

Abstract
Global hypoxia-ischemia interrupts oxygen delivery and blood flow to the entire brain. Previous studies of global brain hypoxia-ischemia have primarily focused on injury to the cerebral cortex and to the hippocampus. Susceptible neuronal populations also include inhibitory neurons in the thalamic reticular nucleus. We therefore investigated the impact of global brain hypoxia-ischemia on the thalamic circuit function in the somatosensory system of young rats. We used single neuron recordings and controlled whisker deflections to examine responses of thalamocortical neurons to sensory stimulation in rat survivors of 9 min of asphyxial cardiac arrest incurred on postnatal day 17. We found that 48–72 h after cardiac arrest, thalamocortical neurons demonstrate significantly elevated firing rates both during spontaneous activity and in response to whisker deflections. The elevated evoked firing rates persist for at least 6–8 weeks after injury. Despite the overall increase in firing, by 6 weeks, thalamocortical neurons display degraded receptive fields, with decreased responses to adjacent whiskers. Nine minutes of asphyxial cardiac arrest was associated with extensive degeneration of neurites in the somatosensory nucleus as well as activation of microglia in the reticular nucleus. Global brain hypoxia-ischemia during cardiac arrest has a long-term impact on processing and transfer of sensory information by thalamic circuitry. Thalamic circuitry and normalization of its function may represent a distinct therapeutic target after cardiac arrest.
Introduction
Global hypoxia-ischemia interrupts oxygen delivery and blood flow to the entire brain. It occurs during cardiac arrest, which affects >350,000 people in the United States each year (Centers for Disease Control and Prevention, 2002). Untreated, cardiac arrest rapidly leads to brain death. If treated with cardiopulmonary resuscitation, however, cardiac arrest is survivable, but survivors often show evidence of injury in selectively vulnerable regions of the brain. In humans, vulnerable regions include cerebellum, basal ganglia, hippocampus, frontoparietal cortex, and thalamus (Graham, 1977; Ng et al., 1989; Horn and Schlote, 1992; Fujioka et al., 1994; Arbelaez et al., 1999; Hausmann et al., 2007; Choi et al., 2010). In animals, vulnerable cell populations include cerebellar Purkinje cells (Myers and Yamaguchi, 1977; Pulsinelli and Brierley, 1979; Kumar et al., 1988; Radovsky et al., 1997), hippocampal CA1 pyramidal neurons (Myers and Yamaguchi, 1977; Pulsinelli and Brierley, 1979; Radovsky et al., 1997; Böttiger et al., 1998; Fink et al., 2004), cortical pyramidal neurons [in layers III, V, VI in dogs and primates (Graham, 1977; Myers and Yamaguchi, 1977; Kumar et al., 1988), and layer V in rodents (Pulsinelli and Brierley, 1979; Fink et al., 2004)], and GABAergic inhibitory neurons in the thalamic reticular nucleus (RT; Radovsky et al., 1997; Böttiger et al., 1998; Geocadin et al., 2000). As initial survival from cardiac arrest improves, reaching 40% in some locales (Nichol et al., 2008), functional correlates of neuronal injury emerge as the paramount determinants of neurological outcome among survivors (Graves et al., 1997).
Functional correlates of injury to the cerebellum and the basal ganglia (motor impairment; Venkatesan and Frucht, 2006) and to the hippocampus (amnesia; Volpe and Hirst, 1983; Cummings et al., 1984; Volpe et al., 1986; Maryniak et al., 2008; Peskine et al., 2010) are well characterized in cardiac arrest survivors (Khot and Tirschwell, 2006; Mateen et al., 2011). In contrast, functional consequences of injury to the thalamus are poorly understood but may be crucially important. For instance, RT regulates attention (Crick, 1984; McAlonan et al., 2008), the sleep–wake state (McCormick and Bal, 1997), and transmission of sensory information from the periphery to the cerebral cortex (Hartings and Simons, 2000; McAlonan et al., 2008). Injury to RT may impact both general arousal and sensory perception. Furthermore, injury to thalamic somatosensory and visual nuclei, the ventral posterior medial (VPm) and the lateral geniculate, respectively, can further degrade sensory processing. Whether thalamic injury contributes to post-cardiac arrest neurological deficits is unknown.
To understand how thalamic injury affects post-cardiac arrest neurological function, we examined responses of thalamocortical VPm neurons to light touch in rat survivors of cardiac arrest and resuscitation early in life. Results indicate that cardiac arrest-associated global hypoxia-ischemia leads to elevated spontaneous and evoked activity after resuscitation, significantly disrupting thalamic circuit function. Physiologic changes are associated with histologic evidence of injury in RT and in VPm. Thus, thalamic dysfunction likely contributes to deficits in sensory processing after cardiac arrest, and thalamic circuitry may represent an under-appreciated therapeutic target.
Materials and Methods
Animals.
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Male Sprague Dawley rats were used for all experiments and were housed with their mother until postnatal day 30 (P30). Food and water were provided ad libitum. A total of 32 rats were used in this study. For neurophysiologic experiments, the rats underwent either cardiac arrest (n = 7) or sham (n = 9) intervention at P17–19 (see below, Cardiac arrest and resuscitation in Materials and Methods). After cardiac arrest, the rats were divided into the early group, which was assessed 48–72 h after arrest, and the late group, assessed at least 6 weeks after arrest. The early group consisted of 6 sham and 3 injured rats, the late group of 3 sham and 4 injured rats. The experimenter was blind to the injury status of the rats at the time of the recordings and data analyses. There were no grossly observable differences in appearance or behavior between injured and sham rats in either the early or the late groups. Specifically, all rats in the study groomed, whisked, ate, and drank. There were no differences in posture or gross locomotion in the cage, and no obvious differences in anesthetic requirement or physiologic parameters during the recordings. In addition to rats used for neurophysiologic recordings, 16 rats were used for histochemical analyses of neurodegeneration and microglial abundance using amino cupric silver staining and Iba1, respectively (Imai et al., 1996; Switzer, 2000). These rats were divided into groups as follows: two male and two female sham rats perfused 72 h after cardiac arrest, three male and three female injured rats perfused at 24 h after cardiac arrest, and three male and three female injured rats perfused at 72 h after cardiac arrest.
Cardiac arrest and resuscitation.
The model of developmental asphyxial cardiac arrest has been described in detail previously (Fink et al., 2004). Briefly, P17–19 rats underwent tracheal intubation and placement of femoral arterial and venous lines under isoflurane/nitrous oxide anesthesia. The rats were mechanically ventilated under neuromuscular blockade. Arterial blood pressure, electroencephalogram, and electrocardiogram were continuously monitored and recorded. Core body temperature was maintained constant at 37°C with a servo-controlled heating blanket. The anesthetic was then briefly washed out with room air, and rats were disconnected from the ventilator. Asystole ensued within 60–90 s of apnea and was allowed to continue for 9 min. The rat was then resuscitated with mechanical ventilation using 100% oxygen, intravenous epinephrine, and sodium bicarbonate, and manual chest compressions. Upon return of spontaneous circulation, the rats additionally received a 20 ml/kg bolus of 5% dextrose in normal saline intravenously to prevent dehydration. After ∼1 h, mechanical ventilation was discontinued, the rats were extubated, arterial and venous lines were removed, and wound margins were sutured. The rats were observed for an additional 1 h in a chamber with 100% oxygen to mimic a clinical scenario and then returned to the mother.
Surgical preparation for electrophysiologic recordings.
