

Abstract
Pharmacological approaches are available to medically-managed patients with symptomatic BPH before surgical intervention is required. These include daily treatment with alpha-blockers and 5-alpha-reductase inhibitors alone or in combination. These medical approaches have two major problems. First, treatments are chronic and must be taken daily. Second, there are significant financial costs and quality of life issues for such chronic treatments. Is it possible to develop effective acute therapy for symptomatic BPH without the long-term androgen deprivation-induced side effects? Two seminal but rarely cited studies of Walsh [Peters, Walsh: N Engl J Med 317:599–604, 1987] and Coffey et al. [Sufrin et al.: Invest Urol 13:418–423, 1976], combined with the growing understanding of the stem cell organization of the prostate stromal (S) and epithelial (E) compartments and their reciprocal paracrine and autocrine interactions provides the rationale for an acute approach.
The Walsh study documents that: (1) androgen deprivation disrupts the reciprocal interaction between the prostate S and E thereby decreasing the weight of both compartments and (2) once BPH develops, androgen deprivation does not decrease the number of stem cell units in either the S or E compartments since subsequent androgen restoration fully restores the enlarged gland. The Coffey study documents that acute androgen deprivation sensitizes S–E interactions to radiation induced disruptions so that following radiation, androgen restoration does not induce full gland regrowth. Therefore, effective therapy for symptomatic BPH should be achievable by acute treatment with reversible androgen deprivation for a limited period followed by a single dose of conformal external beam radiation before allowing the man to recovery his normal serum testosterone.
Keywords: prostate inflammation, BPH, epithelial stem cells, mesenchymal stem cells, stromal-epithelial interactions, paracrine/autocrine loops
INTRODUCTION: PRESENT MEDICAL THERAPIES
BPH is a progressive condition of the aging male characterized by prostate growth accompanied by lower urinary tract symptoms (LUTS). The presence of LUTS in association with an enlargement of the prostate, as detected on digital rectal examination and routine laboratory evaluation of serum Prostate Specific Antigen (PSA) levels form the clinical basis for diagnosing BPH [1]. PSA levels serve as a surrogate measure of prostate volume, with a prostate equal to or larger than 30 cm3 usually associated with a PSA value of equal to or greater than 1.5 ng/ml [2]. With the increased use of PSA measurements particularly by primary care physicians, more men are being diagnosed with initially asymptomatic BPH. Risk factors associated with progression of BPH to clinical symptoms include prostate enlargement of equal to or greater than 30 cm3, moderate to severe symptoms (AUA-Symptom index score equal to or greater than 8), and PSA values equal to or greater than 1.5 ng/ml [1]. If untreated, BPH can result in both quality of life issues and acute urinary retention requiring surgical intervention. Presently, there are several pharmacological approaches to medically manage patients before surgical intervention is required. These include daily treatment with alpha-blockers (i.e., alfuzosin, doxazosin, tamsulosin, and terazosin) and 5-alpha-reductase inhibitors [5ARI] (i.e., finasteride and dutasteride) used alone or in combination.
Alpha-blockers target the adrenergic alpha1 receptors on smooth muscle cells within prostate stromal compartment inhibiting their tonic contraction. Clinical trials of daily alpha blockers have demonstrated a 10–20% improvement in total symptom scores. However, chronic alpha-blocker therapy does not reduce the risk of disease progression or long -term complications and does not reduce the size of the prostate [3,4]. In contract to alpha-blockers, chronic treatment with 5-alpha reductase inhibitors (5ARI) does cause a decrease in prostate volume. This occurs because 5ARI retards the irreversible conversion of testosterone to dihydrtestosterone (DHT). DHT is 10 times more potent an androgen due to its higher affinity binding for the androgen receptor than testosterone [5]. Thus 5ARI treatment lowers prostate tissue DHT without lowering tissue levels of testosterone inducing prostate epithelial apoptosis thus causing decrease prostate volume [6].
There are at least two isotypes of 5-alpha reductase with type I being expressed in many tissues including the prostate and type II having a more limited tissue expression including the prostate. Finasteride is an excellent type I inhibitor while dutasteride is an effective dual I/II inhibitor [6]. There have been several large finasteride clinical trials including the PROSPECT and PLESS studies. The PROSPECT trial was a 2-year study while the PLESS trial was initially a 4-year study which was extended for two additional years (i.e., 6 years total). These trials document that chronic finasteride treatment reduces prostate volume about 18–21% and improves urinary flow rate by ~1.4–1.9 ml/sec and lowers AUA-SI score by 2.1–3.3 [1]. The response to chronic treatment with the dual type I/II 5ARI, dutasteride in similar 2–4 year trials appears slightly better with prostate volume being reduced ~25%, urinary flow rate being increased by 2.2–2.7 ml/sec, and AUA-SI scores being reduced by 4.5–6.5 [1].
Based upon these results from single agent trials, it was logical to test the response to a combinational treatment with alpha-blocker plus 5ARI. The first large-scale placebo controlled trial of such a combinational approach was the MTOPS study. In this MTOPS trial, BPH patients were given either a combination of doxazosin plus finasteride or each agent alone or placebo for 4.5 years. The results of the MTOPS trial documented that the greatest benefit in terms of decreasing the long-term risk of symptom progression is achieved with the combinational treatment. This trial was followed by the SMART-1 trial which evaluated the combination of the dual 5ARI, dutasteride plus the alpha-blocker, tamsulosin for 6 months. This trial was designed to determine whether the alpha-blocker could be discontinued after 6 months of combination therapy without negatively affecting urinary symptoms. This study found that in the majority of men started on combination therapy, the alpha-blocker could be stopped and 5ARI continued for long-term control of disease progression [1].
