

Abstract
CheY, a response regulator protein in bacterial chemotaxis, serves as a prototype for the analysis of response regulator function in two-component signal transduction. Phosphorylation of a conserved aspartate at the active site mediates a conformational change at a distal signaling surface that modulates interactions with the flagellar motor component FliM, the sensor kinase CheA, and the phosphatase CheZ. The objective of this study was to probe the conformational coupling between the phosphorylation site and the signaling surface of CheY in the reverse direction by quantifying phosphorylation activity in the presence and absence of peptides of CheA, CheZ, and FliM that specifically interact with CheY. Binding of these peptides dramatically impacted autophosphorylation of CheY by small molecule phosphodonors, which is indicative of reverse signal propagation in CheY. Autodephosphorylation and substrate affinity, however, were not significantly affected. Kinetic characterization of several CheY mutants suggested that conserved residues Thr-87, Tyr-106, and Lys-109, implicated in the activation mechanism, are not essential for conformational coupling. These findings provide structural and conceptual insights into the mechanism of CheY activation. Our results are consistent with a multistate thermodynamic model of response regulator activation.
Two-component regulatory systems, composed of a sensor kinase and its cognate response regulator, are widely used to accomplish signal transduction in bacteria, archaea, lower eukaryotes, and plants (1). CheY, a response regulator protein in bacterial chemotaxis, serves as a prototype to study the function of this class of proteins. In its active, phosphorylated form, CheY exhibits enhanced binding to a switch component, FliM, at the flagellar motor, which, in Escherichia coli, induces a change from counterclockwise to clockwise (CW) flagellar rotation. Ultimately, the phosphorylation status of CheY determines the swimming behavior of the bacterial cell. CheY is phosphorylated at Asp-57 with phosphoryl groups from the sensor kinase CheA and subsequently is dephosphorylated with the assistance of CheZ. CheY also exhibits an intrinsic autodephosphorylation activity and can autophosphorylate by using small molecule phosphodonors such as acetyl phosphate (AcP) or phosphoramidate (PAM; for review, see ref. 2). Genetic analysis as well as NMR and x-ray crystallographic studies identified overlapping binding surfaces for CheA, FliM, and CheZ on α-helix 4, β-sheet 5, and α-helix 5 of CheY (3–12).
A series of genetic and biochemical studies indicate that conserved
residues Thr-87, Tyr-106, and Lys-109 play major roles in the
phosphorylation-mediated conformational change of CheY. CheY mutants
87TA (13), 106YL (14), and 109KR (15), for example, can be
phosphorylated in vitro but fail to generate a CW signal
in vivo when introduced into a strain lacking wild-type
cheY. The recently solved crystal structures of
beryllofluoride (BeF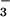 )-activated CheY (16) and two
other phosphorylated response regulators, FixJ and SpoOA (17, 18),
indicate that the conserved threonine and lysine (corresponding to
Thr-87 and Lys-109 in CheY) directly coordinate the phosphoryl group at
the active site. These structures, together with the NMR structure of
BeF
)-activated CheY (16) and two
other phosphorylated response regulators, FixJ and SpoOA (17, 18),
indicate that the conserved threonine and lysine (corresponding to
Thr-87 and Lys-109 in CheY) directly coordinate the phosphoryl group at
the active site. These structures, together with the NMR structure of
BeF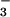 -activated CheY (19), implicate the conserved
threonine and tyrosine (corresponding to Tyr-106 in CheY) as key
residues in an activation mechanism in which relocation of the
threonine to directly interact with the aspartyl phosphate causes a
coordinated reorientation of the tyrosine side chain from an outside,
solvent-accessible conformation to an inside, solvent-inaccessible
conformation. The crystal structures of nonphosphorylated CheY bound to
a domain of CheA (9, 10) and of BeF
-activated CheY (19), implicate the conserved
threonine and tyrosine (corresponding to Tyr-106 in CheY) as key
residues in an activation mechanism in which relocation of the
threonine to directly interact with the aspartyl phosphate causes a
coordinated reorientation of the tyrosine side chain from an outside,
solvent-accessible conformation to an inside, solvent-inaccessible
conformation. The crystal structures of nonphosphorylated CheY bound to
a domain of CheA (9, 10) and of BeF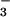 -activated CheY
bound to a peptide of FliM (12) provide further evidence that the
positioning of the Tyr-106 side chain at the signaling surface of CheY
is a major determinant for the differential recognition of the
phosphorylated and nonphosphorylated forms of CheY by its partner
proteins. Whereas phosphorylation increases the affinity of CheY for
FliM (20) and CheZ (21), phosphorylation decreases the affinity of CheY
for CheA (22).
-activated CheY
bound to a peptide of FliM (12) provide further evidence that the
positioning of the Tyr-106 side chain at the signaling surface of CheY
is a major determinant for the differential recognition of the
phosphorylated and nonphosphorylated forms of CheY by its partner
proteins. Whereas phosphorylation increases the affinity of CheY for
FliM (20) and CheZ (21), phosphorylation decreases the affinity of CheY
for CheA (22).
The objective of this study was to investigate the aforementioned conformational coupling between the phosphorylation site and the signaling surface of CheY in the reverse direction. There is evidence from the two-domain response regulator OmpR that DNA binding at the C-terminal transcriptional activation domain stimulates phosphorylation in the N-terminal (“CheY-like”) receiver domain (23). Accordingly, we set out to probe a conformational change of the single-domain response regulator CheY by measuring its phosphorylation activity in the presence and absence of peptides of CheA, CheZ, and FliM that specifically interact with CheY. We found that peptide binding dramatically impacts autophosphorylation activity, providing evidence for reverse information flow in CheY. Results with several CheY mutants showed that conserved residues 87, 106, and 109 are individually not essential for conformational coupling.
