

Abstract
Background.
Hyperhomocysteinemia is associated with increased venous thrombosis and cardiovascular disease (CVD). Mutations in the human methylenetetrahydrofolate reductase (MTHFR) gene have been associated with increased homocysteine levels and risks of CVD in various populations including those with kidney disease. Here, we evaluated the influence of MTHFR variants on progressive loss of kidney function.
Methods.
We analyzed 821 subjects with hypertensive nephrosclerosis from the longitudinal National Institute of Diabetes and Digestive and Kidney Diseases African-American Study of Kidney Disease and Hypertension (AASK) Trial to determine whether decline in glomerular filtration rate (GFR) over ∼4.2 years was predicted by common genetic variation within MTHFR at non-synonymous positions C677T (Ala222Val) and A1298C (Glu429Ala) or by MTHFR haplotypes. The effect on GFR decline was then supported by a study of 1333 subjects from the San Diego Veterans Affairs Hypertension Cohort (VAHC), followed over ∼4.5 years. Linear effect models were utilized to determine both genotype [single-nucleotide polymorphism (SNP)] and genotype (SNP)-by-time interactions.
Results.
In AASK, the polymorphism at A1298C predicted the rate of GFR decline: A1298/A1298 major allele homozygosity resulted in a less pronounced decline of GFR, with a significant SNP-by-time interaction. An independent follow-up study in the San Diego VAHC subjects supports that A1298/A1298 homozygotes have the greatest estimated GFR throughout the study. Haplotype analysis with C677T yielded concurring results.
Conclusion.
We conclude that the MTHFR-coding polymorphism at A1298C is associated with renal decline in African-Americans with hypertensive nephrosclerosis and is supported by a veteran cohort with a primary care diagnosis of hypertension. Further investigation is needed to confirm such findings and to determine what molecular mechanism may contribute to this association.
Keywords: AASK, glomerular filtration rate, hypertension, kidney disease, MTHFR
Introduction
Methylenetetrahydrofolate reductase (MTHFR) is a key enzyme for intracellular folate homeostasis and metabolism which irreversibly catalyzes the conversion of 5,10-methylenetetrahydrofolate to 5-metyltetrahydrofolate (5-MTHF), a cosubstrate for homocysteine remethylation to methionine. Mutations in the MTHFR gene, found on human chromosome 1p36.3, have been associated with increased homocysteine levels [1, 2], which are in turn linked to increased venous thrombosis and cardiovascular disease (CVD) [3–5]. Two common nonsynonymous single-nucleotide polymorphisms (SNPs) have been noted in the MTHFR gene, which functionally alter the protein product [6]. C677T, found in Exon 4, (rs1801133, Ala222Val) results in a reduced-activity thermolabile variant, which has decreased stability and specificity of action [7] and is associated with decreased MTHFR activity and increased homocysteine levels [1]. A1298C (rs1801131, Glu429Ala), found in Exon 7, also reduces MTHFR activity, though seemingly less severely than C677T [8]. There may also be a functional interaction between these two polymorphisms which affect MTHFR enzyme activity [8]. These polymorphisms have previously been associated with a number of diseases including neural tube defects [1, 8], malignancies [9, 10], peripheral vascular disease [11] and CVD [7, 12].
Hyperhomocysteinemia is a long recognized problem in kidney disease patients, particularly in those with end-stage renal disease (ESRD) on dialysis, and thus may play a role in their increased CVD risk [13]. Combined as a risk factor with fibrinogen, homocysteine may explain almost 40% of the attributable mortality in chronic kidney disease (CKD) [14]. MTHFR polymorphisms have also been associated with CVD in ESRD subjects [15, 16] and increased risk of diabetic nephropathy [17, 18]. Whether common MTHFR variation is associated with accelerated chronic decline in renal function has not been systematically studied in a large at risk cohort.
Our study aims to evaluate two longitudinal hypertensive cohorts, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) sponsored multicenter randomized trial African-American Study of Kidney Disease and Hypertension (AASK) [19] and a local San Diego Veterans Affairs Hypertension Cohort (VAHC) [20] to determine if common MTHFR polymorphisms predict decline in renal function over time [by glomerular filtration rate (GFR) or estimated glomerular filtration rate (eGFR) slope].
Materials and methods
Subjects
AASK study subjects.
The first cohort included subjects from the AASK study, a 21 center randomized controlled prospective trial which has been described previously [19, 21]. Briefly, participants were 18- to 70-year-old self-identified African-Americans with hypertension and a clinical diagnosis of hypertensive renal disease, documented by an initial GFR (by [125I]-iothalamate clearance) between 20 and 65 mL/min/1.73 m2, urine protein:creatinine ratio (UPCR) <2.5 g/g and no other identifiable causes of renal insufficiency, particularly excluding subjects with diabetes mellitus. Based on a 3 × 2 factorial design, participants were randomized to one of two goal blood pressure (BP) ranges (‘usual’ mean arterial pressure goal of 102–107 mmHg or a lower mean arterial pressure goal of ≤92 mmHg) and to double-blinded treatment with one of three antihypertensive drug classes (40% to beta-blockade with metoprolol, 50–200 mg/day; 40% to angiotensin converting enzyme (ACE) inhibition with ramipril, 2.5–10 mg/day or 20% to calcium channel blockade with amlodipine, 5–10 mg/day). GFR was assessed by renal clearance of [125I]-iothalamate at baseline twice, at 3 and 6 months, and then every 6 months thereafter, for a mean of 4.3 ± 1.3 years (range 3.0–6.4 years) [22]. The primary endpoint in the current study was chronic GFR change over time, defined as that beginning 3 months after randomization, in order to better reflect long-term disease progression [22]. This was necessary given that the medical interventions (particularly calcium channel blockade) were noted to have acute effects on GFR which may differ from their long-term effects on disease progression [23, 24]. Time to event analyses were performed with a combined endpoint of ESRD, death or decrease of GFR by 50% [25].
Genomic DNA from blood leukocytes was ascertained and prepared at the time of enrollment from 830 of the original 1094 participants in the AASK study, who consented for genetic evaluation. Each subject gave informed, written consent to the local institutional review boards. The genotyped AASK subset (n = 830) did not differ from the complete AASK cohort (n = 1094) [19, 22, 26] in age, sex, body mass index (BMI), duration of hypertension, systolic blood pressure (SBP), diastolic blood pressure, serum creatinine, baseline GFR, baseline proteinuria or overall chronic rate of progression of renal disease (by normal and high proteinuria groups). Analysis was limited to the 821 of the subjects with genotype data at the MTHFR loci. Anthropometric descriptive values of the AASK study are presented in Table 1, participants were predominantly male (60.1%), had a mean age of 53.9 ± 10.7 years and a baseline GFR of 47.6 ± 13.5 mL/min/1.73 m2. Mean follow-up was 51.1 ± 15.3 months (4.3 ± 1.3 years).
Table 1.