While these results are encouraging, there are two major problems with these medical approaches. First, the treatment is chronic and has to be taken on a daily basis. Second, due to the requirement for daily treatment with the 5ARI, there are significant side effects [i.e., ~7–8% of the patients become impotent and 5–6% had a decreased libido within the first year of treatment] [1]. This raises the issue of whether it is possible to develop effective non-chronic therapy for symptomatic BPH without the long term androgen deprivation induced side effects. Two seminal but unfortunately rarely cited older studies of Walsh [7] and Coffey et al. [8] provide the rationale for such an approach. In order to appreciate the importance of these two older studies in providing the rationale for developing such an approach, an understanding of the mechanism of action of androgen in the normal prostate is required.
MECHANISM OF ANDROGEN ACTION IN THE PROSTATE
Androgen is the major growth factor for normal and BPH prostate tissue. For androgen to function, it must bind and activate signaling from Androgen Receptor (AR) protein [5]. In order to understand the mechanism for such AR dependent signaling in prostate, an understanding of the two phase development of the prostate is needed. The first ontogeny phase is initiated during embryonic life and continues into the early neonate period. During embryonic life, the prostate develops from the urogenital sinus (UGS) anlagen in males under the stimulation of systemic androgen from the testes. Such ontogeny requires reciprocal paracrine interactions between UGS stromal cells derived from the mesoderm and UGS epithelial cells derived from the ectoderm driven by a critical level of circulating androgen, Figure 1. Prostate ontogeny is initiated by an epithelial to stroma paracrine interaction permanently committing mesoderm UGS mesenchymal stem cells, denoted #1 in Figure 1, to producing a subset of differentiated progeny, denoted #2 in Figure 1, which mature into smooth muscle (SM) cells, denoted #3 in Figure 1, expressing both 5α-alpha reductase enzyme and AR protein [9,10].
Fig. 1.
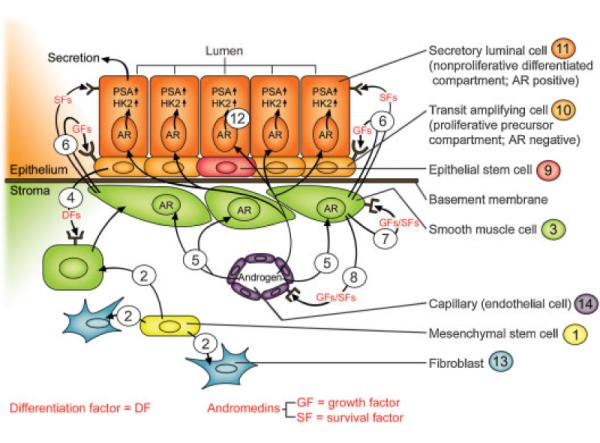
Reciprocal stromal epithelial interactions in the prostate.
The commitment of UGS stromal stem cells to produce SM cells expressing both of these proteins is dependent upon paracrine factors (i.e., Sonic Hedgehog, etc.) secreted by ectoderm UGS epithelial cells, denoted #4 in Figure 1 [9,11]. This induction is mandatory since genetically inherited defects in either 5-alpha reductase or AR prevent prostate development [5]. This is because expression of 5α-reductase by these SM cells allows them to irreversibly convert testosterone (T) to dihydrotestosterone (DHT). DHT is 10 times more potent than T in its ability to stimulate AR dependent transcription [5]. Thus, this 5-alpha reductase activity amplifies the low levels of circulating androgen secreted by the embryonic testes to produce a sufficient level of DHT to bind and activate AR signaling in these SM cells, denoted #5 in Figure 1. This AR signaling stimulates synthesis and secretion of soluble paracrine and autocrine growth factors (GF) and survival factors (SF), termed andromedins (e.g., IGF-1, FGF-7 and -10, and VEGF) by these SM cells, denoted #6 in Figure 1 [5,12,13]. Once secreted by the SM cells, andromedins diffuse and bind to their cognate receptors on specific cells types. Within the stromal compartment, such andromedin binding stimulates myogenesis, denoted #7 in Figure 1, and vasculo-genesis, denoted #8 in Figure 1. Within the epithelial compartment, andromedins stimulate epithelium to mature from medullary cords of undifferentiated epithelium which contain epithelial stem cells, denoted #9 in Figure 1, with no canalization into a simple stratified glandular epithelium composed of a basal layer of cuboidal cells, denoted #10 in Figure 1, upon which resides a second layer of columnar secretory-luminal cells, denoted #11 in Figure 1, adjacent to a patent lumen.
During embryonic and the early post-natal period, circulating level of T is sufficient to allow the normal S to E interaction for the initial ontogeny of the prostate. The circulating level of T decreases within the first year of life, however such that the prostate does not continue to grow. A second phase of prostate growth is initiated during puberty inducing the prostate to reach its normal adult size by 18–20 years of age. This second prostate growth phase is induced because at puberty, circulating T rises again to a sufficient level to re-stimulate adequate production of stromal andromedins.
Such positive paracrine stimulatory loops, Figure 1, do not go unopposed, however, since the prostate does not normally continue to grow once it has reached it adult size, even though there is no decrease in andromedin levels. Instead, the prostate reaches its normal adult size and then net growth ceases even though circulating levels of androgen are maintained. After reaching its adult size, the prostate epithelial compartment enters a steady state maintenance phase in which the rate of epithelial proliferation balances the rate of death such that neither overgrowth nor regression of the gland normally occurs [5]. At about the age of 40, however, in the vast majority of men, this kinetic balance is disrupted and with further aging and the gland enlarges eventually produces BPH [14]. The realization that both the stromal and epithelial compartments are organized into interactive stem cell units is providing a mechanistic understanding of how the reciprocal positive feedback loops are regulated so that the steady state size of the prostate is maintenance without overgrowth in the young adult. This realization also provides a framework for defining both the cause of BPH development in the aging male and new approaches for its treatment.
