

Abstract
Kaposi sarcoma (KS) tumors often contain a wild-type p53. However, the function of this tumor suppressor in KS tumor cells is inhibited by both MDM2 and latent nuclear antigen (LANA) of Kaposi sarcoma-associated herpes virus (KSHV). Here, we report that MDM2 antagonist Nutlin-3 efficiently reactivates p53 in telomerase-immortalized human umbilical vein endothelial cells (TIVE) that had been malignantly transformed by KSHV as well as in KS tumor cells. Reactivation of p53 results in a G1 cell cycle arrest, leading to inhibition of proliferation and apoptosis. Nutlin-3 inhibits the growth of “KS-like” tumors resulting from xenografted TIVE-KSHV cells in nude mice. In addition, Nutlin-3 strongly inhibits expression of the pro-angiogenic and pro-inflammatory cytokine angiopoietin-2 (Ang-2). It also disrupts viral latency by inducing expression of KSHV lytic genes. these results suggest that Nutlin-3 might serve as a novel therapy for KS.
Key words: Kaposi sarcoma (KS), nutlin-3, p53, cell cycle arrest, apoptosis, angiopoietin-2
Introduction
Mutations or functional loss of the tumor suppressor p53 accounts for more than 50% of all cancers.1,2 Kaposi sarcoma (KS), a vascular malignancy of endothelial cell origin, often carries a wild-type p53.3 Most KS tumor cells are latently infected by Kaposi sarcoma-associated herpes virus (KSHV) and express the viral latent nuclear antigen (LANA) encoded by open reading frame 73 (ORF73), which physically interacts with p53 and inhibits its function.4,5 In addition, p53 in KS tumor cells is associated with MDM2, an E3 ligase that targets p53 for proteomic degradation.6,7
Rescue or reactivation of p53 has been sought as a strategy for cancer treatment. Recently, an MDM2 antagonist named Nutlin-3 was developed to specifically bind to MDM2 with a much higher affinity at the site where p53 binds, thus preventing p53 from degradation and re-activating its function.8 This molecule is very effective in inducing p53 reactivation and causing tumor regression in various cancer models, including KSHV-induced primary effusion lymphomas (PEL).9,10 Interestingly, Nutlin-3 also disrupts the association between p53 and KSHV latent protein LANA, leading to a complete reactivation of p53 in PEL cells.9
The efficacy of Nutlin-3 on KS has not been tested. Distinct from PEL, which are KSHV-transformed B cells, KS tumors comprise a variety of cell types, including KSHV-infected “spindle-shaped” KS tumor cells, red blood cells, inflammatory cells such as monocytes, neutrophils, lymphocytes and blood vessel endothelial cells.11,12 The growth of KS tumors depends on the angiogenic and inflammatory activities in the tumors.12,13 In addition, viral replication also plays pivotal roles in tumor development.14 In this study, we investigated the effects of Nutlin-3 on KS tumor growth in a xenograft mouse model that involved a KSHV-transformed human endothelial cell line.
Results
Nutlin-3 reactivates p53 in KS tumor cells and KSHV-infected endothelial cells.
