

Abstract
Novel water-soluble decacationically armed C60 and C70 decaiodide monoadduct, C60- or C70[>M(C3N6+C3)2] were synthesized, characterized, and applied as photosensitizers and potential nano-PDT agents against pathogenic bacteria and cancer cells. A high number of cationic charges per fullerene cage and H-bonding moieties were designed for rapid binding to the anionic residues displayed on the outer parts of bacterial cell walls. In the presence of a high number of electron-donating iodide anions as parts of quaternary ammonium salts in the arm region, we found that C70[>M(C3N6+C3)2] produced more HO• than C60[>M(C3N6+C3)2], in addition to 1O2. This finding offers an explanation of the preferential killing of Gram-positive and Gram-negative bacteria by C60[>M(C3N6+C3)2] and C70[>M(C3N6+C3)2], respectively. The hypothesis is that 1O2 can diffuse more easily into porous cell walls of Gram-positive bacteria to reach sensitive sites, while the less permeable Gram-negative bacterial cell wall needs the more reactive HO• to cause real damage.
Keywords: Decacationic C70 monoadduct, photosensitizer, phototoxicity, PDT effect, bacteria cell-wall binding
INTRODUCTION
Photodynamic therapy (PDT)1–4 employs the combination of non-toxic photosensitizers (PS) with harmless visible to near-IR light to generate the highly reactive oxygen species (ROS), such as singlet oxygen (1O2), superoxide radicals (O2−•), or hydroxyl radicals (HO•) that kill disease-causing cells by a non-specific attack.5,6 Increasing prevalence of multi-antibiotic drug resistance in multiple classes of pathogens7,8 contributes to the difficulty in treating the related infectious diseases. Because these pathogens may be deficient in defenses against ROS, especially, singlet oxygen and hydroxyl radicals that display a non-specific high reactivity toward certain critical chemical moieties of the cell, the PDT approach may serve as a promising alternative treatment method for infections and cancer. The non-specificity nature of PDT also allows its use against a wide range of bacteria, viruses, yeasts, and protozoa by killing these pathogenic agents in the infection.
The reaction mechanism of PDT involves an initial photoexcitation of PS to the singlet excited state upon exposure to a light source, followed by intersystem crossing processes resulting in the formation of a long-lived triplet excited state of PS that can transfer photoenergy to triplet molecular oxygen in the biological system. Accordingly, an effective organic PS should be capable of exhibiting (1) a strong and sharp optical absorption band at the PDT wavelength for a high yield of photoexcitation, (2) a high yield of molecular triplet excited state generation, and (3) effective molecular interactions with the target cell surface to produce ROS in a short distance without undue damage to the surrounding host tissue.
Fullerenes are particularly photostable and manifest little photobleaching compared to many other traditional tetrapyrrole-backbone based PS found in porphyrins and chlorins.9 Rapid photobleaching characteristics of porphyrin-type PS often disrupt its PDT efficacy after a short period of light illumination due mainly to photodegradation of the compound and subsequent alteration of its chromophore structure, resulting in the loss of photosensitizing capability. By contrast, a larger number of interlinked aromatic olefinic bonds available on the fullerene cage can tolerate a certain degree of fullerenyl π-conjugation interruption, induced by the back-attack of singlet oxygen or radical species on the olefin bonds, delaying the loss of their photophysical properties. This chemical nature coupled with a relatively slower reaction rate of C60 and C70 cage toward ROS than that of porphyrin-type PS leads to a photostability that dramatically extends the length of time they can respond to illumination. It will also allow many more effective catalytic PDT cycles to be carried out with a single application of PS.
It is our particular interest in mediating broad-spectrum PDT killing of pathogenic Gram-positive (e.g. Staphylococcus aureus) and Gram-negative (e.g. Escherichia coli) bacterial targets. Owing to specific differences in the physiology, cell wall, and cytoplasmic membrane structures between Gram-positive and Gram-negative bacteria.10–12 it is necessary to significantly revise the functional addends of each fullerene cage for effective targeting selectivity and drug-delivery. By taking chemical functional groups and structures of the cell-wall into account to maximize potential interaction forces of drugs at the cell surface, combination of H-bonding and a high number of cationic charges in one molecular compound are beneficial for inducing strong binding. However, synthesis of fullerenyl monoadducts bearing a well-defined high number of cationic charges remains challenging and has rarely been reported.
In this paper, we describe a rational linkage of water-soluble quaternary alkylammonium multi-salts and ester-amide functional groups into a well-defined decacationic malonate together with an efficient synthetic method for its attachment on either a C60 or C70 nanocage. The synthesis led to the preparation of new amphiphilic [60]fullerene- (1) or [70]fullerene-based (2) nano-photosensitizers, respectively. In these structures, arm moieties each bearing a high number of cationic charges and an amide moiety, capable of inducing the H-bonding in the vicinity of the fullerene cage, are designed for cell-targeting. Accordingly, they may allow us to study specifically the feasibility as nano-PDT agents toward both Gram-positive and Gram-negative bacterial species.2,13 The approach also gives a systematic structure–function relationship of fullerene monoadducts on the influence of their biological activity, aiming to achieve the optimized photodynamic activation and enhancement of the PDT efficiency.
RESULTS AND DISCUSSION
Surface binding interactions of fullerene derivatives with –D-Ala-D-Ala residues of the bacteria cell wall may be increased by the incorporation of polar moieties on C60/C70 capable of inducing multiple H-bondings and static charge interactions with amino acid functional residues. Based on this consideration, we applied a malonate precursor arm, as that shown in the structure of C60[>M(C3N6+C3)2] 1 and C70[>M(C3N6+C3)2] 2 (Scheme 1), to include two esters and two amide moieties for a sufficient number of carbonyl and –NH groups in a short length of ~20 Ǻ for making effective multi-binding sites. More importantly, the use of a well-defined water-soluble pentacationic moiety N6+C3 at each side of the arm may increase the crucial select-targeting specificity on the bacterial cell wall. Thus, N6+C3 become a common synthon for the structural modification of PDT nanomedicines. It can be derived from the quarternization of N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amine precursor N6C3. Attachment of N6+C3 to two ester moieties of a malonate forms two extended bipolar pentacationic side-arms capable of forming strong static-charge binding interactions to anionic carboxylate end-groups of the terminal D-Ala-D-Ala dipeptide of the bacterial cell wall.