Surgical procedures in developing animals have been described previously (Shoykhet et al., 2003). Either 2–3 d (early) or at least 6 weeks (late) after asphyxial cardiac arrest, rats were anesthetized with isoflurane, and underwent (1) tracheotomy, (2) placement of an external jugular venous and femoral arterial catheters, and (3) craniotomy. A steel post was affixed to the skull with dental acrylic to allow holding the rat's head without pressure points for the remainder of the experiment. The craniotomy (∼2 mm2) was performed through the skull overlying VPm, 2–3 mm caudal to bregma and 2–3 mm lateral to midline. Dura mater was left intact. A ground screw was placed through the skull and fixed with dental acrylic. After completions of all surgical procedures, the rat was transferred to the vibration isolation table and placed in a custom-made head holder. Mechanical ventilation using a volume-controlled Inspira ventilator (Harvard Apparatus) was initiated using tidal volumes of ∼8 ml/kg and rates of 100 and 70 breaths/min in younger and older rats, respectively. Neuromuscular blockade was initiated with a bolus dose of pancuronium bromide (∼1 mg/kg) and maintained with a continuous infusion of pancuronium (0.8 mg · kg−1 · h−1) in 5% dextrose/0.9% sodium chloride for the remainder of the experiment. Isoflurane anesthesia was then discontinued, and the rat was transitioned to fentanyl analgesia using continuous fentanyl infusion at ∼8–10 μg · kg−1 · h−1. At these doses, the rats enter a state of dissociative analgesia without compromise of thalamocortical network dynamics observed under anesthesia (Simons et al., 1992).
The rat's physiologic state during the recording session was continuously monitored as described previously (Shoykhet and Simons, 2008). Briefly, temperature was maintained at 37°C using a servo-controlled heating blanket (Harvard Apparatus) and, in young rats, a 20W DC lamp. Intra-arterial pressure and heart rate were monitored using a blood pressure monitor (WPI Inc) connected to the arterial line via a pressure transducer. If mean arterial pressure (MAP) did not remain in the developmentally appropriate range, experiments were discontinued. Adequate analgesia was assured throughout the recording session by monitoring pupillary constriction and by maintaining lack of MAP and HR elevation in response to gentle touch. At the end of the recording session, the rats were deeply anesthetized with >5% isoflurane in room air and perfused transcardially for cytochrome oxidase (CO) histochemistry.
Electrophysiologic recordings.
Extracellular recordings were obtained using stainless steel microelectrodes (6–8 MΩ impedance at 1 kHz; FHC). The electrode was advanced perpendicularly to the pia using a hydraulic micropositioner (Kopf Instruments). Entry into VPm was signaled by an abrupt increase in spontaneous and evoked neural activity both in sham and in injured animals. In both groups, VPm was somatotopically organized, individual units were easily isolated, and there were no silent zones. Signals were initially bandpass filtered between 300 Hz and 10 KHz and amplified 10- to 100-fold using a DC-powered preamplifier (Grass Instruments). The resulting signal was further filtered using a 60 Hz notch and a bandpass filter (BAK Electronics) and digitized using a PCI-MIO-16E4 data acquisition board (National Instruments) connected to a personal computer.
Whisker stimulation and data acquisition.
Facial vibrissae were deflected using a multiangle piezoelectric stimulator (Simons, 1983) controlled by custom-written LabView software. The stimulator was attached to the whisker ∼5 mm from the face in young rats and ∼10 mm from the face in adults. The stimuli consisted of a hold-ramp-hold pattern, delivered over a total of 500 ms. The stimulator was calibrated to deflect the whisker at ∼125 mm/s; the deflection amplitude was 0.5 mm in young rats and 1 mm in adults, resulting in equivalent angular deflection (∼5.7°) in all age groups. The whisker was deflected in eight standard directions, with deflection in each direction repeated 10 times for a total of 80 stimuli per whisker. Deflections were interleaved in a random manner. For each neuron, data were collected at least from its principle whisker (PW), defined as the whisker that evokes the most robust response. An attempt was also made to collect data from all four of the immediately adjacent whiskers (AWs)—rostral, caudal, dorsal, and ventral.
Data were collected for 500 ms; action potential time stamps were collected with 100 μs resolution. Action potential waveforms were digitized at 32 kHz and stored for off-line analyses. Off-line, the action potential waveforms were sorted using custom written software that allowed for isolation of single-unit recordings based on a combination of waveform parameters, including principal component analysis, deflection slopes, and amplitude of peaks and valleys. To confirm further the single-unit nature of the recordings, sorted spikes were examined using an interspike interval histogram; only recordings with <1% of the sorted spikes within the absolute refractory period (1 ms) were used in the analyses.
Histology and immunohistochemistry.
Rats used in neurophysiologic recordings were perfused with 4% paraformaldehyde, their brains were extracted, postfixed for at least 24 h, and cryoprotected in 30% w/v sucrose solution. Brains were then blocked, snap-frozen in liquid nitrogen, and stored at −20°C until processing. Brains were sectioned in a coronal plane into 30-μm-thick sections for glutamic acid decarboxylase (GAD) immunohistochemistry and CO staining, and into 10-μm-thick sections for Fluoro-Jade B (FJB) staining. The sections were collected sequentially such that a given 70-μm-thick block of tissue generated tissue sections for all three stains. For GAD immunohistochemistry, free-floating sections were collected into PBS and incubated for 1 h at room temperature in PBS containing 1.5% goat serum and 0.2% Triton-X. Mouse anti-GAD65 antibody (1:5000, MAB351, Millipore) was added to the above solution, and the tissue was incubated overnight a 4°C. The primary antibody was visualized with the Vectastain ABC kit (PK-4002, Vectorlabs) using manufacturer's protocol. The tissue was then mounted on slides, dried overnight, dehydrated in alcohol, and coverslipped with Permount. The number of GAD65+ neuronal somata in each 30 μm section was counted using ImageJ software by two independent observers who were blind to the experimental status of the animal. Soma was counted if it clearly possessed a nucleus and a visible nucleolus contained within the slide. If counts from the two observers disagreed by >20%, a third, experienced observer recounted the cells. At least 7 slides per animal were examined, and the counts were averaged across the slides. No attempt was made to determine the true density of GAD65+ neurons in RT.
CO staining was performed in a previously described manner (Wong-Riley et al., 1978; Land and Simons, 1985). Electrode recording locations were confirmed on CO-stained tissue counterstained with thionin. FJB staining was performed as described previously (Schmued et al., 1997; Fink et al., 2004).
In a separate series of experiments, the collected brains were processed for aminocupric silver staining (Switzer, 2000). At the designated time after intervention, rats were deeply anesthetized with isoflurane and perfused transcardially with 0.034% NaCacodylate buffer followed by 1.4% NaCacodylate fixative in 4% paraformaldehyde/4% sucrose solution. The heads were surgically removed and postfixed for 48 h; brain manipulation was deferred to minimize staining artifact. The brains were then removed, placed in buffer, and shipped for commercially available staining (NeuroScience Associates).
Data analyses.
Spike time data were converted into peristimulus time histograms (PSTHs) with 1 ms resolution. Response magnitudes evoked by whisker deflection onset (ON) and offset (OFF) were defined as the number of action potentials discharged during a 25-ms-long window encompassing the corresponding transient components on the population PSTH. Spontaneous activity was defined as the firing rate during a 100-ms-long period of data collection preceding whisker deflection. Plateau activity was defined as the firing rate during a 100-ms-long window in the second half of the 200 ms period of sustained whisker deflection in the direction evoking the largest sustained response. Only responses from neurons with a statistically significant PW-evoked ON response were included in the analyses. ON responses were considered statistically significant if the firing rate during the 25-ms-long ON window examined at the deflection angle evoking the maximum ON response (ONmax) exceeded the spontaneous firing rate (one-tailed t test, p < 0.025). The proportion of VPm neurons with significant PW-evoked responses among all recorded VPm neurons is similar between sham and cardiac arrest (CA) animals both in the early and in the late groups (early sham 41/45 vs early CA 28/34, Fisher exact p = 0.3; late sham 29/32 vs late CA 46/52, Fisher exact p = 1). Neurons were classified as slowly adapting if plateau activity exceeded spontaneous firing (one-tailed t test, p < 0.025). Response latency was computed from each neuron's PSTH accumulated over all deflection angles and was defined as the time of the 1 ms bin in which the spike count exceeded spontaneous firing using a Poisson distribution (p < 0.05; Shoykhet and Simons, 2008). Angular tuning of the ON response was examined using the ratio of ONmax to the ON response averaged over all deflection angles.