To examine the effects of Nutlin-3 on KS tumor growth, we used a KS tumor cell line and a KSHV-transformed endothelial cell line as models. The KS-SLK1 cell line was originally derived from an oral KS lesion of an HIV-1-negative kidney transplant recipient receiving immunosuppressive therapy15 and exhibits features of malignant transformation. However, upon multiple passages of culture in vitro, the cell line has lost the viral genomes. The second model was a telomeraseimmortalized human umbilical vein endothelial cell line (TIVE) that had been malignantly transformed upon KSHV infection and grows “KS-like” tumors in nude mice.16 We treated both cell lines in culture with 5 nM Nutlin-3, a dose that was previously shown to reactivate p53 in KSHV-infected PEL cell lines.9,10
Based on nucleotide sequence of RT-PCR products representing the entire p53 ORF from KS-SLK1 and TIVE-KSHV, both cell lines carry a wild-type p53 gene (data not shown). This is consistent with the fact that both cell lines responded well to Nutlin-3 treatment. As shown in Figure 1A, Nutlin-3 treatment led to a 7.2- and 8.7-fold increase in p21 transcript and a 5.8- and 5.4-fold increase in MDM2 transcript in KS-SLK1 and TIVE-KSHV cell lines, respectively. Because both p21 and MDM2 are direct targets for transcriptional induction by p53, this data suggests that Nutlin-3 reactivates p53 in both cell lines. This was further confirmed by western blot analysis demonstrating substantial increases in p53, p21 and MDM2 proteins in both cell lines upon Nutlin-3 treatment (Fig. 1B). Nutlin-3 induction of p21 in these cells implies a negative regulation of cell cycle by this agent. To test this hypothesis, we conducted a DNA content-based cell cycle analysis by performing propidium iodide (PI) staining and flow cytometry of TIVE-KSHV cells treated with Nutlin-3 and DMSO (placebo). Compared with placebo, Nutlin-3 significantly decreases cell population in S phase, while it increases cell population in G1 phase (Fig. 1C). Together, these results confirm that Nutlin-3 is highly effective in reactivating p53 in both cell lines.
Figure 1.

Nutlin-3 reactivates p53 to induce a G1 cell cycle arrest. (A) Real-time RT-PCR data showing relative levels of p21 and MDM2 mRNAs from KS-SLK1 and TIVE-KSHV cells at 48 h post-treatment (hpt) with placebo (DMSO) and Nutlin-3, respectively; (B) western blot detection of p21, MDM2 and p53 proteins from KS-SLK1 and TIVE-KSHV cells at 48 hpt with placebo (DMSO) and Nutlin-3, respectively; (C) flow cytometry analysis of PI-stained TIVE-KSHV cells at 48 hpt. Numbers of cells at different cell cycle phases were determined based on DNA contents of the different populations.
Nutlin-3 inhibits proliferation and induces apoptosis of KS tumor cells.
We next examined if Nutlin-3 could inhibit proliferation and induce apoptosis of KS-SLK1 and TIVE-KSHV cells through p53 reactivation and induction of p21. As a control, we included primary human umbilical vein endothelial cells (HUVEC). Western blot analysis indicates that Nutlin-3 reactivates p53 in HUVEC (data not shown). As shown in Figure 2A, Nutlin-3 inhibits proliferation of all three cell lines; however, this inhibitory effect is much more dramatic in the two tumor cell lines when compared with HUVEC. Moreover, the number of TIVE-KSHV cells was notably lower on day 3 post-treatment with Nutlin-3 than what was initially seeded on day 0, suggesting that Nutlin-3 induces cell death or apoptosis. The growth of KS-SLK1 cells was not completely blocked by Nutlin-3 during the first two days. However, the fact that the number of cells on day 3 post-treatment was much lower than that on day 1 posttreatment indicates that Nutlin-3 induces apoptosis in these cells as well. In contrast, the reduction of cells by Nutlin-3 is not significant in HUVEC. To confirm this selective induction of apoptosis to tumor cells by Nutlin-3, we performed 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining to examine apoptosis. As shown in Figure 2B, Nutlin-3 substantially increases the numbers of apoptotic cells in TIVE-KSHV and KS-SLK1, which is featured by nuclear breakage or fragmentation. We detect an average of 18.1% and 19.2% of apoptotic cells from Nutlin-3-treated TIVE-KSHV and KS-SLK1 respectively, compared with 3.4% and 3.3% from placebo-treated cells (Fig. 2C). In contrast, we hardly find any apoptotic cells from Nutlin-3- and placebo-treated HUVEC. To further confirm Nutlin-3 induction of apoptosis in tumor cells, we purified genomic DNA from HUVEC, TIVE-KSHV and KS-SLK1 cells following treatment with Nutlin-3 or placebo for 3 d and analyzed by electrophoresis in agarose gel. As shown in Figure 2D, treatment with Nutlin-3 notably results in much more fragmented DNA than treatment with placebo in TIVE-KSHV and KS-SLK1 cells. In contrast, Nutlin-3 induces very little DNA fragmentation in HUVEC. Collectively, these results strongly suggest that Nutlin-3 not only inhibits proliferation, but also selectively induces apoptosis of KS tumor cells.
