

Abstract
The pharmacological inhibition or genetic ablation of cyclophilin-D (CypD), a critical regulator of the mitochondrial permeability transition pore (mPTP), confers myocardial resistance to acute ischemia-reperfusion injury, but its role in post-myocardial infarction (MI) heart failure is unknown. The aim of this study was to determine whether mitochondrial CypD is also a therapeutic target for the treatment of post-MI heart failure. Wild-type (WT) and CypD–/– mice were subjected to either sham surgery or permanent ligation of the left main coronary artery to induce MI, and were assessed at either 2 or 28 days to determine the long-term effects of CypD ablation. After 2 days, myocardial infarct size was smaller and left ventricular (LV) function was better preserved in CypD–/– mice compared to WT mice. After 28 days, when compared to WT mice, in the CypD–/– mice, mortality was halved, myocardial infarct size was reduced, LV systolic function was better preserved, LV dilatation was attenuated and in the remote non-infarcted myocardium, there was less cardiomyocyte hypertrophy and interstitial fibrosis. Finally, ex vivo fibroblast proliferation was found to be reduced in CypD–/– cardiac fibroblasts, and in WT cardiac fibroblasts treated with the known CypD inhibitors, cyclosporin-A and sanglifehrin-A. Following an MI, mice lacking CypD have less mortality, smaller infarct size, better preserved LV systolic function and undergo less adverse LV remodelling. These findings suggest that the inhibition of mitochondrial CypD may be a novel therapeutic treatment strategy for post-MI heart failure.
Keywords: cyclophilin-D, ischemia, heart failure, fibrosis
Introduction
Despite recent advances in medical and device therapy, ischemic heart failure remains one of the leading causes of morbidity and mortality worldwide. In the United States alone, it has been estimated that 5.7 million people are currently suffering with heart failure, resulting in healthcare costs of $37.2 billion [1]. Ischemic heart failure is a complex chronic syndrome which is characterized by both structural and functional changes in both the infarcted and the non-infarcted remote myocardium. In response to a myocardial infarction (MI), the heart undergoes a complex intrinsic reparative process of ventricle remodelling which results in collagen scar tissue formation in the infarcted myocardium, and hypertrophy and fibrosis of the non-infarcted myocardium, resulting in ventricular dilatation, contractile dysfunction and subsequent heart failure [2, 3]. Therefore, novel treatment strategies that are capable of limiting myocardial infarct size, preventing adverse left ventricular (LV) remodelling and preserving LV systolic function are required to improve clinical outcome in this patient group.
In this respect, the mitochondrial permeability transition pore (mPTP), a critical mediator of cell death, has emerged as an important therapeutic target for limiting acute ischemia-reperfusion injury. The genetic ablation [4, 5] and pharmacological inhibition [6–8] of mitochondrial cyclophilin-D (CypD), a key regulatory component of the mPTP, has been reported to reduce myocardial infarct size in animal studies. However, it must be appreciated that CypD-deficient hearts are still susceptible to mPTP opening induced through mechanisms which are independent of CypD. More recently, cyclosporin-A (CsA), a pharmacological inhibitor of CypD has been reported to limit myocardial infarct size and improve LV function in ST-elevation MI patients undergoing primary percutaneous coronary intervention when given at reperfusion [9, 10]. Whether targeted inhibition of mitochondrial CypD is also beneficial in non-reperfused post-MI heart failure is currently unknown.
Materials and methods
A detailed Methods section is available in the online data supplement.
Animals
Experiments using animals were carried out in accordance with the United Kingdom Home Office Guide on the Operation of Animal (Scientific Procedures) Act of 1986 and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). B6Sv129 mice were obtained from Harlan (Derby, UK) and mice deficient in mitochondrial CypD–/– were generated as previously described [4].
Murine model of myocardial infarction
Wild-type (WT) and CypD–/– mice (male, 9–11 weeks old) were anaesthetized and subjected to MI by permanent ligation of the left descending coronary artery. Sham-operated animals underwent a similar surgical procedure but without coronary artery ligation. Experimental analyses were performed at either 2 or 28 days after surgery.
Echocardiographic assessment
Transthoracic two-dimensional echocardiography was performed under light anaesthesia with spontaneous respiration using isoflurane to evaluate changes in heart function following the sham procedure or MI at day 2 or 28 using a VingMed Vivid 7 Dimension™ ultrasound imaging machine (GE Healthcare, Bedford, UK). Internal dimensions of the LV cavity were measured from a parasternal short axis view by M-mode scan of the LV at the mid-papillary muscle level. All cardiac measurements were averaged over four to five consecutive cardiac cycles.