Demographic characteristics at the start of the study for subjects from each of the two cohortsa
| Cohort | AASK | VAHC |
| N | 821 | 1333 |
| Means | ||
| Age (years) | 53.9 ± 10.7 | 64.0 ± 12.7 |
| BMI (kg/m2) | 31.0 ± 6.5 | 30.2 ± 5.9 |
| SBP (mmHg) | 150.0 ± 23.8 | 140.5 ± 19.4 |
| DBP (mmHg) | 95.8 ± 14.3 | 76.3 ± 12.8 |
| Pulse (mmHg) | 72.0 ± 12.2 | 72.7 ± 14.1 |
| Serum creatinine (mg/dl) | 2.0 ± 0.71 | 1.09 ± 0.31 |
| Baseline GFR (mL/min/1.73m2) | 47.6 ± 13.5 (iothalamate) | 80.0 ± 23.8 (eGFR by MDRD) |
| GFR slope over the study (mL/min/1.73m2/year) | −1.92 ± 2.4 | −1.86 ± 1.4 |
| Follow-up—months (years) | 51.1 ± 15.3 (4.3 ± 1.3) | 53.9 ± 22.8 (4.5 ± 1.9) |
| Percentages (N) | ||
| Ethnicity | 100% African-American | 68.2% Caucasian (909) |
| 15.2% African-American (202) | ||
| 5.8% Hispanic (77) | ||
| 10.9% Other (139) | ||
| Males/females | 60.1/39.8% (494/327) | 95.3/4.7% (1270/63) |
| Smokers (never/current/past) | 42.9/26.8/29.9% (352/220/245) | 25.5/20.0/54.5% (330/259/705) |
| Randomized BP goal | Low 50.2% (412)/usual 49.8% (409) | N\A |
| Randomized anti-hypertensive regimen | ACEI/beta blocker/calcium channel blocker: 41.0% (336)/39.5% (324)/19.2% (157) | N\A |
| % On beta-blocker | N\A | 41.6% (555) |
| % On ACE or angiotensin II receptor blocker | N\A | 52.4% (699), 8.2% (109) |
| % On calcium channel blocker | N\A | 32.4% (432) |
| Genotypes (N) | ||
| C677T: CC, CT, TT | 645, 164, 11 | 649, 551, 131 |
| (χ2 = 0.025, P = 0.88) | (χ2 = 1.01, P = 0.31) | |
| A1298C: AA, AC, CC | 582, 217, 21 | 687, 531, 115 |
| (HWE χ2 = 0.021, P = 0.89) | (HWE χ2 = 0.74, P = 0.39) | |
Means reported ± standard deviations. Randomized BP goal and anti hypertensive regimen are only available in the clinical trial AASK. HWE, Hardy–Weinberg equilibrium; DBP, Diastolic blood pressure.
Follow-up study: San Diego VAHC.
The San Diego VAHC is a multiethnic, noninterventional/observational cohort study of veterans recruited from San Diego Veterans Affairs Healthcare System primary care internal medicine clinics in 2003–04 [20, 27]. By self report, 68.2% were Caucasian, 15.2% African-American, 5.8% Hispanic and 10.9% other (Asian/Pacific Islander, American Indian, etc). Subjects had been diagnosed by their primary care physicians with essential hypertension or were on BP lowering medications, excluding subjects with known secondary causes of hypertension. The cohort utilizes the comprehensive VA complete electronic medical record (EMR) known as [Veterans Health Information Systems and Technology Architecture (VISTA); http://www.va.gov/VISTA_MONOGRAPH/] [28], which includes all vital signs, laboratory data, medical diagnoses, pharmacy records and procedure codes in digital format. EMR data were extracted from Microsoft SQL-Server tables for study participants from 1 year prior to their date of consent (October 2003–04) to October 2009. Subjects were excluded if they had prevalent or incident ESRD during this period (n = 30), including patients with a history of kidney transplantation, due to the inability to precisely determine when ESRD was reached. eGFR was estimated by the Modification in Diet and Renal Disease (MDRD) equation [29].
Genomic DNA was acquired from buccal cells at the time of enrollment into the study. In total, 1333 subjects were genotyped at the MTHFR locus and were included in this follow-up study as described in Table 1; 95.3% were men, reflecting the veteran population with 68.2% self-identified as white and 15.2% as black. At entry, 18.4% (245/1333) had evidence of CKD, defined by eGFR ≤60 mL/min/1.73 m2 [30]. By the end of the study, 38.4% had a diagnosis of type-2 diabetes with <2% of them coded for renal complications of diabetes. The mean age at the start of the study was 64.0 ± 12.7 years (range 21.7–91.4) with mean follow-up of 53.9 ± 22.8 months (4.5 ± 1.9 years), similar follow-up as the AASK study. The primary endpoint in this study was change in eGFR over time.
Genomics
SNP genotyping at MTHFR.
Two common polymorphisms at MTHFR were genotyped (C677T and A1298C). SNPs were obtained from the public database (http://www.ncbi.nlm.nih.gov/snp). Genomic DNA, from blood leukocytes in the AASK cohort and buccal cells in the VAHC group, was typed by an immobilized probe approach [31]. Reproducibility of genotyping was verified with 50 blinded replicate samples. Both SNPs evaluated in this study were in Hardy–Weinberg equilibrium (Table 1; P > 0.05) and were in partial linkage disequilibrium (for African-American subjects in each cohort D′ = 0.99, whereas r2 = 0.029 in VAHC and r2 = 0.0.024 in AASK), as determined by the Hill algorithm [32]. Minor allele frequency of A1298C and C677T were 15.7 and 11.3%, respectively in AASK and 16.6 and 12.9%, respectively in African-American subjects in VAHC. Online supplemental Table 1 shows the minor allele frequencies for each ethnic group in VAHC. Excluded subjects with incident or prevalent ESRD in VAHC had similar MTHFR A1298C and C677T distributions of genotypes compared to the study cohort.
Haplotype inference.
Two common variants were genotyped at MTHFR, allowing the inference of haplotypes from unphased diploid genotypes by the HAP imperfect phylogeny method (version 3.0; http://research.calit2.net/hap/Instructions.htm) [33]. Three common haplotypes (C677/A1298, 677T/A1298 and C677/1298C) were observed in the AASK study and VAHC study, shown in Online Supplemental Table 2, with the fourth haplotype (1298C/677T) absent in AASK and <1% in VAHC (0% in African-Americans). Due to the multiethnic nature, in the VAHC cohort, haplotype inference were performed in white and African-American subjects only and then combined for analysis. The other ethnic groups were too small for accurate inference to be determined. Haplotype analyses were determined by subjects with greater than or equal to one copy of each haplotype.
Statistical analyses
Descriptive statistics.
Means ± standard deviations are reported except as noted. All variables were normally distributed with the exception of UPCR in the AASK cohort, thus values are log transformed for analysis of association with the genetic polymorphisms.
Progression of GFR loss in AASK and VAHC using mixed models.
Linear mixed effects models (PROC MIXED) in SAS 9.2 (Cary, NC) [34] were utilized to assess the influence of the MTHFR polymorphisms on longitudinal GFR [23] (R. M. Salem, M. M. Fung, V. Bhatnagar, M. S. Lipkowitz et al., in preparation). This mixed model approach tests for both SNP main and SNP-by-time interaction effects and can be applied to unstructured and unbalanced longitudinal data [35], particularly to the repeated measurements taken at unequal time intervals of the VAHC study, which utilizes observational information in the EMR [34, 36, 37]. As noted, GFR in AASK was determined by renal clearance of [125I]-iothalamate assessed at baseline twice, at 3 and 6 months and then every 6 months thereafter, for a mean of 4.3 ± 1.3 years. Model 1 of the AASK cohort includes age at the start of the study, sex and randomization BP goal and antihypertensive regimen as covariates. In VAHC, the GFR was estimated by the MDRD equation and Model 1 includes age at date of enrollment, sex and ethnicity as covariates. Additionally, principal components were calculated using 77 additional SNPs from unrelated loci in order to adjust for possible admixture due to an ethnically diverse sampling in the VAHC cohort. Four principal components were then used to capture differences due to ethnicity and were entered as covariates.