Figure 2.

Nutlin-3 inhibits proliferation and induces apoptosis of KS tumor cells. (A) Growth curves of HUVEC, KS-SLK1 and TIVE-KSHV cells cultured in the presence of Nutlin-3 (5 nM) or placebo (DMSO). Identical amounts (5 × 104) of cells were initially seeded in 1 ml media, which was changed daily with fresh Nutlin-3 or DMSO. Results represent the mean values from three replicates; (B) DAPI nuclear staining of TIVE-KSHV cells after a 4-d treatment with Nutlin-3 and DMSO, respectively; (C) percentages (%) of apoptotic HUVEC, TIVE-KSHV and KS-SLK1 cells detected by DAPI staining (from B); (D) agarose (0.7%) gel electrophoresis analysis of total genomic DNA purified from HUVEC, TIVE-KSHV and KS-SLK1 cells that were treated with Nutlin-3 and DMSO, respectively, for 3 d.
Nutlin-3 inhibits “KS-like” tumor growth in nude mice.
To test if Nutlin-3 inhibits KS tumor growth, we subcutaneously inoculated TIVE-KSHV cells into nude mice. When the average tumor size reached 0.2 cm3, we began treating the mice with daily intra-peritoneal (IP) injection of Nutlin-3 (50 mg/kg of mice, in PBS) or placebo (DMSO in PBS). As shown in Figure 3A and B, Nutlin-3 effectively inhibits tumor growth. In the five mice that were treated with placebo, all tumors grew to over 1 cm3 within 40 d post-inoculation. However, in the five mice that were treated with Nutlin-3, tumors are limited to smaller than 0.25 cm3. All Nutlin-3- and placebo-treated mice were healthy without manifesting any signs of side effects. These results suggest that Nutlin-3 effectively inhibits tumor growth in vivo without causing obvious side effects.
Figure 3.

Nutlin-3 inhibits tumor growth in nude mice. (A) examples of tumors resulting from xenografted TIVE-KSHV cells in nude mice that were treated with daily IP injection of Nutlin-3 or placebo; (B) average tumor volumes from a total of 10 tumors in five Nutlin-3-treated mice and 10 tumors in placebo-treated mice over a period of 52 d. Treatment started on day 10 post-inoculation when average tumor size was about 0.2 cm3.
Nutlin-3 inhibits expression of angiopoietin-2 (Ang-2).
KS tumors strongly express the pro-angiogenic and pro-inflammatory cytokine Ang-2,13,14 which is induced by KSHV infection.15 Since p53 also regulates angiogenesis, we investigated if Nutlin-3 treatment could have any effects on the expression of this cytokine. As demonstrated in Figure 4, both KS-SLK1 and TIVE-KSHV cell lines express Ang-2, although the mRNA level of Ang-2 in TIVE-KSHV cells is 4.9 times higher than in KS-SLK1 cells. Upon treatment with Nutlin-3, Ang-2 mRNA level is reduced to 12% and 2.4% of that in placebo-treated KS-SLK1 and TIVE-KSHV cells, respectively (Fig. 4A and B). Results from western blot analysis demonstrate that Ang-2 protein is substantially reduced in Nutlin-3-treated KS-SLK1 and TIVE-KSHV cells (Fig. C). To examine if Ang-2 levels are reduced in Nutlin-3-treated mice, we performed ELISA on blood collected from the two groups of mice. The average blood level of Ang-2 in Nutlin-3-treated mice is 2.9 ng/ml compared with 7.2 ng/ml in the control group of mice (Fig. 4D). Collectively, these results consistently suggest that Nutlin-3 inhibits tumor angiogenesis and inflammation by suppressing Ang-2 expression.