Magnetic resonance imaging (MRI)
Myocardial infarct size at 2 days after MI was determined by cardiac MRI using late gadolinium enhancement because the infarcted tissue could not be clearly defined by Masson-trichrome histological staining. Mice were anaesthetized with isoflurane and placed in a 9.4T MRI scanner (VNMRS, Varian, Inc., Santa Clara, CA, USA). For late gadolinium contrast enhancement, 0.6 mmol/kg of Gadolinium (III)-diethyltriaminepentaacetic acid (Magnevist, Schering AG, Germany) was injected intraperitoneally. Infarct and LV volumes were analysed by two observers blinded to the treatment allocation (S.Y.L. and A.N.P.) using Segment software (http://segment.heiberg.se/) and infarct size was presented as a percentage of the left ventricle myocardial volume.
Histomorphometric analysis
At 28 days and following echocardiographic assessment, mice were killed. Heart weight was measured and normalized to body weight. The hearts were divided into four equal short-axis transverse ventricular slices and subjected to routine histological procedure. To determine the infarct size, Masson-trichrome stained histology images were captured with Epson perfection V100 photo scanner (Epson Ltd., Herts, UK). Myocardial interstitial fibrosis was assessed in the post-eroseptal non-infarcted myocardium where five random histological fields from each heart were captured with a SPOT RT camera and software v3.2 (Diagnostic Instruments, Inc., Sterling Heights, MI, USA) at 400 times magnification. Mean cardiomyocyte cross-sectional area was determined in a LV mid-section stained with Alexa Fluor® 488 conjugated wheat germ agglutinin (Invitrogen, Paisley, UK) and approximately 200 cardiomyocytes at 630 times magnification were sampled from each heart. An investigator (S.Y.L.), blinded to the treatment protocols performed the above analyses using NIH Image J 1.63 software.
Western blot analysis of myocardial apoptosis
The expression of cleaved caspase-3 and uncleaved caspase-3 (Cell Signaling Technology, Danvers, MA, USA) was analysed in infarcted LV myocardium by Western blotting at 2 days after surgery. Band intensity was determined by computerized densitometry using NIH Image J 1.63 software and expressed as arbitrary units after normalized with α-tubulin (Abcam Plc, Cambridge, UK).
Cardiac fibroblast isolation and proliferation assay
Cardiac fibroblasts were isolated from WT and CypD–/– mice using a modification of a previously published method [11] to determine the effect of CypD inhibition on cardiac fibroblast proliferation. Discoidin domain receptor 2 (DDR2, 4 μg/ml, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to stain the cardiac fibroblasts and confirm the purity of the isolation [12]. Cardiac fibroblasts from 1–3 passages were seeded at 2.5 × 104 cells/12-well plate and synchronized by serum deprivation in 0.1% serum overnight. Cells were then incubated in 10% serum-containing fibroblast growth medium for 48 hours in the presence of either ethanol (0.1%), CsA (0.4 μM) or sanglifehrin-A (SfA, 1 μM). Changes in cell number were calculated using a haemocytometer and expressed as a percentage change over the starting cell number (2.5 × 104 cells).
Statistical analysis
Data are presented as mean ± S.E.M. The mean values were analysed using unpaired t-test or one-way anova followed by a Tukey’s multiple comparison post hoc test where appropriate. A P < 0.05 was considered to be statistically significant. Mortality was analysed by the Kaplan–Meier method and comparison of survival curves was performed with log-rank test.
Results
Mortality following myocardial infarction
Cumulative survival of WT and CypD–/– mice was recorded for 28 days following the induction of MI or sham surgery. No deaths were observed in sham-operated animals. In the animals subjected to MI, the survival rate at 28 days after MI was significantly higher in CypD–/– than WT mice (83%[10/12]versus 44%, [10/23], respectively: P < 0.01) (Fig. 1). Interestingly, the deaths only occurred within a time window of days 3–5 after MI, and autopsy performed on the deceased animals revealed hemothorax, the presence of blood clot around the heart and in the chest cavity, indicating acute infarct rupture as the probable cause of death.
Fig 1.
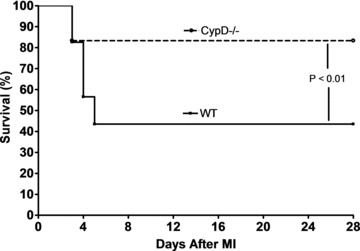
Kaplan–Meier survival curve analysis for WT (n= 23) and CypD–/– (n= 12) mice after MI. CypD–/– mice experienced significantly better survival after MI when compared to WT mice. Note that all deaths occur between 3 and 5 days after MI. Sham-operated WT and CypD–/– mice have 100% survival rate (not shown in figure).