Scheme 1.

Synthetic pathway for the preparation of decacationic C60[>M(C3N6 +C3)2] 1 and C70[>M(C3N6 +C3)2] 2.
Synthetically, large solubility-incompatibility between water-soluble pentacationic intermediate N6+C3 side-arms and highly hydrophobic fullerene cage incurred much challenge and difficulty in controlling the reaction homogeneity and overall processes for the exclusive monoadduct formation, the efficiency and yield, and the product purification and characterization. Following many attempts using different synthetic routes, we found that the best approach for the preparation of decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate methyl quaternary ammonium salt 1 and its C70 analogue compound 2 is to begin with a simple well-defined fullerene monoadduct derivatives, such as di(tert-butyl)fullerenyl malonates C60[>M(t-Bu)2] 3 and C70[>M(t-Bu)2] 4, respectively, capable of undergoing the chemical conversion reaction with N6+C3-derived intermediates, as shown in Scheme 1. The alternative approach to synthesize first the water-soluble N6+C3-malonate-N6+C3 intermediate arm followed by its attachment to a fullerene cage resulted in much complication in the product purification and characterization, even though the same compound was obtained. One potentially plausible synthetic route of using the hydrophobic N6C3-malonate-N6C3 intermediate arm was found to be problematic owing to its partial charge-transfer complexation with the [70]fullerene cage.
The structural verification of 4 was made by various spectroscopic techniques, including 13C NMR spectrum (supporting information) displaying one peak at δ 162.39 corresponding to the chemical shift of carbonyl carbon and 35 peaks in different intensities (33 peaks each with 2C and two peaks each with 1C) in the region of δ 125–155 that can be accounted for all 68 fullerenyl sp2 carbons. The data is consistent with a C70 monoadduct structure having a Cs-symmetrical ellipsoidal structure with a plane of symmetry across the cyclopropane-bridged [6,6] bond. In general, Bingel monoaddition reaction14 of malonate esters on C70 may lead to four regioisomers of methano-[70]fullerenes each with a differently bridged [6,6]-fullerenyl sp3 bonding location, defined as the α-type bonding at the C(1)–C(2), the β-type bonding at the C(5)–C(6), the ε-type bonding at the C(7)–C(21), and the κ-type bonding at the C(20)–C(21).15,16 These regioisomers exhibit a molecular symmetry of Cs, C2v, C1, and C2v, respectively. Chemical reactivity of all these bonding locations was found to be the highest at the most strain region of the cage pole area giving the order of C(1)–C(2) > C(5)–C(6) > C(7)–C(21) > C(20)–C(21) (lying on the flat equator of the C70 spheroid).17 Therefore, it is reasonable for us to predict the major monomalonate-addition product C70[>M(t-Bu)2] 4 being the α-type analogue. In fact, the 13C spectrum of 4 showing the pattern and number of carbon peaks, consisting with the compound having a molecular Cs-symmetry, should provide the unambiguous support for this structural assignment.
Subsequent facile transesterification reaction of 4 with the well-characterized tertiary-amine precursor arm moiety, 4-hydroxy-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl) butanamide 5 (C3N6C3-OH) was carried out under acidic conditions using trifluoroacetic acid as the catalytic reagent (Scheme 1). The reaction was found to result in the formation of decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate, protonated quaternary ammonium trifluoroacetate salt, C70[>M(C3N6+C3H)2] 7 in 71% yield. The structure of 7 was verified by various spectroscopic techniques (supporting information) including 13C NMR spectrum showing two peaks at δ 170.45 and 165.50 corresponding to the chemical shift of a carbonyl carbon of the amide (–HN-C=O) and the ester (–O-C=O) moieties, respectively. It also displayed a total of 68 peaks in the region of δ 125–155 accounted for 68 fullerenyl sp2 carbons. The spectral data indicated clearly a C1-symmetry of the compound 7. As the simple transesterification condition should not complicate the structural configuration of methano[70]fullerene cage moiety, the decrease of molecular symmetry from that (Cs) of 4 may be indicative of strong interactions between flexible hexapropyl-hexa(aminoethyl) butanamide arms and the C70 cage. These interactions may induce a nonsymmetrical environment among cage carbons. It also revealed the likelihood of two pentacationic C3N6C3-O– arms to be located at the vicinity of the cage surface, as shown in the cluster form of Fig. 1, presumably, by the interactive binding of multiple hydrophobic n-propyl groups.
Figure 1.
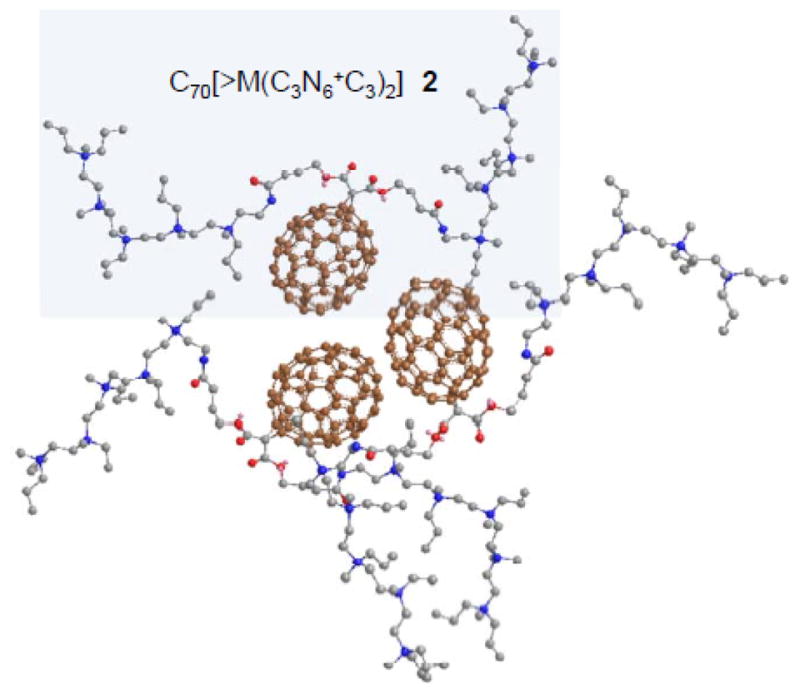
Proposed schematic molecular packing of C70[>M(C3N6 +C3)2] 2 and 2′ by taking the high water-solubility of the compound, packing tendency of C70 cages, and the analysis of their 13C NMR spectra giving the C1 symmetry of the cage moiety into consideration. The drawing was made by using the Chem 3D program for the energy minimization.