Materials and Methods
Bacterial Strains, Plasmids, and Mutant Constructions.
The E. coli Δ(cheYm60-21) strain K0641recA and the ptrp cheYZ plasmid pRBB40 have been described (24). Derivatives of pRBB40 carrying mutant cheY alleles 87TA (13) and 109KR (25) have been reported. cheY allele 106YA was constructed by splicing-by-overlap-extension polynucleotide chain reaction (26). The mutation generated was confirmed by sequencing the entire cheY gene of the resulting pRBB40 plasmid.
Behavioral Assay.
The rotational phenotype of strain KO641recA carrying cheY106YA on plasmid pRBB40 was determined as described (27).
Protein Purification and Peptide Synthesis.
E. coli CheY (wild type and mutant) and CheA proteins were purified from overexpressing strains as described (28). Protein concentrations were determined with a Lowry colorimetric assay (Bio-Rad). CheA124–257, which includes the P2 domain of CheA, was purified from strain RP3098 carrying plasmid pTM22 (29), as described (5). Because of the lack of tryptophan residues in CheA-P2, its concentration was determined by using a bicinchoninic acid assay (Pierce). The FliM peptide, corresponding to the N-terminal 16 residues of FliM (MGDSILSQAEIDALLN) and the CheZ peptides (wild type and 205VE), corresponding to the C-terminal 19 residues of CheZ (AGVVASQDQVDDLLDSLGF and AGVVASQDQEDDLLDSLGF, respectively) were obtained from Macromolecular Resources (Fort Collins, CO) and from the University of North Carolina Program in Molecular Biology and Biotechnology Micro Protein Chemistry Facility. Both peptides were purified by reverse-phase HPLC to a chromatographic homogeneity of at least 90% to remove residual fluorescent contaminants.
Fluorescence Measurements of CheY Phosphorylation.
Fluorescence measurements were made on a Perkin–Elmer LS-50B spectrofluorimeter; Perkin–Elmer fl winlab v.1.1 software was used to operate the instrument and analyze data. Tryptophan fluorescence of CheY was measured at an excitation wavelength of 292 nm and an emission wavelength of 346 nm. Slit widths were adjusted to accommodate the respective protein concentrations and varying fluorescence intensities of the different CheY mutant proteins. The minimum response time of the instrument is 20 ms. All samples were maintained at 24.5–25.5°C with a circulating water bath.
Time courses of approach to steady-state phosphorylation were monitored by following the decrease of intrinsic CheY fluorescence that results from phosphorylation. To initiate the reaction, CheY (wild type or mutant at 5 μM final concentration) was mixed with an equal volume of the phosphodonors PAM or AcP (final concentration of 5–100 mM or 0.5–10 mM, respectively) by using an Applied Photophysics (Surrey, U.K.) RX2000 stopped-flow accessory (dead time = 6 ms). All reactions were carried out in 100 mM Hepes, pH 7.0/10 mM MgCl2. At PAM concentrations below 100 mM, KCl was added to the phosphodonor solution at the appropriate concentrations to maintain constant ionic strength. Peptides were added to both CheY and phosphodonor solutions at equal concentrations before initiating the reaction. The final concentrations were 1 mM for CheZ or FliM peptide and 15 μM for CheA-P2. At these ligand concentrations, ≈80% of CheY is bound to FliM or CheZ peptide and ≈90% is bound to CheA-P2. Observed first-order rate constants (kobs) were obtained by exponential analysis of the resulting time courses. To determine the steady-state phosphorylation properties of CheY, the relative fluorescence quenching at steady state, taken from the individual stopped-flow experiments, was plotted against PAM concentration.
For CheY mutants 87TA, 106YA, and 109KR, the phosphorylation time courses in the absence of peptide were followed in a standard cuvette; phosphodonor was added manually with a Hamilton syringe through an injection port. The final protein concentrations were 1.6 μM. Because the phosphorylation of these mutants proceeded very slowly (for several minutes), the high temporal resolution achieved by stopped-flow methods was unnecessary. In addition, measurements performed by the stopped-flow technique over longer time scales yielded time courses that did not strictly obey first-order kinetics. This discrepancy seems to be attributable to diffusion processes between the reaction chamber and the reactants present in the connecting tubing of the stopped-flow cuvette.
Results from pre-steady-state kinetics were analyzed according to a reaction scheme of Lukat et al. (30). CheY phosphorylation by AcP or PAM can be treated as a first-order reaction with respect to phosphodonor concentration, and there is no indication of saturation even at high substrate concentrations (31, 32). Therefore, kobs can be expressed as follows:
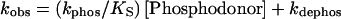 |
1 |
The quantity (kphos/Ks) [Phosphodonor] represents an effective rate constant for autophosphorylation. The ratio of (kobs − kdephos) to kobs thus yields the fraction of phosphorylated CheY at a given phosphodonor concentration.
Binding Assay.