Statistical treatment of the data.
Alpha-trimmed means were used to compare average values among populations. An α-trimmed mean is defined as the sample mean derived from the set of observations n from which κ largest and smallest values have been removed, and where κ is the next largest integer of α × n (Fisher and van Belle, 1993). α-trimmed means are more robust with respect to the effect of outliers, and statistical analyses using α-trimmed means become more conservative due to reduction in the degrees of freedom and inclusion of only the most frequently observed values in the calculations. As done previously in developing animals (Shoykhet and Simons, 2008), means for the current study were computed with α = 0.05. Means were compared using either parametric (Student's t test or ANOVA) or nonparametric (Mann–Whitney or Kruskal–Wallace ANOVA) tests as indicated. Differences were considered statistically significant if two-tailed probability values were ≤0.05 unless otherwise indicated. Data are presented as mean ± SEM.
Results
We examined responses of thalamocortical neurons to controlled whisker deflections in four groups: (1) early sham (n = 41 neurons); (2) early cardiac arrest (n = 28); (3) late sham (n = 29); and (4) late cardiac arrest (n = 46). The most striking difference between sham rats and those that have recovered from 9 min of cardiac arrest is the firing rate of thalamocortical neurons both at baseline and in response to sensory stimuli. Figure 1 summarizes spontaneous and principal whisker-evoked firing rates of thalamocortical neurons in the early group, i.e., 48–72 h after cardiac arrest. Population PSTHs demonstrate that the firing rates are higher in injured animals (Fig. 1A). Results are quantified in Figure 1B. Spontaneous firing rates of thalamocortical neurons are 6.0 ± 0.7 Hz in the early sham group and 8.9 ± 0.8 Hz in the early cardiac arrest group (p = 0.001). Onset of movement of the principal whisker from rest in the maximally effective direction (ONmax, see Materials and Methods, Data analyses) evokes on average 1.7 ± 0.1 spikes per stimulus in thalamocortical neurons in the early sham animals, consistent with previously published literature in developing rats (Shoykhet and Simons, 2008). In the early cardiac arrest group, ONmax responses are increased by 50% compared with shams (Fig. 1B), as principal whisker deflection onset evokes on average 2.7 ± 0.2 spikes per stimulus (p = 0.004, corrected for unequal variance). Similarly, firing rates during the sustained portion of the whisker deflection (plateau) and those evoked by the whisker's return to rest (OFF) in early cardiac arrest animals exceed control firing rates in early sham animals by 60% and 50%, respectively (plateau: 15 ± 1.8 Hz in early sham vs 25 ± 1.5 Hz in early cardiac arrest, p < 0.001; OFF: 1.6 ± 0.1 spikes/stimulus in early sham vs 2.4 ± 0.2 spikes/stimulus in early cardiac arrest; Fig. 1B). Thus, thalamocortical neurons demonstrate significantly elevated spontaneous and principal whisker-evoked firing rates 48–72 h after cardiac arrest.
Figure 1.

Responses of thalamocortical neurons 48–72 h after CA or sham insult (Sh). A, Population PSTHs of action potentials discharged by thalamocortical neurons in the Early Sh (n = 41) and the Early CA group (n = 28 neurons). Data are presented in 1 ms bins. PSTHs are collapsed across all neurons and all deflection angles. The whisker deflection during the 500 ms period of data collections is represented schematically below the PSTH. The various time epochs used to compare responses quantitatively are shown above the PSTH. B, Quantitative comparison of spontaneous and evoked responses between Early Sh and Early CA groups. Spont, spontaneous. Data are shown as mean ± SEM, *p < 0.05.
Firing rates of thalamocortical neurons remain elevated, albeit to a lesser extent, for at least 6 weeks after cardiac arrest. Figure 2 shows responses of thalamocortical neurons to principal whisker deflections in the late sham and the late cardiac arrest groups. Response magnitudes in late sham animals are comparable to those observed in prior studies in adult rats (Simons and Carvell, 1989; Kwegyir-Afful et al., 2005). Because spontaneous and evoked response magnitudes, as well as receptive field size, in adult animals are known to differ from those in developing rats (Shoykhet and Simons, 2008), responses in late injured animals are compared only to age-appropriate control values, which were obtained in late sham rats. Although population PSTHs are qualitatively similar between the late sham and the late injured groups (Fig. 2A), quantitative differences persist. Responses to the dynamic portions of the sensory stimulus, i.e., ON and OFF responses, remain elevated by 20–30% in the late cardiac arrest group compared with the late sham group (ON: late sham 2.3 ± 0.2 spikes/stimulus vs late cardiac arrest 2.8 ± 0.2 spikes/stimulus, p = 0.05; OFF: late sham 1.9 ± 0.2 spikes/stimulus vs late cardiac arrest 2.3 ± 0.3 spikes/stimulus, p = 0.01, corrected for unequal variances; Fig. 2B). Spontaneous firing rates and plateau responses are similar between the late sham and the late cardiac arrest groups (Fig. 2B; spontaneous activity: late sham 12.1 ± 1.1 Hz vs late cardiac arrest 12.5 ± 1.2 Hz, p = 0.7; plateau activity: late sham 42.1 ± 5.4 Hz vs late cardiac arrest 49.3 ± 6.0 Hz, p = 0.2). These data demonstrate that abnormally elevated firing rates of thalamocortical neurons persist for some, but not for all, response components for at least 6 weeks after cardiac arrest.
Figure 2.

Responses of thalamocortical neurons >6 weeks after CA or sham insult (Sh). A, Population PSTHs of thalamocortical neurons in the Late Sh (n = 29) and the Late CA group (n = 46). Whisker deflection schematic is below the PSTH. B, Quantitative comparison of spontaneous and evoked responses between the Late Sh and the Late CA groups. Spont, spontaneous. Data are shown as mean ± SEM, *p < 0.05.
Receptive fields
In normal rats, thalamocortical neurons recorded under conditions identical to those used here have multiwhisker receptive fields (Simons, 1983; Simons and Carvell, 1989). We examined receptive field organization of thalamocortical neurons early and late after cardiac arrest. Figure 3 shows that 48–72 h after cardiac arrest, AW-evoked responses appear smaller both in absolute magnitude (Fig. 3A) and when examined as a ratio of AW-evoked to PW-evoked responses (AW/PW ratio, Fig. 3B). These apparent differences in the early group, however, are not statistically significant (p = 0.38 for AW ON; p = 0.1 for AW/PW ratio). When AW-evoked responses are examined as a function of the AW's position with respect to the PW, it becomes clear that the apparent reduction in AW-evoked response magnitude results from a significant reduction in responses evoked by the caudal AW in early cardiac arrest rats, compared with the early shams (caudal AW/PW ratio: 0.6 ± 0.08 in early sham vs 0.28 ± 0.08 in early cardiac arrest, p = 0.008, corrected for unequal variances and for multiple comparisons, Fig. 3C).
Figure 3.

