

Abstract
Genome-wide gene deregulation and oxidative stress appear to be critical factors determining the high variability of phenotypes in Down's syndrome (DS). Even though individuals with trisomy 21 exhibit a higher survival rate compared to other aneuploidies, most of them die in utero or early during postnatal life. While the survivors are currently predicted to live past 60 years, they suffer higher incidence of age-related conditions including Alzheimer's disease (AD). This paper is centered on the mechanisms by which mitochondrial factors and oxidative stress may orchestrate an adaptive response directed to maintain basic cellular functions and survival in DS. In this context, the timing of therapeutic interventions should be carefully considered for the successful treatment of chronic disorders in the DS population.
1. Introduction
Down's syndrome (DS) or trisomy 21 is a prevalent genetic cause of intellectual disability due to full or partial triplication of chromosome 21 (HSA21). The presentation varies greatly between individuals. The molecular bases of this variation is “the gene dosage effect” caused by the extra chromosome 21, which leads to a global imbalance on gene expression [1]. However, the molecular mechanisms by which such gene dosage imbalance causes DS-specific abnormalities remain unclear.
Albeit trisomy 21 is the most common aneuploidy that infants can survive, the rate of miscarriage of fetuses with DS during the first trimester is almost 50% [2]. The survival rate for the first 18 years of life of DS individuals is 50.3% of the total DS population, and the greatest percent of deaths is observed during the first 5 years of life (35.9%). The death rate drops to 13.1% between 19 and 40 years, and DS individuals of 40+ years have a greater chance to live beyond 60 years of age in developed countries, especially those without congenital heart disease [3].
A remarkable feature of the syndrome is the presence of Alzheimer's disease (AD) neuropathology in the brain of nearly all DS individuals, the majority of which develop dementia with age [4]. Besides dementia, other aging features appear prematurely such as cataracts, diabetes, hair graying, leukemia, and hearing and visual impairment. Together, they define DS as a “segmental progeroid syndrome” [5–7]. Mitochondria represent both a principal source as well as a target of free radicals, which in turn cause structural damage and activate signaling pathways associated with ageing and age-related diseases [8–10]. Both oxidative stress and mitochondrial dysfunction are prominent features of DS [11–14]. The relation between oxidative stress, genome imbalances, specific HSA21 genes, and the DS phenotype has been discussed elsewhere [11, 14–17]. In this paper, we will primarily focus on mitochondrial deregulation, oxidative stress, and the emergence of an adaptive response, which may influence the timing and extent of clinical manifestations in DS.
2. Mitochondria and Oxidative Stress
Mitochondria have three major functions: generation of ATP, production of reactive oxygen species (ROS) and initiation of apoptosis. NADH and FADH2 formed in glycolysis, fatty acid oxidation, and the citric acid cycle are used to reduce oxygen to water by a series of electron carriers located in the inner mitochondrial membrane. The flow of electrons leads to the pumping of protons out of the mitochondrial matrix and the formation of a proton gradient across the inner membrane, which provides the driving force used by ATP synthase to produce ATP. This process is known as oxidative phosphorylation (OXPHOS) [18, 19]. Most of the cellular ROS are produced by electrons escaping from the electron transport chain (ETC) which are captured by O2. Some studies suggest that as much as 2–5% of the total O2 intake ends up forming superoxide radicals. These are scavenged by antioxidant enzymes such as mitochondrial superoxide dismutase (SOD2) and glutathione peroxidase (Gpx) [20]. Mitochondrial DNA (mtDNA) encodes 37 genes: 13 mRNAs for subunits of ETC complexes, 22 tRNAs, and 2 rRNAs operating protein translation in the mitochondrial matrix [18]. Since mtDNA is in close proximity to the ETC, it can suffer mutations under excessive ROS production, leading to impaired gene expression and further reductions in ETC efficiency. Mitochondria eventually become dysfunctional beyond repair and lose their electrochemical membrane potential (MMP). The loss of MMP activates the permeability transition pore, releasing mitochondrial material to the cytoplasm. Ultimately, this triggers the execution phase of the apoptotic process [18], which has been implicated in multiple conditions including mitochondrial diseases, DS, and age-related neurodegeneration [21, 22].