Figure 4.
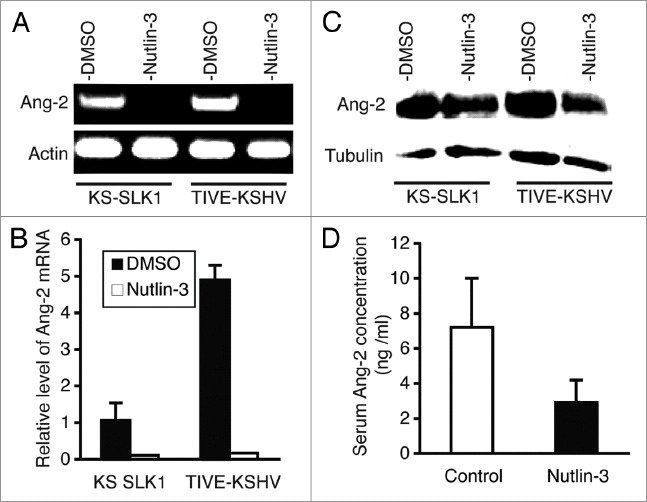
Nutlin-3 inhibits expression of Ang-2. (A) Agarose (1.2%) gel electrophoresis analysis of real-time RT-PCR products of Ang-2 and actin transcripts from KS-SLK1 and TIVE-KSHV cells at 48 hpt with Nutlin-3 and DMSO; (B) relative levels of Ang-2 mRNA from KS-SLK1 and TIVE-KSHV cells at 48 hpt with Nutlin-3 and DMSO; (C) western blot detection of Ang-2 protein from KS-SLK1 and TIVE-KSHV cells at 48 hpt with Nutlin-3 and DMSO. The level of tubulin was used for calibration of sample loading; (D) average serum levels of Ang-2 from five Nutlin-3-treated and five placebo-treated mice as determined by ELISA.
Nutlin-3 disrupts KSHV latency.
Since KSHV replication plays important roles in KS tumor development, we next examined the effect of Nutlin-3 on viral replication. We found that Nutlin-3 treatment efficiently induces KSHV lytic gene expression. As shown in Figure 5A, the transcripts of KSHV lytic genes RTA (ORF50), ORF-K2, ORF-K1, ORF59, ORF65 and ORF-K8.1 increase by 16.2-, 2.6-, 4.6-, 2.2-, 3.3- and 4.0-fold in TIVE-KSHV cells following Nutlin-3 treatment for 3 d, respectively (Fig. 5A). Results from immunofluorescence antibody staining with a monoclonal antibody against KSHV ORF65 demonstrate higher expression of this viral small capsid protein in most of the Nutlin-3-treated cells than in placebo-treated cells (Fig. 5B). Thus, Nutlin-3 treatment disrupts viral latency by inducing expression of KSHV lytic genes.
Figure 5.

Nutlin-3 disrupts KSHV latency by inducing viral lytic gene expression. (A) KSHV lytic transcripts RTA (ORF50), ORF-K2, ORF-K1, ORF59, ORF65 and ORF-K8.1 and latent transcript vFLIP (ORF71) from TIVE-KSHV cells at 72 hpt with Nutlin-3 or placebo were measured by real-time PCR; (B) IFA staining of KSHV small capsid protein ORF65 with an anti-ORF65 monoclonal antibody on TIVE-KSHV cells at 96 hpt with Nutlin-3 or placebo. DAPI staining was used to reveal all cells in the fields.