Fluorescence measurements of the binding affinities between CheY (wild type or mutant) and FliM or CheZ peptide were performed as described (27). Dissociation constants were determined by fitting the titration data to a quadratic binding equation with the dissociation constant and total fluorescence change as adjustable parameters (33). A single binding site was assumed in the equation, and ligand concentration was corrected for the amount bound to CheY. The standard error given by the graphing software is an indicator for the accuracy of fit to a single data set. Background fluorescence associated with CheZ or FliM peptides was subtracted from the observed fluorescence signal. Measurement of peptide binding to phosphorylated CheY mutants 87TA and 109KR was complicated by the fact that a significant fraction of CheY was present in the nonphosphorylated form even after the addition of 100 mM PAM. Based on the measured rate constants, kobs at 100 mM PAM (see Fig. 2) and kdephos (Table 1), the phosphorylated fraction was estimated to be 63% for CheY87TA and 75% for CheY109KR (Eq. 1). Therefore, the resulting fluorescence change upon titration with FliM peptide contained a contribution resulting from initial FliM peptide binding to nonphosphorylated CheY and subsequent phosphorylation of this species (complete phosphorylation was assumed). These fluorescence contributions could be calculated based on measurements of the respective binding and phosphorylation reactions and were subtracted from the overall fluorescence signal.
Figure 2.

Phosphorylation kinetics of CheY mutants by PAM. Reactions with CheY87TA (○,●), CheY106YA (□,■), and CheY109KR (⋄,♦) were performed in the absence (open symbols) and the presence (closed symbols) of 1 mM FliM peptide. Observed first-order rate constants (kobs) were determined from individual phosphorylation time courses at the indicated phosphodonor concentrations. Constant ionic strength was maintained.
Table 1.
Phosphorylation rate constants determined in this study
| CheY variant | Ligand | kphos/KS (M−1 ⋅ s−1)* | kdephos (s−1)† |
|---|---|---|---|
| Wild type | None | 8.0 | 0.044 |
| CheA-P2 | 1.4 | 0.038 | |
| CheZ peptide | 100 | 0.045 | |
| CheZ205VE peptide | 19 | n/d | |
| FliM peptide | 230 | 0.068 | |
| 87TA | None | 0.055 | 0.0033 |
| FliM peptide | 1.4 | 0.0082 | |
| 106YA | None | 0.14 | 0.046 |
| FliM peptide | 4.4 | 0.038 | |
| 109KR | None | 0.30 | 0.0097 |
| FliM peptide | 1.8 | 0.019 |
n/d, not determined.
Determined using PAM as phosphodonor.
Determined by radiolabeling.
For binding measurements between wild-type CheY and
BeF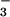 , 1.6 μM CheY in the absence of peptide and 0.4
μM CheY in the presence of 1 mM FliM peptide were titrated with
increasing amounts of BeCl2 in 100 mM Hepes, pH
7.0/10 mM MgCl2/10 mM NaF. Under these
conditions, the most likely species to interact with CheY is
BeF
, 1.6 μM CheY in the absence of peptide and 0.4
μM CheY in the presence of 1 mM FliM peptide were titrated with
increasing amounts of BeCl2 in 100 mM Hepes, pH
7.0/10 mM MgCl2/10 mM NaF. Under these
conditions, the most likely species to interact with CheY is
BeF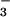 (34).
(34).
Dephosphorylation Assays.
Measurement of the rates of autodephosphorylation of phosphorylated CheY proteins was based on a published assay (35). Purified [32P]CheA (28 pmol) was incubated with 400 pmol of CheY (wild type or mutant) in a 100-μl reaction containing 100 mM Hepes, pH 7.0/20 mM MgCl2 at 25°C. KCl was added to maintain a level of ionic strength identical to the stopped-flow reaction conditions. Where appropriate, CheA-P2 (15 μM final concentration), FliM (1 mM final concentration), or CheZ peptide (1 mM final concentration) was added a few seconds after CheY had been incubated with [32P]CheA. Aliquots of 10 μl were removed at various times, added to 2× Laemmli denaturing buffer, and separated by SDS/PAGE. Relative amounts of [32P]CheY were determined by phosphorimaging. Exponential analysis of the time courses yielded first-order rate constants. To assess the sensitivity of CheY (wild type or 106YA) to CheZ, 15 or 150 pmol CheZ was added to the reaction.
Results
Phosphorylation Kinetics of Wild-Type CheY.
The effect of CheA, CheZ, and FliM on the phosphorylation properties of CheY was assessed by following the phosphorylation-associated decrease in tryptophan fluorescence. Instead of full-length protein, only fragments of CheA, CheZ, and FliM that interact with CheY were used in the respective phosphorylation and binding reactions. Full-length proteins would obscure fluorescence measurements because of the presence of additional tryptophan residues, and in the case of CheZ, dephosphorylation activity would interfere with the effect of binding on the phosphorylation properties of CheY. Peptides of CheZ (36) and FliM (37), corresponding to the C-terminal 19 aa of CheZ and the N-terminal 16 aa of FliM, respectively, and a single domain of CheA (termed CheA-P2; refs. 7 and 29) have been shown to bind specifically to CheY in a phosphorylation-dependent fashion.
We measured the pre-steady-state kinetics of CheY phosphorylation by the small molecule phosphodonor PAM in the presence and absence of peptide ligand. The associated time-dependent decrease in CheY fluorescence was monitored by using a stopped-flow apparatus with a resolution in the millisecond range, and the values of the corresponding observed phosphorylation rate constants (kobs) were plotted as a function of the PAM concentration (Fig. 1). The data in both the presence and absence of ligand indicated a linear dependence on phosphodonor concentrations with no indication of substrate saturation at concentrations up to 100 mM, consistent with another study (32). These results suggested that the binding affinity of CheY for phosphodonor is very low (KS ≫ 100 mM) even in the presence of CheZ or FliM peptide. On the other hand, the slope of the rate of CheY phosphorylation was increased by at least an order of magnitude in the presence of either peptide and decreased ≈6-fold in the presence of CheA-P2. The slope corresponds to the catalytic efficiency of CheY, which is given by the second-order rate constant of the reaction between CheY and phosphodonor, kphos/Ks (Eq. 1; see Table 1 for a summary of rate constants determined in this study). In the absence of any evidence for altered substrate affinity in the presence of peptide, our data are consistent with the simple interpretation that peptide binding primarily affects autophosphorylation activity (i.e., kphos). Very similar results were obtained from experiments with the phosphodonor AcP (data not shown). A control experiment with a mutant CheZ peptide that binds CheY with greatly decreased affinity (38) showed that the mere presence of peptide in the phosphorylation reaction is not sufficient to affect CheY phosphorylation significantly (Fig. 1).