Besides mtDNA, nuclear DNA (nDNA) encodes approximately 1600 mitochondrial genes [18, 21]. Because of the split location of mitochondrial genes, mitochondrial genetics does not follow Mendelian rules. While mitochondria and mtDNA are maternally inherited [21], nuclear encoded mitochondrial genes (NEMGs) are inherited from both parents. Since each cell has thousands of mitochondria and mtDNA copies; individual differences in the ratio of normal and mutant mtDNA lead to heteroplasmy. Variations in heteroplasmy and in the energy requirements of specific cells and tissues dictate the variability in the presentation of mitochondrial diseases, not unlike what is observed in DS. The proportion of mutated mtDNAs varies spatially (depending on the cell and tissue) and temporally (over the individual's life). Thus, a particular mtDNA mutation may cause variable phenotypes [18, 23]. For example, the mtDNA mutation tRNALeu A3243G has been associated with mitochondrial myopathy, encephalopathy, lactic acidosis, stroke-like episodes (MELAS), diabetes mellitus, Leigh's disease, and progressive external ophthalmoplegia (PEO) [24]. This variability in phenotypes may also be relevant to DS, where there is a high rate of mtDNA mutations and several mitochondrial genes are disproportionally expressed.
3. Mitochondria in DS and DSAD
In addition to a handful of mitochondrial genes in HSA21 whose deregulation may impair mitochondrial function, the evidence suggests that cytoplasmic inheritance of deleterious mtDNA mutations in maternal mitochondria can influence the frequency of DS in families or increase DS incidence in pregnancies from older age females [25, 26]. Mitochondrial activity is essential for spindle formation and chromosome segregation during meiosis and early embryogenesis [27]. Age appears to influence mitochondrial function in oocytes and follicular cells, and mtDNA mutations in oocytes have been found to be age related [23, 27]. Dysfunctional mitochondria have been implicated in the predisposition to chromosomal nondisjunction during the first and second meiotic divisions, in mitotic errors in embryos, and in the reduced quality and developmental potential of aged oocytes and embryos [23, 27, 28]. Thus, variable levels of mtDNA mutations in maternal mitochondria would be present in different DS individuals. Since mtDNA mutations accumulate with age, individuals starting their lives with higher mtDNA mutation rates would be more predisposed to age-related dementia. In fact, DS with Alzheimer's disease (DSAD) exhibit higher rates of mtDNA mutations in frontal cortex compared to DS and age-matched controls (Figure 1(a)).
Figure 1.
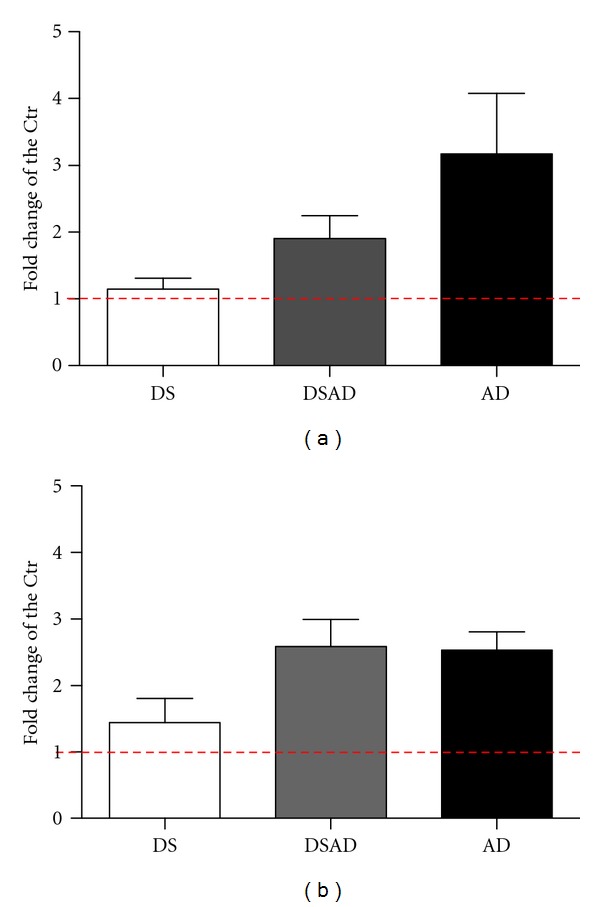
Accumulation of mitochondrial DNA mutations in DS, DSAD, and AD frontal cortex (a) and lymphoblastoid cell lines (LCL) (b). The graph was plotted as fold difference with respect to age-matched controls. DS brains age group: 0–40, DSAD age group: 45–68, and AD age group: 65–90. For each group 6 to 16 samples were analyzed. LCL lines for all groups (DS, DSAD, DAD, and control) were obtained from 40–60 years old donors, 6–8 samples per group. The red line shows the baseline mutation level for the control group.