Interestingly, we found that the use of C70-compatible deuterated solvents, such as CDCl3 and CS2, for enhancing the detectability of [70]fullerenyl carbon peak signals has led to the full suppression of pentacationic arm moieties in the spectrum. Contrarily, the application of water-compatible deuterated solvents, such as D2O and DMF-d7, reduced significantly the signal intensity of [70]fullerenyl carbon peaks, likely owing to the cage aggregation.
Conversion of the compound 7 to decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl) [70]fullerenyl malonate methyl quaternary ammonium salt, C70[>M(C3N6+C3)2] 2, was accompanied by multiple iodide anions. Infrared spectrum of 2 clearly showed four sharp characteristic bands at 794.33, 723.08, 578.72, and 530.19 cm−1 corresponding to the absorption of a C70 half-cage, revealing no bismalonate adduct formation (with an additional malonate addend attaching at the α-bond of the opposite pole). Unexpectedly, we were not able to detect any fullerenyl sp2 carbon signals in the 13C NMR spectrum of 2 under various deuterated solvent mixtures. However, these carbon signals re-appeared when all iodide anions were replaced by trifluoroacetate anions, leading to the corresponding salt 2′. Accordingly, the 13C NMR spectrum of 2′ displayed a peak at δ 170.45 corresponding to the chemical shift of an amide carbonyl carbon (–HN-C=O), similar to that of 7. It also displayed a total of 68 peaks in the region of δ 125–155 accounted for 68 fullerenyl sp2 carbons, indicating a similar C1-symmetry as that of the compound 7. The observation led to our assumption on the possibility of reversible partial electron-transfer from the iodide (I−) donors to C70 cages forming a low degree of radicalized cage moieties that reduced significantly the resolution of 13C signals.
The mass spectroscopic data collection of C70[>M(C3N6+C3)2] was proven to be difficult due to its polycationic nature and facile fragmentations occurring at the conjunction of the C70 cage and the decacationic malonate arm, giving mainly the highly detectable C70 ion mass at m/z 841, as displayed in MALDI–TOF mass spectra (supporting information). Fortunately, we are able to acquire several spectra in the high mass region showing the molecular ion mass (M+) at m/z 3340 in low intensity accompanied with several identifiable fragmentation ion mass peaks at m/z 1355, 1584, 1654, 1682, 1753, 2043, and 2079. By variation of applied laser intensity, spectra containing many ion mass peaks in significant signal intensity in the medium mass region can be obtained. Structural elucidation of these ion fragments (the supporting information) in these two spectra allowed us to conclude (1) a clear monoadduct structure of 2 by the sole detection of an ion fragment mass of C70(-CH3)+ at m/z 855 without the corresponding ion fragment mass of the bisadduct C70(-CH3)(>CH2)+ at m/z 869, (2) a facile process of nearly full de-quaternization of 2 under MALDI–MS conditions giving mostly detectable monocationic mass fragments, and (3) the bond cleavage of n-propyl group on the nitrogen atom is faster than that of the methyl group and the main chain moiety. These data strongly support the molecular mass of 2 as a (C3N6+C3)2-malonate-C70 structure with many consistent (C3N6Cn)2-malonate-C70 (Cn: methyl or n-propyl group) fragments.
The molecular aggregation characteristics of amphiphilic nanostructures 1 and 2 in water were investigated under the preparation condition in close resemblance to that used in the in vitro PDT studies. Typically, the sample was dissolved in DMA under ultrasonication for a period of ~5.0 min, giving a master solution in a concentration of 2.0 mM. A portion of this solution was then diluted by H2O to pre-determined concentrations of 1.0, 10, and 100 μM for transmission electron microscopy (TEM) measurements. As a result, TEM micrographs of C60[>M(C3N6+C3)2] displayed no obvious bilayered fullerosome18–20 formation at either 1.0 or 10 μM. A sporadic formation of fullerosome vesicles each in a larger particle diameter of 60–100 nm were observed at a higher concentration of 100 μM. In the case of C70[>M(C3N6+C3)2] compound, a similar observation of no clearly detectable bilayered fullerosome nanovesicles was found at all concentrations of 1.0, 10, and 100 μM. There was only a small quantity of irregularly shaped nanospherical particles of 2 found at concentrations of 10 and 100 μM. This implied the hydrophilic strength of two pentacationic arms overwhelm the hydrophobic character of C60 and C70 cages, leading to high water-solubility of 1 and 2 with much suppression of their tendency to form a sizable particles under PDT experimental conditions. In the light of a slightly higher ability of C70 moiety, with ten more carbon atoms in one cage, to give molecular cluster formation than that of C60, we proposed a likely molecular cluster packing configuration of 2 and 2′ in water, as shown in Fig. 1. It includes unsymmetrical hydrophobic malonate ester arm moiety interactions with the C70 cage that induces the loss of molecular cage symmetry observed in its 13C NMR spectrum. Partial blocking of the cage surface by the arms allows only a very limit number of hydrophobic cages to undergo irregular coalescence into a nanosphere in H2O.