Receptive field properties of thalamocortical neurons after CA or sham insult (Sh). A, AW ON responses in Early Sh and Early CA groups. Responses are averaged across all deflection directions and across all AWs studied. B, Average ratio of AW-evoked to PW-evoked ON responses in Early Sh and Early CA groups (see Materials and Methods, Animals). C, Average AW/PW ratios in Early Sh and Early CA groups as a function of AW position with respect to the PW. R, rostral; C, caudal; D, dorsal; V, ventral. Data were collected from 34 rostral, 18 caudal, 23 dorsal, and 25 ventral AWs in the early sham group, and from 17 rostral, 15 caudal, 13 dorsal, and 15 ventral AWs in the early CA group. D, AW-evoked ON responses are decreased in the Late CA group compared with the Late Sh group. E, AW/PW ratio is smaller in the Late CA versus the Late Sh group. Responses are averaged across all deflection directions and across all AWs studied. F, Reduced rostral and caudal AW/PW ratios in the Late CA group compared with the Late Sh group. n = 16, 29, 21, and 24 for R, C, D, and V AWs, respectively, in the Late Sh group. n = 37, 36, 35, and 21 for R, C, D, and V AWs, respectively, in the Late CA group. All data are shown as mean ± SEM, *p < 0.05.
As the rats recover from cardiac arrest, the differences between AW-evoked responses in sham and injured rats become more pronounced. Figure 3D shows that AW-evoked responses are significantly smaller in absolute magnitude in late cardiac arrest rats compared with late shams (0.40 ± 0.03 spikes/stimulus in late cardiac arrest vs 0.56 ± 0.06 spikes/stimulus in late sham, p = 0.02). Similarly, the average AW/PW ratio is decreased in late cardiac arrest rats (0.32 ± 0.03 in late cardiac arrest vs 0.46 ± 0.04 in late sham, p = 0.01, Fig. 3E). Examination of the data based on the AW's position with respect to the PW reveals that reductions in the responses to the rostral and caudal AWs are responsible for the overall diminished AW responses in injured rats (Fig. 3F). Thus, AW-evoked responses of thalamocortical neurons are smaller in rats after cardiac arrest. These data indicate that in injured rats, receptive fields of thalamocortical neurons become less multiwhisker and, thus, more focused on the principal whisker.
Response latency
We examined response latency of thalamocortical neurons early and late after cardiac arrest. In the early group, the average response latency is 5.0 ± 0.28 ms in sham rats, comparable to prior reports (Shoykhet and Simons, 2008). The average latency is increased by 20% to 6.1 ± 0.24 ms in rats after cardiac arrest (Fig. 4A, p = 0.003, corrected for unequal variances). Figure 4B,C shows the distribution of latencies among thalamocortical neurons in sham and cardiac arrest rats, respectively. These stem-and-leaf diagrams demonstrate that in sham rats, >50% of the neurons respond to whisker deflection with a latency of ≤4 ms (Fig. 4B). In contrast, only 15% of neurons respond with a latency of ≤4 ms 48–72 h after cardiac arrest (Fig. 4C). By 6 weeks after arrest, the average latencies and their distributions are equivalent between sham and injured rats (data not shown). Therefore, the increase in average latency early after injury results from a dearth of short latency responses to sensory stimuli. With time, rats recover from this deficit.
Figure 4.

Prolonged response latency among thalamocortical neurons 48–72 h after CA or sham insult (Sh). A, Average response latency in the Early Sh and the Early CA groups. B, Distribution of response latencies in the Early Sh group. Latency values are plotted in 1 ms bins. The bimodal latency distribution at this age (P19–21) is consistent with previous data (Shoykhet and Simons, 2008). C, Distribution of response latencies in the Early CA group. Note the decrease in short latency responses of thalamocortical neurons to whisker deflection.
Histologic evidence of neuronal injury after cardiac arrest
Elevated firing rates among thalamocortical neurons in rats after cardiac arrest suggest possible injury to thalamic inhibitory neurons. In rodents, the majority of intrathalamic GABAergic interneurons reside in the RT (Barbaresi et al., 1986; Harris and Hendrickson, 1987). We therefore examined RT inhibitory neurons for histological evidence of injury. Figure 5 shows FJB staining, which identifies degenerating neurons. FJB readily identified degenerating layer V cortical (Fig. 5B) and CA1 hippocampal neurons after cardiac arrest (Fig. 5F) but demonstrated only faint staining of neurites in VPm (Fig. 5D) and failed to identify neuronal degeneration in RT (data not shown). We then identified inhibitory RT neurons using an antibody to GAD65, an isoform of the GABA synthetic enzyme GAD. Figure 6 shows that the RT appears qualitatively similar in sham and in injured rats. Furthermore, the number of GAD65+ neurons in RT does not differ between sham and injured animals (Fig. 6C). Finally, we indirectly examined activation of microglia in RT after cardiac arrest using Iba1 immunohistochemistry. Figure 7 shows that activated microglia, which express the Ca2+-binding protein Iba1 (Imai et al., 1996), invade the RT within 72 h after cardiac arrest. Together, FJB, GAD65 and Iba1 staining patterns suggest that inhibitory RT neurons likely suffer a nonlethal injury in this model of asphyxial cardiac arrest.
Figure 5.
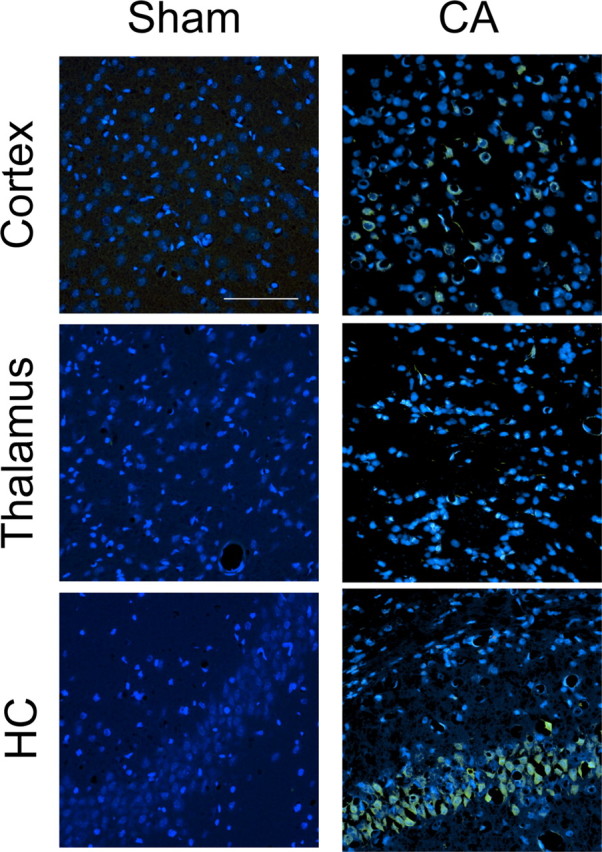
FJB staining in rat brains 72 h after 9 min CA. In the cortex (top row), Layer V neurons are FJB+ after CA. In the thalamus (middle row), FJB staining in VPm reveals faintly positive neuronal processes but no soma. In the hippocampus (HC; bottom row), CA1 hippocampal neurons are FJB+ after CA. All images are double-stained with DAPI (blue) and FJB (green). Scale bar: 100 μm (applies to all images).
Figure 6.

Preserved cellular architecture and number of GABAergic neurons in the thalamic RT 6 weeks after CA compared with sham insult (Sh). A, Coronal section through the somatosensory thalamus demonstrating GAD65+ GABAergic neurons in the RT of a sham-operated rat. B, RT architecture is preserved after CA. C, GAD65+ neurons in RT were counted in sham (n = 5 rats, 7–9 slices/rat) and CA rats (n = 4 rats, 7–9 slices/rat). Data represent the average number of GAD65+ RT cells/30 μm slice. No attempt was made to estimate true cell density. VPm, ventral posterior medial nucleus; IC, internal capsule. Scale bars: A (for A, B), 500 μm (low-power images); 100 μm (high-power images).