ORGANIZATION OF PROSTATE EPITHELIAL STEM CELL UNITS
Stem cells (SCs) are defined by their capacity for self-renewal, multi-lineage differentiation and replicative quiescence. Pluripotent embryonic SCs possess the most plasticity and can give rise to all tissues of an organism. During embryogenesis, there is a developmental process which results in the creation of tissue restricted stem cells, termed adult SCs which loss their pluripotency, but retain ability to self-renew and undergo multi-lineage differentiation to maintain the tissue. Adult SCs are generally quiescent and reside in a specialized cellular location known as a niche [15]. The niche provides a microenvironment that maintains the balance between quiescence and self-renewal of the stem cell population. The concept of adult SCs in the prostate first emerged to explain the profound capacity of this tissue for cyclic regeneration using a large cohort of adult age-matched (6-month-old at start of experiment) male rats [16].
Initially, the starting number of ventral prostate cells (i.e., 14.7±1.9×107) in these 6-month-old rats was determined on a group of these intact (i.e., control) rats and the remaining animals were castrated. One week of regression was allowed to reduce the total number of ventral prostate cells in these castrated animals to a value (i.e., 3.6±0.6×107) of only 25% of that of the intact control animals. The histology of these regressed glands documented the nearly complete loss of the secretory-luminal cells coupled with a major reduction in the volume of each glandular lumen. After this week of regression, the androgen deprived animals were subsequently implanted subcutaneously with a testosterone-filled silastic capsule, which restored the serum testosterone to intact control values (i.e., 2–3 ng/ml). One week after this testosterone replacement, the total number of ventral prostate cells increased by more than fourfold (i.e., two population doublings per cycle) back to a value identical with that of the intact control animals. After 1 week of exogenous androgen, the testosterone implant was surgically removed from each castrated animal (i.e., the 2-week involution/restoration cycle completed). The castrasted animals were then begun on a second cycle consisting of 1 week without exogenous androgen to induce prostatic regression followed by 1 week with exogenous androgen to induce two additional population doublings. This cycling was continued and after 10, 20, and 30 of such involution/restoration cycles, the total number of ventral prostate cells determined. These results documented that even after 60 additional population doublings (i.e., 30 cycles) the ventral prostate is completely able to repopulate itself normally [16]. This remarkable prostatic regenerative capacity documents that the self renewal of stem cells within the adult prostate is androgen independence. This conclusion is further demonstrated by the fact that if 1-year-old intact male rats with 16.8±0.9×107 total ventral prostate cells are castrated, the total number of prostate cell decreases to less than half within 1 month. If 1 year of androgen deprivation is allowed before restoring serum testosterone via silastic implant, the total number of ventral prostate cells in these long term castrates is fully restored to a value of 15.0±2.4×107 cells within 1 month.
Since our original studies 20 years ago, a large number of independent groups have added to the knowledge of how the prostate epithelial stem cell units are organized [17–23]. The results of these combined efforts are summarized in Figure 2. Prostate epithelial stem cells, denoted #9 in Figure 1, are present in niches within the basal layer of the epithelial compartment at a very low frequency (i.e., 0.5–1%). A defining characteristic of a prostate epithelial stem cell is that it does not express the AR or p63 proteins. This is consistent with the fact that embryonic UGS epithelial cells from either AR or p63 knock-out mice undergo prostatic ontogeny and glandular renewal when transplanted in combination with wild-type UGS mesenchymal cells [20,24,25]. While an individual prostate stem cell possess high self-renewal capacity, it proliferates infrequently to renew it selves and simultaneously generate progeny for two distinct cell lineages, Figure 2. The first and much less frequent lineage commitment is for terminally differentiation into a proliferatively quiescent neuroendocrine cell (NE in Fig. 2) which secretes a series of peptide growth factors [5]. The second and much more common lineage commitment is for differentiation into a progenitor that undergoes a limited number of proliferative replications (i.e., amplifications), before transiting a maturation process of terminal differentiation [5,19]. This progenitor is termed a transit-amplifying (TA) cell, denoted #10 in Figure 1. A TA cell does not express AR protein and is dependent for proliferation, but not survival, on andromedins produced by stromal cells [5,19]. One of the defining characteristics of a TA cell is its obligatorily expression of p63, as well as other basal markers such as cytokeratins 5 and 14, Jagged-1 and Notch-1 [5,19].
Fig. 2.
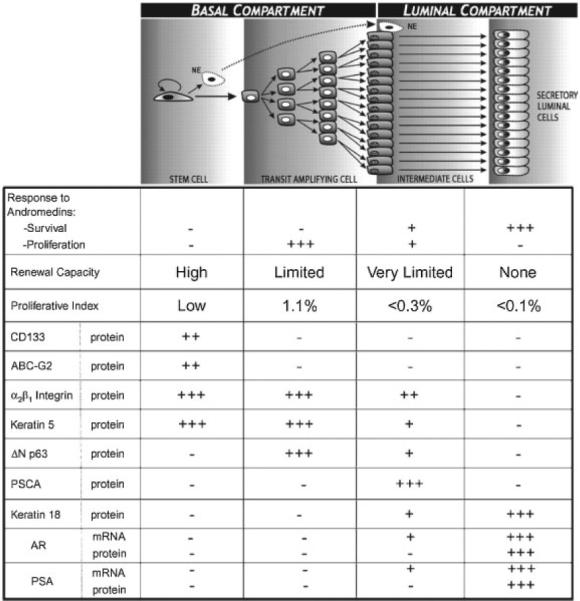
Overview of the phenotypic characteristics of the various cell subtypes with a prostate epithelial stem cell unit.
An individual TA progenitor cell undergoes a limited number of amplifying cell divisions expanding the cell population derived from a single stem cell before maturing into intermediate cell. This maturation involves down-regulation of p63, Jagged-1, Notch-1 and basal cytokeratins 5 and 14 expression [5,17,19,26]. A defining characteristic of an intermediate cell is its unique expression of prostate stem cell antigen (PSCA). This cell is termed Intermediate because it expresses both luminal lineage specific cytokeratins 8 and 18 as well as the basal lineage specific cytokeratins 5 and 15, and AR mRNA, but not AR protein. As an intermediate cell migrates upward from the basal layer to form the luminal layer, it stops expressing PSMA and now expresses AR protein [5,17,19,26]. Engagement of the AR pathway within a luminal cell, denoted #12 in Figure 1, induces its differentiation from a cuboidal into a columnar secretory-luminal cell expressing prostate specific markers like PSA.