Joint modeling.
In AASK, to address the potential confounding effects of reaching ESRD death, loss to follow-up or decrease in 50% of GFR (n = 192) on the longitudinal GFR change, joint modeling of longitudinal and time to event data was implemented [34]. In VAHC, participants with incident or prevalent ESRD were excluded from this analysis due to the EMR nature of the cohort and difficulty in discerning precisely when ESRD was reached, thus only death (n = 210) or loss to follow-up were included in the joint modeling. All VAHC subjects were noninformatively censored at their last serum creatinine and eGFR measurement. In brief for both cohorts, a flexible joint model framework has been formulated [38], allowing a broad range of dependencies between the longitudinal responses and event endpoints through a latent bivariate Gaussian process without specifying a class variable [39]. The joint model combines longitudinal and time to event information, allowing the covariance structure to be adjusted for censoring and event data [34, 38].
Plots/figures.
To illustrate the influence of the MTHFR A1298C variant on longitudinal GFR decline, chronic longitudinal GFR was plotted by SNP genotype over time (Figures 1 and 2). For demonstrative purposes, the longitudinal GFR profile by SNP genotype was generated using an adaptive regression cubic spline approach [40], with 95% confidence intervals for fitted spline function of the covariate-adjusted GFR values (from the joint model). The haplotype figures (Figures 1b and 2b) show presence of the MTHFR C677/1298C (one or two copies) versus absence (zero copies) due to the low frequency of two copies (3.0% in AASK and 8.0% in VAHC).
Fig. 1.
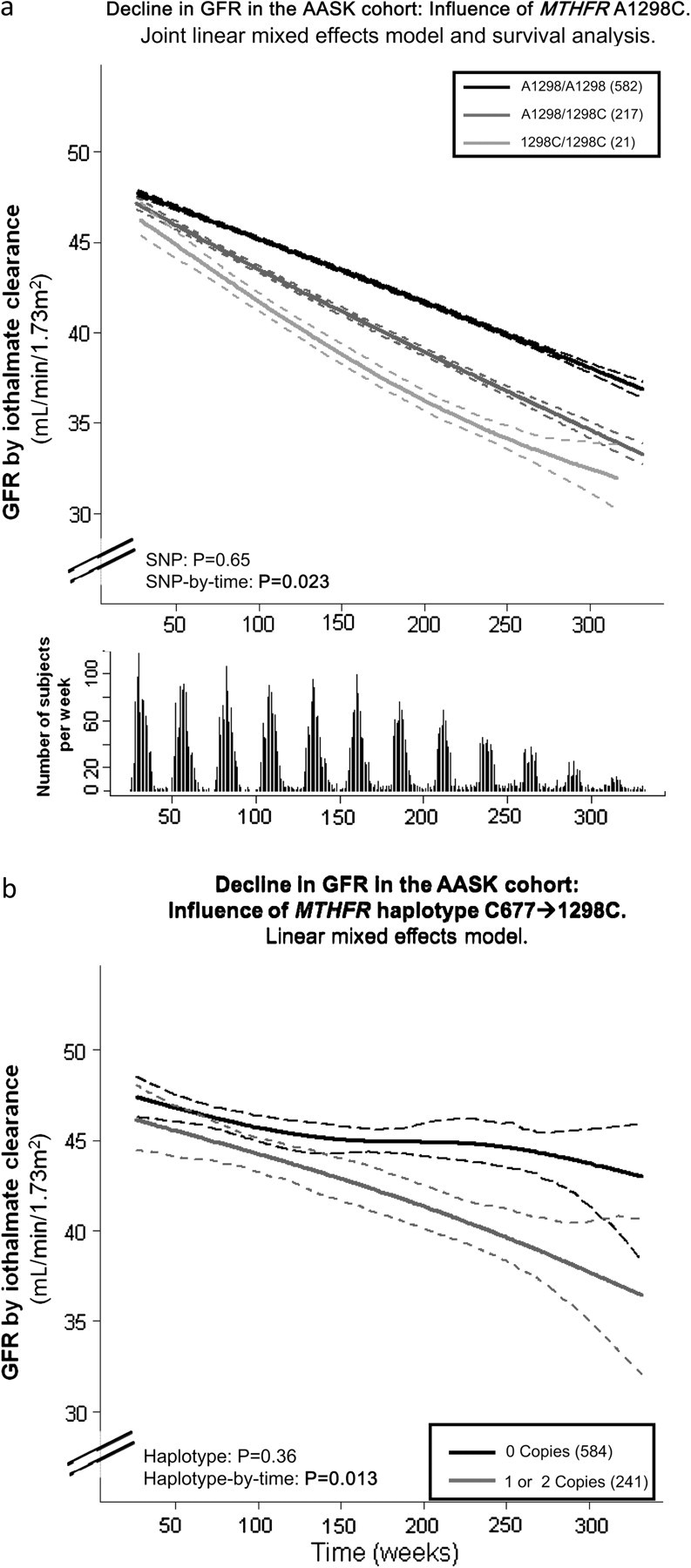
MTHFR polymorphism A1298C and haplotype with C677T predict decline in renal function over time in the AASK Trial. GFR decline (mL/min/1.73 m2/year) is plotted on the Y-axis and time in weeks from start of study is plotted on the X-axis. (a) SNP (genotype). The influence of MTHFR polymorphism A1298C on longitudinal GFR decline is shown. Adjusted GFR values from the joint analysis model, which combines longitudinal and time to event analysis are plotted using an adaptive regression cubic spline with 95% confidence intervals (dashed lines) for fitted curves. AASK subjects that were homozygous for the wild-type allele (A1298/A1298, light gray line) had the slowest decline in renal function (measured by iothalamate clearances over ∼4.2 years). Heterozygotes (dark gray line) and homozygotes for the minor allele (1298C/1298C, black line) had greater declines, showing the significant SNP-by-time interaction, P = 0.023 (main SNP effect P = 0.65), with adjustment for age, sex, drug and BP goal randomization groups. Below the graph indicates the number of GFR measurements that are included by week at each time point. (b) Haplotype. The influence of MTHFR haplotype 677C/1298C on longitudinal GFR decline are plotted for the AASK Study. Adjusted GFR values from the linear mixed effects model are plotted using an adaptive regression cubic spline with 95% confidence intervals (dashed lines) for fitted curves. Haplotype 677C/1298C had a haplotype-by-time interaction on GFR (by iothalamate clearance) as determined by linear mixed model analysis, such that those with at least one copy (dark gray line) had accelerated decline (P = 0.013; main haplotype effect P = 0.36) compared to those with no copies of the haplotype (solid black line).
Fig. 2.