Figure 7.
Microglia are activated in the hippocampus and in the Reticular Nucleus 72 h after cardiac arrest. Activated microglia were stained for Iba1. A, Absent Iba1 staining in the hippocampus of sham-operated rats. B, Iba1+ microglia are present in the CA1 region of the hippocampus 72 h after cardiac arrest (DG, dentate gyrus). C, Absent Iba1 staining in the thalamus of sham-operated rats. D, Iba1-stained microglia are present in the thalamic Reticular Nucleus 72 h after cardiac arrest. Scale bar: (in A) A–D, 500 μm.
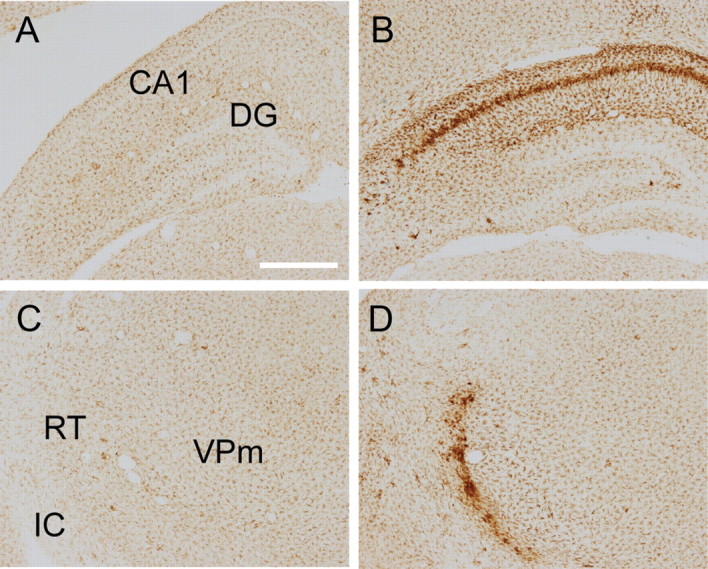
We further characterized degeneration of neurons and neurites in injured animals using De Olmos's amino cupric silver stain (De Olmos and Ingram, 1971). This variant of the silver stain provides excellent signal-to-noise ratio and, regardless of cell death mechanism, stains all degenerating neuronal components (soma, dendrites, axons, and synaptic boutons) an intense black (Switzer, 2000). Figure 8 highlights the extent of cardiac arrest-induced injury in the cerebral cortex, in the thalamus, and in the hippocampus. In the cerebral cortex, we observed selective degeneration of pyramidal neurons and their apical axons in Layer V of the primary somatosensory cortex. We also noted degeneration of long-range corticocortical fibers in Layer I. In the thalamus, the most salient feature is degeneration of neurites in RT and in VPm, the latter of which contains the thalamocortical neurons analyzed above. We did not observe somatic degeneration either in RT or in VPm. Finally, degeneration of hippocampal CA1 neurons, a well documented phenomenon in hypoxic-ischemic brain injury, occurred 72 h after cardiac arrest. Interestingly, we also observed neuronal degeneration in CA2, CA3, and dentate gyrus regions of the hippocampus, with injury becoming notable within 24 h of arrest. Indeed, all six rats examined at 24 h after arrest demonstrated degeneration in the hippocampus outside of the CA1 region.
Figure 8.

Neurodegeneration in the rat brain after a 9 min CA or sham insult. Degenerating neurons and neurites were visualized with Amino Cupric silver stain. Top row: in the cortex, degeneration of Layer V pyramidal neurons and their apical dendrites is apparent at 72 h. Middle row: in the thalamus, neurites in VPm and in RT also degenerate 72 h after CA. Bottom row: in the hippocampus, neuronal degeneration in CA2, CA3, and the dentate gyrus peaks at 24 h after CA, whereas degeneration in CA1 becomes more pronounced 72 h after CA. Scale bars: 500 μm (low-power images); 100 μm (high-power images).
Discussion
The current data reveal significant, persistent abnormalities in the function of thalamocortical neurons after cardiac arrest in the developing rat. Forty-eight hours after injury, spontaneous and stimulus-evoked firing rates of thalamocortical neurons are increased ≈50–60%. Six weeks after injury, thalamocortical neurons continue to demonstrate abnormally elevated firing rates in response to principal whisker deflection. Yet, adjacent whisker-evoked responses show a persistent reduction, resulting in abnormally small receptive fields. Histological findings in the thalamus after cardiac arrest reveal (1) injury to RT without overt cell death; and (2) neurite degeneration in VPm. Together, the functional and histological data suggest that disruption in thalamocortical circuitry may contribute to functional deficits observed in cardiac arrest survivors.
Increased neuronal activity in the thalamus after cardiac arrest is consistent with recent description of increased cerebral blood flow (CBF) in the thalamus for at least 1 h after similar injury (Manole et al., 2009). In human survivors of global brain hypoxia-ischemia, arterial spin labeling revealed cerebral hyperperfusion ≈5 d after injury (Pollock et al., 2008). Interestingly, hyperperfusion was greater in younger patients, perhaps reflecting age-dependent CBF regulation (Wintermark et al., 2004). Whether neuronal activity correlates with CBF after global hypoxia-ischemia remains unknown. Blood oxygenation level-dependent (BOLD) MRI signal may reflect aggregate neuronal activity over time in a given brain region, although it may not directly link neuronal activity to CBF (Logothetis et al., 2001). The magnitude of sensory stimulation-evoked change in the BOLD signal in the somatosensory cortex correlates with outcome in human cardiac arrest survivors (Gofton et al., 2009). Similar studies focused on the thalamus are lacking but may contribute to prognostic evaluation of comatose cardiac arrest survivors.
Pathophysiology of increased thalamocortical activity likely involves, but may not be limited to, injury to the RT inhibitory neurons. Both physiologic and histological evidence support the hypothesis that intrathalamic inhibition is disrupted after cardiac arrest. From a physiologic perspective, RT neurons exert a tonic inhibitory influence over thalamocortical neurons under normal conditions (Jones, 2002). Pharmacologic blockade of RT-mediated inhibition using GABA antagonists results in increased spontaneous and evoked firing rates among thalamocortical neurons (Hartings and Simons, 2000). Indeed, responses of thalamocortical neurons after cardiac arrest qualitatively resemble their responses during GABA receptor blockade (Hartings and Simons, 2000). Functional disinhibition of VPm neurons may result either from decreased firing of RT neurons and/or from degeneration of RT-to-VPm connections in injured animals. Both mechanisms are readily amenable to future study. From the histological perspective, prior anatomic studies have demonstrated injury to RT neurons after a global hypoxic-ischemic insult (Radovsky et al., 1995, 1997; Hossmann et al., 2001). In the present study, the injury to RT neurons appears sublethal because neither amino cupric silver nor FJB staining demonstrated frank neuronal degeneration in RT. Yet, the presence of activated Iba1-positive microglia in RT within 24 h of cardiac arrest indicates that RT neurons are sufficiently injured to evoke an inflammatory response. Thus, available evidence continues to suggest that RT neurons are more sensitive to a hypoxic-ischemic insult than VPm neurons. Furthermore, injury to RT neurons appears to contribute to thalamic circuit dysfunction after cardiac arrest.
