Biochemistry. 2011;50:1556–1566. doi: 10.1021/bi1015474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai TY, Yang CY, Shih HL, Wang AH, Chou SH. Xanthomonas campestris PqqD in the pyrroloquinoline quinone biosynthesis operon adopts a novel saddle-like fold that possibly serves as a PQQ carrier. Proteins. 2009;76:1042–1048. doi: 10.1002/prot.22461. [DOI] [PubMed] [Google Scholar]
- 18.Toyama H, Nishibayashi E, Saeki M, Adachi O, Matsushita K. Factors required for the catalytic reaction of PqqC/D which produces pyrroloquinoline quinone. Biochem Biophys Res Commun. 2007;354:290–295. doi: 10.1016/j.bbrc.2007.01.001. [DOI] [PubMed] [Google Scholar]
- •19.Wecksler SR, Stoll S, Iavarone AT, Imsand EM, Tran H, Britt RD, Klinman JP. Interaction of PqqE and PqqD in the pyrroloquinoline quinone (PQQ) biosynthetic pathway links PqqD to the radical SAM superfamily. Chem Commun (Camb) 2010;46:7031–7033. doi: 10.1039/c0cc00968g. [DOI] [PubMed] [Google Scholar]; This study hints at the probable complexity of PqqD's role as the field tries to identify the first enzyme in the PQQ biosynthetic pathway.
- 20.Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu SP, King D, Britt RD, Klinman JP. Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-L-methionine enzyme. Biochemistry. 2009;48:10151–10161. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saini P, Eyler DE, Green R, Dever TE. Hypusine-containing protein eIF5A promotes translation elongation. Nature. 2009;459:118–121. doi: 10.1038/nature08034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park MH. The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A) J Biochem. 2006;139:161–169. doi: 10.1093/jb/mvj034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •23.Park MH, Nishimura K, Zanelli CF, Valentini SR. Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids. 2010;38:491–500. doi: 10.1007/s00726-009-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this review, a number of deletion and mutation studies involving eIF5A as well as DHS and DOHH are summarized, defining the role of eIF5A and its hypusine modification in cell growth and protein synthesis.
- 24.Cano VS, Medrano FJ, Park MH, Valentini SR. Evidence for conformational changes in the yeast deoxyhypusine hydroxylase Lia1 upon iron displacement from its active site. Amino Acids. 2010;38:479–490. doi: 10.1007/s00726-009-0407-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leahy JG, Batchelor PJ, Morcomb SM. Evolution of the soluble diiron monooxygenases. FEMS Microbiol Rev. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- ••26.Vu VV, Emerson JP, Martinho M, Kim YS, Munck E, Park MH, Que L., Jr. Human deoxyhypusine hydroxylase, an enzyme involved in regulating cell growth, activates O2 with a nonheme diiron center. Proc Natl Acad Sci U S A. 2009;106:14814–14819. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides the first extensive spectroscopic characterization of DOHH, revealing a diiron-peroxo complex of unprecedented stability. Further, this enzyme is active toward its deoxyhypusine eIF5A substrate, demonstrating the relevance of the peroxo complex to the catalytic cycle of DOHH
- •27.Krebs C, Bollinger JM, Jr., Booker SJ. Cyanobacterial alkane biosynthesis further expands the catalytic repertoire of the ferritin-like 'di-iron-carboxylate' proteins. Curr Opin Chem Biol. 2011;15:291–303. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review gives a brief yet comprehensive description of the so-called carboxylate di-iron proteins and the mechanism of oxygen activation in these enzymes, before going on to describe potential new members of this family.
- 28.Wilmot CM, Davidson VL. Uncovering novel biochemistry in the mechanism of tryptophan tryptophylquinone cofactor biosynthesis. Curr Opin Chem Biol. 2009;13:469–474. doi: 10.1016/j.cbpa.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Fu R, Lee S, Krebs C, Davidson VL, Liu A. A catalytic di-heme bis-Fe(IV) intermediate, alternative to an Fe(IV)=O porphyrin radical. Proc Natl Acad Sci U S A. 2008;105:8597–8600. doi: 10.1073/pnas.0801643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ling Y, Davidson VL, Zhang Y. Unprecedented Fe(IV) Species in a Diheme Protein MauG: A Quantum Chemical Investigation on the Unusual Mössbauer Spectroscopic Properties. J Phys Chem Lett. 2010;1:2936–2939. doi: 10.1021/jz101159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••31.Jensen LM, Sanishvili R, Davidson VL, Wilmot CM. In crystallo posttranslational modification within a MauG/pre-methylamine dehydrogenase complex. Science. 2010;327:1392–1394. doi: 10.1126/science.1182492. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports the crystal structure of the TTQ biosynthesis enzyme MauG in complex with its protein substrate, and the demonstration that the complex is fully active in the crystal. The surprise was the realization that the 6-electron oxidation is taking place via an activated oxygen species that is ~ 40 Å distant from the site of TTQ formation.
- 32.Lee S, Shin S, Li X, Davidson VL. Kinetic mechanism for the initial steps in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2009;48:2442–2447. doi: 10.1021/bi802166c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin S, Abu Tarboush N, Davidson VL. Long-range electron transfer reactions between hemes of MauG and different forms of tryptophan tryptophylquinone of methylamine dehydrogenase. Biochemistry. 2010;49:5810–5816. doi: 10.1021/bi1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin S, Lee S, Davidson VL. Suicide inactivation of MauG during reaction with O2 or H2O2 in the absence of its natural protein substrate. Biochemistry. 2009;48:10106–10112. doi: 10.1021/bi901284e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu R, Liu F, Davidson VL, Liu A. Heme iron nitrosyl complex of MauG reveals an efficient redox equilibrium between hemes with only one heme exclusively binding exogenous ligands. Biochemistry. 2009;48:11603–11605. doi: 10.1021/bi9017544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yukl ET, Goblirsch BR, Davidson VL, Wilmot CM. Crystal structures of CO and NO adducts of MauG in complex with pre-methylamine dehydrogenase: implications for the mechanism of dioxygen activation. Biochemistry. 2011;50:2931–2938. doi: 10.1021/bi200023n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abu Tarboush N, Jensen LM, Feng M, Tachikawa H, Wilmot CM, Davidson VL. Functional importance of tyrosine 294 and the catalytic selectivity for the bis-Fe(IV) state of MauG revealed by replacement of this axial heme ligand with histidine. Biochemistry. 2010;49:9783–9791. doi: 10.1021/bi101254p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •38.Tarboush NA, Jensen LM, Yukl ET, Geng J, Liu A, Wilmot CM, Davidson VL. Mutagenesis of tryptophan199 suggests that hopping is required for MauG-dependent tryptophan tryptophylquinone biosynthesis. Proc Natl Acad Sci U S A. 2011;108:16956–16961. doi: 10.1073/pnas.1109423108. [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript demonstrates that the oxidation of TTQ, effected by the enzyme MauG, requires a radical hopping mechanism to overcome the large distance between the MauG bis-Fe(IV) oxidant and the TTQ synthetic site.