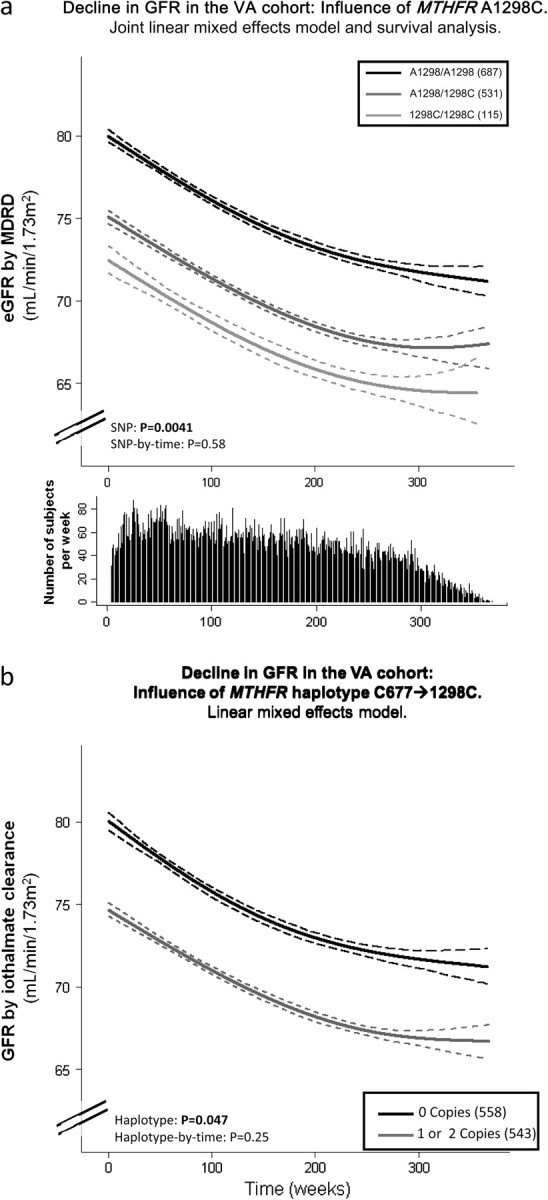
MTHFR polymorphism A1298C and haplotype with C677T was associated with renal function in the San Diego VAHC. GFR decline (mL/min/1.73 m2/year as estimated by the MDRD equation) is plotted on the Y-axis and time in weeks from start of study is plotted on the X-axis. (a) SNP (genotype). The influence of MTHFR polymorphism A1298C on longitudinal GFR decline is plotted for the VAHC Study. Adjusted GFR values from the joint model which combines longitudinal and time to event analysis are plotted using an adaptive regression cubic spline with 95% confidence intervals (dashed lines) for fitted curves. The mean curve for A1298/A1298 (solid black line), A1298/1298C (dark gray line) and 1298C/1298C (light gray line) are plotted. Subjects who were homozygous for the wild-type allele (A1298/A1298, light gray line) had the greatest eGFR. The difference in eGFR was statistically significant P = 0.0041 for the main SNP effect (SNP-by-time interaction, P = 0.58), adjusted for age, sex and ethnicity. Adjusting for possible confounding due to ethnic admixture using a principal components analysis yielded similar results. Below the graph indicates the number of subjects per week at each time point. (b) Haplotype. The influence of MTHFR haplotype 677C/1298C on longitudinal eGFR decline is plotted for the VAHC Study. Adjusted eGFR values from the linear mixed effects model are plotted using an adaptive regression cubic spline with 95% confidence intervals (dashed lines) for fitted curves. Subjects with at least one copy of the haplotype 677C/1298C (dark gray line) had worse renal function as determined by linear mixed model analysis compared to those with 0 copies (solid black line), main haplotype effect P = 0.047 adjusted for sex, ethnicity and baseline age (haplotype-by-time interaction P = 0.25).
Power analyses.
Statistical power was determined using the online instrument G*Power 3 (www.psycho.uni-duesseldorf.de/abteilungen/aap/gpower3/literature) [41]. More than 95% power to detect an effect size of 0.15 would be achieved with a sample size of 800, assuming alpha of 0.025.
All analyses were performed by the study investigators and not by the AASK data-coordinating center.
Results
Primary studies in NIDDK AASK subjects with hypertensive nephrosclerosis
MTHFR polymorphism contributes to change in GFR over time.
This genomic study cohort of 821 AASK study participants genotyped at the MTHFR loci (Table 1) had a mean age of 53.9 ± 10.7 years and initial GFR of 47.6 ± 13.5 mL/min/1.73 m2. Table 2 reports the results of association testing of MTHFR genetic variants on chronic GFR decline.
Table 2.
Effects of MTHFR genetic variants on GFR slopea
| Position | RefSNP | Minor allele frequency by cohort | SNP effect |
SNP-by-time effect |
|||
| Model 1 | Joint model | Model 1 | Joint model | ||||
| MTHFR variant | C677T | rs1801133 | AASK: T (0.11); | 0.54 | 0.47b | 0.99 | 0.97b |
| VAHC: T (0.31) | 0.16 | 0.81 | |||||
| A1298C | rs1801131 | AASK: C (0.16); | 0.68 | 0.65 | 0.021 | 0.023 | |
| VAHC: C (0.29) | 0.0065c | 0.0041 | 0.24 | 0.58 | |||
Effects of MTHFR polymorphisms and haplotypes are shown. In NIDDK AASK subjects, slope is chronic (after 3 months in the trial), determined by iothalamate clearance, with Model 1 results adjusted for sex, baseline age and randomized BP goal and antihypertensive treatment as covariates. In VAHC, the GFR was estimated by the MDRD equation, and Model 1 includes age at date of enrollment, sex and ethnicity as covariates. Bold P-values denote significance after Bonferroni correction.
The joint model for VAHC did not converge, and thus results are not reported.
After additional adjustment from principal component analysis of admixture in VAHC, P = 0.0055. The Joint Model combines the linear mixed model and time to event analysis, allowing estimation of the longitudinal analysis adjusted for time to event information and considers the same covariates as Model 1. In AASK, this includes time to death, dialysis, loss to follow-up or decrease of GFR by 50% and in VAHC includes the time to death or loss to follow-up.
MTHFR A1298C affected change in GFR: heterozygotes A1298/1298C and 1298C/1298C minor allele homozygosity predicted a greater decline of GFR with a significant SNP-by-time interaction (P = 0.021 in Model 1 which includes enrollment age, sex and BP and medication randomization groups as covariates; Table 2). Slope estimates are shown in Online Supplemental Table 2, with subjects with A1298/A1298 having a decline per week of 0.034 mL/min/1.73 m2 versus 0.048 and 0.042 mL/min/1.73 m2 in heterozygotes and 1298C/1298C subjects, respectively. The association was also not attenuated in a fully adjusted model which included sex, baseline age and UPCR, randomization group for BP control and medication, a moving average of SBP over the study period and history of CVD or stroke (P = 0.029, results not shown). Furthermore, when adjusting the models additionally for baseline GFR, results were not significantly altered. A joint analysis of this linear mixed model and time to event analysis accounting for death, dialysis, loss to follow-up and decrease of GFR by 50%, shown in Figure 1a, continued to show this association, P = 0.023 (with the Model 1 covariates). C677T alone did not show an individual effect, but haplotypes shown in Online Supplemental Table 3 suggest a potential interaction between C677 and the 1298C, such that the presence of the C677/1298C haplotype was associated with increased decline in GFR over time (P = 0.013 in Model 1; Figure 1b). No independent main effects of either of the polymorphisms or haplotypes were observed.
Neither of the MTHFR polymorphisms (A1298C or C677T) associated with drug or BP-goal randomization groups, sex, age, GFR or BMI at the start of the study. Baseline UPCR ratio (log transformed for normalization) was associated with MTHFR A1298C such that those with major allele homozygosity had the least amount of UPCR at enrollment (P = 0.014 adjusted for sex and age and P = 0.022 adjusted for sex, age, initial GFR and randomized drug and BP goal; results not shown).