Altered thalamic input after cardiac arrest likely has significant implications for experience-dependent plasticity in the developing cortex. In the rodent visual cortex, neonatal hypoxia-ischemia impairs ocular dominance plasticity following monocular deprivation (Failor et al., 2010). Concurrently, it alters the phenotype of inhibitory parvalbumin-expressing neurons and reduces overall activity levels. Thus, it has been suggested that neonatal hypoxia-ischemia impairs cortical plasticity by altering development of inhibition in the visual cortex (Failor et al., 2010). The present data suggest an additional mechanism whereby hypoxic-ischemic injury during development may impair cortical plasticity. Namely, hypoxia-ischemia may alter thalamic input either via direct injury to thalamic circuitry or via disruption of the corticothalamic feedback loop (Ergenzinger et al., 1998). Abnormal thalamic input, in turn, may contribute to dysfunctional cortical plasticity. In the rodent somatosensory system, altered sensory input during development disrupts maturation of inhibitory and excitatory cortical circuitry (Simons and Land, 1987; Shoykhet et al., 2005) and results in permanent behavioral deficits (Carvell and Simons, 1996). By analogy, abnormal thalamic input after cardiac arrest may impact functional maturation of cortical circuitry and result in long-lasting deficits.
Current data also suggest that CNS injury after cardiac arrest involves neuronal fibers. Physiologically, 48–72 h after cardiac arrest we observed prolongation of the response latency. The difference in latency (>1 ms) exceeds resolution of the spike time-stamp (100 μs) tenfold and resolution of the digitized spike waveform (32 kHz) 32-fold. Thus, technical details of spike-sorting cannot account for the magnitude of the observed difference. Two lines of reasoning suggest that prolonged latency reflects hypoxic-ischemic injury to subcortical long-range myelinated fibers, rather than altered temporal integration of trigeminothalamic inputs by VPm neurons. First, injury to RT likely renders VPm neurons more depolarized and thus more likely to cross threshold sooner (Hartings et al., 2003). If trigeminothalamic input were unaffected by cardiac arrest, then VPm neurons should respond at shorter, not longer, latencies. Yet, we observed the opposite. Second, neurons in the principal trigeminal nucleus (PR5), which carry primary trigeminothalamic input to VPm, synapse onto VPm neurons on proximal dendrites and soma, providing for a potent and high-fidelity synaptic drive (Deschênes et al., 2003). Most VPm neurons receive inputs from ≤2 PR5 neurons (Deschênes et al., 2003). Given such potent and focused input, changes in its temporal integration by VPm neurons are unlikely to account fully for a 1-ms-long latency delay after cardiac arrest.
Histologic evidence for injury to long-range myelinated fibers derives from amino cupric silver staining, which demonstrated degeneration of neurites in the somatosensory thalamus. The exact origin of these processes remains to be determined, which will likely require transmission electron microscopic analysis as well as neuroanatomic tracing studies of degenerating fibers and associated synapses. Injury to myelinated fibers has been demonstrated previously in pigs after cardiac arrest (Sharma et al., 2011). The importance of white matter injury in development of neuroprotective therapies has been recognized in stroke and traumatic brain injury (TBI). In stroke, failure to account for white matter injury in animal models possibly contributed to failure of human clinical trials (Gladstone et al., 2002). In TBI models, axonal injury constitutes a major pathophysiologic mechanism, and its amelioration, a key therapeutic strategy (Garman et al., 2011; Greer et al., 2011b; Oda et al., 2011). In contrast, in cardiac arrest models, neuroprotection studies have focused primarily on preserving neuronal somata, not on protecting neuronal fibers. Yet, MRI data in humans confirm that white matter injury occurs after global hypoxia-ischemia (Bianchi and Sims, 2008; Greer et al., 2011a). The present data suggest that cardiac arrest neuropathology includes white matter injury.
An additional subset of degenerating neurons after asphyxial cardiac arrest—cortical layer V pyramidal neurons—while observed previously (Fink et al., 2004), has been further delineated by the amino cupric silver staining. Layer V in rodents contains a heterogeneous set of pyramidal neurons. Superficial Layer Va neurons project to the striatum and to the contralateral cortex, whereas deeper Layer Vb neurons project to the posteromedial thalamic nucleus, superior colliculus, tectum, and spinal cord (Molnár and Cheung, 2006). Neurons projecting to distinct targets demonstrate distinct physiologic and, to some extent, morphological features (Hattox and Nelson, 2007). Degenerating silver-stained neurons in this study are Layer Va pyramidal cells with a single large apical dendrite extending to the pia. Although the precise identity of degenerating neurons remains to be determined, this morphology is consistent with that of corticostriatal and/or corticocortical pyramidal neurons (Hattox and Nelson, 2007).
The specific impact of dysfunctional thalamic circuitry on development of sensory processing mechanisms may depend both on the animal's age at the time of hypoxic-ischemic insult and on specific details of the insult itself. The present pediatric cardiac arrest model uses P17 rats in contrast to models of neonatal hypoxia-ischemia, which mostly use P7 animals (Rice et al., 1981). The age difference is significant because rodent P7 and P17 neurons differ in several physiologic processes. For example, GABAergic currents are excitatory at P7 but inhibitory at P17 due to developmentally regulated expression of the K+-Cl− cotransporter KCC2 (Rivera et al., 2005). In humans, the developmental increase in KCC2 expression in the prefrontal cortex occurs primarily in utero (Hyde et al., 2011). Behaviorally, at P7 the eyelids are fused, and active whisking, which rodents use to explore their environment (Welker, 1964), has yet to emerge. By P17, the eyes are open, and animals have begun whisking. The changes in neuronal activity associated with this behavioral development impact how visual and somatosensory systems respond to sensory deprivation (Stern et al., 2001; Desai et al., 2002) and, similarly, may impact how they respond to injury.
In summary, the present work indicates that asphyxial cardiac arrest disrupts thalamic circuitry in the developing brain. After insult, activity of thalamocortical neurons is abnormally elevated with disorganized receptive field structure, leading to altered sensory input into the cerebral cortex. Hyperactive, disorganized thalamic input may contribute to cortical pathology and behavioral abnormalities observed after cardiac arrest and may represent a readily modifiable therapeutic variable.
Footnotes
This work was supported by NIH Grants NS19950 (to D.J.S.), ND045968 (to R.S.B.C.), NS07003 and NS30318 (to P.M.K.), and by the Advanced Research Fellowship from the Children's Hospital of Pittsburgh, Pediatric Critical Care Scientist Development Program (5K12-HD047349–08, University of Utah), Children's Discovery Institute of the St. Louis Children's Hospital, McDonnell Center for Systems Neuroscience at Washington University School of Medicine, and by the Child Health Research Center of Excellence in Developmental Biology at Washington University School of Medicine (K12-HD01487, to M.S.).