Myocardial infarct size
All the mice that were subjected to MI showed a discrete transmural infarct at the LV free wall and apex on late gadolinium enhancement cardiac MRI at 2 days after MI (Fig. 2A). At 2 days after MI, the myocardial infarct size (as measured by the volume of late gadolinium enhancement and expressed as a percentage of the LV volume) was significantly smaller in CypD–/– mice compared to WT mice (27.6 ± 4.7%versus 40.4 ± 1.5%, respectively: P < 0.05) (Fig. 2B). At 28 days after MI, the myocardial infarct size (as measured by histology) was significantly smaller in CypD–/– mice compared to WT mice, when expressed as a percentage of either the LV area (21.3 ± 1.3%versus 33.9 ± 4.3%, respectively: P < 0.05) (Fig. 2C) or the LV internal circumference (37.8 ± 3.5%versus 50.7 ± 4.8%, respectively: P < 0.05) (Fig. 2D).
Fig 2.
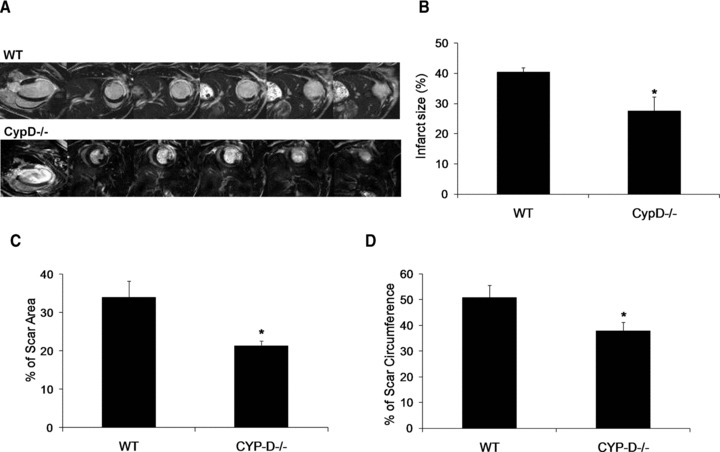
(A) Representative two-chamber and short-axis MRI late gadolinium enhancement images of the WT and CypD–/– mice at 2 days after MI. (B) Graph showing a smaller myocardial infarct size expressed as a percentage of LV volume at 2 days after MI in CypD–/– mice (n= 6) when compared to WT (n= 5). Infarct size was expressed as (C) percentage of scar area and (D) percentage of scar circumference in WT (n= 10) and CypD–/– (n= 10) mice 28 days after MI. *P < 0.05 versus WT.
Myocardial apoptosis
To evaluate the effect of CypD ablation on myocardial apoptosis 2 days after MI, myocardial cleaved and uncleaved caspase-3 protein expression was determined by Western blot analysis. In the WT mice there was a significant increase in both cleaved (2.7 ± 0.5 MI versus 0.7 ± 0.3 sham: P < 0.05) and uncleaved (1.8 ± 0.2 MI versus 1.0 ± 0.3 sham: P < 0.05) caspase-3 protein expression following MI (Fig. 3). However, in the CypD–/– mice the increase in both cleaved (1.2 ± 0.2 MI versus 0.7 ± 0.3 sham: P > 0.05) and uncleaved (1.0 ± 0.1 MI versus 0.7 ± 0.1 sham: P > 0.05) caspase-3 protein expression following MI was not significant (Fig. 3). Therefore, the increase in both cleaved (2.7 ± 0.5 WT versus 1.2 ± 0.2 CypD–/–: P < 0.05) and uncleaved caspase-3 protein expression (1.8 ± 0.2 WT versus 1.0 ± 0.1 CypD–/–: P < 0.05) induced by MI was significantly attenuated in CypD–/– mice (Fig. 3).
Fig 3.
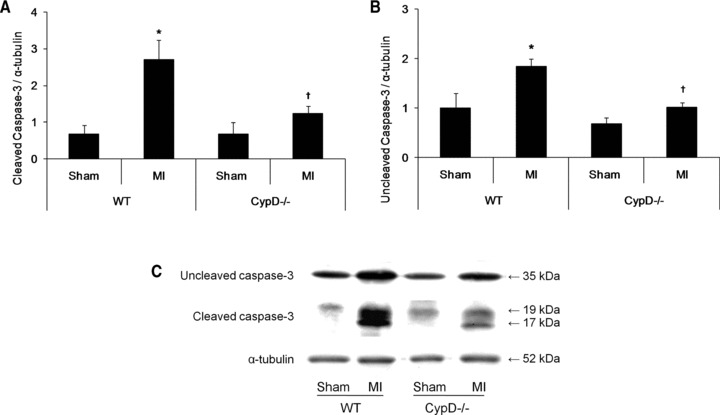
Myocardial apoptosis in CypD–/– and WT animals after MI. Western blot analysis of (A) cleaved caspase-3 (19 kD) and (B) uncleaved caspase-3 (35 kD) in the infarcted myocardium of WT and CypD–/– mice (n= 3–6) collected at 2 days after MI. (C) Representative Western blots of the myocardial caspase-3 proteins expression in WT and CypD–/– mice. *P < 0.05 versus respective sham; †P < 0.05 versus WT MI.