Follow-up study: San Diego VAHC.
The supportive follow-up study consisted of 1333 veterans genotyped at the two common non-synonymous MTHFR loci as described in Table 1. In total, 95.3% were male and 68.2% self identified as Caucasian.
In VAHC, MTHFR A1298C predicted differences in eGFR over a mean follow-up of 4.5 ± 1.9 years, adjusted in Model 1 by sex, ethnicity and baseline age, though in a main SNP effect and not an SNP-by-time effect (Table 2). Slope estimates for the SNP-by-time effect, similar to those seen in AASK, are shown in Online Supplemental Table 2. Subjects homozygous for the minor allele (1298C/1298C) had a lower eGFR compared to heterozygotes (A1298/1298C) or wild-type homozygotes (A1298/A1298; P = 0.0065) throughout the study. Results were not changed when adjusting for evidence of AKI, SBP (a moving average value), smoking and comorbid conditions [diabetes, CVD, cancer and stroke (P = 0.034, data not shown)]. A joint model, which combines longitudinal and time to event analysis adjusting for censoring (loss to follow-up) and death, yielded similar results, with a main SNP effect for eGFR (P = 0.0041, Model 1, shown in Figure 2a). To adjust for the potential confounding effect of admixture, these analyses were further adjusted for four principal components, which capture ethnic differences within the VAHC cohort; results were statistically similar (for the main SNP effect, Model 1 P = 0.0055). However, given the main effect of the association, further adjustment for baseline eGFR reduced the significance of the association with the SNP.
Haplotype analyses (shown in Online Supplemental Table 3) of combined white and African-American subjects revealed a similar effect, such that subjects with presence of the 1298C allele, in conjunction with the C677 allele had a lower eGFR (P = 0.047, Figure 2b) adjusted for age, sex and ethnicity. No polymorphism or haplotype was associated SNP-by-time with eGFR decline.
Consistent with the main effect noted, eGFR at baseline was associated with the A1298C polymorphism, such that the homozygotes with the minor allele (1298C/1298C) had a lower eGFR at the start of the study (P = 0.008 after age, sex and ethnicity adjustment) than those with homozygote major alleles (A1298/A1298). The variant was not associated with sex or BMI in the VAHC study, though the participants differed in age at the date of consent (P < 0.001), with A1298/A1298 homozygotes being younger (62.7 ± 12.3 years) than A1298/1298C (65.8±12.7 years) or 1298C/1298C (65.9 ± 12.9 years) subjects.
Discussion
Overview
We present here evidence that the MTHFR polymorphism A1298C is associated with decline of GFR and GFR in two different populations, African-Americans with hypertensive nephrosclerosis and ethnically mixed veterans with hypertension, respectively. Previous studies have indicated that this gene is associated with a number of morbid conditions including cardiovascular and cerebrovascular disease. This is the first study to our knowledge that evaluates two common polymorphisms within the gene and associates them with longitudinal continuous renal function decline from either change in iothalamate-derived GFR or eGFR over time.
MTHFR genetic variation and the kidney
Few studies have evaluated MTHFR polymorphisms associations with renal disease. Consistent with our study, two case–control studies showed increased incidence of ESRD in subjects with 1298C/1298C [16, 42]. A study that divided patients by diagnostic categories of renal failure found that the C677T polymorphism alone and in combination with the A1298C polymorphism was associated with hypertensive nephrosclerosis [43]. One study of subjects with diabetes noted that the C677T mutation may have increased risk of diabetic nephropathy, though the A1298C polymorphism showed no association [18]. Two other studies reported the same association with C677T, though did not evaluate the A1298C polymorphism [44, 45]. The AASK trial excluded subjects with diabetes at the start although ∼14% developed diabetes by the end of the study and 38.4% of the VAHC cohort had diabetes by the end of the study. Thus, our study was not powered to study such an association.
In renal failure, associations of these MTHFR polymorphisms and CVD has had mixed results [15, 46, 47]. One case–control study failed to find an association with these polymorphisms and angiographic evidence of coronary artery disease [48]. A recent genome wide association study of BP with >34 000 subjects implicated a MTHFR polymorphism (rs17367504, in partial LD with A1298C and C677T with r2 = 0.36 and 0.064, respectively, from http://hapmap.ncbi.nlm.nih.gov/, which is located in an intron [49]), and a pharmacogenetic study suggested that the C677T polymorphism may influence response to an ACE inhibitor [50]. Thus, there may be unknown mechanisms in which MTHFR gene influences BP and drug response which may be either the cause or the result of decline in renal function either directly or indirectly.
The two common genetic mutations studied here, C677T and A1298C, have both been noted to decrease MTHFR enzyme activity [8]. Increased plasma homocysteine levels have then been associated with the C677T polymorphism alone and in combination with the A1298C mutation [8, 51]. As these polymorphisms [52] and elevated homocysteine levels have been associated with CVD morbidity and mortality, it was hypothesized that the pathophysiologic mechanism might be mediated through the elevated homocysteine levels. Yet effectively lowering the homocysteine levels with combination vitamin treatment, such as folate, B6 and B12, has not been shown to reduce mortality in advanced renal failure patients [53], who may have too large of a burden of disease to be successfully modified. Additionally, it has been suggested that homocysteine may be a marker of vascular disease rather than a causative agent [54]. A recent report notes that the C677T polymorphism may be a marker of 5-MTHF, the circulating metabolite of folic acid participating in homocysteine metabolism which appears to be the key regulator of endothelial nitric oxide synthase coupling and nitric oxide bioavailability in human vessels [55]. This may then encourage formation of oxygen-free radicals, contributing to elevated vascular oxidative stress, leading to a number of alterations in endothelial function and thrombogenicity and ultimately resulting in atherothrombosis [56]. Thus, plasma homocysteine, often associated with cerebrovascular and CVD, may be an indirect marker of 5-MTHF rather than a primary regulator of endothelial function. Consistent with this hypothesis are reports that elevated homocysteine levels may not be predictive of GFR progression in patients with CKD [57] nor their CVD mortality [58].
It is uncertain whether the MTHFR A1298C polymorphism also increases homocysteine levels. Previous studies have noted conflicting reports [16, 42, 48]. We were unable to evaluate plasma homocysteine levels since they were not measured at the start of the AASK trial and unavailable in the VAHC (an entirely observational cohort, utilizing the clinical EMR).
Most studies have focused on the C677T polymorphism, which is in incomplete linkage disequilibrium with A1298C [6, 59]. The C677T polymorphism has been hypothesized to have greater effects on MTHFR activity and influence on phenotypic outcomes than A1298C because of its location within the catalytic region. The C677T polymorphism is in Exon 4, within the N-terminal catalytic domain of the enzyme, whereas the A1298C polymorphism is in Exon 7, within the C-terminal regulatory domain [8]. The A1298C polymorphism may thus affect enzyme regulation, possibly by influencing S-adenosylmethionine, which is an allosteric inhibitor of MTHFR that binds in the C-terminal region [60].
Advantages and limitations of this study
Advantages of this study include use of the AASK trial with an independent supporting follow-up study. This AASK trial had accurately measured GFR by different techniques at multiple time points with very close follow-up. We were able to demonstrate the importance of the MTHFR gene by supportive results in a diverse observational cohort, VAHC, with detailed data from an EMR (further discussed below).