Discussion
The advent of small chemicals such as Nutlin-3 for reactivation of p53 provides a novel strategy for cancer therapy. Acting as an antagonist of the p53 negative regulator MDM2 by binding with higher affinity to its p53-association motif,8 Nutlin-3 is highly efficient in causing tumor cell death in various tumor models. Nutlin-3 has been found to be effective for treating KSHV-induced PEL in “xenograft”-based mouse models.9,10 In this study, we examined the therapeutic effects of Nutlin-3 on KS tumors, which differ from PEL and other types of lymphomas in many ways. For instances, unlike PEL, which consists mainly of KSHV-transformed B cells, KS tumors comprise a variety of cell types, including KSHV-infected “spindle-shaped” tumor cells, red blood cells, infiltrating inflammatory cells and blood vessel cells. The progression of KS tumors not only depends on the proliferation and survival of KSHV-infected tumor cells but also relies heavily on angiogenesis, inflammation and viral replication. Nutlin-3 might affect each of these activities to slow down KS tumor growth.
Using KS tumor cell line KS-SLK1 and KSHV-transformed endothelial cell line TIVE-KSHV as models, we found that Nutlin-3 effectively reactivates p53 to induce expression of p21 and elicit a G1 cell cycle arrest, resulting in reduced proliferation and increased apoptosis of these cells. Consistent with the in vitro data, Nutlin-3 effectively inhibits the growth of “KS-like” tumors resulting from xenografted TIVE-KSHV cells in nude mice. Intriguingly, with the concentration we used, Nutlin-3 inhibits proliferation of primary endothelial cells in culture but does not induce apoptosis. Further supporting this observation, Nutlin-3 causes little to no observable side effects, while it effectively inhibits tumor growth in mice. While the mechanism of such selective induction of apoptosis to KS tumor cells remains to be determined, it is certainly due to differences between the cell types.
Reactivation of p53 can lead to cell death, reversible quiescence or irreversible senescence.16,17 It is well established that p53 and the mammalian target of rapamycin (mTOR) pathway crosstalk to regulate cellular senescence.16,18 Under certain circumstances, p53 suppresses senescence in favor of inducing quiescence by inhibiting the mTOR pathway, and Nutlin-3 has been shown to cause quiescence in certain cell types.19 Interestingly, KSHV activates the mTOR pathway.20 Consequently, KS tumor cells likely have activated or higher levels of mTOR activity than primary endothelial cells. When treated with the same concentration of Nutlin-3, the latter might be more sensitive to this agent for suppression of senescence and induction of quiescence. In contrast, with higher levels of mTOR activity, KS tumor cells might be less sensitive to Nutlin-3 for induction of quiescence but more in favor of senescence and apoptosis. On the other hand, the mTOR pathway also plays important roles in KSHV pathogenesis,21,22 and Rapamycin has been proven effective in inhibiting KS tumor growth and KSHV virion production.23 Although KS tumor cells are likely less sensitive to Nultin-3 for suppression of senescence and induction of quiescence when compared with primary endothelial cells, this agent may still inhibit the mTOR pathway in these cells to some extent. Therefore, it would be interesting to further investigate whether and how Nutlin-3, in conjunction with reactivating p53, inhibits the mTOR pathway to suppress KS tumor growth.
Angiogenesis and inflammation play pivotal roles in KS tumor development. Previously, Nutlin-3 was found to inhibit expression of VEGF by disrupting the association between MDM2 and hypoxia-inducible factor-1α (HIF-1α) by competing for the same binding motif that allows association between MDM2 and p53.24 We found that Nutlin-3 strongly inhibits expression of Ang-2, a pro-angiogenic and pro-inflammatory cytokine that is highly expressed in KS tumors. The mechanism of Nutlin-3 downregulation of Ang-2 remains to be elucidated. However, since transcription of Ang-2 can also be induced by hypoxia and is regulated by HIF-1α,25,26 it is highly possible that the same mechanism by which Nutlin-3 downregulates VEGF is used to inhibit Ang-2 expression. Regardless of the mechanisms involved, our results support that Nutlin-3 negatively impacts tumor angiogenesis by inhibiting the expression of important pro-angiogenic and pro-inflammatory factors, such as VEGF and Ang-2. Furthermore, senescent cells contribute to tumor growth by secreting multiple pro-angiogenic and pro-inflammatory factors.17,27 By suppressing senescence via inhibition of the mTOR pathway, Nutlin-3 might further reduce the levels of pro-angiogenic and pro-inflammatory factors in KS tumors.