Left ventricular volumes and function
LV volumes and function were assessed by echocardiography in both WT and CypD–/– mice following either sham surgery or MI (Table 1). At 2 days, there was a significant reduction in LV function in the WT mice following MI as demonstrated by reductions in LV fractional shortening, LV ejection fraction and cardiac output. Interestingly, this impairment in LV function at 2 days after MI was significantly attenuated in CypD–/– mice. At 28 days, there was a significant increase in LV chamber dimensions in the WT mice following MI as demonstrated by the LV end diastolic diameter and LV end systolic diameter. In addition, there was a significant reduction in LV function in the WT mice following MI as demonstrated by reductions in LV fractional shortening and LV ejection fraction. Interestingly, both the LV dilatation and impairment in LV function at 28 days after MI was significantly attenuated in CypD–/– mice.
Table 1.
Cardiac measurements in WT and CypD–/– mice assessed at 2 and 28 days following sham surgery or MI
| 2 days | Sham | MI | ||
|---|---|---|---|---|
| WT | CypD–/– | WT | CypD–/– | |
| n number | 3 | 4 | 11 | 10 |
| Heart rate (bpm) | 564.1 ± 26.8 | 565.6 ± 9.1 | 552.5 ± 20.1 | 554.3 ± 16.7 |
| LVEDD (cm) | 0.37 ± 0.03 | 0.35 ± 0.01 | 0.40 ± 0.01 | 0.41 ± 0.02 |
| LVESD (cm) | 0.24 ± 0.02 | 0.23 ± 0.01 | 0.30 ± 0.01 | 0.28 ± 0.02 |
| FS (%) | 35.8 ± 1.4 | 33.7 ± 1.4 | 24.8 ± 1.0* | 31.8 ± 1.9† |
| EF (%) | 71.6 ± 1.4 | 70.2 ± 1.4 | 55.8 ± 1.6* | 66.4 ± 2.5† |
| 28 days | Sham | MI | ||
| WT | CypD–/– | WT | CypD–/– | |
| n number | 7 | 7 | 10 | 10 |
| Body weight, BW (g) | 28.2 ± 0.5 | 28.4 ± 0.3 | 29.6 ± 0.6 | 27.3 ± 0.7† |
| Heart weight, HW (mg) | 153.2 ± 4.9 | 163.2 ± 4.9 | 250.5 ± 16.4* | 223.4 ± 10.1* |
| HW/BW (mg/g) | 5.44 ± 0.2 | 5.7 ± 0.2 | 8.4 ± 0.4* | 8.2 ± 0.4* |
| Heart rate (bpm) | 550.7 ± 24.3 | 485.4 ± 26.6 | 501.7 ± 21.4 | 497.1 ± 21.0 |
| LVEDD (cm) | 0.38 ± 0.02 | 0.38 ± 0.02 | 0.47 ± 0.02* | 0.40 ± 0.02 |
| LVESD (cm) | 0.25 ± 0.02 | 0.24 ± 0.01 | 0.39 ± 0.02* | 0.30 ± 0.02† |
| FS (%) | 34.3 ± 3.1 | 36.0 ± 2.0 | 17.8 ± 1.3* | 26.8 ± 1.8*† |
| EF (%) | 69.1 ± 4.1 | 72.4 ± 2.3 | 41.0 ± 2.8* | 58.0 ± 2.8*† |
LVEDD: left ventricular end-diastolic diameter; LVESD: left ventricular end-systolic diameter; FS: fractioning shortening; EF: ejection fraction.
P < 0.05 versus respective sham, †P <0.05 versus MI WT.
Reactive ventricular hypertrophy and myocardial interstitial fibrosis in remote myocardium
At 28 days after MI, although there was a significant increase in heart weight/body weight following MI indicating post-MI LV hypertrophy, there were no significant differences between CypD–/– and WT mice (Table 1). In the remote remodelled LV myocardium of WT animals, there was a significant increase in both cardiomyocyte cross-sectional area (196.6 ± 4.1 μm2 sham, n= 7 versus 328.6 ± 15.9 μm2 MI, n= 10: P < 0.05) (Fig. 4A, B) and myocardial interstitial fibrosis (1.1 ± 0.19% sham, n= 7 versus 4.9 ± 0.58% MI, n= 10: P < 0.05) (Fig. 5A, B). Interestingly, both the increase in cardiomyocyte cross-sectional area (328.6 ± 15.9 μm2 WT versus 244.0 ± 6.7 μm2 CypD–/–, n= 10: P < 0.05) and myocardial interstitial fibrosis (4.9 ± 0.57% WT versus 1.5 ± 0.19% CypD–/–, n= 10: P < 0.05) in the remote myocardium were significantly attenuated in CypD–/– mice after MI. In addition, CypD ablation also significantly reduced the interstitial fibrosis-to-infarct area ratio (0.17 ± 0.03 WT versus 0.07 ± 0.01 CypD–/–: P < 0.05) (Fig. 5C).