Figure 1.

Phosphorylation kinetics of wild-type CheY by PAM. Reactions were performed in the absence of peptide (○), the presence of 1 mM FliM peptide (□), the presence of 1 mM CheZ peptide (⋄), the presence of 15 μM CheA-P2 (▵), or the presence of 1 mM CheZ205VE peptide (×). Measurements were done with a stopped-flow instrument, and constant ionic strength was maintained. Observed first-order rate constants (kobs) were determined from individual phosphorylation time courses at the indicated phosphodonor concentrations. For clarity, kobs values obtained in the absence of peptide (○) and in the presence of CheA-P2 (▵) are replotted in Inset with a different y-axis scale.
The y-axis intercepts on the plot in Fig. 1 correspond to the rate constants of dephosphorylation of phosphorylated CheY, kdephos (Eq. 1). They seemed to be similar in all cases, which was confirmed by measuring the decay of radiolabeled, phosphorylated CheY in the presence and absence of ligand (Table 1). In conclusion, it appeared that binding of CheA-P2, CheZ, or FliM peptide alters the autophosphorylation activity of CheY but has no significant effect on its autodephosphorylation activity nor on the affinity of CheY for its phosphodonor. Our results with CheA-P2 confirmed and extended initial measurements by Mayover et al. (31).
The effects of ligands on pre-steady-state kinetics were also reflected in steady-state measurements of CheY phosphorylation (data not shown). Increasing PAM concentrations led to increasing steady-state levels of phosphorylated CheY as assayed by fluorescence quenching. Saturation was observed when essentially all of the CheY present had been converted to the phosphorylated form. As expected, the presence of FliM or CheZ peptides greatly reduced the amount of phosphodonor needed to convert CheY fully from the nonphosphorylated to the phosphorylated form, whereas the presence of CheA-P2 increased the amount of phosphodonor needed.
Phosphorylation Kinetics of CheY Mutants.
The results described above suggested that ligand binding at the signaling surface of CheY mediates a conformational change that can lead to altered phosphorylation activity. The simplest assumption is that this conformational change involves the same conserved residues as the phosphorylation-mediated conformational change, principally Thr-87, Tyr-106, and Lys-109. As a possible mechanism, FliM or CheZ peptide binding to nonphosphorylated CheY might mediate a coordinated reorientation of the tyrosine and threonine side chains toward the active site. Therefore, one might expect that ligand binding has no appreciable impact on the phosphorylation activity of CheY mutants that harbor amino acid substitutions at the described positions. To test this hypothesis, we chose previously characterized CheY mutants 87TA and 109KR, which can be phosphorylated in vitro but do not promote CW flagellar rotation in vivo in a ΔcheY host (13, 15). In addition, we generated a third CheY mutant with a tyrosine-to-alanine substitution at position 106. The cheY106YA allele, introduced into a ΔcheY host on a plasmid, also did not support CW flagellar rotation.
To assess the degree of conformational coupling in these mutants, the pre-steady-state phosphorylation kinetics were measured in the presence and absence of FliM peptide, as described above for wild-type CheY (Fig. 2 and Table 1). All mutants could be phosphorylated by PAM in vitro, albeit more slowly than wild-type CheY, and phosphorylation activity (expressed as kobs) increased linearly with phosphodonor concentration. Unexpectedly, all mutant proteins exhibited a considerable increase in kobs values in the presence of FliM peptide, indicative of significant conformational coupling between the signaling surface and the phosphorylation site. By contrast, the effect of FliM binding on autodephosphorylation was relatively minor (Table 1), suggesting that, as was observed for wild-type CheY, peptide binding primarily affects autophosphorylation activity in the mutant proteins.
It should be noted that, for CheY106YA in the absence of peptide, the analysis of phosphorylation time courses that yielded kobs contained a large margin for error because of very low fluorescence quenching upon addition of phosphodonor. The small decrease in fluorescence intensity also indicated that only a marginal fraction of CheY106YA is present in the phosphorylated form, which is consistent with the relatively low values obtained for kobs (Fig. 2) compared with kdephos (Table 1).
Peptide Binding.
Communication between the signaling surface and the phosphorylation site in CheY mutants with substitutions at positions 87, 106, and 109 should also be reflected in their ability to bind to FliM with increased affinity upon phosphorylation. A previous bead-binding assay, however, indicated that the phosphorylated and unphosphorylated forms of CheY mutant 109KR bind to full-length FliM with similar affinities (39). To obtain more quantitative results, we measured binding of this mutant as well as CheY87TA and CheY106YA to FliM peptide by fluorescence spectroscopy (Fig. 3). Indeed, phosphorylation resulted in a 4.5-fold increase in the affinity of CheY87TA and a 2-fold increase in the affinity of CheY109KR to FliM peptide compared with a 10-fold increase for wild-type CheY. However, the absolute affinity of the phosphorylated species to peptide was lower for the mutants than for wild-type CheY. It was not feasible to determine the affinity of phosphorylated CheY106YA to FliM peptide accurately, because even in the presence of high concentrations of PAM, only a small fraction of CheY106YA was present in the phosphorylated form (see above).