References
- Arbelaez A, Castillo M, Mukherji SK. Diffusion-weighted MR imaging of global cerebral anoxia. AJNR Am J Neuroradiol. 1999;20:999–1007. [PMC free article] [PubMed] [Google Scholar]
- Barbaresi P, Spreafico R, Frassoni C, Rustioni A. GABAergic neurons are present in the dorsal column nuclei but not in the ventroposterior complex of rats. Brain Res. 1986;382:305–326. doi: 10.1016/0006-8993(86)91340-5. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Sims JR. Restricted diffusion in the splenium of the corpus callosum after cardiac arrest. Open Neuroimag J. 2008;2:1–4. doi: 10.2174/1874440000802010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttiger BW, Schmitz B, Wiessner C, Vogel P, Hossmann KA. Neuronal stress response and neuronal cell damage after cardiocirculatory arrest in rats. J Cereb Blood Flow Metab. 1998;18:1077–1087. doi: 10.1097/00004647-199810000-00004. [DOI] [PubMed] [Google Scholar]
- Carvell GE, Simons DJ. Abnormal tactile experience early in life disrupts active touch. J Neurosci. 1996;16:2750–2757. doi: 10.1523/JNEUROSCI.16-08-02750.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. State-specific mortality from sudden cardiac death—United States, 1999. MMWR Morb Mortal Wkly Rep. 2002;51:123–126. [PubMed] [Google Scholar]
- Choi SP, Park KN, Park HK, Kim JY, Youn CS, Ahn KJ, Yim HW. Diffusion-weighted magnetic resonance imaging for predicting the clinical outcome of comatose survivors after cardiac arrest: a cohort study. Crit Care. 2010;14:R17. doi: 10.1186/cc8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–4590. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Tomiyasu U, Read S, Benson DF. Amnesia with hippocampal lesions after cardiopulmonary arrest. Neurology. 1984;34:679–681. doi: 10.1212/wnl.34.5.679. [DOI] [PubMed] [Google Scholar]
- De Olmos JS, Ingram WR. An improved cupric-silver method for impregnation of axonal and terminal degeneration. Brain Res. 1971;33:523–529. doi: 10.1016/0006-8993(71)90130-2. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Deschênes M, Timofeeva E, Lavalee P. The relay of high-frequency sensory signals in the whisker-to-barreloid pathway. J Neurosci. 2003;23:6778–6787. doi: 10.1523/JNEUROSCI.23-17-06778.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergenzinger ER, Glasier MM, Hahm JO, Pons TP. Cortically induced thalamic plasticity in the primate somatosensory system. Nat Neurosci. 1998;1:226–229. doi: 10.1038/673. [DOI] [PubMed] [Google Scholar]
- Failor S, Nguyen V, Darcy DP, Cang J, Wendland MF, Stryker MP, McQuillen PS. Neonatal cerebral hypoxia-ischemia impairs plasticity in rat visual cortex. J Neurosci. 2010;30:81–92. doi: 10.1523/JNEUROSCI.5656-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink EL, Alexander H, Marco CD, Dixon CE, Kochanek PM, Jenkins LW, Lai Y, Donovan HA, Hickey RW, Clark RS. Experimental model of pediatric asphyxial cardiopulmonary arrest in rats. Pediatr Crit Care Med. 2004;5:139–144. doi: 10.1097/01.pcc.0000112376.29903.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher LD, van Belle G. Biostatistics: a methodology for the health sciences. New York: Wiley; 1993. [Google Scholar]
- Fujioka M, Okuchi K, Sakaki T, Hiramatsu K, Miyamoto S, Iwasaki S. Specific changes in human brain following reperfusion after cardiac arrest. Stroke. 1994;25:2091–2095. doi: 10.1161/01.str.25.10.2091. [DOI] [PubMed] [Google Scholar]
- Garman RH, Jenkins LW, Switzer RC, 3rd, Bauman RA, Tong LC, Swauger PV, Parks SA, Ritzel DV, Dixon CE, Clark RS, Bayir H, Kagan V, Jackson EK, Kochanek PM. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma. 2011;28:947–959. doi: 10.1089/neu.2010.1540. [DOI] [PubMed] [Google Scholar]
- Geocadin RG, Muthuswamy J, Sherman DL, Thakor NV, Hanley DF. Early electrophysiological and histologic changes after global cerebral ischemia in rats. Mov Disord. 2000;15(Suppl 1):14. doi: 10.1002/mds.870150704. [DOI] [PubMed] [Google Scholar]
- Gladstone DJ, Black SE, Hakim AM. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- Gofton TE, Chouinard PA, Young GB, Bihari F, Nicolle MW, Lee DH, Sharpe MD, Yen YF, Takahashi AM, Mirsattari SM. Functional MRI study of the primary somatosensory cortex in comatose survivors of cardiac arrest. Exp Neurol. 2009;217:320–327. doi: 10.1016/j.expneurol.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Graham DI. Pathology of hypoxic brain damage in man. J Clin Pathol Suppl (R Coll Pathol) 1977;11:170–180. doi: 10.1136/jcp.s3-11.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JR, Herlitz J, Bång A, Axelsson A, Ekström L, Holmberg M, Lindqvist J, Sunnerhagen K, Holmberg S. Survivors of out of hospital cardiac arrest: their prognosis, longevity and functional status. Resuscitation. 1997;35:117–121. doi: 10.1016/s0300-9572(97)00035-x. [DOI] [PubMed] [Google Scholar]
- Greer D, Scripko P, Bartscher J, Sims J, Camargo E, Singhal A, Furie K. Serial MRI changes in comatose cardiac arrest patients. Neurocrit Care. 2011a;14:61–67. doi: 10.1007/s12028-010-9457-8. [DOI] [PubMed] [Google Scholar]
- Greer JE, McGinn MJ, Povlishock JT. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci. 2011b;31:5089–5105. doi: 10.1523/JNEUROSCI.5103-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RM, Hendrickson AE. Local circuit neurons in the rat ventrobasal thalamus—a GABA immunocytochemical study. Neuroscience. 1987;21:229–236. doi: 10.1016/0306-4522(87)90335-6. [DOI] [PubMed] [Google Scholar]
- Hartings JA, Simons DJ. Inhibition suppresses transmission of tonic vibrissa-evoked activity in the rat ventrobasal thalamus. J Neurosci. 2000;20 doi: 10.1523/JNEUROSCI.20-19-j0001.2000. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Temereanca S, Simons DJ. State-dependent processing of sensory stimuli by thalamic reticular neurons. J Neurosci. 2003;23:5264–5271. doi: 10.1523/JNEUROSCI.23-12-05264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattox AM, Nelson SB. Layer V neurons in mouse cortex projecting to different targets have distinct physiological properties. J Neurophysiol. 2007;98:3330–3340. doi: 10.1152/jn.00397.2007. [DOI] [PubMed] [Google Scholar]
- Hausmann R, Seidl S, Betz P. Hypoxic changes in Purkinje cells of the human cerebellum. Int J Legal Med. 2007;121:175–183. doi: 10.1007/s00414-006-0122-x. [DOI] [PubMed] [Google Scholar]
- Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol. 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- Hossmann KA, Oschlies U, Schwindt W, Krep H. Electron microscopic investigation of rat brain after brief cardiac arrest. Acta Neuropathol. 2001;101:101–113. doi: 10.1007/s004010000260. [DOI] [PubMed] [Google Scholar]
- Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, Straub RE, Ye T, Colantuoni C, Herman MM, Bigelow LB, Weinberger DR, Kleinman JE. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Jones EG. Thalamic circuitry and thalamocortical synchrony. Philos Trans R Soc Lond B Biol Sci. 2002;357:1659–1673. doi: 10.1098/rstb.2002.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khot S, Tirschwell DL. Long-term neurological complications after hypoxic-ischemic encephalopathy. Semin Neurol. 2006;26:422–431. doi: 10.1055/s-2006-948323. [DOI] [PubMed] [Google Scholar]
- Kumar K, White BC, Krause GS, Indrieri RJ, Evans AT, Hoehner TJ, Garritano AM, Koestner A. A quantitative morphological assessment of the effect of lidoflazine and deferoxamine therapy on global brain ischaemia. Neurol Res. 1988;10:136–140. doi: 10.1080/01616412.1988.11739830. [DOI] [PubMed] [Google Scholar]