We have recently documented that an additional function of the ligand occupied AR in prostatic secretory-luminal cells is to suppress the ability of these secretory cells to proliferate even in the presence of continuously high andromedin levels [27]. This active suppression involves an AR dependent pathway which up regulates expression of p21 and p27 Cdk inhibition proteins resulting in proliferative quiescence and terminal differentiation of these secretory-luminal cells. It is this ligand occupied AR suppression of proliferation of the secretory-luminal cells which prevents the androgen stimulated S to E positive feed-forward loop, Figure 1, from inducing continuous net prostatic growth in the presence of a continuous supply of andromedins.
Since secretory-luminal cells are the terminal stage of maturation of a hierarchical expanding population of cells comprising an individual stem cell unit, Figure 2, these secretory-luminal cells are quantitatively the major epithelial phenotype present in the gland even though they are proliferatively quiescent (i.e., terminally differentiated). Unlike their proliferating precursors, terminally differentiated secretory-luminal cells, denoted #11 in Figure 1, acquire a dependence on stromally derived andromedins for their survival. Hence, androgen deprivation induces apoptosis of these secretory-luminal cells due to a decrease in andromedin levels. It is the death of these secretory-luminal cells which predominately accounts for the regression of the prostate induced by androgen deprivation.
ORGANIZATION OF PROSTATE STROMAL STEM CELL UNITS
Besides prostate epithelial stem cells, there are also mesenchymal stem cells within prostate stroma. This conclusion is based upon the demonstration that there is a self-renewing subpopulation of prostate stromal cells from BPH tissue which express: (1) mesenchymal stem cell markers; (2) strong proliferative potential; and (3) ability to differentiate to fibroblastic, myogenic, adipogenic, and osteogenic lineages [28]. Of these potential lineages, the most characteristic commitment of progeny from prostate mesenchymal stem cell is to differentiate into a fibroblast, denoted #13 in Figure 1, or a smooth muscle cell, denoted #3 in Figure 1. This latter commitment, as discussed earlier, is due to embryonic paracrine effects of prostate epithelial cells, denoted #4 in Figure 1, which permanently commit the mesenchymal stem cells to give rise to a subset of progeny which can mature into AR expressing SM cells [9]. In the adult, the maturation of these SM requires paracrine factors secreted by prostate epithelial cells resulting in their expression of both 5-alpha reductase and AR proteins [10]. Once these SM cells mature, androgen binding to AR induces their secretion of andromedins, denoted #6 in Figure 1, including insulin-like growth factor-1 [IGF-1] [12], FGF-7 and -10 [5], and VEGF [13].
IGF-1 is not only a paracrine factor for prostate epithelial cells but also a critical autocrine factor, denoted #7 in Figure 1, for prostate stromal cells. In IGF-1 homozygous null mice, the prostate is smaller than in wild-type mice having less stromal SM cells as well as epithelial cells. If these IGF-1 null mice are castrated and then given androgen replacement, the restored gland is still smaller than in comparably treated wild type mice. When IGF-1 replacement is given alone to these castrated IGF-1null mice, there is a major increase in prostate smooth muscle volume with a smaller increase in prostate epithelial content. When androgen is combined with IGF-1 replacement to these castrated IGF-1 null mice, the prostate is maximally restored [29]. In wild-type hosts, including humans, IGF-1 production by SM is highly androgen stimulated [12,30]. These results document that IGF-1 is an androgen dependent autocrine factor produced on prostate SM, denoted #7 in Figure 1, and also a paracrine factor for prostate epithelial cells, denoted #6 in Figure 1. Another androgen dependent paracrine factor produced by SM, denoted #8 in Figure 1, is vascular endothelial growth factor [VEGF] [13]. VEGF maintains the endothelial cells, denoted #14 in Figure 1, and thus the blood supply in the prostate stromal compartment affecting both the stromal and epithelial cells [31].
INFLAMMATORY CHANGES IN BPH
During the development of symptomatic BPH, there is an increase in the ratio of prostate stromal to epithelial area going from a 3:1 ratio in non-hyperplastic normal prostates of young men to a ratio of 5:1 in BPH tissue [31]. This change is associated with an increase in cellular turnover in the stromal compartment of BPH versus normal prostate tissue [32]. Two thirds of this stromal compartment is composed of smooth muscle (SM) [33]. It has been suggested that the pathogenesis of BPH is associated with subtle changes in the phenotype of these SM cells in BPH [34]. This is based upon the demonstration that in BPH tissue, the SM is characterized by phenotypic changes including downregulation of SM myosin heavy chain (i.e., SMMHC) m-RNA expression [34] and upregulation of alpha2 macroglobulin [35]. The mechanism for such changes are not fully known but it is known that SM cells switch phenotype from contractive to proliferative in response to extrinsic and/or intrinsic stimuli, also know as SM phenotype modulation [36].
There is a growing literature supporting the concept that in BPH, such SM phenotype modulation occurs due to a chronic immune inflammatory process [37]. This idea is based upon the fact that nearly all BPH specimens contain inflammatory infiltrates but no bacterial or foreign antigens have been identified. Recognition of prostate secretory produces, like prostate specific antigen (PSA) and human Glandular Kallikrien-2 (hK2), by autoreactive T cells and animal models of experimental prostatitis demonstrate an autoimmune component to chronic prostate inflammation. The infiltrate consists predominantly of chronically activated CD4+ T lymphocytes, which are permanently recruited to prostate tissue via elevated expression of interleukin 15 (IL-15) and interferon gamma (IFN-gamma), proinflammatory cytokines produced by prostate SM and infiltrating T-cells, respectively. Disregulation of the immune response in BPH is further compounded by elevated expression of the proinflammatory IL-17 by autoreactive T cells, which stimulates enhanced production of IL-6 and IL-8 which stimulate stromal growth further increasing IL-15 levels. This combination of events, thus initiates an inflammation process [37]. Such an initial inflammation reaction eventually disrupts the barrier functions of the epithelial tight junctions allowing more autoantigens like PSA and hK2 to be released into the stromal compartment.