Figure 3.

Binding of FliM peptide to CheY mutants. CheY87TA (○,●), CheY106YA (□), CheY109KR (⋄,♦), and wild-type CheY (▵,▴), were titrated with FliM peptide in the absence (open symbols) or the presence (closed symbols) of 100 mM PAM. The fraction of CheY bound to peptide is given by the relative decrease in fluorescence intensity (ΔI/ΔImax). The dissociation constants in the absence and presence of PAM are, respectively (in μM), 260 ± 13 and 25 ± 1 for wild-type CheY, 181 ± 16 and 40 ± 3 for CheY87TA, 508 ± 39 for CheY106YA, and 211 ± 9 and 100 ± 3 for CheY109KR.
Discussion
Stimulation of phosphorylation activity upon binding to a target was first noted in the two-domain response regulator OmpR (23). In this study, we provide evidence suggesting that this observation may be a general phenomenon that affects all response regulators, even those with a single domain such as CheY. Our results indicate that the interaction of CheY with parts of CheA, CheZ, and FliM dramatically affects autophosphorylation activity. For most kinetic assays, we used fluorescence spectroscopy, in which the phosphorylation activity of CheY is inferred from changes in the environment of the fluorophore, Trp-58, at the active site. Although this technique is indirect, agreement of the data with the current kinetic model for CheY phosphorylation strongly suggested that measurements obtained by fluorescence indeed reflect altered phosphorylation activity of CheY in the presence of ligand: the measured time courses strictly obeyed first-order kinetics, the deduced rate constants showed a first-order dependence with respect to the phosphodonor concentration (Figs. 1 and 2) and the autodephosphorylation rate constants, extrapolated from fluorescence measurements (Figs. 1 and 2 and AcP data not shown), agreed well with results obtained from radiolabeling (Table 1).
A Phenomenon Preordained by Thermodynamics?
Results from pre-steady-state kinetics implied that ligand binding primarily affected autophosphorylation, whereas autodephosphorylation as well as binding of phosphodonor to CheY remained unaltered (Fig. 1 and Table 1). Autophosphorylation was increased ≈10-fold and ≈30-fold by CheZ and FliM peptides, respectively, but was reduced 6-fold by CheA-P2. This differential effect correlated well with the binding affinities of these ligands to CheY: FliM and CheZ peptide bound ≈10-fold more tightly to the phosphorylated form of CheY (Fig. 3 and data not shown), and CheA-P2 bound ≈6-fold more tightly to the nonphosphorylated form (7, 22). The apparent coupling between ligand binding and phosphorylation activity can be described by a four-state model (23) representing a thermodynamic box of equilibria (Fig. 4A). CheY exists in two phosphorylation equilibria, one in the absence and one in the presence of ligand, and two binding equilibria, one with the nonphosphorylated and one with the phosphorylated species of CheY. In such a system, the ratio of the two phosphorylation equilibrium constants must equal the ratio of the two binding equilibrium constants, because the free energy of a reaction is directly proportional to the logarithm of the equilibrium constant, and the free energy difference between two species must be the same regardless of the path taken between the two species. The equilibrium constants for the phosphorylation reactions are simply the ratios of the rate constants of the forward and reverse reaction. For wild-type CheY, the ratio of the forward phosphorylation rate constants in the presence and absence of ligand is similar to the ratio of the equilibrium constants for peptide binding in the presence and absence of phosphorylation. Thus, we can infer that peptide binding must not significantly affect the rate of the reverse phosphorylation reaction, i.e., transfer of a phosphoryl group from CheY to ammonia or acetate to form PAM or AcP, respectively. To our knowledge, no attempts have been made to quantify this extremely unfavorable reaction.
Figure 4.

Models of CheY activation. (A) Four-state model. The model describes a thermodynamic box of equilibria for coupling of the binding of a ligand to the phosphorylation of CheY. “P” denotes a phosphoryl group and “L” denotes a peptide ligand. Colored arrows indicate a shift of the respective equilibria in the presence of FliM or CheZ peptide (green) or CheA-P2 (red). See the text for further explanation. (B) Eight-state model. The described four-state model is expanded by equilibria that couple phosphorylation and ligand binding to a protein conformational change. Ellipses denote an inactive, and rectangles denote an active conformation. Equilibria are thought to be distributed as follows. ① is favored in the absence of phosphorylation and ligand. Phosphorylation stabilizes ② (blue). Binding of CheA-P2 stabilizes ③ (red), whereas binding of FliM or CheZ peptide stabilizes ④ (green). Already in active conformations, ② binds CheZ or FliM peptide with increased affinity, and ④, with CheZ or FliM peptide as ligands, phosphorylates rapidly to favor ➎. On the other hand, ② is in an unfavorable conformation to bind CheA-P2, and ③, with CheA-P2 as ligand, is in an unfavorable conformation to autophosphorylate.
A related thermodynamic box can be constructed if the phosphorylation
reactions with small molecule phosphodonors are replaced by binding
reactions with the phosphate analog BeF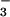 .
BeF
.