Similar differences were observed when mtDNA mutations were analyzed in lymphoblastoid cells (LCL) (Figure 1(b)) [29], indicating a systemic increase in mtDNA mutations in DSAD. Specific mtDNA nucleotides were mutated at higher rates in DSAD and sporadic AD than in controls, and mutations in replication and transcription regulatory sequences resulting in reduced mtDNA levels and light strand gene expression were found in brains of DSAD and AD individuals [29, 30]. Consistent with these studies, a previous report indicates defective repair of mtDNA damage in DS [31]. Interestingly, DS brains without AD exhibited a slight increase in mtDNA levels, suggesting a compensatory upregulation of mitochondrial biogenesis, which disappeared in DSAD subjects. This decrease in mitochondrial biogenesis with dementia correlates with increased Aß levels and deposition, suggesting Aβ-related toxic mechanisms affecting mitochondrial biogenesis (Figure 2) [29].
Figure 2.
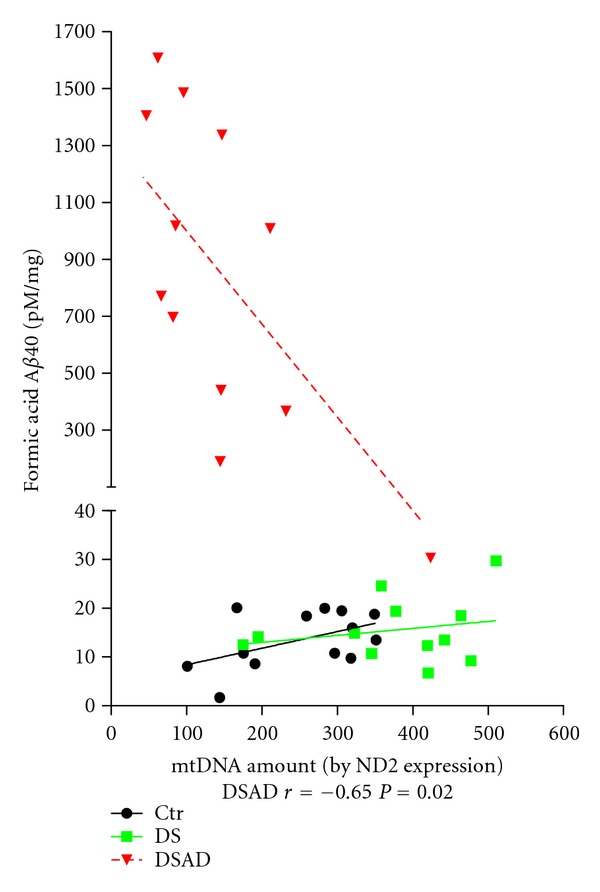
Levels of Aβ correlated with mitochondrial biogenesis represented as mtDNA amount. There was a significant inverse correlation between insoluble Aβ and mitochondrial DNA amount only in DSAD cases. Results reprinted from [29].
4. Oxidative Stress and Mitochondrial Alterations
Increased oxidative stress in DS and AD correlates with a decrease in several mitochondrial components including complex IV nuclear encoded subunit IV, mtDNA encoded subunit I [32], complex I nuclear encoded 24 and 75 kDa subunits [33], complex V nuclear encoded β subunit and complex III nuclear encoded core protein I [23], and mitochondrial ATPase6 and mitochondrial transcription factor A (Tfam) [34]. A recent study in DS fibroblasts found a specific deficiency in complex I, increased levels of several ETC components, and increased porin levels, further suggesting that mitochondrial biogenesis is upregulated in DS. The defect in complex I was associated with decreased cAMP-dependent phosphorylation of complex I 18 kDa subunit, reduced protein kinase A activity and low basal levels of cAMP. Mitochondrial superoxide production and oxidative stress were found to be 3 times higher in DS fibroblast, which were rescued by treatment with a cAMP analog [35]. The general changes in expression of mitochondrial enzymes correlate with a downregulation of the major mitochondrial heat shock protein, HSP60 [36, 37], which is critical to prevent protein aggregation during thermal and ROS stress. In addition, a number of mitochondrial proteins are elevated in DS including mitochondrial aconitase, NADP-linked isocitrate dehydrogenase [38], and the mitochondria-targeted ES1 protein homologue [39], all of which may be part of a compensatory antioxidant response to increased mitochondrial ROS production.