Our study also has limitations. In genetic studies, we must reduce the possibility of false-positive findings. Thus, we reduced the target P-value (by Bonferroni correction P = 0.025) and performed haplotype analyses, which continued to yield significant GFR predictions, with results that were congruent across the mixed model approach.
An exact replication of the AASK trial is unlikely, given the scope of its duration, size, intensity, complexity and expense. Though randomized clinical trials are considered the gold standard for clinical research, detailed comparison with observational studies via meta-analysis provides evidence that well-designed observational studies can produce valid and similar results [61, 62]. The VAHC has many characteristics which make it an ideal study population: primary care ascertainment basis, detailed and comprehensive EMR, nearly complete picture of medical use from a single provider, and the ability to track subjects longitudinally. Though, a limitation is that accuracy and completeness is reliant upon the EMR. Advanced statistical methods (including linear mixed models; see Methods) were employed to best utilize and transform the ambulatory setting data into models which may emulate a clinical trial.
The VAHC cohort consists of all veteran subjects, who are primarily male and with heterogeneous biogeographic ancestry. A principal component analysis was performed in order to adjust for potential confounding effect of admixture. This population was generally elderly (at 64.0 ± 12.7 years at the start of the study) with a corresponding modest decrease in eGFR (at 80.0 ± 23.8 mL/min/1.73 m2), though not so low as GFR in subjects with nephrosclerosis from the AASK study (Table 1).
Because the VAHC cohort is based upon the EMR, iothalamate clearance results, which were utilized in the AASK study, were not available. Rather, eGFR was determined by the MDRD equation [29], using ambulatory serum creatinine measurements all performed and reported by the same San Diego Veterans Affairs clinical laboratory. Such eGFR values are most accurate in subjects with GFR <60 mL/min/1.73 m2, tending to underestimate GFR in healthy individuals [29]. Though our sample included healthy subjects as well as those with CKD Stages 1 through 4 [30], imprecision of eGFR determination by underestimation in healthy individuals would be expected to bias the results toward the null, in contrast to the significant findings in our analyses. In AASK, eGFR and iothalamate clearance yielded similar results [63]. Further adjusting the results of the MTHFR A1298C association for baseline GFR in AASK did not significantly change the results. However, in VAHC given that a main SNP effect is noted, adjustment for baseline eGFR reduced the significance of the result. In longitudinal analyses, inclusion of the main effect as a covariate has been shown to result in an attenuation of a true signal and lead to an erroneous result [64].
Additionally, MTHFR A1298C was associated with UPCR in AASK as a potential confounder to the association with GFR decline over time. However, UPCR was available in few VAHC subjects at baseline and thus, we were unable to confirm the association or statistically adjust for it. As proteinuria is an important risk factor for kidney disease progression that may provide a potential link to the genotype, future study is needed to investigate this relationship.
Though MTHFR genetic variation at the A1298C locus influenced GFR progression in both cohorts, an SNP-by-time interaction is noted that in the AASK cohort, whereas a main SNP effect was noted in the VAHC cohort (Figures 1 and 2; Table 2). One potential reason may be that the influence of the variant may change over time as the kidney disease progresses. The AASK cohort, all with both hypertension and CKD, had a lower mean GFR at the start of the study and greater progression with more variability throughout the study, thus illustrating an SNP-by-time interaction (Figure 1a). Whereas the VAHC cohort, all with hypertension but only partially with CKD, had higher GFR at the start of the study illustrated only the main SNP effect (Figure 2a). The two cohorts studied here also differ in other substantive ways (Table 1): entry age (AASK less than VAHC), entry level of hypertension (AASK greater than VAHC), sex (more females in AASK than VAHC) and perhaps most importantly, biogeographic ancestry (the AASK cohort entirely African-American compared with the multiethnic VAHC). African-Americans with ‘hypertensive nephrosclerosis’ may be victims of more specific genetic predispositions to progressive azotemia, arising from genetic susceptibility variation at the MYH9 [65]/ApoL1 [66] loci. Treatment of BP also differed between the cohorts such that the AASK participants had well controlled BP monitored by the trial regularly and VAHC patients received standard care by their primary care physicians.
Conclusions and perspectives
Our study indicates that the MTHFR-coding polymorphism at A1298C is associated with renal decline. This was shown in African-Americans with hypertensive nephrosclerosis and supported by a veteran cohort with a primary care diagnosis of hypertension. Though polymorphisms from this gene appear to be associated with homocysteine levels and have been associated with cardiovascular risk factors including hypertension and here renal function, results should be confirmed in other populations and the precise pathophysiologic mechanism will require further investigation.
Supplementary data
Supplementary Tables 1–3 are available online at http://ndt.oxfordjournals.org.
Acknowledgments
We appreciate the support of the National Institutes of Health/National Center on Minority Health and Health Disparities-sponsored (MD000220) EXPORT minority health center as well as the National Institutes of Health/National Center for Research Resources-sponsored (RR00827) General Clinical Research Center.
Funding. This work was supported by the Department of Veterans Affairs and the National Institutes of Health: HL58120 (D.T.O.C.); RR00827 (UCSD General Clinical Research Center); MD000220 (UCSD Comprehensive Research Center in Health Disparities; CRCHD); DK048689 (AASK); RR000071 (Mount Sinai General Clinical Research Center), DK057867 (M.S.L.). N.J.S. is supported in part by the Scripps Translational Sciences Institute Clinical Translational Science Award (U54 RR0252204-01). This work was also supported by the CVD Genetics team and the High-Throughput Genotyping Group at Roche Molecular Systems, Inc.
Conflict of interest statement. At the time of research, M.M.F. had grant support from Forest Laboratories; currently she owns stock and is employed by Amgen. No other conflicts of interest. The results presented in this paper have not been published previously in whole or part, except in abstract format. This paper was presented as a poster at the American Society of Nephrology Renal Week 2009.