The light-induced PDT efficiency can be correlated partially to optical absorption extinction coefficients of the PS in the wavelength range applied. Accordingly, we studied UV-vis spectra of C60[>M(C3N6+C3)2] and C70[>M(C3N6+C3)2] in DMA at the concentration of 1.0 × 10−5 M, as shown in Fig. 2A, in comparison with the parent monoadducts C60[>M(t-Bu)2] 3 and C70[>M(t-Bu)2] 4 in CHCl3. Interestingly, the optical absorption profile of 4 showing peaks with the maxima (λmax) located at 355, 370, 403, 462, and 539 (shoulder band) nm matches well with those reported for the α-type C70 monoadducts21,22 that provided further confirmation of our structural assignment of 4. Upon the structural conversion of 4 with decacationic di(C3N6C3-O)–malonate arm, the intensity of absorption of C70[>M(C3N6+C3)2] increases at wavelengths <450 nm with the disapperance of all characteristic peaks of the monoadduct 4 into a featureless curve. Since the cage moiety was not modified from 4, the difference may imply the occurrence of cage cluster formation, consistent with that proposed in Fig. 1. The change leads to a higher total visible absorption of 2 than that of the C60 analogue 1 in a ratio of 1.99 (ε2/ε1) over 400–700 nm (Fig. 2A). However, an opposite relationship was found in the UVA region (320–400 nm) giving a ε2/ε1 ratio of 0.79, indicating a stronger optical absorption of C60 monoadduct cage in these wavelength range. It is also interesting to note a higher ε value for the protonated quaternary ammonium trifluoroacetate salt, C70[>M(C3N6+C3H)2] 7, than 2 over all wavelengths (300–800 nm). To correlate the light absorption efficiency of 2 under PDT experimental conditions, we compared its visible spectrum profile taken in DMAc–H2O (1:19) to that in DMA (Figs. 2Ba and 2Bb) and found a nearly identical ε value over the range of 400–700 nm. Photostability of C60[>M(C3N6+C3)2] and C70[>M(C3N6+C3)2] in aqueous solution was evaluated under a constant irradiation using a LED light source equipped with a fiber optical lens (200 mW/cm2 at 400 ± 5 nm) over a period of 5.0 h. Consequently, high retention of a UV-vis absorption profile of fullerene derivatives was observed.
Figure 2.

UV-vis spectra of (A) C60[>M(t-Bu)2] 3 in CHCl3, C60[>M(C3N6 +C3)2] 1 in DMAc, C70[>M(t-Bu)2] 4 in CHCl3, C70[>M(C3N6 +C3)2] 2 in DMAc, and C70[>M(C3N6 +C3H)2] 7 in DMAc and (B) C70[>M(C3N6 +C3)2] 2 in (a) DMAc, (b) DMAc–H2O (1:19), and (c) DMAc-CHCl3-CS2 (1:1:2). The concentration of all samples is 1.0 × 10−5 M.
ROS Probe Experiments
Illumination of C60 or C70 monoadduct derivatives with visible or UVA light promotes its transition to a long-lived trriplet excited state, via intersystem crosing processes, and subsequent intermolecular energy transfer to the molecular oxygen yielding a highly reactive singlet oxygen (1O2) in Type-II reactions.23 In the presence of physiological concentration of reductants, such as NADH, more reactive ROS species, such as superoxide anion (O2−•) and hydroxyl radical (HO•), may be produced in polar solvent and water.24 In this study, a comparative singlet oxygen production efficiency study of the compounds 1 and 2 in DMSO–H2O (1:520, 1.0 × 10−6 M) was carried out by many increamental exposures of the sample to a UVitron visible-enhanced lamp (200 W, 250–600 nm with λmax at 420 nm) in an exposure interval period of 5.0 sec (the light dose measured to be ~5.0 mJ/cm2 per period) followed by the flourescence emission measurement of the probe. For highly efficent in situ selective trapping of singlet oxygen, we applied water-compatible 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABMA, 8.0 × 10−6 M) as a fluorescent probe which exhibits the emission maximum at 429 nm. The corresponding most effective excitation wavelengths were found to be 379 and 400 nm. Accordingly, the later one closer to the visible range was used. At this wavelength, the optical absorption extinction coefficient value of 2 is slightly higher than that of 1, as shown by the blue mark in Fig. 2A. The chemical trapping of 1O2 by highly fluorecent ABMA leads to the formation of non-fluorescent 9,10-endoperoxide product. The conversion allows us to follow the loss of fluorescece emission intensity at 429 nm for its correlation to the proportional quantity of 1O2 produced due to the high kinetic rate constant of the trapping reaction in aqueous media,25 assuming the internal decay of 1O2 in the solvent system is identical for both experiments of 1 and 2. As a result, we observed a slightly higher 1O2 production rate of C60[>M(C3N6+C3)2], even though the difference is not statistically significant, as shown in Fig. 3a. The use of a lower irradiation dose for the similar measuremnt directly in the UV-vis-NIR spectrometer, operated at λex 400 nm (single wavelength excitation) with the monochrometor, gave a much slower production rate of 1O2 for both cases that displyed a clearly higher rate for C60[>M(C3N6+C3)2] at this excitation wavelength (Fig. 3b).
Figure 3.

Singlet oxygen production efficient comparison of the compounds 1 and 2 in DMSO–H2O (1:520, 1.0 × 10−6 M), using anthracene tetracarboxylic acid ABMA (8.0 × 10−6 M) as the fluorescent 1O2-trapping agent at λex 400 nm and λem 429 nm for detection with the irradiation source of (a) Uvitron lamp (200 W) and (b) spectrometer lamp/monchromator operated with a single wavelength at λex 400 nm.
A similar trend was also confirmed by using 1O2 probe singlet oxygen sensor green (SOG) reagent. Upon the tapping of 1O2 by the anthracene moiety of SOG, the resulting 9,10-endoperoxide-linked fluorescein product SOG-EP is highly green fluorescent (excitation/emission maxima ~504/525 nm) that can be used for the correlation of relative 1O2 quantity produced. It is worthwhile to mention that the SOG probe chemistry is potentially more complicated than the statement above due to the fact that both SOG and SOG-EP are capable of sensitizing 1O2 production and undergoing photobleaching processes themselves, resembling those of fluorescein derivatives.26 This complication can be lessened by using blue light for fullerene excitation that is not absorbed by the probe. However, if we assume the amount of the additional 1O2 molecules derived from SOG and SOG-EP and the photobleaching rate being nearly identical among experiments under the same conditions, the fluorescence emission intensity of SOG-EP detected will still be valuable for the relative comparison purpose. Accordingly, Fig. 4a showed the results from the SOG-based fluorescence probe assay experiments of 1 and 2 in 96-well black-sided plates under incremental irradiation with a blue LED light source (415 ± 15 nm) under the total fluence intensity range of 0–6.0 J/cm2. It is evident that C60[>M(C3N6+C3)2] gave a significantly higher rate of 1O2 detected than that of C70[>M(C3N6+C3)2]. The marked difference in the 1O2 generation rate of 1 and 2 between the data of Fig. 3 (without PBS) and 4a (with PBS) led to our consideration of potential involvement of the Type-I photoreaction processes in electrolyte solution giving reactive radical species in the latter experimental conditions.