BeF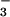 has been shown to form a stable complex with
CheY that mimics its activated state (19, 40). In this case, therefore,
it is possible to measure all four equilibria. We measured the binding
affinity of CheY to BeF
has been shown to form a stable complex with
CheY that mimics its activated state (19, 40). In this case, therefore,
it is possible to measure all four equilibria. We measured the binding
affinity of CheY to BeF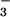 in the presence and absence
of FliM peptide (Fig. 5). CheY bound
≈10-fold more tightly to BeF
in the presence and absence
of FliM peptide (Fig. 5). CheY bound
≈10-fold more tightly to BeF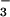 when associated with
FliM peptide. This result correlated very well with the 10-fold tighter
binding of CheY to FliM peptide in the presence of
BeF
when associated with
FliM peptide. This result correlated very well with the 10-fold tighter
binding of CheY to FliM peptide in the presence of
BeF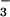 (40) than in its absence (Fig. 3).
(40) than in its absence (Fig. 3).
Figure 5.

Binding of BeF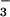 to CheY. Wild-type CheY in reaction
buffer containing 10 mM NaF was titrated with BeCl3 in the
absence (○) or presence (□) of 1 mM FliM peptide.
The fraction of CheY bound to peptide is given by the relative decrease
in fluorescence intensity
(ΔI/ΔImax). The
dissociation constants are 7.7 ± 0.7 μM in the absence of
peptide and 0.81 ± 0.04 μM in the presence of peptide.
to CheY. Wild-type CheY in reaction
buffer containing 10 mM NaF was titrated with BeCl3 in the
absence (○) or presence (□) of 1 mM FliM peptide.
The fraction of CheY bound to peptide is given by the relative decrease
in fluorescence intensity
(ΔI/ΔImax). The
dissociation constants are 7.7 ± 0.7 μM in the absence of
peptide and 0.81 ± 0.04 μM in the presence of peptide.
Structural Implications.
The impact of ligand binding on autophosphorylation but not on
autodephosphorylation (Fig. 1 and Table 1) suggested that ligand
binding changes the conformation of nonphosphorylated CheY but not that
of phosphorylated CheY. The notion of a propagating conformational
change in nonphosphorylated CheY upon ligand binding is supported by
recent structural studies. Chemical shift changes in NMR spectra of
CheY in the presence of CheZ or FliM peptides were observed not only in
C-terminal regions of the protein that constitute the putative
ligand-binding surface, but also at the distally located
phosphorylation site (11). Asp-57, Thr-87, Tyr-106, and Lys-109 were
among the residues affected, consistent with the concept that this
conformational change involves the same structural elements as the
conformational change mediated by phosphorylation. Moreover, in two
crystal structures of a complex of CheY and the P2 domain of CheA, the
CheY active site was found in a more “open” conformation compared
with apo-CheY (9, 10). According to our results, this conformation
seems to reduce rather than increase reactivity toward small molecule
phosphodonors (Fig. 1). The enormous rate enhancement in CheA-mediated
phosphotransfer may be caused, therefore, simply by an increase in the
local concentration of phosphodonor (31). To explain the dramatic
effect on the phosphorylation kinetics of CheY, ligand binding may
induce or stabilize a conformation at the phosphorylation site of CheY
that stabilizes (or, in case of CheA-P2, destabilizes) a transition
state during autophosphorylation. The conserved threonine and lysine
(as well as the Mg2+) likely participate in this
process, because they have been shown to coordinate the fluorines in
BeF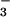 -activated CheY (16) and the phosphoryl oxygens
in other phosphorylated response regulators (17, 18). Our conclusion
that ligand binding does not significantly alter the conformation of
phosphorylated CheY is supported by the high similarity of the recently
solved structures of BeF
-activated CheY (16) and the phosphoryl oxygens
in other phosphorylated response regulators (17, 18). Our conclusion
that ligand binding does not significantly alter the conformation of
phosphorylated CheY is supported by the high similarity of the recently
solved structures of BeF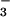 -activated CheY alone (16,
19) and in complex with a peptide of FliM (12).
-activated CheY alone (16,
19) and in complex with a peptide of FliM (12).
Interesting insights are obtained when the concept of an equilibrium between active and inactive states is applied to the described conformational changes in CheY. In the original model, phosphorylation is coupled to a protein conformational change in the absence of any peptide ligand (1, 41, 42). This equilibrium favors the inactive conformation in the absence of phosphorylation but favors the activated conformation in the presence of phosphorylation. Our results imply the existence of additional equilibria that also couple the binding of ligand to a protein conformational change. Consequently, not only phosphorylation but also binding of CheZ or FliM peptides would stabilize the active conformation of CheY. It follows that phosphorylation increases peptide binding, and peptide binding enhances phosphorylation activity. CheA-P2, in contrast, would stabilize the inactive conformation of CheY rather than induce a new conformation. This idea is supported by the structure of CheY bound to CheA-P2 as determined by Welch et al. (10). In this complex, CheY is virtually identical to the structure of CheY87TI (43), a mutant presumably locked in an inactive conformation. Based on Fig. 4A, the equilibria described here may be integrated into a model of CheY activation that recognizes eight individual states (Fig. 4B).
A recent crystallographic study suggests that the interaction between the conserved active site lysine and the aspartyl phosphate is directly involved in the activation of response regulators in addition to the emphasized coordinated rearrangement of the conserved threonine and tyrosine/phenylalanine residues (16). This “branched” mechanism can account for our observation that CheY mutant proteins lacking either Thr-87, Tyr-106, or Lys-109 exhibited mostly reduced—but in all cases significant—conformational coupling in both directions (Figs. 2 and 3). In these mutants, conformational coupling would predominantly occur through the respective alternate branch that is still functional, i.e., through Thr-87/Tyr-106 or Lys-109. The extent of coupling, although substantial, was apparently not sufficient to promote CW flagellar rotation in a ΔcheY host, which implied that all three residues function synergistically to activate CheY fully. It would appear that the modest decrease in binding observed for CheY87TA and CheY109KR compared with wild-type CheY (Fig. 3) is sufficient to account for their loss-of-function phenotype (13, 15), because the interaction of CheY with the flagellar switch is highly cooperative (44). This explanation is likely to be true for CheY106YA as well, although the affinity of this mutant protein to FliM peptide could not be measured. The phenotype of these mutants is probably not caused by a decrease in the cellular concentration of phosphorylated CheY, as inferred from the in vitro measurements of CheA- and CheZ-mediated phosphorylation reactions (refs. 13, 15, and 35 and data not shown), which would be the primary route of phosphoryl group flow in vivo.