ROLE OF PSA-HK2 IN BPH
In normal prostate tissue, high concentrations of PSA are stored in the prostatic ductal network [38]. A very small quantity of PSA leaks out of the prostatic ductal network to generate the low ng/ml level that is measured in the circulation [38]. In contrast, in BPH, there is a disruption of normal tissue architecture in the prostate resulting in the leakage of increased amounts of PSA into the tissue interstitium and then into the circulation [38]. This enhanced release of autoantigens amplifies the problem producing a chronic inflammatory casade which “activates” the SM cells enhancing their abilities to produce andromedins. Besides being an autoantigen for initiating such an immune cascade, PSA is also an enzyme which when released into the stroma can subsequently degrade extracellular matrix proteins such as fibronectin and laminin. PSA can also release growth factors bound within the matrix structure. Previous in vitro studies have documented PSA's ability to cleave Insulin-like growth factor binding protein-3 (IGFBP3) leading to release of the prostatic mitogen, insulin-like growth factor-1 (IGF-1) [39] and to specifically activate the small latent form of TGFβ2 [40].
PSA can also form complexes with alpha-2macroglobulin (A2M) produced by smooth muscle within the stromal compartment of BPH tissue. Upon complex formation with proteases such as PSA, A2M undergoes a dramatic conformational change such that the A2M-protease complex is now recognized as a ligand by the A2M/LRP receptor. This receptor is expressed in the liver where it is involved in clearance of A2M-proteases complexes from the circulation. However, is also present in other tissue compartments and is highly expressed by macrophages. A number of laboratories have demonstrated that conformationally altered A2M can activate or inhibit signal transduction pathways through binding to membrane receptors [41,42]. A2M binding to A2M receptor/LRP activates G-protein activated pathways [42]. For instance binding of protease activated A2M to A2M/LRP receptors stimulates macrophage to increase cellular and nuclear COX-2 [43,44]. This COX-2 stimulation could be blocked with inhibitors of Protein kinase C, phospholipase A2, MAP kinase and phosphoinositide-3-kinase (PI 3-kinase) consistent with A2M receptor mediated activation of these downstream signal transduction pathways [41]. In addition to its role as a general protease inhibitor, increasing evidence points to a role for A2M in the regulation of cytokines in serum and tissue. Several cytokines, including interleukin-4, IL -10, TGF-β and nerve growth factor-α (NGF-α), bind covalently to A2M via disulfide bonds [45,46]. A2M can also bind interferon, tumor necrosis factor (TNF) and VEGF [47,48]. Interestingly, these interactions only occur when A2M is in its conformationally altered protease-bound state [48]. The significance of A2M binding to growth factors is unclear but evidence indicates its role for increased sequestration and degradation of growth factor/A2M complexes.
RATIONALE FOR ACUTE ANDROGEN DEPRIVATION PLUS SINGLE DOSE RADIATION COMBINATIONAL THERAPY FOR BPH
It is a common misconception that androgen deprivation therapy targets the only prostate epithelial compartment. As discussed earlier, the epithelial response induced by androgen deprivation is initiated by the decrease in production of andromedins by prostate stromal cells. In addition, androgen deprivation also results in a loss of VEGF production by stromal SM cells resulting in a decrease in prostate blood vessel density and blood flow [24]. This means that androgen deprivation therapy for BPH actually targets the stromal compartment to disrupt the reciprocal stromal-epithelial paracrine interactions and thereby restricting the hierarchical expansion of both the stromal and epithelial stem cells. This conclusion is documented by a classic study performed nearly 20 years ago by Dr. Patrick C. Walsh at Hopkins. Dr. Walsh treated a small series (i.e., n=9) of patients with symptomatic BPH with reversible androgen deprivation using an LHRH agonist for 6 months [7]. In all patients, serum testosterone decreased to castrate levels. Associated with this testosterone decrease was a decrease in enlarged prostate size of 25 % (i.e., mean prostate volume going from 48 g reaching a plateau in 4 months of androgen deprivation of 36 g). Morphological analysis of biopsies before and at 6 months of androgen deprivation documented a decrease of 40% in epithelial and 20% in stromal weight. Such androgen deprivation also resulted in 2/3 of the patients having an improvement in symptom scores and 1/3 having improvement in urinary flow rates [7]. These therapeutic effects were lost, however, when the LHRH agonist was stopped. Such loss of efficacy was associated with a return of the serum testosterone to normal physiolological levels and a corresponding return to the enlarged pretreatment prostate weight for both the stromal and epithelial compartments within 6 months of cessation of daily LHRH treatment [7].
Three conclusions can be made based upon this study. First, androgen deprivation disrupts the reciprocal interaction between the prostate stroma and epithelium and thereby decreases the weight of both compartments. Second, once BPH develops, androgen deprivation does not decrease the number of stem cell units in either the stromal and epithelial compartments since subsequent androgen restoration fully restored the enlarged gland. Third, once BPH develops, androgen ablation must be given chronically to remain therapeutic. Such chronic treatment, however, produces unwanted side effects.