Figure 4.

Activation of fluorescence probes for detecting the ROS generation by 1 and 2 (5.0 μM in each well) using different fluorescent probes (a) SOG (5.0 μM) and (b) HPF (5.0 μM), followed by delivery of the stated fluence of blue light.
Indeed, under the similar experimental procedures relevant to PDT as those used for the data collection of Fig. 4a, but with the application of the fluorescent probe of 3′-p-(hydroxyphenyl)-fluorescein27 (HPF) instead, a reverse relationship was observed with higher fluorescence emission intensity of fluorescein for C70[>M(C3N6+C3)2] than that for C60[>M(C3N6+C3)2] under the total fluence intensity range of 0–5.0 J/cm2, as shown in Fig. 4b. Since the HPF probe is an essential tool for the selective detection of HO• and peroxynitrite, via quinone formation detached from the fluorescein moiety (excitation/emission maxima ~490/515 nm), with the detection sensitivity reported to be roughly 145- and 90-folds higher for HO• than 1O2 and O2−•, respectively,27 the measured fluorescence intensity can be correlated roughly to the yield of hydroxyl radicals produced. Besides, O2−• is the precusor species to the formation of HO•, directly monitoring the presence of HO• will also serve the confirmation purpose of superoxide radical formation, i.e. higher yield for C70[>M(C3N6+C3)2] than C60[>M(C3N6+C3)2].
It is well-recognized that 1O2 can give rise to several other types of ROS, including O2−•, H2O2, and HO•, in a sequential reaction with iodide anion (I−),28 In this case, the yield of O2−• or HO• should be proportional to the quantity of 1O2. Therefore, our observation of a higher 1O2 production yield of C60[>M(C3N6+C3)2] in PBS solution (Fig. 4a) giving a lower production yield of radical species (Fig. 4b) revealed no direct correlation of 1O2 to O2−• or HO• yields, or the latter species may not derive directly from the former one in these experiments. Contrarily, they may be in a competitive production process to each other in either the triplet energy-transfer (Type-II) or electron-transfer (Type-I) mechanism involving the electron-donating iodide anion (I−) and the electron-accepting fullerene cage. This mechanistic path can be realized by the facile photoexcitation of C60/C70 cages to their singlet excited state followed by the quantitative intersystem crossing to their corresponding excited triplet transient states, 3C60*[>M(C3N6+C3)2] and 3C70*[>M(C3N6+C3)2]. Apparently, the latter molecule exhibited a higher electron-accepting capability to allow electron-transfer from the iodide anion leading to the formation of C70−•[>M(C3N6+C3)2] prior to the further transfer this electron to O2 that gave a higher yield of O2−• and, thus, HO•.
Antimicrobial Effect of Fullerene-mediated PDT
Two highly water-soluble decacationic fullerenes 1 and 2 were first applied for comparison in the PDT-killing of the Gram-positive S. aureus. In general, it is known that Gram-positive bacteria are more sensitive and susceptible to PDT destruction than Gram-negative bacteria. This allowed us to use a lower concentration (up to 10 μM) of fullerene derivatives for S. aureus killing than that (up to 100 μM) needed for effective E. coli killing. Our previous PDT experiments have indicated a fast binding process of PS to microbial cells.29 Thus, a short incubation time of 30 min was applied for both species. As a result shown in Fig. 5, neither decacationic fullerene derivative 1 nor 2 gave any detectable toxicity to both bacteria in the dark in a concentration up to 10 μM. A pronounced PDT effect was observed after illumination on the 1/2-incubated cells using a broad-band white light source with total fluence intensity of 100 J/cm2 with a sharp cytotoxicity trend even at low fullerene concentration of 1.0 μM, giving 3 logs killing for C60[>M(C3N6+C3)2] and 2 logs killing for C70[>M(C3N6+C3)2]. As the administered dose increased to a concentration of 3.0 μM, a 5-log killing effectiveness was observed for the compound 1 and a corresponding 3-log for the compound 2. Interestingly, C60[>M(C3N6+C3)2] was able to eradicated the cells at higher concentrations than 3.0 μM. In the case of C70[>M(C3N6+C3)2], a 5-log effective killing was achieved at a concentration of 10 μM. Two killing curves of 1 and 2 were significantly different from each other (p<0.01).
Figure 5.
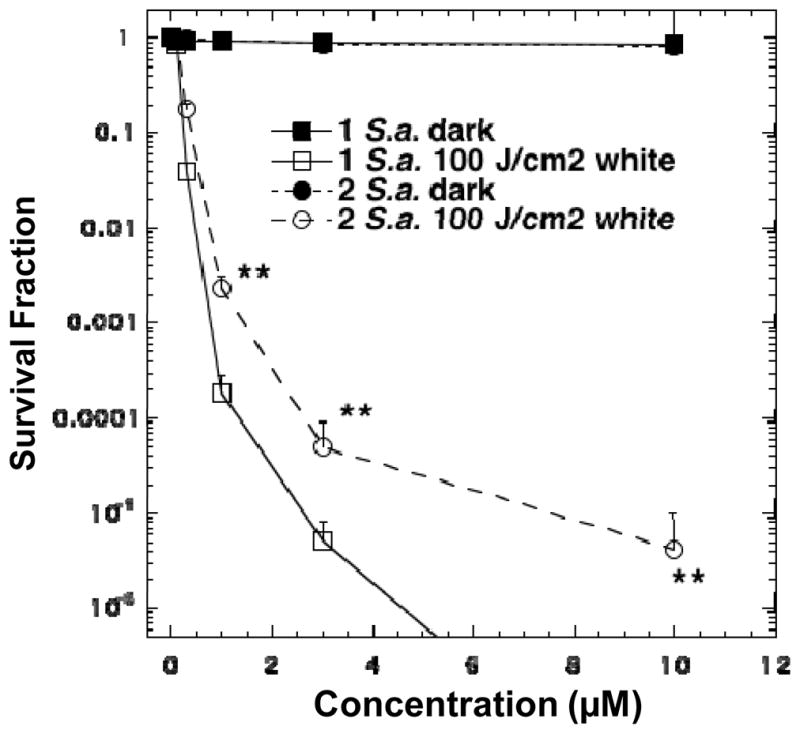
PDT killing of Gram-positive bacteria, S. aureus. Bacteria [10(8) cells/mL] were incubated with stated concentration of C60[>M(C3N6 +C3)2] 1 or C70[>M(C3N6 +C3)2] 2 for 30 min followed by delivery or not of 100 J/cm2 of white light. ** P<0.01.