- Kwegyir-Afful EE, Bruno RM, Simons DJ, Keller A. The role of thalamic inputs in surround receptive fields of barrel neurons. J Neurosci. 2005;25:5926–5934. doi: 10.1523/JNEUROSCI.1360-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land PW, Simons DJ. Cytochrome oxidase staining in the rat SmI barrel cortex. J Comp Neurol. 1985;238:225–235. doi: 10.1002/cne.902380209. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A. Neurophysiological investigation of the basis of the fMRI signal. Nature. 2001;412:150–157. doi: 10.1038/35084005. [DOI] [PubMed] [Google Scholar]
- Manole MD, Foley LM, Hitchens TK, Kochanek PM, Hickey RW, Bayir H, Alexander H, Ho C, Clark RS. Magnetic resonance imaging assessment of regional cerebral blood flow after asphyxial cardiac arrest in immature rats. J Cereb Blood Flow Metab. 2009;29:197–205. doi: 10.1038/jcbfm.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryniak A, Bielawska A, Walczak F, Szumowski £, Bieganowska K, Rekawek J, Paszke M, Szymaniak E, Knecht M. Long-term cognitive outcome in teenage survivors of arrhythmic cardiac arrest. Resuscitation. 2008;77:46–50. doi: 10.1016/j.resuscitation.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Mateen FJ, Josephs KA, Trenerry MR, Felmlee-Devine MD, Weaver AL, Carone M, White RD. Long-term cognitive outcomes following out-of-hospital cardiac arrest: a population-based study. Neurology. 2011;77:1438–1445. doi: 10.1212/WNL.0b013e318232ab33. [DOI] [PubMed] [Google Scholar]
- McAlonan K, Cavanaugh J, Wurtz RH. Guarding the gateway to cortex with attention in visual thalamus. Nature. 2008;456:391–394. doi: 10.1038/nature07382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Cheung AF. Towards the classification of subpopulations of layer V pyramidal projection neurons. Neurosci Res. 2006;55:105–115. doi: 10.1016/j.neures.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Myers RE, Yamaguchi S. Nervous system effects of cardiac arrest in monkeys. Preservation of vision. Arch Neurol. 1977;34:65–74. doi: 10.1001/archneur.1977.00500140019003. [DOI] [PubMed] [Google Scholar]
- Ng T, Graham DI, Adams JH, Ford I. Changes in the hippocampus and the cerebellum resulting from hypoxic insults: frequency and distribution. Acta Neuropathol. 1989;78:438–443. doi: 10.1007/BF00688181. [DOI] [PubMed] [Google Scholar]
- Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, Rea T, Lowe R, Brown T, Dreyer J, Davis D, Idris A, Stiell I. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300:1423–1431. doi: 10.1001/jama.300.12.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Gao G, Wei EP, Povlishock JT. Combinational therapy using hypothermia and the immunophilin ligand FK506 to target altered pial arteriolar reactivity, axonal damage, and blood-brain barrier dysfunction after traumatic brain injury in rat. J Cereb Blood Flow Metab. 2011;31:1143–1154. doi: 10.1038/jcbfm.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskine A, Rosso C, Picq C, Caron E, Pradat-Diehl P. Neurological sequelae after cerebral anoxia. Brain Inj. 2010;24:755–761. doi: 10.3109/02699051003709581. [DOI] [PubMed] [Google Scholar]
- Pollock JM, Whitlow CT, Deibler AR, Tan H, Burdette JH, Kraft RA, Maldjian JA. Anoxic injury-associated cerebral hyperperfusion identified with arterial spin-labeled MR imaging. AJNR Am J Neuroradiol. 2008;29:1302–1307. doi: 10.3174/ajnr.A1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- Radovsky A, Safar P, Sterz F, Leonov Y, Reich H, Kuboyama K. Regional prevalence and distribution of ischemic neurons in dog brains 96 hours after cardiac arrest of 0 to 20 minutes. Stroke. 1995;26:2127–2133. doi: 10.1161/01.str.26.11.2127. discussion 2133–2134. [DOI] [PubMed] [Google Scholar]
- Radovsky A, Katz L, Ebmeyer U, Safar P. Ischemic neurons in rat brains after 6, 8, or 10 minutes of transient hypoxic ischemia. Toxicol Pathol. 1997;25:500–505. doi: 10.1177/019262339702500512. [DOI] [PubMed] [Google Scholar]
- Rice JE, 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol. 2005;562:27–36. doi: 10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Miclescu A, Wiklund L. Cardiac arrest-induced regional blood-brain barrier breakdown, edema formation and brain pathology: a light and electron microscopic study on a new model for neurodegeneration and neuroprotection in porcine brain. J Neural Transm. 2011;118:87–114. doi: 10.1007/s00702-010-0486-4. [DOI] [PubMed] [Google Scholar]
- Shoykhet M, Simons DJ. Development of thalamocortical response transformations in the rat whisker-barrel system. J Neurophysiol. 2008;99:356–366. doi: 10.1152/jn.01063.2007. [DOI] [PubMed] [Google Scholar]
- Shoykhet M, Shetty P, Minnery BS, Simons DJ. Protracted development of responses to whisker deflection in rat trigeminal ganglion neurons. J Neurophysiol. 2003;90:1432–1437. doi: 10.1152/jn.00419.2003. [DOI] [PubMed] [Google Scholar]
- Shoykhet M, Land PW, Simons DJ. Whisker trimming begun at birth or on postnatal day 12 affects excitatory and inhibitory receptive fields of layer IV barrel neurons. J Neurophysiol. 2005;94:3987–3995. doi: 10.1152/jn.00569.2005. [DOI] [PubMed] [Google Scholar]
- Simons DJ. Multi-whisker stimulation and its effects on vibrissa units in rat SmI barrel cortex. Brain Res. 1983;276:178–182. doi: 10.1016/0006-8993(83)90561-9. [DOI] [PubMed] [Google Scholar]
- Simons DJ, Carvell GE. Thalamocortical response transformation in the rat vibrissa/barrel system. J Neurophysiol. 1989;61:311–330. doi: 10.1152/jn.1989.61.2.311. [DOI] [PubMed] [Google Scholar]
- Simons DJ, Land PW. Early experience of tactile stimulation influences organization of somatic sensory cortex. Nature. 1987;326:694–697. doi: 10.1038/326694a0. [DOI] [PubMed] [Google Scholar]
- Simons DJ, Carvell GE, Hershey AE, Bryant DP. Responses of barrel cortex neurons in awake rats and effects of urethane anesthesia. Exp Brain Res. 1992;91:259–272. doi: 10.1007/BF00231659. [DOI] [PubMed] [Google Scholar]
- Stern EA, Maravall M, Svoboda K. Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron. 2001;31:305–315. doi: 10.1016/s0896-6273(01)00360-9. [DOI] [PubMed] [Google Scholar]
- Switzer RC., 3rd Application of silver degeneration stains for neurotoxicity testing. Toxicol Pathol. 2000;28:70–83. doi: 10.1177/019262330002800109. [DOI] [PubMed] [Google Scholar]
- Venkatesan A, Frucht S. Movement disorders after resuscitation from cardiac arrest. Neurol Clin. 2006;24:123–132. doi: 10.1016/j.ncl.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Volpe BT, Hirst W. The characterization of an amnesic syndrome following hypoxic ischemic injury. Arch Neurol. 1983;40:436–440. doi: 10.1001/archneur.1983.04050070066017. [DOI] [PubMed] [Google Scholar]
- Volpe BT, Holtzman JD, Hirst W. Further characterization of patients with amnesia after cardiac arrest: preserved recognition memory. Neurology. 1986;36:408–411. doi: 10.1212/wnl.36.3.408. [DOI] [PubMed] [Google Scholar]
- Welker WI. Analysis of sniffing in the albino rat. Behaviour. 1964;22:223–244. [Google Scholar]
- Wintermark M, Lepori D, Cotting J, Roulet E, van Melle G, Meuli R, Maeder P, Regli L, Verdun FR, Deonna T, Schnyder P, Gudinchet F. Brain perfusion in children: evolution with age assessed by quantitative perfusion computed tomography. Pediatrics. 2004;113:1642–1652. doi: 10.1542/peds.113.6.1642. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT, Merzenich MM, Leake PA. Changes in endogenous enzymatic reactivity to DAB induced by neuronal inactivity. Brain Res. 1978;141:185–192. doi: 10.1016/0006-8993(78)90629-7. [DOI] [PubMed] [Google Scholar]