Relevance in Vivo.
Ames et al. (23) have suggested that the response regulator OmpR may be phosphorylated while it is bound to its target, which can be envisioned for CheY as well. In this case, small molecule phosphodonors would remain the sole relevant phosphodonor in vivo, because the kinase CheA, as part of the chemotaxis receptor complex, is physically separated from the flagellar switch (2). Assuming a cytoplasmic concentration of 35 μM for nonphosphorylated CheY at steady state (45) and a switch-binding affinity of 100 μM (which is ≈10-fold weaker than that estimated for phosphorylated CheY; refs. 45 and 46), ≈30% of switch-binding sites would be occupied by nonphosphorylated CheY. Cellular AcP concentrations can reach up to 1.5 mM (47), at which point a significant fraction of CheY molecules would be phosphorylated while bound to the flagellar switch. AcP levels, as a physiological indicator, might thus be integrated into the chemotaxis response by modulating the prestimulus swimming behavior of the bacterium.
Acknowledgments
We thank Sydney Kustu for the initial idea that primed this study, Sandy Parkinson for providing E. coli strains, Chris Halkides for help with the analysis of binding data, Gerald Guanga for performing the behavioral assay, and the University of North Carolina Program in Molecular Biology and Biotechnology Micro Protein Chemistry Facility for peptide purification. This work was supported by National Institutes of Health Grant GM50860 (to R.B.B.).
Abbreviations
- AcP
acetyl phosphate
- CW
clockwise
- PAM
phosphoramidate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Stock A M, Robinson V L, Goudreau P N. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 2.Bren A, Eisenbach M. J Bacteriol. 2000;182:6865–6873. doi: 10.1128/jb.182.24.6865-6873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roman J S, Meyers M, Volz K, Matsumura P. J Bacteriol. 1992;174:6247–6255. doi: 10.1128/jb.174.19.6247-6255.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sockett H, Yamaguchi S, Kihara M, Irikura V M, MacNab R M. J Bacteriol. 1992;174:793–806. doi: 10.1128/jb.174.3.793-806.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McEvoy M M, Zhou H, Roth A F, Lowry D F, Morrison T B, Kay L E, Dahlquist F W. Biochemistry. 1995;34:13871–13880. doi: 10.1021/bi00042a019. [DOI] [PubMed] [Google Scholar]
- 6.Shukla D, Matsumura P. J Biol Chem. 1995;270:24414–24419. doi: 10.1074/jbc.270.41.24414. [DOI] [PubMed] [Google Scholar]
- 7.Swanson R V, Lowry D L, Matsumura P, McEvoy M M, Simon M I, Dahlquist F W. Nat Struct Biol. 1995;2:906–910. doi: 10.1038/nsb1095-906. [DOI] [PubMed] [Google Scholar]
- 8.Zhu X, Volz K, Matsumura P. J Biol Chem. 1997;272:23758–23764. doi: 10.1074/jbc.272.38.23758. [DOI] [PubMed] [Google Scholar]
- 9.McEvoy M M, Hausrath A C, Randolph G B, Remington S J, Dahlquist F W. Proc Natl Acad Sci USA. 1998;95:7333–7338. doi: 10.1073/pnas.95.13.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welch M, Chinardet N, Mourey L, Birck C, Samama J-P. Nat Struct Biol. 1998;5:25–29. doi: 10.1038/nsb0198-25. [DOI] [PubMed] [Google Scholar]
- 11.McEvoy M M, Bren A, Eisenbach M, Dahlquist F W. J Mol Biol. 1999;289:1423–1433. doi: 10.1006/jmbi.1999.2830. [DOI] [PubMed] [Google Scholar]
- 12.Lee S-Y, Cho H S, Pelton J G, Yan D, Henderson R K, King D S, Huang L-S, Kustu S, Berry E A, Wemmer D E. Nat Struct Biol. 2001;8:52–56. doi: 10.1038/83053. [DOI] [PubMed] [Google Scholar]
- 13.Appleby J L, Bourret R B. J Bacteriol. 1998;180:3563–3569. doi: 10.1128/jb.180.14.3563-3569.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu X, Amsler C D, Volz K, Matsumura P. J Bacteriol. 1996;178:4208–4215. doi: 10.1128/jb.178.14.4208-4215.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukat G S, Lee B H, Mottonen J M, Stock A M, Stock J B. J Biol Chem. 1991;266:8348–8354. [PubMed] [Google Scholar]
- 16.Lee, S.-Y., Cho, H. S., Pelton, J. G., Yan, D., Berry, E. A. & Wemmer, D. E. (2001) J. Biol. Chem. in press. [DOI] [PubMed]
- 17.Birck C, Mourey L, Gouet P, Fabry P, Schumacher P, Rousseau P, Kahn D, Samama J-P. Structure (London) 1999;7:1505–1515. doi: 10.1016/s0969-2126(00)88341-0. [DOI] [PubMed] [Google Scholar]