In the PDT experiments against E. coli, the need to use 8-times higher concentrations of fullerene drugs led to the observation of some dark toxicity (1–2 logs) at the higher concentrations of 60 and 80 μM. Surprisingly, the order of PDT effectiveness of these two fullerene drugs was opposite to that seen with S. aureus. In this case, C70[>M(C3N6+C3)2] was most effective for E. coli killing with 3 logs at 40 μM, 5 logs at 60 μM, and eradicating the cells at 80 μM, as shown in Fig. 6. On the other hand, C60[>M(C3N6+C3)2] only killed 1 log at 40 μM, 2 logs at 60 μM and 3 logs at 80 μM. These two curves were significantly different (p<0.01).
Figure 6.
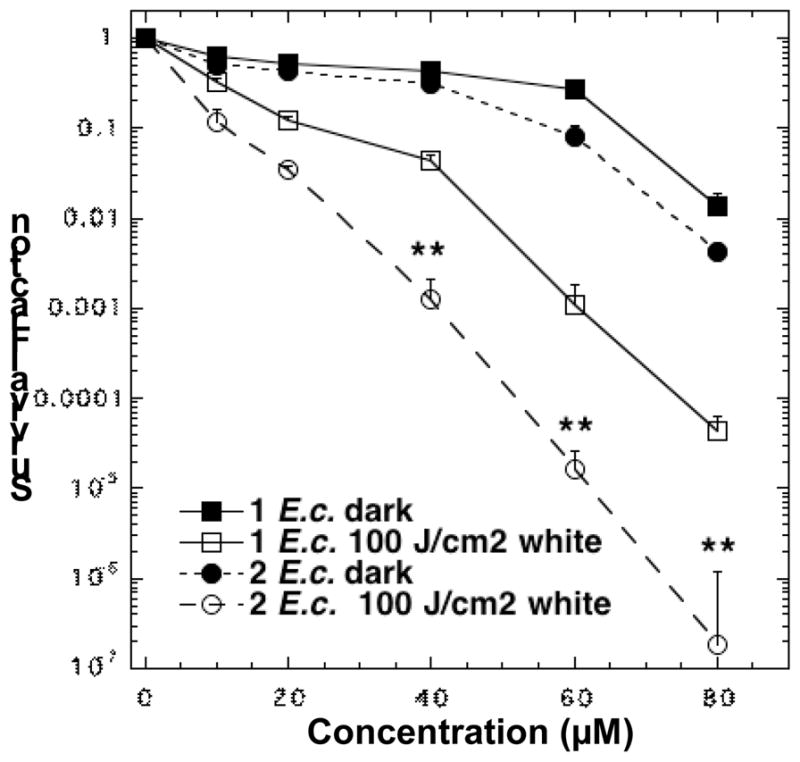
PDT killing of Gram-negative bacteria, E. coli. Bacteria [10(8) cells/mL] were incubated with stated concentration of 1 or 2 for 30 min followed by delivery or not of 100 J/cm2 of white light. ** P<0.01.
Anti-cancer Effect of Fullerene-mediated PDT
A longer incubation period of 24 h was applied for the experiments with cancer cells owing to their relatively slow process to uptake fullerene derivatives. Furthermore as it appears that cancer cells are more sensitive to the dark toxicity effects of both fullerene derivatives and DMA organic solvent, we kept the concentration to a single low value of 2.0 μM. The irradiation was made by a white light source (400–700 nm,) delivered at the intensity of 100 mW/cm2 with a variable fluence dose of 0, 10, 20, 40, and 80 J/cm2 for giving the demonstration of a light-dose dependence of cell killing. As depicted in Fig. 7, C70[>M(C3N6+C3)2] was apparently more effective at cancer-cell killing than C60[>M(C3N6+C3)2], with 1 log of cells killing at 40 J/cm2 and 2 logs at 80 J/cm2. Since the MTT assay cannot reliably measure more than 2 logs of killing, therefore, the latter light-dose effectiveness is equivalent to eradication. By contrast, the fullerene drug 1 killed less than 1 log at all fluences up to 80 J/cm2. These two curves were significantly different (p<0.05-0.001).
Figure 7.

PDT killing of human cancer cells. HeLa cells were incubated with 2.0 μM of C60[>M(C3N6 +C3)2] 1 or C70[>M(C3N6 +C3)2] 2 for 24 h, followed by delivery of stated fluence of white light, returned to incubator for 24 h, and then carried out the MTT assay. * P<0.05; ** P<0.01; *** P<0.001.