5. DS and Hormesis
Based on the considerations above, it is conceivable that oxidative stress and redox changes play a dual role in DS. At low levels, they promote cellular proliferation, while at higher levels, they produce oxidative damage and initiate apoptosis [40]. Adaptive response signaling, also known as hormesis, is triggered by sublethal stress, which stimulates cellular functional changes to protect against a subsequent exposure to more severe stress [41]. Consequently, compensatory mechanisms can prepare the cell to resist higher stress levels [42].
Since mitochondria are the main source of ROS production, their role is essential in age-related oxidative damage. While abundant research supports the idea that reduced oxidative stress is associated with increased life span [43–46] several experiments showed inconsistent or even contradictory results in human studies when interventions aimed to lower ROS level [47] were unable to produce health beneficial effects [48, 49]. In a recent example, which is relevant to DS, a 2-year randomized placebo-controlled daily oral antioxidant supplementation did not improve cognitive functioning nor it stabilized cognitive decline in DSAD [50]. These findings suggest that mitochondrial ROS production could indeed trigger cellular processes that promote health and longevity. Such signaling events, or adaptive response, are observed in the context of caloric restriction (CR), one of the best intervention strategies to increase life span from yeast to mammals. In fact, CR induces mitochondrial hormesis (mitohormesis) [51] by increasing mitochondrial respiration and elevating mitochondrial ROS production without changing ATP production [52].
A prominent sensor related to hormesis is the Keap1-Nrf2-ARE signaling complex. Under normal redox conditions, the transcription factor NFE2-related factor 2 (Nrf2) binds to Kelch-like ECH-associated protein 1 (Keap1) in the cytosol leading to its proteasomal degradation [53]. Keap1 is a cysteine-rich protein that senses redox changes in the cell. Under oxidative stress, conformational changes in Keap1 lead to its dissociation from the Nrf2-Keap1 complex and to the translocation of free Nrf2 into the nucleus, where it binds to antioxidant response element (ARE) regions in the genome, and activates the expression of stress response genes [54, 55]. So far, there is no complete information on Nrf2/Keap1 genes, protein levels or activities in DS. However, a recent study comparing gene expression profiles in DS and euploid astrocytes found that Nrf-2-associated oxidative stress response genes were differentially regulated in DS, supporting the presence of hormesis in DS [56]. Additional evidence of hormesis in DS comes from experiments showing increased activity of mitogen-activated protein kinases (MAPKs), including ERK1/2, SAPKs, and p38 in DS and AD brains [57]. MAPKs phosphorylate Nrf2 enabling its dissociation from the Nrf2/Keap1 complex [58]. GPx and catalase are also Nrf2 target genes carrying ARE sequences in their promoters [59]. Interestingly, higher intellectual function in DS correlated with increased expression of GPx, which could be part of the adaptive response in those individuals [60].
Nrf2 also interacts with PPARγ, PGC1α, and PI3K/Akt, all of which participate in mitochondrial biogenesis [61]. Thus, these factors may underlie mtDNA increase [29] and mitochondrial biogenesis [35] in DS cells. Finally, a generalized downregulation of mitochondrial activity has been observed in different DS cell types including neurons, astrocytes, pancreatic β cells, endothelial cells, and fibroblasts, which is consistent with a cellular adaptation to reduce ROS production and prevent cellular injury [56]. However, additional stressors and/or challenges in the form of infections, seizures, age-related loss of function, and so forth can eventually exhaust the capacity of the functional adaptations to avert cellular damage. In this context, chronic respiratory infections and multiple signs of early senescence such as cataracts, skin atrophy, seizures, leukemia, and AD-type neuropathology may be the result of oxidative stress, mitochondrial dysfunction, and additional factors acting systemically or in specific organs and tissues. Thus, the severity of the DS phenotype may be the result of the initial level of mitochondrial mutations, the accumulation of oxidative damage, and the magnitude of the cellular adaptations triggered by these changes. Activation of an early adaptive response by initial sublethal levels of stress may translate in a longer survival. However, the combination of chronic stress and age-related changes would result in the premature and accelerated development of age-related conditions such as dementia and AD pathology.