Fig 4.
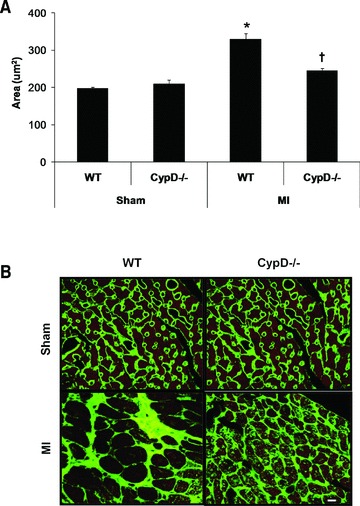
Cardiomyocyte cross-sectional area in remote myocardium of CypD–/– and WT mice 28 days after MI. (A) In WT mice but not CypD–/– mice, cardiomyocyte cross-sectional area increased significantly following MI, such that post-MI cross-sectional area was significantly greater in WT mice when compared to CypD–/– ones. (B) Representative high power photomicrographs of wheat germ agglutinin-stained LV cross-sections of non-infarcted myocardium. Bar = 10 μm. n= 7–10. *P < 0.05 versus respective WT sham. †P < 0.05 versus WT MI.
Fig 5.
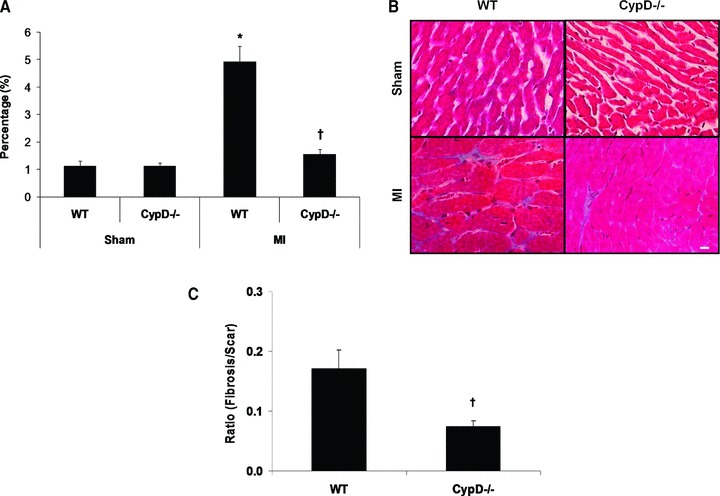
Myocardial interstitial fibrosis in remote myocardium of CypD–/– and WT mice 28 days after MI. (A) In WT mice but not CypD–/– mice, myocardial interstitial fibrosis increased significantly following MI, such that the extent of post-MI myocardial interstitial fibrosis was significantly greater in WT mice when compared to CypD–/– mice. (B) Representative high power photomicrographs of Masson trichrome-stained LV cross-sections of non-infarcted myocardium. Bar = 10 μm. (C) Interstitial fibrosis expressed as a ratio to percentage of scar area. n= 7–10. *P < 0.05 versus respective WT sham. †P < 0.05 versus WT MI.
Ex vivo cardiac fibroblast proliferation
Cardiac fibroblasts isolated from WT and CypD–/– mice were identified by DDR2 immunostaining, and demonstrated to co-localize with wheat germ agglutinin (Fig. 6A). The degree of fibroblast proliferation, when cultured in 10% serum, was significantly lower in cardiac fibroblasts isolated from CypD–/– mice compared to WT mice (111.0 ± 8.0% WT versus 43.5 ± 5.2% CypD–/–: P < 0.05) (Fig. 6B). Importantly, this effect of CypD ablation on fibroblast proliferation in the WT cardiac fibroblasts was reproduced by the known pharmacological mPTP inhibitors, CsA and SfA (11.0 ± 8.0% control versus 42.5 ± 5.4% CsA and 39.0 ± 4.4% SfA: P > 0.05) (Fig. 6B).
Fig 6.