Finally, most KS tumor cells are latently infected by KSHV, suggesting that viral latency is established and required for tumor growth.28 Although a subset of tumor cells undergoes spontaneously lytic replication, which contributes to KS tumor development through different mechanisms,14,24 disruption of latency in the majority of tumors cells could be detrimental, as productive viral lytic replication ultimately kills the cells. In this study, we found that Nutlin-3 induces expression of KSHV lytic genes, suggesting that this agent might inhibit KS tumor growth by disrupting viral latency in tumor cells.
In summary, our data strongly support Nutlin-3 as a highly effective agent in inhibiting proliferation and inducing apoptosis of KS tumors cells. Furthermore, it suppresses expression of angiogenic factors and disrupts viral latency to limit tumor growth. These findings highlight great potentials of this novel agent for treating KS tumors.
Materials and Methods
Cells, media and reagents.
Telomerase-immortalized human umbilical vein endothelial cells (TIVE) that had been latently infected and malignantly transformed with KSHV was a gift from Dr. Rolf Renny, University of Florida, Gainesville. TIVE-KSHV and KS tumor cell line KS-SLK1 were cultured in Dulbecco's modified Eagle medium (DMEM) plus 10% fetal bovine serum (FBS). Primary human umbilical vein endothelial cells (HUVEC) were purchased from Cell Application, Inc. and cultured in endothelial cell media provided by the manufacturer. Nutlin-2 (Sigma-Aldrich) was used at a final concentration of 5 nM for cultured cells. Dimethyl Sulfoxide (DMSO), which was used to dissolve and make stock solution of Nutlin-3, was used as placebo (control). All cells were cultured at 37°C with 5% CO2.
Proliferation and cell cycle analysis.
For proliferation assay, identical amounts (5 × 104) of cells were seeded in each well of 24-well plates with 1 ml of culture media and cultured in the presence of Nutlin-3 (5 nM) or DMSO. Cells from each well were trypsinized, re-suspended in 1 ml of phosphate buffered saline (PBS) and counted under microscope by using a hemocytometer. For cell cycle analysis, cells treated with Nutlin-3 or DMSO were collected and fixed with cold ethanol. Upon staining with propidium iodide (40 ug/ml), followed by washing with PBS, the cells were subjected to flow cytometric analysis by using the BD FACSCanto™ flow cytometer (BD Biosciences). Numbers of cells in different cell cycle phases were determined based on DNA content.
Real-time RT PCR.
Total RNA was isolated by using a RNA purification kit (Promega). Reverse transcription (RT) of total RNA was performed by using Superscript Transcriptase II (Invitrogen). Primers and condition for quantification of all KSHV transcripts by real-time PCR was conducted as reported previously in references 25 and 26. The following primers were used for quantification of host transcripts of (1) 5′-ACT CTC AGG GTC GAA AAC GG-3′ (p21 forward); (2) 5′-CCT CGC GCT TCC AGG ACT G-3′ (p21 reverse); (3) 5′-CTG TGT TCA GTG GCG ATT GG-3′ (MDM2 forward); (4) 5′-AGG GTC TCT TGT TCC GAA GC-3′ (MDM2 reverse); (5) 5′-GTT TGC TAC TGG AAA AAG AGG AAA GAG-3′ (Ang-2 forward); (6) 5′-AGG GCT GCT ACG CTG CC-3′ (Ang-2 reverse); (7) 5′-CAA TGA CCC CTT CAT TGA CC-3′ [GAPDH (glyceraldehyde-3-phosphate dehydrogenase) forward] and (8) 5′-GAT CTC GCT CCT GGA AGA TG-3′ (GAPDH reverse). Each reaction was repeated three times. All transcripts were normalized to transcript of the “house-keeping” gene GAPDH.