References
- 1.van der Put NM, Steegers-Theunissen RP, Frosst P, et al. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet. 1995;346:1070–1071. doi: 10.1016/s0140-6736(95)91743-8. [DOI] [PubMed] [Google Scholar]
- 2.Goyette P, Frosst P, Rosenblatt DS, et al. Seven novel mutations in the methylenetetrahydrofolate reductase gene and genotype/phenotype correlations in severe methylenetetrahydrofolate reductase deficiency. Am J Hum Genet. 1995;56:1052–1059. [PMC free article] [PubMed] [Google Scholar]
- 3.Den Heijer M, Lewington S, Clarke R. Homocysteine, MTHFR and risk of venous thrombosis: a meta-analysis of published epidemiological studies. J Thromb Haemost. 2005;3:292–299. doi: 10.1111/j.1538-7836.2005.01141.x. [DOI] [PubMed] [Google Scholar]
- 4.Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002;288:2015–2022. doi: 10.1001/jama.288.16.2015. [DOI] [PubMed] [Google Scholar]
- 5.Boushey CJ, Beresford SA, Omenn GS, et al. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA. 1995;274:1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- 6.Martin YN, Salavaggione OE, Eckloff BW, et al. Human methylenetetrahydrofolate reductase pharmacogenomics: gene resequencing and functional genomics. Pharmacogenet Genomics. 2006;16:265–277. doi: 10.1097/01.fpc.0000194423.20393.08. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher O, Kessling AM. MTHFR association with arteriosclerotic vascular disease? Hum Genet. 1998;103:11–21. doi: 10.1007/s004390050776. [DOI] [PubMed] [Google Scholar]
- 8.van der Put NM, Gabreels F, Stevens EM, et al. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62:1044–1051. doi: 10.1086/301825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pereira P, Stanton V, Jothy S, et al. Loss of heterozygosity of methylenetetrahydrofolate reductase in colon carcinomas. Oncol Rep. 1999;6:597–599. doi: 10.3892/or.6.3.597. [DOI] [PubMed] [Google Scholar]
- 10.Campbell IG, Baxter SW, Eccles DM, et al. Methylenetetrahydrofolate reductase polymorphism and susceptibility to breast cancer. Breast Cancer Res. 2002;4:R14. doi: 10.1186/bcr457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khandanpour N, Willis G, Meyer FJ, et al. Peripheral arterial disease and methylenetetrahydrofolate reductase (MTHFR) C677T mutations: A case-control study and meta-analysis. J Vasc Surg. 2009;49:711–718. doi: 10.1016/j.jvs.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Morita H, Taguchi J, Kurihara H, et al. Genetic polymorphism of 5,10-methylenetetrahydrofolate reductase (MTHFR) as a risk factor for coronary artery disease. Circulation. 1997;95:2032–2036. doi: 10.1161/01.cir.95.8.2032. [DOI] [PubMed] [Google Scholar]
- 13.Bostom AG, Lathrop L. Hyperhomocysteinemia in end-stage renal disease: prevalence, etiology, and potential relationship to arteriosclerotic outcomes. Kidney Int. 1997;52:10–20. doi: 10.1038/ki.1997.298. [DOI] [PubMed] [Google Scholar]
- 14.Shishehbor MH, Oliveira LP, Lauer MS, et al. Emerging cardiovascular risk factors that account for a significant portion of attributable mortality risk in chronic kidney disease. Am J Cardiol. 2008;101:1741–1746. doi: 10.1016/j.amjcard.2008.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haviv YS, Shpichinetsky V, Goldschmidt N, et al. The common mutations C677T and A1298C in the human methylenetetrahydrofolate reductase gene are associated with hyperhomocysteinemia and cardiovascular disease in hemodialysis patients. Nephron. 2002;92:120–126. doi: 10.1159/000064485. [DOI] [PubMed] [Google Scholar]
- 16.Poduri A, Mukherjee D, Sud K, et al. MTHFR A1298C polymorphism is associated with cardiovascular risk in end stage renal disease in North Indians. Mol Cell Biochem. 2008;308:43–50. doi: 10.1007/s11010-007-9610-7. [DOI] [PubMed] [Google Scholar]
- 17.Sun J, Xu Y, Zhu Y, et al. Genetic polymorphism of methylenetetrahydrofolate reductase as a risk factor for diabetic nephropathy in Chinese type 2 diabetic patients. Diabetes Res Clin Pract. 2004;64:185–190. doi: 10.1016/j.diabres.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 18.Moczulski D, Fojcik H, Zukowska-Szczechowska E, et al. Effects of the C677T and A1298C polymorphisms of the MTHFR gene on the genetic predisposition for diabetic nephropathy. Nephrol Dial Transplant. 2003;18:1535–1540. doi: 10.1093/ndt/gfg211. [DOI] [PubMed] [Google Scholar]
- 19.Wright JT, Jr, Kusek JW, Toto RD, et al. Design and baseline characteristics of participants in the African American Study of Kidney Disease and Hypertension (AASK) Pilot Study. Control Clin Trials. 1996;17(4 Suppl):3S–16S. doi: 10.1016/s0197-2456(96)00081-5. [DOI] [PubMed] [Google Scholar]
- 20.Salem RM, Pandey B, Richard E, et al. Electronic medical records in the post-genomic era: The VA Hypertension Primary Care Longitudinal Cohort. Health Informatics J. 2010;16:274–286. doi: 10.1177/1460458210380527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sika M, Lewis J, Douglas J, et al. Baseline characteristics of participants in the African American Study of Kidney Disease and Hypertension (AASK) Clinical Trial and Cohort Study. Am J Kidney Dis. 2007;50:78–89. doi: 10.1053/j.ajkd.2007.03.004. 89 e71. [DOI] [PubMed] [Google Scholar]
- 22.Wright JT, Jr, Bakris G, Greene T, et al. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA. 2002;288:2421–2431. doi: 10.1001/jama.288.19.2421. [DOI] [PubMed] [Google Scholar]
- 23.Agodoa LY, Appel L, Bakris GL, et al. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA. 2001;285:2719–2728. doi: 10.1001/jama.285.21.2719. [DOI] [PubMed] [Google Scholar]
- 24.Hall WD, Kusek JW, Kirk KA, et al. Short-term effects of blood pressure control and antihypertensive drug regimen on glomerular filtration rate: the African-American Study of Kidney Disease and Hypertension Pilot Study. Am J Kidney Dis. 1997;29:720–728. doi: 10.1016/s0272-6386(97)90125-6. [DOI] [PubMed] [Google Scholar]
- 25.Gassman JJ, Greene T, Wright JT, Jr., et al. Design and statistical aspects of the African American Study of Kidney Disease and Hypertension (AASK) J Am Soc Nephrol. 2003;14(7 Suppl 2):S154–S165. doi: 10.1097/01.asn.0000070080.21680.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fogo A, Breyer JA, Smith MC, et al. Accuracy of the diagnosis of hypertensive nephrosclerosis in African Americans: a report from the African American Study of Kidney Disease (AASK) Trial. AASK Pilot Study Investigators. Kidney Int. 1997;51:244–252. doi: 10.1038/ki.1997.29. [DOI] [PubMed] [Google Scholar]
- 27.Fung MM, Chen Y, Lipkowitz MS, et al. Adrenergic beta-1 receptor genetic variation predicts longitudinal rate of GFR decline in hypertensive nephrosclerosis. Nephrol Dial Transplant. 2009;24:3677–3686. doi: 10.1093/ndt/gfp471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown SH, Lincoln MJ, Groen PJ, et al. VistA–U.S. Department of Veterans Affairs national-scale HIS. Int J Med Inform. 2003;69:135–156. doi: 10.1016/s1386-5056(02)00131-4. [DOI] [PubMed] [Google Scholar]
- 29.Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 30.Levey AS, Stevens LA, Coresh J. Conceptual model of CKD: applications and implications. Am J Kidney Dis. 2009;53(3 Suppl 3):S4–S16. doi: 10.1053/j.ajkd.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 31.Cheng S, Grow MA, Pallaud C, et al. A multilocus genotyping assay for candidate markers of cardiovascular disease risk. Genome Res. 1999;9:936–949. doi: 10.1101/gr.9.10.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill W. Estimation of linkage disequilibrium in randomly mating populations. Heredity. 1974;33:229–239. doi: 10.1038/hdy.1974.89. [DOI] [PubMed] [Google Scholar]