One consequence of the considerations above is that not only the compounds but also the timing of treatment options should be carefully considered in DS patients. For example, long-term treatments designed to reduce oxidative stress may not add any incremental benefit on top of the changes driven by hormesis. Interventions would be more effective if introduced at the very onset of stress or disease. In fact, recent findings indicate that exercise-induced oxidative stress ameliorates insulin resistance and generates an adaptive response enhancing the endogenous antioxidant defense capacity [62]. However, supplementation with antioxidants may preclude the health-promoting effects of exercise in humans [62]. Thus, under normal conditions, antioxidants may not help and may even interfere with hormesis. According to this hypothesis, they would be most effective when an additional stressor is present.
In conclusion, DS is the result of a whole genome imbalance caused by triplication of HSA21 genes. The severity and spectrum of the syndrome vary greatly. Besides oxidative damage, mtDNA mutations and mitochondrial dysfunction emerge as important modulators of DS phenotypes. This variability is further influenced by an adaptive cellular response to stress. A comprehensive and detailed analysis of signature pathways unique to hormesis will be required to fully assess the role of the adaptive response in DS (Figure 3). Key elements of hormesis may be valuable predictors of disease onset and treatment outcomes in DS individuals.
Figure 3.
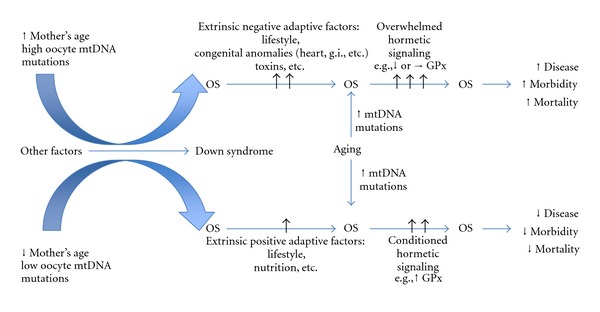
Modulation of DS phenotypes by oxidative stress and mitochondrial factors. Fetal oxidative stress (OS) levels could be determined by the mother's age and initial mtDNA mutation levels in oocytes. Besides the genetic/intrinsic factors that create the genomic instability in DS, environmental factors and lifestyle modulate the initial OS further. Since all these factors that play a role in the level of OS differ individually, the OS-related changes will also be observed in variation. Simply, while the low level of OS could initiate the positive adaptive response by activating proper defense signaling, high level of OS will start destructive signaling where the adaptive response could not be able to accommodate the clearance of the damage. More positive factors (e.g., lifestyle, advantageous genetic background—mitochondrial haplotype, APOE, BDNF genotype, etc.—and nutrition) will feed the adaptive response positively, while negative factors (e.g., congenital defects, sedentary lifestyle, genotypes, etc.) will increase the OS further. In both low and high levels of initial OS conditions, aging will affect this process negatively by increasing OS, such as increasing mtDNA mutation accumulation and decline in mitochondrial functions. Under increasing OS conditions with aging, individuals with DS will be prone to develop more morbid conditions and prone to death depending on their initial adaptive response signaling. In other words, negative factors will lead to earlier clinical manifestations of age-related conditions, while positive adaptations (e.g., conditioned hormetic signaling) may support normal cellular and systemic functions for longer periods of time.
Acknowledgments
This paper is supported by Grants from the National Institutes of Health (HD38466) (J. Busciglio) and Alzheimer's Disease Research Center (AG16573) (Pilot Project awarded to P. Coskun).