Discussion of PDT Results
The data showed interesting differences between the photoactivity of decacationic fullerene compounds that differ only in the number of carbon atoms in the fullerene cage. The decacationic arms attached to these fullerenes served to give the molecules rapid binding to the anionic residues displayed on the outer parts of bacterial cell walls. The large number of ionic groups attached to the fullerene also provided enhanced water solubility, which is a necessary requirement for biological activity. Even though these fullerenes displayed a rapid ability to photoinactivate bacteria, they were also efficient in mediating the PDT killing of cancer cells although a longer incubation time was needed. A large molecular weight of these fullerenes (around 3300) means that they are too large to diffuse through the plasma membrane of mammalian cells, which is a rapid process, and are therefore taken up by the slower process of adsorptive endocytosis. The differences in effectiveness we found between C60 and C70 was interesting. For Gram-positive bacteria C60[>M(C3N6+C3)2] was better at photokilling than C70[>M(C3N6+C3)2], while for Gram-negative bacteria and for cancer cells the opposite was the case, in that C70[>M(C3N6+C3)2] was better at photokilling than C60[>M(C3N6+C3)2]. We have previously reported30 that Type-II reactive oxygen species (ROS), i.e. singlet oxygen, 1O2, are better at killing Gram-positive bacteria than Type-I ROS, i.e. hydroxyl radicals, HO•, while the reverse is true for Gram-negative bacteria (HO• is better at killing than 1O2). Therefore we decided to test the type of ROS (HO• or 1O2) produced when 1 and 2 were illuminated in simple PBS solution. To do this it was necessary to employ blue light to excite the fullerene, as if white light was used the light alone would activate the probe to some extent through the green absorption peak. The results demonstrated that C60[>M(C3N6+C3)2] produced more 1O2 while C70[>M(C3N6+C3)2] produced more HO•. This finding offers an explanation of the preferential killing of Gram-positive bacteria by 1 and the preferential killing of Gram-negative bacteria by 2. The hypothesis is that 1O2 can diffuse more easily into porous cell walls of Gram-positive bacteria to reach sensitive sites, while the less permeable Gram-negative bacterial cell wall needs the more reactive HO• to cause real damage31,32 It has not been reported whether HO• or 1O2 is more efficient in killing cancer cells, but our data would suggest that HO• may be more effective in this case as well.
CONCLUSION
A novel water-soluble decacationic armed C70 monoadduct decaiodide C70[>M(C3N6+C3)2] was synthesized, characterized, and applied as a PS and potential nano-PDT agent against pathological bacteria and cancer cells. The arm structure was designed to include a large high number of cationic charges per C70 and H-bonding moieties were designed for rapid binding to the anionic residues displayed on the outer parts of bacterial cell walls and high water-solubility. The use of propyl groups on quaternary ammonium salt moieties was aimed to provide an interaction balance between the arm moieties and the cage in the presence of water. These structural characterisatics were found to overwhelming the hydrophobicity of the fullerene cage moiety that largely prohibited the sizable cage aggregation in water.
In the presence of a high number of electron-donating iodide anions as parts of quaternary ammonium salts in the arm region, we found that C70[>M(C3N6+C3)2] produced more HO• than C60[>M(C3N6+C3)2], in addition to 1O2. Both ROS were detected using several fluorescence probes. This finding offers an explanation of the preferential killing of Gram-positive and Gram-negative bacteria by C60[>M(C3N6+C3)2] and C70[>M(C3N6+C3)2], respectively. The data are consistent with the hypothesis that 1O2 can diffuse more easily into porous cell walls of Gram-positive bacteria to reach sensitive sites, while the less permeable Gram-negative bacterial cell wall needs the more reactive HO2• to cause real damage. Since the production rate of 1O was detected in a similar range for 1 and 2 in the absence of PBS, the differences shown in the presence of PBS electrolyte salts led to our suggestion that the observed production of HO• may arise partially from the photoinduced electron-transfer (Type-I) mechanism involving the electron-donating iodide anions (I−) and the fullerene cages at their excited triplet transient states, 3C60*[>M(C3N6+C3)2] and 3C70*[>M(C3N6+C3)2] having different electron-accepting strength. The presence of electrolyte salts makes the ion exchange and I−-dissociation possible and increases the efficiency of electron transport that forms the corresponding C60−•[>M(C3N6+C3)2] and C70−•[>M(C3N6+C3)2], respectively, as the source for the O2−• and the derived ROS radicals generation.
We previously reported33 that antimicrobial PDT using a cationic functionalized fullerene and white light of mice with wounds that were heavily contaminated with bacteria could prevent the death of mice due to the development of sepsis. The possible clinical application of fullerenes as PDT-agents is to some degree limited by the need to use shorter wavelength light for excitation, but we have also recently reported34 that this drawback can actually be an advantage when shorter penetration into tissue is desired. The temporal selectivity (short times give binding to bacteria, while long times allow uptake into mammalian cells) suggests the same agents could be used to treat both infections and cancer.35 When fullerene mediated is PDT used with a long incubation time it can kill multiple types of cancer cells including head and neck, breast and esophageal cancer that respond poorly to alternative cancer therapies.
EXPERIMENTAL SECTION
Materials
Reagents of γ-butyrolactone (GBL), BF3·Et2O, triethylamine, 1,8-diazabicyclo[5,4,0]-undec-7-ene (DBU), carbon tetrabromide (CBr4), trifluoroacetic acid, potassium carbonate, and iodomethane were purchased from Aldrich Chemicals and used without further purification. The chemical di-tert-butyl malonate was purchased from Tokyo Chemical Industry Co., Ltd. A C60 sample with a purity of 99.0% and a C70 sample with a purity of 98.0% were purchased from either Nano-C, Inc. or Term USA, Inc. 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABMA) was either purchased from Santa Cruz Biotechnology, Inc. or synthesized according to the literature procedure.36 Sodium sulfate was employed as a drying agent. Solvents were routinely distilled before use. Singlet oxygen sensor green (SOG) and 3′-p-(hydroxyphenyl) fluorescein (HPF) were obtained from Invitrogen, Ltd. as solutions in either methanol (SOG) or dimethyl formamide (HPF).
Spectroscopic Measurements
Infrared spectra were recorded as KBr pellets on a Thermo Nicolet Avatar 370 FT-IR spectrometer. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance Spectrospin–500 spectrometer. UV-vis spectra were recorded on a Perkin Elmer Lambda 750 UV-Vis-NIR Spectrometer. MALDI-mass spectra were recorded on a WATERS Micromass MALDI-TOF mass spectrometer. Elemental analysis was taken by Galbraith Laboratories, Inc. These spectroscopic methods were applied for all compounds synthesized in conjunction with elemental analysis of some compounds to confirm the purity of samples at least ≥95%.