Is there a way to decrease the reciprocal S to E interactions even in the presence of physiologically normal levels of circulating androgen, so that chronic androgen deprivation is not needed to maintain the lowered BPH prostate size? Experimentally, the answer is yes based upon the studies of Dr. Donald S Coffey et al. reported more than 30 years ago [8]. In these studies, male rats were androgen deprived (i.e., castrated) and allowed to regress over several days due to the death of the androgen dependent differentiated stem cell progeny. After this regression period, the ventral prostate was subsequently exposed to a single dose of gamma irradiation varying from 0 to 18 Gy. After this irradiation, the animals were allowed to go untreated for several days before the animals were given physiological androgen replacement for 10 days. In animals castrated and given no radiation or testosterone treatment, the ventral prostate regressed to only 20% of their starting size in intact non-castrated, non-irradiated hosts. The histology of the ventral prostates from these castrate only animals was atrophic with glandular acini composed of a dramatically reduced number of total cells and a complete lack of secretory-luminal cells. When castrated non-irradiated rats were treated for 10 days with exogenous testosterone replacement, the ventral prostates were fully restored to their starting size in intact non-castrated, non-irradiated hosts. Histologically, this restoration resulted in the regeneration of the normal high cellularity of each acini and the reappearance of secretory-luminal cells. In contract, when rats were castrated, irradiated and then subsequently given 10 days of androgen treatment, there was a dose dependent inhibition of the full ventral prostate restoration [i.e., 75% reduction in the androgen restoration induced regrowth of the ventral prostate by a single radiation dose of 18 Gy] [8]. Also the histology of these androgen stimulated ventral prostates in these irradiated animals was atrophic with glandular acini composed of a dramatically reduced number of total cells with a complete lack of secretory-luminal cells as similarly seem in the animals not given androgen replacement. Paradoxically, Coffey et al. further documented that when the ventral prostate of intact males was irradiated with a similar single exposure to 18 Gy, there was less than a 10% decrease in ventral prostate weight over the subsequent 10 day observation period and there was no loss of secretory-luminal cells [8].
These results document that there is a radiation sensitivity for disrupting prostate S–E interactions induced by acute androgen deprivation. This is consistent with the destruction of these S–E interactions being enhanced by the decreased levels of andromedins, like IGF-1 and VEGF, within the prostate of acutely androgen deprived hosts. Apparently, once disrupted by radiation, prostate S–E interactions are not fully recovered by androgen restoration. These results lead to the hypothesis that an effective therapy for men with symptomatic BPH is to treated with reversible androgen deprivation (i.e., LHRH or 5ARI) for ~2 months and then give a single dose of conformal external beam radiation before allowing the man to recovery his normal serum testosterone (i.e., acute androgen deprivation/radiation combinational approach).
There are precedents for such radiation sensitivity for permanently disrupting S–E interactions induced by hormonal deficiency in human. For example, similar S–E interactions are involved in the mechanism of estrogen action in the breast [49]. In adult males, breast tissue is present but the mammary glands are atrophic. When men with metastatic prostate cancer are given estrogens, they develop gynecomastia. If they are given a single exposure to as low as 9 Gy of radiation before such estrogen treatment is begun, the gynecomastia is prevented even up to 4 years post-radiation [50]. If the radiation is given 1–4 months after the beginning of estrogen treatment, however, it is much less able to prevent gynecomastia [51].
REFERENCES
- 1.Nasland MJ, Miner M. A Review of the Clinical Efficacy and Safety of 5alpha- Reductase Inhibitors for the Enlarged Prostate. Clin Ther. 2007;29:17–25. doi: 10.1016/j.clinthera.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 2.Roehrborn CG, McConnell JD, Lieber M, Kaplan S, Geller J, Malek GH, Castellanos R, Coffield S, Saltzman B, Resnick M, Cook TJ, Waldstreicher J. Serum prostate-specific antigen concentration is a powerful predictor of acute urinary retension and need for surgery in men with clinical benign prostatic hyperplasia. Urology. 1999;53:473–480. doi: 10.1016/s0090-4295(98)00654-2. [DOI] [PubMed] [Google Scholar]
- 3.Boyle P, Roehrborn C, Harkaway R, Logie J, de la Rosette J, Emberton M. %-Alpha reductase inhibition provides superior benefits to alpha blockade by preventing AUR and BPH-related surgery. Eur Urol. 2004;45:620–626. doi: 10.1016/j.eururo.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Souverein PC, Erkens JA, de la Rosette JJ, Leufkens HG, Herings RM. Drug treatment of benign prostatic hyperplasia and hospital admission for BPH-related surgery. Eur Urol. 2003;43:528–534. doi: 10.1016/s0302-2838(03)00089-7. [DOI] [PubMed] [Google Scholar]
- 5.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles's heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88:2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 6.Xu Y, Dalrymple SL, Becker RE, Denmeade SR, Isaacs JT. Pharmacological basis for the enhanced efficacy of Dutasteride against prostate cancer. Clin Cancer Res. 2006;12:4072–40796. doi: 10.1158/1078-0432.CCR-06-0184. [DOI] [PubMed] [Google Scholar]