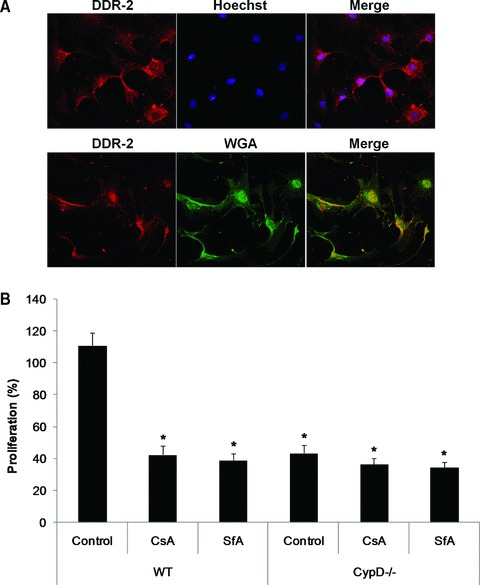
Proliferation of cardiac fibroblasts isolated from WT and CypD–/– hearts. (A) Representative confocal micrographs of DDR2-stained (red) cardiac fibroblasts isolated from WT mice. Cells were counterstained with Hoechst-33258 (blue) for nuclei or wheat germ agglutinin (green) for cell membranes. (B) The proliferation of cardiac fibroblasts isolated from WT mice was significantly increased in response to 10% serum when compared to those isolated from CypD–/– mice. This increased in proliferation observed in the WT cardiac fibroblasts was prevented by CsA (0.4 μM) or SfA (1.0 μM). n= 4. *P < 0.05 versus WT control.
Discussion
The major finding of the present study is that the heart deficient in mitochondrial CypD is more resistant to the detrimental effects of heart failure following an MI. Following MI, CypD–/– mice were demonstrated to have less mortality, smaller myocardial infarct size, less myocardial apoptosis, better preserved LV systolic function, reduced LV dilatation and in the remote non-infarcted myocardium less cardiomyocyte hypertrophy and interstitial fibrosis.
The CypD-regulated mPTP has been previously investigated in the context of heart failure using a known pharmacological inhibitor of CypD, CsA. The probability of mPTP opening in viable cardiomyocytes was reported to be increased in viable cardiomyocytes isolated from dogs with chronic heart failure [13, 14] and in phenylephrine-induced neonatal cardiomyocyte hypertrophy [15]. Furthermore, CsA has been reported to preserve mitochondrial morphology in the heart following ischemia [16]. However, as well as inhibiting CypD, CsA also inhibits other cyclophilins and calcineurin. The inhibition of calcineurin-mediated dephosphorylation of the apoptogenic protein Bcl-2-associated death promoter (BAD) can, in itself, result in cardioprotection [17]. In addition, calcineurin inhibition has been recently demonstrated to inhibit mitochondrial fragmentation which may also have cytoprotective effects [18]. Deletion of the CypD gene has been shown to prevent heart failure caused by doxorubicin-induced cardiotoxicity, or as a result of calcium dysregulation by the overexpression of the β2a subunit of L-type calcium channels [19]. However, the role of CypD in the setting of post-MI heart failure has not been previously investigated.
In the present study, WT mice had a post-MI mortality of ∼56%, and this was found to occur within the time window of 3 to 5 days after MI and appeared due to infarct rupture. Our findings concur with previous studies implicating infarct rupture occurring in response to reduced tensile strength of the infarcted myocardium as a major cause of mortality [20–22]. Interestingly, in mice lacking CypD–/–, mortality was almost one third of that in WT mice, a finding which may in part be attributable to the smaller myocardial infarct size observed at 2 days after MI resulting in reduced risk of infarct rupture. In the present study, myocardial interstitial fibrosis was increased following MI.
The increase in myocardial interstitial fibrosis in this region of the heart results in an increase in stiffness of the heart and reduced LV distensibility, resulting in further LV dysfunction [23–25]. Compared to WT mice, those lacking CypD had better LV systolic function after MI in terms of increased fractional shortening and ejection fraction. These findings may be attributed to a smaller myocardial infarct size, less cardiomyocyte hypertrophy and reduced interstitial fibrosis in the remote myocardium of CypD–/– hearts after MI. This finding was also supported by an observation in cardiac-specific transgenic mice overexpressing CypD showing cardiac hypertrophy and worsening of cardiac function, such as fractional shortening, heart rate and LV dimensions, over time [4]. Interestingly, there was no difference in the ratio between heart/body weight despite the fact that WT mice exhibited a larger cardiomyocyte cross-sectional area than CypD–/– mice after MI, although there was a difference in the increase in heart size after MI between WT and CypD–/– mice. This discrepancy may have been due to the smaller body weight after MI in CypD–/– mice which may have masked the differences, but this is only speculation. Therefore, cardiac hypertrophy may have been more accurately quantified if normalized with tibial length, which remains constant after maturity [26].