Western blot analysis and enzyme-linked immunosorbent assay (ELISA). Total cell lysates were prepared in lysis buffer [100 mM Tris, pH 7.4, 1 mM EDTA, 100 mM NaCl, 1% sodium dodecyl sulfate (SDS) and proteinase inhibitors] and used for SDS-PAGE (PAGE) and western blot analysis. Antibodies specific for p53 (R&D Systems), MDM2 (R&D), p21 (R&D), Ang-2 (Santa Cruz Biotechnology, Inc.) and β-tubulin (Sigma) were used as primary antibodies to react with the blots first. After washing with PBS plus 0.2% tween-20, the blots were then incubated with either a horse radish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG (Santa Cruz) and subjected to chemiluminescence's detection. To measure the relative levels of Ang-2 in mice, two drops of blood were collected from the tail vein of each mouse. Upon dilution (20 µl blood in 180 µl PBS), all samples were subjected to a standard procedure for “double-sandwich” ELISA; a rabbit anti-Ang-2 antibody (Santa Cruz) for coating the plates and capturing Ang-2 and a mouse anti-Ang-2 antibody (R&D Systems) for detecting Ang-2. An alkaline phosphatase-conjugated goat anti-mouse-IgG (Santa Cruz) was used to measure the detecting antibody. A standard curve was established by following the same procedure to measure the optical density (OD) values of a series of dilutions of a recombinant Ang-2 (R&D Systems). Each sample was run six times, and the average OD value of each sample was used for calculating Ang-2 levels in mouse blood.
Immunofluorescence antibody (IFA) and 4′,6-diamidino-2-phenylindole (DAPI) staining. Nutlin-3- or DMSO-treated cells were fixed with 1% paraformaldehyde (PFA). IFA staining of cells expressing KSHV lytic protein ORF65 was conducted as described in the text, using a monoclonal anti-ORF65 antibody. DAPI was performed for nuclear staining.
Xenografting KSHV-TIVE cells into nude mice and treatment with nutlin-3.
Xenografts of TIVE-KSHV cells were injected into 6-week-old female nude mice. Two injections, one on either side of the abdominal midline, consisting of identical numbers of cells (5 × 106 per injection site) per injection were administered per mouse. A total of 10 mice were used. Ten days post-inoculation, the mice were randomly split into two groups: one treated with a daily intra-peritoneal (IP) injection of Nutlin-3 (50 mg/kg of mice) and the other treated with placebo (DMSO). Tumor volume (length × width × height) was measured on a weekly basis with a caliper.
Acknowledgments
This study was supported from grants DE017333, CA096512, CA124332 and CA119889 from National Institute of Health to ShouJiang Gao, and from start-up funds from Case western Reserve University, School of Dental Medicine to FengChun Ye. We thank Dr. Rolf Renny from University of Florida for providing the TIVE-KSHV cells. We are also grateful to Jennifer Rebeles at the Greehey Children's Cancer Research Institute, University of Texas Health Science Center at San Antonio, Texas, for technical assistance in flow cytometry and cell cycle analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Suzuki K, Matsubara H. Recent advances in p53 research and cancer treatment. J Biomed Biotechnol. 2011;2011:978312. doi: 10.1155/2011/978312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soussi T. Tp53 mutations in human cancer: database reassessment and prospects for the next decade. Adv Cancer Res. 2011;110:107–139. doi: 10.1016/B978-0-12-3864697.00005-0. [DOI] [PubMed] [Google Scholar]
- 3.Tornesello ML, Biryahwaho B, Downing R, Hatzakis A, Alessi E, Cusini M, et al. Tp53 codon 72 polymorphism in classic, endemic and epidemic Kaposi's sarcoma in African and Caucasian patients. Oncology. 2009;77:328–334. doi: 10.1159/000260905. [DOI] [PubMed] [Google Scholar]
- 4.Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 5.Si H, Robertson ES. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J Virol. 2006;80:697–709. doi: 10.1128/JVI.80.2.697-709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piette J, Neel H, Maréchal V. Mdm2: keeping p53 under control. Oncogene. 1997;15:1001–1010. doi: 10.1038/sj.onc.1201432. [DOI] [PubMed] [Google Scholar]