References
- 1.Pritchard MA, Kola I. The “gene dosage effect” hypothesis versus the “amplified developmental instability” hypothesis in Down syndrome. Journal of Neural Transmission, Supplement. 1999;(57):293–303. [PubMed] [Google Scholar]
- 2.Leporrier N, Herrou M, Morello R, Leymarie P. Fetuses with Down’s syndrome detected by prenatal screening are more likely to abort spontaneously than fetuses with Down’s syndrome not detected by prenatal screening. Journal of Obstetrics and Gynaecology. 2003;110(1):18–21. [PubMed] [Google Scholar]
- 3.Bittles AH, Bower C, Hussain R, Glasson EJ. The four ages of Down syndrome. European Journal of Public Health. 2007;17(2):221–225. doi: 10.1093/eurpub/ckl103. [DOI] [PubMed] [Google Scholar]
- 4.Coppus A, Evenhuis H, Verberne GJ, et al. Dementia and mortality in persons with Down’s syndrome. Journal of Intellectual Disability Research. 2006;50(10):768–777. doi: 10.1111/j.1365-2788.2006.00842.x. [DOI] [PubMed] [Google Scholar]
- 5.Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects. 1978;14(1):5–39. [PubMed] [Google Scholar]
- 6.Colvin L, Jurenka SB, Van Allen MI. Down Syndrome. New York, NY, USA: Marcel Dekker; 2003. [Google Scholar]
- 7.Pallardó FV, Lloret A, Lebel M, et al. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: ataxia-telangiectasia, Down syndrome, fanconi anaemia and werner syndrome. Biogerontology. 2010;11(4):401–419. doi: 10.1007/s10522-010-9269-4. [DOI] [PubMed] [Google Scholar]
- 8.Harman D. Free radical theory of aging: dietary implications. American Journal of Clinical Nutrition. 1972;25(8):839–843. doi: 10.1093/ajcn/25.8.839. [DOI] [PubMed] [Google Scholar]
- 9.Harman D. The biologic clock: the mitochondria? Journal of the American Geriatrics Society. 1972;20(4):145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 10.Miquel J, Fleming JE. A two-step hypothesis on the mechanisms of in vitro cell aging: cell differentiation followed by intrinsic mitochondrial mutagenesis. Experimental Gerontology. 1984;19(1):31–36. doi: 10.1016/0531-5565(84)90029-9. [DOI] [PubMed] [Google Scholar]
- 11.Cenini G, Dowling AL, Beckett TL, et al. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochimica et Biophysica Acta. 2012;1822(2):130–138. doi: 10.1016/j.bbadis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perluigi M, Butterfield DA. The identification of protein biomarkers for oxidative stress in Down syndrome. Expert Review of Proteomics. 2011;8(4):427–429. doi: 10.1586/epr.11.36. [DOI] [PubMed] [Google Scholar]
- 13.Busciglio J, Pelsman A, Wong C, et al. Altered metabolism of the amyloid β precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33(5):677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 14.Shukkur EA, Shimohata A, Akagi T, et al. Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Human Molecular Genetics. 2006;15(18):2752–2762. doi: 10.1093/hmg/ddl211. [DOI] [PubMed] [Google Scholar]
- 15.Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and down syndrome. Current Alzheimer Research. 2006;3(5):521–528. doi: 10.2174/156720506779025305. [DOI] [PubMed] [Google Scholar]
- 16.Tiano L, Busciglio J. Mitochondrial dysfunction and Down's syndrome: is there a role for CoQ10? BioFactors. 2011;37(5):386–392. doi: 10.1002/biof.184. [DOI] [PubMed] [Google Scholar]
- 17.Porta S, Serra SA, Huch M, et al. RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: apotential pathogenic process in neurodegeneration. Human Molecular Genetics. 2007;16(9):1039–1050. doi: 10.1093/hmg/ddm049. [DOI] [PubMed] [Google Scholar]
- 18.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10(1):12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sjodin B, Hellsten Westing Y, Apple FS. Biochemical mechanisms for oxygen free radical formation during exercise. Sports Medicine. 1990;10(4):236–254. doi: 10.2165/00007256-199010040-00003. [DOI] [PubMed] [Google Scholar]