Apoptosis is known to contribute to the progression of heart failure following an MI [24, 27–29], but the role of the mPTP as a mediator of apoptotic cell death is controversial [4, 5, 16, 30–32]. In the current study, the extent of apoptosis, as determined by the myocardial expression of cleaved caspase-3 was significantly reduced in the CypD–/– mice compared to WT mice at 2 days after MI. It would have been preferable, if these findings had been confirmed using an additional method for detecting apoptotic cell death such as terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining. Whether this attenuation in myocardial apoptotic activity was the result of a smaller myocardial infarct size or actually related to the absence of a CypD-regulated mPTP is unclear. In fact, cardiac-specific transgenic mice overexpressing CypD have been shown to have increase TUNEL staining, increased cytochrome C release and increased caspase 9 cleavage in the heart [4]. In addition, several studies have suggested that mitochondrial CypD may actually have an anti-apoptotic effect, interacting with Bcl-2 to prevent mitochondrial cytochrome c release independent of the mPTP [30]. In this regards, further studies evaluating the release of various apoptotic effectors from the mitochondria after MI may provide more insight as to the anti-apoptotic effect of CypD inhibition, in particular the involvement of caspase-dependent (cytochrome c, Apaf-1, Smac/Diablo and HtrA2/Omi) and caspase-independent [apoptosis-inducing factor (AIF), endonuclease G] cell death.
A previous study has reported that the pharmacological inhibition of CypD using Debio-025 at the time of myocardial reperfusion did preserve LV systolic function and reduce mortality in mice, beneficial effects which may be attributable to the infarct size reduction mediated by CypD inhibition [33]. In addition, recent clinical studies have showed that CsA, administered at the time of reperfusion, significantly reduced infarct size and improved LV function in patients undergoing primary percutaneous coronary intervention for an acute MI [9, 10]. A major limitation of our study, which was a model of permanent coronary artery occlusion, is whether the beneficial effects on post-MI remodelling observed in CypD-deficient mice was simply the result of an acute reduction in myocardial infarct size. We do not believe that this is the case for three reasons: (1) the reduction in myocardial interstitial fibrosis was more than expected for the reduction in infarct size (i.e. the smaller interstitial fibrosis-to-infarct area ratio in CypD–/– mice); (2) CypD ablation would be expected to limit myocardial infarct size in a reperfused infarct and (3) the proliferative potential of CypD-deficient fibroblasts is reduced in our cell culture experiment. These findings suggest that CypD ablation may be having a direct effect on post-MI remodelling but of course this needs to be investigated in further studies. In this respect, a previous study also reported less myocardial interstitial fibrosis in CypD–/– mice subjected to heart failure induced by doxorubicin, or by calcium overload [19]. Interestingly, in some previous non-cardiac studies [34, 35] but not all [36], CsA has been reported to inhibit fibroblast proliferation in a calcineurin-independent manner [35]. Whether the impaired cardiac fibroblast proliferation in the CypD–/– mice can be used to explain the reduction in interstitial fibrosis observed in the remote myocardium remains to be determined.
In conclusion, the current study has demonstrated that following a non-reperfused MI CypD–/– mice have improved survival, smaller myocardial infarct size, better preserved LV systolic function and less adverse LV remodelling. These findings suggest that the mitochondrial CypD may be a novel therapeutic target for the treatment of post-MI heart failure. As such, new safe and specific pharmacological inhibitors of mitochondrial CypD need to be identified and developed as novel pharmaceutical agents for post-MI heart failure.
Acknowledgments
This work was supported by British Heart Foundation programme grants (RG/08/015/26411), a Department of Health’s National Institute of Health Research (NIHR) Biomedical Research Centres funding scheme, and Welcome Trust project grant (WT081285 to S.Y.L.). We sincerely thank Dr. C.P. Baines and Dr. J.D. Molkentin (Department of Pediatrics, Children’s Hospital Medical Center, Cincinnati, OH, USA) for the provision of the CypD–/– mice.