- 7.Brooks CL, Gu W. Dynamics in the p53-Mdm2 ubiquitination pathway. Cell Cycle. 2004;3:895–899. doi: 10.4161/cc.3.7.997. [DOI] [PubMed] [Google Scholar]
- 8.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 9.Sarek G, Kurki S, Enbäck J, Iotzova G, Haas J, Laakkonen P, et al. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J Clin Invest. 2007;117:1019–1028. doi: 10.1172/JCI30945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petre CE, Sin SH, Dittmer DP. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J Virol. 2007;81:1912–1922. doi: 10.1128/JVI.01757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francès C, Lebbé C. Kaposi's sarcoma. Cancer Treat Res. 2009;146:299–309. doi: 10.1007/978-0-387-78574-5_24. [DOI] [PubMed] [Google Scholar]
- 12.Douglas JL, Gustin JK, Dezube B, Pantanowitz JL, Moses AV. Kaposi's sarcoma: a model of both malignancy and chronic inflammation. Panminerva Med. 2007;49:119–138. [PubMed] [Google Scholar]
- 13.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 14.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNFalpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12:235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 15.Ye FC, Blackbourn DJ, Mengel M, Xie JP, Qian LW, Greene W, et al. Kaposi's sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J Virol. 2007;81:3980–3991. doi: 10.1128/JVI.02089-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galluzzi L, Kepp O, Kroemer G. Tp53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) 2010;2:535–537. doi: 10.18632/aging.100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Keizer PL, Laberge RM, Campisi J. p53: Pro-aging or pro-longevity? Aging (Albany NY) 2010;2:377–379. doi: 10.18632/aging.100178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA. 2010;107:9660–9664. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010;2:344–352. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montaner S. Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: emerging insights into the molecular oncogenesis and treatment of Kaposi's sarcoma. Cell Cycle. 2007;6:438–443. doi: 10.4161/cc.6.4.3843. [DOI] [PubMed] [Google Scholar]
- 21.Stallone G, Infante B, Grandaliano G, Schena FP, Gesualdo L. Kaposi's sarcoma and mTOR: a crossroad between viral infection neoangiogenesis and immunosuppression. Transpl Int. 2008;21:825–832. doi: 10.1111/j.1432-2277.2008.00697.x. [DOI] [PubMed] [Google Scholar]
- 22.Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, et al. Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med. 2005;352:1317–1323. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]
- 23.Nichols LA, Adang LA, Kedes DH. Rapamycin blocks production of KSHV/HHV8: insights into the antitumor activity of an immunosuppressant drug. PLoS One. 2011;6:14535. doi: 10.1371/journal.pone.0014535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaRusch GA, Jackson MW, Dunbar JD, Warren RS, Donner DB, Mayo LD. Nutlin3 blocks vascular endothelial growth factor induction by preventing the interaction between hypoxia inducible factor 1alpha and Hdm2. Cancer Res. 2007;67:450–454. doi: 10.1158/0008-5472.CAN-06-2710. [DOI] [PubMed] [Google Scholar]
- 25.Krikun G, Schatz F, Finlay T, Kadner S, Mesia A, Gerrets R, et al. Expression of angiopoietin-2 by human endometrial endothelial cells: regulation by hypoxia and inflammation. Biochem Biophys Res Commun. 2000;275:159–163. doi: 10.1006/bbrc.2000.3277. [DOI] [PubMed] [Google Scholar]
- 26.Yamakawa M, Liu LX, Date T, Belanger AJ, Vincent KA, Akita GY, et al. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res. 2003;93:664–673. doi: 10.1161/01.RES.0000093984.48643.D7. [DOI] [PubMed] [Google Scholar]
- 27.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 28.Ensoli B, Sgadari C, Barillari G, Sirianni MC, Stürzl M, Monini P. Biology of Kaposi's sarcoma. Eur J Cancer. 2001;37:1251–1269. doi: 10.1016/S0959-8049(01)00121-6. [DOI] [PubMed] [Google Scholar]