- 21.Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? insights from evolutionary medicine. Annual Review of Biochemistry. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- 22.Helguera P, Pelsman A, Pigino G, Wolvetang E, Head E, Busciglio J. ets-2 promotes the activation of a mitochondrial death pathway in down’s syndrome neurons. Journal of Neuroscience. 2005;25(9):2295–2303. doi: 10.1523/JNEUROSCI.5107-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schon EA, Kim SH, Ferreira JC, et al. Chromosomal non-disjunction in human oocytes: is there a mitochondrial connection? Human Reproduction. 2000;15(2):160–172. doi: 10.1093/humrep/15.suppl_2.160. [DOI] [PubMed] [Google Scholar]
- 24.Koga Y, Akita Y, Takane N, Sato Y, Kato H. Heterogeneous presentation in A3243G mutation in the mitochondrial tRNALeu(UUR) gene. Archives of Disease in Childhood. 2000;82(5):407–411. doi: 10.1136/adc.82.5.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arbuzova S, Cuckle H, Mueller R, Sehmi I. Familial down syndrome: evidence supporting cytoplasmic inheritance. Clinical Genetics. 2001;60(6):456–462. doi: 10.1034/j.1399-0004.2001.600609.x. [DOI] [PubMed] [Google Scholar]
- 26.Arbuzova S, Hutchin T, Cuckle H. Mitochondrial dysfunction and Down’s syndrome. BioEssays. 2002;24(8):681–684. doi: 10.1002/bies.10138. [DOI] [PubMed] [Google Scholar]
- 27.Eichenlaub-Ritter U, Wieczorek M, Lüke S, Seidel T. Age related changes in mitochondrial function and new approaches to study redox regulation in mammalian oocytes in response to age or maturation conditions. Mitochondrion. 2011;11:783–796. doi: 10.1016/j.mito.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Eichenlaub-Ritter U. Reproductive semi-cloning respecting biparental origin. Reconstitution of gametes for assisted reproduction. Human Reproduction. 2003;18(3):473–475. doi: 10.1093/humrep/deg080. [DOI] [PubMed] [Google Scholar]
- 29.Coskun PE, Wyrembak J, Derbereva O, et al. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. Journal of Alzheimer’s Disease. 2010;20(supplement 2):S293–S310. doi: 10.3233/JAD-2010-100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(29):10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Druzhyna N, Nair RG, Ledoux SP, Wilson GL. Defective repair of oxidative damage in mitochondrial DNA in Down’s syndrome. Mutation Research. 1998;409(2):81–89. doi: 10.1016/s0921-8777(98)00042-1. [DOI] [PubMed] [Google Scholar]
- 32.Nagy Z, Esiri MM, LeGris M, Matthews PM. Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathologica. 1999;97(4):346–354. doi: 10.1007/s004010050997. [DOI] [PubMed] [Google Scholar]
- 33.Kim SH, Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G. The reduction of NADH—ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer’s disease. Life Sciences. 2001;68(24):2741–2750. doi: 10.1016/s0024-3205(01)01074-8. [DOI] [PubMed] [Google Scholar]
- 34.Lee SH, Lee S, Jun HS, et al. Expression of the mitochondrial ATPase6 gene and Tfam in down syndrome. Molecules and Cells. 2003;15(2):181–185. [PubMed] [Google Scholar]
- 35.Valenti D, Manente GA, Moro L, Marra E, Vacca RA. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: involvement of the cAMP/PKA signalling pathway. Biochemical Journal. 2011;435(3):679–688. doi: 10.1042/BJ20101908. [DOI] [PubMed] [Google Scholar]
- 36.Bozner P, Wilson GL, Druzhyna NM, et al. Deficiency of chaperonin 60 in Down’s syndrome. Journal of Alzheimer’s Disease. 2002;4(6):479–486. doi: 10.3233/jad-2002-4604. [DOI] [PubMed] [Google Scholar]
- 37.Martin J, Horwich AL, Hartl FU. Prevention of protein denaturation under heat stress by the chaperonin Hsp60. Science. 1992;258(5084):995–998. doi: 10.1126/science.1359644. [DOI] [PubMed] [Google Scholar]
- 38.Bajo M, Fruehauf J, Kim SH, Fountoulakis M, Lubec G. Proteomic evaluation of intermediary metabolism enzyme proteins in fetal down’s syndrome cerebral cortex. Proteomics. 2002;2(11):1539–1546. doi: 10.1002/1615-9861(200211)2:11<1539::AID-PROT1539>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 39.Shin JH, Weitzdoerfer R, Fountoulakis M, Lubec G. Expression of cystathionine β-synthase, pyridoxal kinase, and ES1 protein homolog (mitochondrial precursor) in fetal Down syndrome brain. Neurochemistry International. 2004;45(1):73–79. doi: 10.1016/j.neuint.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Martin KR, Barrett JC. Reactive oxygen species as double-edged swords in cellular processes: low-dose cell signaling versus high-dose toxicity. Human and Experimental Toxicology. 2002;21(2):71–75. doi: 10.1191/0960327102ht213oa. [DOI] [PubMed] [Google Scholar]