- 33.Halperin E, Eskin E. Haplotype reconstruction from genotype data using imperfect phylogeny. Bioinformatics. 2004;20:1842–1849. doi: 10.1093/bioinformatics/bth149. [DOI] [PubMed] [Google Scholar]
- 34.Littell RC, Milliken GA, Stroup WW, et al. SAS for Mixed Models. 2nd edn. Cary, NC: SAS Institute, Inc.; 2006. [Google Scholar]
- 35.Holden JE, Kelley K, Agarwal R. Analyzing change: a primer on multilevel models with applications to nephrology. Am J Nephrol. 2008;28:792–801. doi: 10.1159/000131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Littell RC, Pendergast J, Natarajan R. Modelling covariance structure in the analysis of repeated measures data. Stat Med. 2000;19:1793–1819. doi: 10.1002/1097-0258(20000715)19:13<1793::aid-sim482>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 37.Wolfinger RD. An example of using mixed models and PROC MIXED for longitudinal data. J Biopharm Stat. 1997;7:481–500. doi: 10.1080/10543409708835203. [DOI] [PubMed] [Google Scholar]
- 38.Henderson R, Diggle P, Dobson A. Joint modelling of longitudinal measurements and event time data. Biostatistics. 2000;1:465–480. doi: 10.1093/biostatistics/1.4.465. [DOI] [PubMed] [Google Scholar]
- 39.Guo Xu, Carlin BP. Separate and joint modeling of longitudinal and event time data using standard computer packages. Am Statistician. 2004;58:16–24. [Google Scholar]
- 40.Denison DGT, Mallick BK, Smith AFM. Bayesian MARS. Stat Comput. 1998;V8:337–346. [Google Scholar]
- 41.Faul F, Erdfelder E, Lang AG, et al. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 42.Tripathi G, Sankhwar SN, Sharma RK, et al. Role of thrombotic risk factors in end-stage renal disease. Clin Appl Thromb Hemost. 2010;16:132–140. doi: 10.1177/1076029609335911. [DOI] [PubMed] [Google Scholar]
- 43.Koupepidou P, Deltas C, Christofides TC, et al. The MTHFR 677TT and 677CT/1298AC genotypes in Cypriot patients may be predisposing to hypertensive nephrosclerosis and chronic renal failure. Int Angiol. 2005;24:287–294. [PubMed] [Google Scholar]
- 44.Neugebauer S, Baba T, Watanabe T. Methylenetetrahydrofolate reductase gene polymorphism as a risk factor for diabetic nephropathy in NIDDM patients. Lancet. 1998;352:454. doi: 10.1016/S0140-6736(05)79188-1. [DOI] [PubMed] [Google Scholar]
- 45.Noiri E, Taguchi J, Nakao A, Fujita T. MTHFR gene polymorphism as an exacerbation factor of diabetic nephropathy in type 2 diabetes. Analysis in Japanese male hemodialysis patients. Diabetes Care. 2000;23:260. doi: 10.2337/diacare.23.2.260b. [DOI] [PubMed] [Google Scholar]
- 46.Pernod G, Bosson JL, Golshayan D, et al. Phenotypic and genotypic risk factors for cardiovascular events in an incident dialysis cohort. Kidney Int. 2006;69:1424–1430. doi: 10.1038/sj.ki.5000312. [DOI] [PubMed] [Google Scholar]
- 47.Ibrahim S, El Dessokiy O. Prevalence of methylenetetrahydrofolate gene (MTHFR) C677T polymorphism among chronic hemodialysis patients and its association with cardiovascular disease: a cross-sectional analysis. Clin Exp Nephrol. 2009;13:501–507. doi: 10.1007/s10157-009-0194-2. [DOI] [PubMed] [Google Scholar]
- 48.Kolling K, Ndrepepa G, Koch W, et al. Methylenetetrahydrofolate reductase gene C677T and A1298C polymorphisms, plasma homocysteine, folate, and vitamin B12 levels and the extent of coronary artery disease. Am J Cardiol. 2004;93:1201–1206. doi: 10.1016/j.amjcard.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Newton-Cheh C, Johnson T, Gateva V, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang S, Yu Y, Venners SA, et al. Effects of MTHFR and MS gene polymorphisms on baseline blood pressure and Benazepril effectiveness in Chinese hypertensive patients. J Hum Hypertens. 2011;25:172–177. doi: 10.1038/jhh.2010.50. [DOI] [PubMed] [Google Scholar]
- 51.Pavarino-Bertelli EC, Sanches de Alvarenga MP, Goloni-Bertollo EM, et al. Hyperhomocysteinemia and MTHFR C677T and A1298C polymorphisms are associated with chronic allograft nephropathy in renal transplant recipients. Transplant Proc. 2004;36:2979–2981. doi: 10.1016/j.transproceed.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 52.Jamison RL, Shih MC, Humphries DE, et al. Effect of the MTHFR C677T and A1298C polymorphisms on survival in patients with advanced CKD and ESRD: a prospective study. Am J Kidney Dis. 2009;53:779–789. doi: 10.1053/j.ajkd.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 53.Jamison RL, Hartigan P, Kaufman JS, et al. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA. 2007;298:1163–1170. doi: 10.1001/jama.298.10.1163. [DOI] [PubMed] [Google Scholar]
- 54.Eikelboom JW, Lonn E, Genest J, Jr., et al. Homocyst(e)ine and cardiovascular disease: a critical review of the epidemiologic evidence. Ann Intern Med. 1999;131:363–375. doi: 10.7326/0003-4819-131-5-199909070-00008. [DOI] [PubMed] [Google Scholar]
- 55.Antoniades C, Shirodaria C, Leeson P, et al. MTHFR 677 C>T Polymorphism reveals functional importance for 5-methyltetrahydrofolate, not homocysteine, in regulation of vascular redox state and endothelial function in human atherosclerosis. Circulation. 2009;119:2507–2515. doi: 10.1161/CIRCULATIONAHA.108.808675. [DOI] [PubMed] [Google Scholar]
- 56.Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med. 1998;338:1042–1050. doi: 10.1056/NEJM199804093381507. [DOI] [PubMed] [Google Scholar]
- 57.Samuelsson O, Lee DM, Attman PO, et al. The plasma levels of homocysteine are elevated in moderate renal insufficiency but do not predict the rate of progression. Nephron. 1999;82:306–311. doi: 10.1159/000045445. [DOI] [PubMed] [Google Scholar]
- 58.Menon V, Sarnak MJ, Greene T, et al. Relationship between homocysteine and mortality in chronic kidney disease. Circulation. 2006;113:1572–1577. doi: 10.1161/CIRCULATIONAHA.105.570127. [DOI] [PubMed] [Google Scholar]
- 59.Sandilands AJ, O'Shaughnessy KM. The functional significance of genetic variation within the beta-adrenoceptor. Br J Clin Pharmacol. 2005;60:235–243. doi: 10.1111/j.1365-2125.2005.02438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rozen R. Molecular genetics of methylenetetrahydrofolate reductase deficiency. J Inherit Metab Dis. 1996;19:589–594. doi: 10.1007/BF01799831. [DOI] [PubMed] [Google Scholar]
- 61.Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med. 2000;342:1887–1892. doi: 10.1056/NEJM200006223422507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kunz R. Randomized trials and observational studies: still mostly similar results, still crucial differences. J Clin Epidemiol. 2008;61:207–208. doi: 10.1016/j.jclinepi.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 63.Wang X, Lewis J, Appel L, et al. Validation of creatinine-based estimates of GFR when evaluating risk factors in longitudinal studies of kidney disease. J Am Soc Nephrol. 2006;17:2900–2909. doi: 10.1681/ASN.2005101106. [DOI] [PubMed] [Google Scholar]
- 64.Fitzmaurice GM, Laird N, Ware JH. Applied Longitudinal Analysis. Hoboken, NJ: Wiley-Interscience; 2004. [Google Scholar]
- 65.Bostrom MA, Freedman BI. The spectrum of MYH9-associated nephropathy. Clin J Am Soc Nephrol. 2010;5:1107–1113. doi: 10.2215/CJN.08721209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African-Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.