Conflict of interest
None declared.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:e21–181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 2.Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res. 2005;66:22–32. doi: 10.1016/j.cardiores.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–8. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 4.Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 5.Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 6.Hausenloy DJ, Maddock HL, Baxter GF, et al. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–43. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 7.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:617–25. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 8.Argaud L, Gateau-Roesch O, Muntean D, et al. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–74. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–81. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 10.Mewton N, Croisille P, Gahide G, et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J Am Coll Cardiol. 2010;55:1200–5. doi: 10.1016/j.jacc.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 11.Kumaran C, Shivakumar K. Calcium- and superoxide anion-mediated mitogenic action of substance P on cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2002;282:H1855–62. doi: 10.1152/ajpheart.00747.2001. [DOI] [PubMed] [Google Scholar]
- 12.Goldsmith EC, Hoffman A, Morales MO, et al. Organization of fibroblasts in the heart. Dev Dyn. 2004;230:787–94. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- 13.Sharov VG, Todor A, Khanal S, et al. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J Mol Cell Cardiol. 2007;42:150–8. doi: 10.1016/j.yjmcc.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharov VG, Todor AV, Imai M, et al. Inhibition of mitochondrial permeability transition pores by cyclosporine A improves cytochrome C oxidase function and increases rate of ATP synthesis in failing cardiomyocytes. Heart Fail Rev. 2005;10:305–10. doi: 10.1007/s10741-005-7545-1. [DOI] [PubMed] [Google Scholar]
- 15.Javadov S, Baetz D, Rajapurohitam V, et al. Antihypertrophic effect of Na+/H+ exchanger isoform 1 inhibition is mediated by reduced mitogen-activated protein kinase activation secondary to improved mitochondrial integrity and decreased generation of mitochondrial-derived reactive oxygen species. J Pharmacol Exp Ther. 2006;317:1036–43. doi: 10.1124/jpet.105.100107. [DOI] [PubMed] [Google Scholar]
- 16.Leshnower BG, Kanemoto S, Matsubara M, et al. Cyclosporine preserves mitochondrial morphology after myocardial ischemia/reperfusion independent of calcineurin inhibition. Ann Thorac Surg. 2008;86:1286–92. doi: 10.1016/j.athoracsur.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minners J, van den Bos EJ, Yellon DM, et al. Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovasc Res. 2000;47:68–73. doi: 10.1016/s0008-6363(00)00069-9. [DOI] [PubMed] [Google Scholar]
- 18.Cereghetti GM, Stangherlin A, dB Martins, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–8. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama H, Chen X, Baines CP, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–44. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tao ZY, Cavasin MA, Yang F, et al. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004;74:1561–72. doi: 10.1016/j.lfs.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 21.Gao XM, Xu Q, Kiriazis H, et al. Mouse model of post-infarct ventricular rupture: time course, strain- and gender-dependency, tensile strength, and histopathology. Cardiovasc Res. 2005;65:469–77. doi: 10.1016/j.cardiores.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 22.Fang L, Gao XM, Moore XL, et al. Differences in inflammation, MMP activation and collagen damage account for gender difference in murine cardiac rupture following myocardial infarction. J Mol Cell Cardiol. 2007;43:535–44. doi: 10.1016/j.yjmcc.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83:1849–65. doi: 10.1161/01.cir.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 24.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–62. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y. Myocardial repair/remodelling following infarction: roles of local factors. Cardiovasc Res. 2009;81:482–90. doi: 10.1093/cvr/cvn333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin FC, Spurgeon HA, Rakusan K, et al. Use of tibial length to quantify cardiac hypertrophy: application in the aging rat. Am J Physiol. 1982;243:H941–7. doi: 10.1152/ajpheart.1982.243.6.H941. [DOI] [PubMed] [Google Scholar]
- 27.Bartling B, Holtz J, Darmer D. Contribution of myocyte apoptosis to myocardial infarction? Basic Res Cardiol. 1998;93:71–84. doi: 10.1007/s003950050065. [DOI] [PubMed] [Google Scholar]
- 28.Sam F, Sawyer DB, Chang DL, et al. Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol Heart Circ Physiol. 2000;279:H422–8. doi: 10.1152/ajpheart.2000.279.1.H422. [DOI] [PubMed] [Google Scholar]
- 29.Cleutjens JP, Blankesteijn WM, Daemen MJ, et al. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res. 1999;44:232–41. doi: 10.1016/s0008-6363(99)00212-6. [DOI] [PubMed] [Google Scholar]
- 30.Eliseev RA, Malecki J, Lester T, et al. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J Biol Chem. 2009;284:9692–9. doi: 10.1074/jbc.M808750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forte M, Bernardi P. The permeability transition and BCL-2 family proteins in apoptosis: co-conspirators or independent agents? Cell Death Differ. 2006;13:1287–90. doi: 10.1038/sj.cdd.4401957. [DOI] [PubMed] [Google Scholar]
- 32.Palma E, Tiepolo T, Angelin A, et al. Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Hum Mol Genet. 2009;18:2024–31. doi: 10.1093/hmg/ddp126. [DOI] [PubMed] [Google Scholar]
- 33.Gomez L, Thibault H, Gharib A, et al. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1654–61. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- 34.Bagci G, Yucel I, Duranoglu Y. The effect of cyclosporin A on cultured rabbit subconjunctival fibroblast proliferation. Ophthalmologica. 1999;213:114–9. doi: 10.1159/000027403. [DOI] [PubMed] [Google Scholar]
- 35.Johnsson C, Gerdin B, Tufveson G. Effects of commonly used immunosuppressants on graft-derived fibroblasts. Clin Exp Immunol. 2004;136:405–12. doi: 10.1111/j.1365-2249.2004.02488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada H, Nishimura F, Naruishi K, et al. Phenytoin and cyclosporin A suppress the expression of MMP-1, TIMP-1, and cathepsin L, but not cathepsin B in cultured gingival fibroblasts. J Periodontol. 2000;71:955–60. doi: 10.1902/jop.2000.71.6.955. [DOI] [PubMed] [Google Scholar]