- 7.Peters CA, Walsh PC. The effect of nafaralin acetate, A luteinizing-hormone-releaseing hormone agonist, on benign prostatic hyperplasia. N Engl J Med. 1987;317:599–604. doi: 10.1056/NEJM198709033171004. [DOI] [PubMed] [Google Scholar]
- 8.Sufrin G, Heston WDW, Hazra T, Coffey DS. The effect of radiation on prostatic growth. Invest Urol. 1976;13:418–423. [PubMed] [Google Scholar]
- 9.Cunha GR, Hayward SW, Daihiya R, Foster BA. Smooth muscle-epithelial interactions in normal and neoplastic prostate development. Acta Anat. 1996;155:63–72. doi: 10.1159/000147791. [DOI] [PubMed] [Google Scholar]
- 10.Bayne CW, Donnelly F, Chapman K, Bollina P, Buck C, Habib F. A novel coculture model for benign prostatic hyperplasia expressing both isoforms of 5alpha-reductase. J Endocrinol Metab. 1998;83:206–213. doi: 10.1210/jcem.83.1.4486. [DOI] [PubMed] [Google Scholar]
- 11.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signaling in prostate regeneration, neoplasia, and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 12.Ohlson N, Bergh A, Stattin P, Wikstrom P. Castration-induced epithelial cell death in human prostate tissue is related to locally reduced IGF-1 levels. Prostate. 2007;67:32–40. doi: 10.1002/pros.20480. [DOI] [PubMed] [Google Scholar]
- 13.Richard C, Kim G, Koikawa Y, Salm SN, Tsujimura A, Wilson EL, Moscatelli D. Androgens modulate the balance between VEGF and angiopoietin expression in prostate epithelial and smooth muscle cells. Prostate. 2002;50:83–91. doi: 10.1002/pros.10035. [DOI] [PubMed] [Google Scholar]
- 14.Rhodes T, Girman CJ, Jacobsen SJ, Roberts RO, Guess HA, Lieber MM. Longitudinal prostate growth rates during 5 years in randomly selected community men 40 to 79 years old. J Urol. 1999;161:1174–1179. [PubMed] [Google Scholar]
- 15.Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: Stem cells and their niche. Cell. 2004;116:769–778. doi: 10.1016/s0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- 16.Isaacs J, Coffey DS. Etiology and disease process of benign prostatic hyperplasia. Prostate Suppl. 1989;2:33–50. doi: 10.1002/pros.2990150506. [DOI] [PubMed] [Google Scholar]
- 17.Tran CP, Lin C, Yamashiro J, Reiter RE. Prostate stem cell antigen is a marker of late intermediate prostate epithelial cells. Mol Cancer Res. 2002;1:113–121. [PubMed] [Google Scholar]
- 18.Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT. CD133, anovel marker for human prostate epithelial stem cells. J Cell Sci. 2004;117:3539–3545. doi: 10.1242/jcs.01222. [DOI] [PubMed] [Google Scholar]
- 19.Litvinov IV, Vander Griend DJ, Xu Y, Antony L, Dalrymple SL, Isaacs JT. Low-calcium serum-free medium selects for growth of normal prostate stem cells. Cancer Res. 2006;66:8598–8607. doi: 10.1158/0008-5472.CAN-06-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Signoretti S, Loda M. Defining cell lineages in prostate epithelium. Cell Cycle. 2006;5:138–141. doi: 10.4161/cc.5.2.2340. [DOI] [PubMed] [Google Scholar]
- 21.Goto K, Salm SN, Coetzee S, Xiong X, Burger PE, Shapiro E, Lepor H, Moscatelli D, Wilson EL. Proximal prostatic stem cells are programmed to regenerate a proximal-distal ductal axis. Stem Cells. 2006;24:1859–1868. doi: 10.1634/stemcells.2005-0585. [DOI] [PubMed] [Google Scholar]
- 22.Xin L, Lukacs RU, Lawson DA, Cheng D, Witte On. Self-renewnal and multilineage differentiation in vitro from prostate stem cells. Stem Cells. 2007;25:2760–2769. doi: 10.1634/stemcells.2007-0355. [DOI] [PubMed] [Google Scholar]
- 23.Barclay WW, Axanova LS, Chen W, Romero L, Maund SL, Soker S, Lees CJ, Cramer SD. Characterization of adult prostate progenitor/stem cells exhibiting self-renewal and multilineage differentiation. Stem Cells. 2007 Nov 29; doi: 10.1634/stemcells.2007-0309. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunha GR, Alarid ET, Turner T, Donjacour AA, Boutin EL, Foster BA. Normal and abnormal development of the male urogenital tract. Role of androgens, mesenchymal-epithelial interactions, and growth factors. J Androl. 1992;13:465–475. [PubMed] [Google Scholar]
- 25.Kurita T, Medina RT, Mills AA, Cunha GR. Role of p63 and basal cells in the prostate. Development. 2004;131:4955–4964. doi: 10.1242/dev.01384. [DOI] [PubMed] [Google Scholar]
- 26.Dalrymple S, Antony L, Xu Y, Uzgare AR, Arnold JT, Savaugeot J, Sokoll LJ, De Marzo AM, Isaacs JT. Role of notch-1 and E-cadherin in the differential response to calcium in culturing normal versus malignant prostate cells. Cancer Res. 2005;65:9269–9279. doi: 10.1158/0008-5472.CAN-04-3989. [DOI] [PubMed] [Google Scholar]
- 27.Vander Griend DJ, Litvinov IV, Gurel B, Antony l, Dalrymple SL, Becker RE, DeMarzo AM, Isaacs JT. Androgen receptor is a cell-context dependent growth suppressor in normal prostate epithelium, but an oncogene in human prostate cancers. in press. [Google Scholar]
- 28.Lin VK, Wang SY, Vasquez DV, Xu CC, Zhang S, Tang L. Prostatic stromal cells derived from benign prostatic hyperplasia specimens posses stem cell like property. Prostate. 2007;67:1265–1276. doi: 10.1002/pros.20599. [DOI] [PubMed] [Google Scholar]
- 29.Kleinberg DL, Ruan W, Yee D, Kovacs T, Vidal S. Insulin-like growth factor (IGF)-1 controls prostate fibromuscular and glandular development in eugonadal mice. Endocrinology. 2007;148::1080–1088. doi: 10.1210/en.2006-1272. [DOI] [PubMed] [Google Scholar]
- 30.Le H, Arnold JT, McFann KK, Blackman MC. DHT and testosterone, but not DHEA or E2, differentially modulate IGF-1, IGFBP-2, and IGFBP-3 in human prostate stromal cells. Am J Physiol Endocrinol Metab. 2006;290:E952–E960. doi: 10.1152/ajpendo.00451.2005. [DOI] [PubMed] [Google Scholar]