- 18.Lewis R J, Brannigan J A, Muchova K, Barak I, Wilkinson A J. J Mol Biol. 1999;294:9–15. doi: 10.1006/jmbi.1999.3261. [DOI] [PubMed] [Google Scholar]
- 19.Cho H S, Lee S-Y, Yan D, Pan X, Parkinson J S, Kustu S, Wemmer D E, Pelton J G. J Mol Biol. 2000;297:543–551. doi: 10.1006/jmbi.2000.3595. [DOI] [PubMed] [Google Scholar]
- 20.Welch M, Oosawa K, Aiziwa S-I, Eisenbach M. Proc Natl Acad Sci USA. 1993;90:8787–8791. doi: 10.1073/pnas.90.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blat Y, Eisenbach M. Biochemistry. 1994;33:902–906. doi: 10.1021/bi00170a008. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Swanson R V, Simon M I, Weis R M. Biochemistry. 1995;34:14626–14636. doi: 10.1021/bi00045a003. [DOI] [PubMed] [Google Scholar]
- 23.Ames S K, Frankema N, Kenney L J. Proc Natl Acad Sci USA. 1999;96:11792–11797. doi: 10.1073/pnas.96.21.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourret R B, Hess J F, Simon M I. Proc Natl Acad Sci USA. 1990;87:41–45. doi: 10.1073/pnas.87.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourret R B, Drake S K, Chervitz S A, Simon M I, Falke J J. J Biol Chem. 1993;268:13089–13096. [PMC free article] [PubMed] [Google Scholar]
- 26.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 27.Schuster M, Zhao R, Bourret R B, Collins E J. J Biol Chem. 2000;275:19752–19758. doi: 10.1074/jbc.M909908199. [DOI] [PubMed] [Google Scholar]
- 28.Hess J F, Bourret R B, Simon M I. Methods Enzymol. 1991;200:188–204. doi: 10.1016/0076-6879(91)00139-n. [DOI] [PubMed] [Google Scholar]
- 29.Morrison T B, Parkinson J S. Proc Natl Acad Sci USA. 1994;91:5485–5489. doi: 10.1073/pnas.91.12.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lukat G S, McCleary W R, Stock A M, Stock J B. Proc Natl Acad Sci USA. 1992;89:718–722. doi: 10.1073/pnas.89.2.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayover T L, Halkides C J, Stewart R C. Biochemistry. 1999;38:2259–2271. doi: 10.1021/bi981707p. [DOI] [PubMed] [Google Scholar]
- 32.Da Re S S, Deville-Bonne D, Tolstykh T, Veron M, Stock J B. FEBS Lett. 1999;457:323–326. doi: 10.1016/s0014-5793(99)01057-1. [DOI] [PubMed] [Google Scholar]
- 33.Eftink M R. Methods Enzymol. 1997;278:221–257. doi: 10.1016/s0076-6879(97)78013-3. [DOI] [PubMed] [Google Scholar]
- 34.Goldstein G. Anal Chem. 1964;36:243–244. [Google Scholar]
- 35.Silversmith R E, Appleby J L, Bourret R B. Biochemistry. 1997;36:14965–14974. doi: 10.1021/bi9715573. [DOI] [PubMed] [Google Scholar]
- 36.Blat Y, Eisenbach M. Biochemistry. 1996;35:5679–5683. doi: 10.1021/bi9530447. [DOI] [PubMed] [Google Scholar]
- 37.Bren A, Eisenbach M. J Mol Biol. 1998;278:207–214. doi: 10.1006/jmbi.1998.1730. [DOI] [PubMed] [Google Scholar]
- 38.Boesch K C, Silversmith R E, Bourret R B. J Bacteriol. 1999;182:3544–3552. doi: 10.1128/jb.182.12.3544-3552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welch M, Oosawa K, Aiziwa S-I, Eisenbach M. Biochemistry. 1994;33:10470–10476. doi: 10.1021/bi00200a031. [DOI] [PubMed] [Google Scholar]
- 40.Yan D, Cho H S, Hastings C A, Igo M M, Lee S-Y, Pelton J G, Stewart V, Wemmer D E, Kustu S. Proc Natl Acad Sci USA. 1999;96:14789–14794. doi: 10.1073/pnas.96.26.14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silversmith R E, Bourret R B. Trends Microbiol. 1999;7:16–21. doi: 10.1016/s0966-842x(98)01409-7. [DOI] [PubMed] [Google Scholar]
- 42.Nohaile M, Kern D, Wemmer D E, Stedman K, Kustu S. J Mol Biol. 1997;273:299–316. doi: 10.1006/jmbi.1997.1296. [DOI] [PubMed] [Google Scholar]
- 43.Ganguli S, Wang H, Matsumura P, Volz K. J Biol Chem. 1995;270:17386–17393. [PubMed] [Google Scholar]
- 44.Cluzel P, Surette M G, Leibler S. Science. 2000;287:1652–1654. doi: 10.1126/science.287.5458.1652. [DOI] [PubMed] [Google Scholar]
- 45.Alon U, Camarena L, Surette M G, Aguera y Arcas B, Liu J, Leibler S, Stock J B. EMBO J. 1998;17:4238–4248. doi: 10.1093/emboj/17.15.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scharf B E, Fahrner K A, Turner L, Berg H C. Proc Natl Acad Sci USA. 1998;95:201–206. doi: 10.1073/pnas.95.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCleary W R, Stock J B, Ninfa A J. J Bacteriol. 1993;175:2793–2798. doi: 10.1128/jb.175.10.2793-2798.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]