- 41.Birringer M. Hormetics: dietary triggers of an adaptive stress response. Pharmaceutical Research. 2011;28(11):2680–2694. doi: 10.1007/s11095-011-0551-1. [DOI] [PubMed] [Google Scholar]
- 42.Calabrese EJ, Baldwin LA. Defining hormesis. Human and Experimental Toxicology. 2002;21(2):91–97. doi: 10.1191/0960327102ht217oa. [DOI] [PubMed] [Google Scholar]
- 43.Schriner SE, Linford NJ, Martin GM, et al. Medecine: extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 44.Harrington LA, Harley CB. Effect of vitamin E on lifespan and reproduction in Caenorhabditis elegans . Mechanisms of Ageing and Development. 1988;43(1):71–78. doi: 10.1016/0047-6374(88)90098-x. [DOI] [PubMed] [Google Scholar]
- 45.Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster . Science. 1994;263(5150):1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 46.Melov S, Ravenscroft J, Malik S, et al. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289(5484):1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- 47.Lott IT, Doran E, Nguyen VQ, Tournay A, Head E, Gillen DL. Down syndrome and dementia: a randomized, controlled trial of antioxidant supplementation. American Journal of Medical Genetics A. 2011;155(8):1939–1948. doi: 10.1002/ajmg.a.34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lapointe J, Hekimi S. When a theory of aging ages badly. Cellular and Molecular Life Sciences. 2010;67(1):1–8. doi: 10.1007/s00018-009-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radical Biology and Medicine. 2011;51(2):327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 50.Lott IT. Antioxidants in Down syndrome. Biochim Biophys Acta. 2012;1822(5):657–663. doi: 10.1016/j.bbadis.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masoro EJ, Austad SN. The evolution of the antiaging action of dietary restriction: a hypothesis. Journals of Gerontology A. 1996;51(6):B387–B391. doi: 10.1093/gerona/51a.6.b387. [DOI] [PubMed] [Google Scholar]
- 52.Sharma PK, Agrawal V, Roy N. Mitochondria-mediated hormetic response in life span extension of calorie-restricted Saccharomyces cerevisiae. Age. 2011;33(2):143–154. doi: 10.1007/s11357-010-9169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate Cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. Journal of Biological Chemistry. 2006;281(34):24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 54.Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, keap1: a historical overview. Antioxidants and Redox Signaling. 2010;13(11):1665–1678. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 55.Kong AN, Owuor E, Yu R, et al. Induction of xenobiotic enzymes by the map kinase pathway and the antioxidant or electrophile response element (ARE/EpRE) Drug Metabolism Reviews. 2001;33(3-4):255–271. doi: 10.1081/dmr-120000652. [DOI] [PubMed] [Google Scholar]
- 56.Helguera P, Seiglie J, Rodriguez J, Busciglio J. Adaptive downregulation of mitochondrial function in Down’s syndrome. doi: 10.1016/j.cmet.2012.12.005. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swatton JE, Sellers LA, Faull RL, Holland A, Iritani S, Bahn S. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. European Journal of Neuroscience. 2004;19(10):2711–2719. doi: 10.1111/j.0953-816X.2004.03365.x. [DOI] [PubMed] [Google Scholar]
- 58.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes to Cells. 2011;16(2):123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Letters. 2005;579(14):3029–3036. doi: 10.1016/j.febslet.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 60.Strydom A, Dickinson MJ, Shende S, Pratico D, Walker Z. Oxidative stress and cognitive ability in adults with Down syndrome. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(1):76–80. doi: 10.1016/j.pnpbp.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Hai CX. ROS acts as a double-edged sword in the pathogenesis of Type 2 diabetes mellitus: is Nrf2 a potential target for the treatment? Mini-Reviews in Medicinal Chemistry. 2011;11(12):1082–1092. doi: 10.2174/138955711797247761. [DOI] [PubMed] [Google Scholar]
- 62.Ristow M, Zarse K, Oberbach A, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(21):8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]