- 31.Lissbrant IF, Lissbrant E, Damber J, Bergh A. Blood vessels are regulators of growth, diagnostic markers, and therapeutic targets in prostate cancer. Scand J Urol Nephrol. 2001;35:437–452. doi: 10.1080/003655901753367532. [DOI] [PubMed] [Google Scholar]
- 32.Shapiro E, Becich MJ, Hartanto V, Lepor H. The relative proportion of stromal and epithelial hyperplasia is related to the development of symptomatic benign prostatic hyperplasia. J Urol. 1992;147:1293–1297. doi: 10.1016/s0022-5347(17)37546-8. [DOI] [PubMed] [Google Scholar]
- 33.Kyprianou N, Huacheng TU, Jacobs SC. Apoptotic versus proliferative activities in human benign prostatic hyperplasia. Hum Pathol. 1996;27:668–675. doi: 10.1016/s0046-8177(96)90396-2. [DOI] [PubMed] [Google Scholar]
- 34.Lin VK, Benaim EA, McConnell JD. Alpha-blockade down-regulates myosin heavy chain expression in human benign prostatic hyperplasia. Urology. 2001;57:170–175. doi: 10.1016/s0090-4295(00)00842-6. [DOI] [PubMed] [Google Scholar]
- 35.Lin VK, Wang D, Lee IL, Vasquez D, Fagelson JE, McConnell JD. Myosin heavy chain gene expression in normal and hyperplastic human prostate tissue. Prostate. 2000;44:193–203. doi: 10.1002/1097-0045(20000801)44:3<193::aid-pros3>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 36.Lin VK, Wang SY, Boetticher NC, Vasquez DV, Saboorian H, McConnell JD, Roehrborn Alpha2 macroglobulin, a PSA binding protein, expressed in human prostate stroma. Prostate. 2005;63:299–308. doi: 10.1002/pros.20183. [DOI] [PubMed] [Google Scholar]
- 37.Owens GK, Kumar MS, Wamhoff BR. Vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 38.Williams SA, Singh P, Isaacs JT, Denmeade SR. Does PSA play a role as a promoting agent during the initiation and/or progression of prostate cancer? Prostate. 2007;67:312–329. doi: 10.1002/pros.20531. [DOI] [PubMed] [Google Scholar]
- 39.Cohen P, Graves HC, Peehl DM, Kamarei M, Giudice LC, Rosenfeld RG. Prostate-specific antigen (PSA) is an insulin-like growth factor binding protein-3 protease found in seminal plasma. J Clin Endocrinol Metab. 1992;75:1046–1053. doi: 10.1210/jcem.75.4.1383255. [DOI] [PubMed] [Google Scholar]
- 40.Dallas SL, Zhao S, Cramer SD, Chen Z, Peehl DM, Bonewald LF. Preferential production of latent transforming growth factor beta-2 by primary prostatic epithelial cells and its activation by prostate-specific antigen. J Cell Physiol. 2005;202:361–370. doi: 10.1002/jcp.20147. [DOI] [PubMed] [Google Scholar]
- 41.Misra UK, Gonzalez-Gronow M, Gawdi G, Hart JP, Johnson CE, Pizzo SV. The role of Grp 78 in alpha 2-macroglobulin-induced signal transduction. Evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP 78-mediated signal transduction. J Biol Chem. 2002;277:42082–42087. doi: 10.1074/jbc.M206174200. [DOI] [PubMed] [Google Scholar]
- 42.Misra UK, Pizzo SV. Activation of Akt/PDK signaling in macrophages upon binding of receptor-recognized forms of alpha2-macroglobulin to its cellular receptor: Effect of silencing the CREB gene. J Cell Biochem. 2004;93:1020–1032. doi: 10.1002/jcb.20233. [DOI] [PubMed] [Google Scholar]
- 43.Misra UK, Pizzo SV. Induction of cyclooxygenase-2 synthesis by ligation of the macrophage alpha(2)-macroglobulin signalling receptor. Cell Signal. 2001;11:801–808. doi: 10.1016/s0898-6568(01)00202-9. [DOI] [PubMed] [Google Scholar]
- 44.Misra UK, Pizzo SV. Regulation of cytosolic phospholipase A2 activity in macrophages stimulated with receptor-recognized forms of alpha 2-macroglobulin: Role in mitogenesis and cell proliferation. J Biol Chem. 2002;277:4069–4078. doi: 10.1074/jbc.M109764200. [DOI] [PubMed] [Google Scholar]
- 45.Garber TR, Gonias SL, Webb DJ. Interleukin-4 and IL-10 bind covalently to activated human alpha2-macroglobulin by a mechanism that requires Cys94. J Interferon Cytokine Res. 2000;20:125–131. doi: 10.1089/107999000312522. [DOI] [PubMed] [Google Scholar]
- 46.Gonias SL, Carmichael A, Mettenburg JM, Roadcap DW, Irvin WP, Webb DJ. Identical or overlapping sequences in the primary structure of human alpha(2)-macroglobulin are responsible for the binding of nerve growth factor-beta, platelet-derived growth factor-BB, and transforming growth factor-beta. J Biol Chem. 2000;275:5826–5831. doi: 10.1074/jbc.275.8.5826. [DOI] [PubMed] [Google Scholar]
- 47.James K, van den Haan J, Lens S, Farmer K. Preliminary studies on the interaction of TNF alpha and IFN gamma with alpha 2-macroglobulin. Immunol Lett. 1992;32:49–57. doi: 10.1016/0165-2478(92)90198-w. [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharjee G, Asplin IR, Wu SM, Gawdi G, Pizzo SV. The conformation-dependent interaction of alpha 2-macroglobulin with vascular endothelial growth factor. A novel mechanism of alpha 2-macroglobulin/growth factor binding. J Biol Chem. 2000;275:26806–26811. doi: 10.1074/jbc.M000156200. [DOI] [PubMed] [Google Scholar]
- 49.Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the estrogen receptor alpha is required for proliferation and morphogenesis in the mammary glands. Proc Natl Acad Sci. 2006;103:2196–2201. doi: 10.1073/pnas.0510974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fass D, Steinfeld A, Brown J, Tessler A. Radiotherapuetic prophylaxis of estrogen-induced gynecomastia: A study of late sequela. Int J Radiat Oncol Biol Phys. 1986;12:407–408. doi: 10.1016/0360-3016(86)90359-7. [DOI] [PubMed] [Google Scholar]
- 51.Alfthan O, Kettunen K. The effect of roentgen ray treatment of gynecomastia in patients with prostate carcinoma treated with estrogenic hormones: A preliminary communication. J Urol. 1965;94:604–606. doi: 10.1016/S0022-5347(17)63684-X. [DOI] [PubMed] [Google Scholar]