Synthesis of Di(tert-butyl)[70]fullerenyl Malonate, C70[>M(t-Bu)2] 4
Finely divided [70]fullerene (2.3 g, 2.7 mmol) was taken into a round bottom flask and added anhydrous toluene (1.6 L) under nitrogen. The solution was stirred for 12 h at ambient temperature to ensure complete dissolution of C70. To the resulting wine-colored solution was added carbon tetrabromide (0.56 g, 1.68 mmol) followed by a solution of di-tert-butyl malonate (0.3 g, 1.39 mmol) in anhydrous toluene (100 mL). The solution was stirred for an additional 30 min and slowly added 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU, 0.52 g, 3.4 mmol) over a period of 15 min. The color of solution slowly turned brown in a reaction period of 8.0 h. The solution was then concentrated on rotary evaporator to roughly 50 mL. Upon the addition of methanol to this concentrated solution, the crude product was precipitated as brown solids which were collected via centrifugation. Purification of di(tert-butyl)[70]fullerenyl malonate, C70[>M(t-Bu)2] 4 was made by column chromatography (silica gel using toluene/hexane (1:1, v/v) as eluent) to afford brown solids in a yield of 65.0% (0.95 g). Spectroscopic data of the compound 4: MALDI–TOF–MS (sinapic acid as the matrix, rel. intensity) m/z 841 (C70H+, 56%), 855 (C70CH3+, 100%), 866 (28%), 882 (13%), 900 (79%), 913 (27%), 926 (28%), 944 (11%), 1023 (8%), 1055 (MH+, 5%) and 1056 (8%); FT-IR (KBr) υmax 3444 (vs, water peak), 2997 (w), 2974 (m, –C-H stretching), 2923 (m, –C-H stretching), 2849 (w), 1741 (vs, malonyl ester –C=O), 1720 (s, malonyl ester –C=O), 1647 (m), 1468 (w), 1451 (m, anti-symmetric deformations of –CH3), 1427 (s), 1391 (m), 1366 (s, symmetric deformations of CH3), 1308 (w), 1273 (s), 1250 (s, asymmetric stretching of –C-C(=O)-O–), 1150 (vs, –C-O-t-Bu stretching and C(C60)-C-C(=O)– deformations), 1093 (s), 1025 (w), 954 (w), 877 (w), 843 (m), 794 (m, C70), 752 (m), 671 (m, C70), 649 (w), 640 (m), 577 (s, C70), 532 (s, C70), and 458 (m, C70) cm−1; UV-vis (CHCl3, cutoff at 245 nm, 1.0 × 10−5 M) λmax 322, 355, 370, 403, 462 and 539 (shoulder band) nm; 1H NMR (500 MHz, CDCl3, ppm) δ 1..68 (s, 18H); 13C NMR (500 MHz, CDCl3–CS2, ppm) δ 162.39 (–C=O), 155.41 (2C), 151.57 (2C), 151.32 (2C), 150.86 (2C), 150.74 (2C), 149.48 (2C), 149.39 (2C), 149.21 (2C), 148.76 (2C), 148.71 (2C), 148.68 (2C), 148.62 (2C), 147.78 (2C), 147.71 (2C), 147.46 (2C), 147.15 (2C), 146.60, 146.09 (2C), 146.05 (2C), 145.06 (2C), 145.01, 144.10 (2C), 144.00 (2C), 143.63 (2C), 143.36 (2C), 142.93 (2C), 142.24 (2C), 141.81 (2C), 140.77 (2C), 137.30 (2C), 133.74 (2C), 132.98 (2C), 131.05 (2C), 130.97 (2C), 130.95 (2C), 84.86 (2C), 28.25 (6C), indicating a molecular Cs-symmetry of 4.
A similar procedure was applied for the preparation of di(tert-butyl)[60]fullerenyl malonate, C60[>M(t-Bu)2] 3 in a yield of 62.0%. Spectroscopic data: MALDI–TOF–MS (sinapic acid as the matrix, rel. intensity) m/z 721 (C60H+, 70%), 734 (C60CH2+, 100%), 747 (C60CHCH2+, 37%), 780 (75%), 794 (21%), 805 [C60C(CO)(CO2H)+, 24%], 824 (20%), 919 (12%), 921 (11%), 935 (MH+, 5%), and 937 (7%); FT-IR (KBr) υmax 3429 (vs, water peak), 2976 (m, –C-H stretching), 2924 (m, –C-H stretching), 2849 (m), 1741 (vs, malonyl ester –C=O), 1633 (m), 1454 (s, anti-symmetric deformations of –CH3), 1427 (m), 1392 (m), 1367 (s, symmetric deformations of CH3), 1273 (vs), 1253 (vs, asymmetric stretching of –C-C(=O)-O–), 1155 (vs, –C-O-t-Bu stretching and C(C60)-C-C(=O)– deformations), 1113 (m), 1059 (w), 1031 (w), 846 (m), 805 (w), 733 (m), 703 (m), 578 (m), 552 (m), and 526 (vs, a characteristic band of C60 monoadduct) cm−1; UV-vis (CHCl3, cutoff at 245 nm, 1.0 × 10−5 M) λmax 257 and 323 (shoulder band) nm; 1H NMR (500 MHz, CDCl3, ppm) δ 1.70 (s, 18H); 13C NMR (500 MHz, CDCl3, ppm) δ 162.28 (–C=O), 145.79, 145.36, 145.26, 145.18, 144.82, 144.72, 144.56, 143.93, 143.17, 143.15, 143.05, 143.03, 142.30, 141.96, 140.95, 139.00, 84.42, and 72.26, consistent with the structure of 3.
Synthesis of 4-Hydroxy-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)-butanamide C3N6C3-OH 5
To a solution of BF3·Et2O (1.98 g, 13.7 mmol) in the anhydrous dichloromethane was added triethylamine (1.60 g, 15.8 mmol, pre-distilled) and stirred at 0 °C for a period of 30 min. In a separate flask, N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)-amine, N5C3-NH2 (1.87 g, 3.9 mmol) and γ-butyrolactone (0.40 g, 4.6 mmol) were dissolved in dichloromethane (10 mL) and added to the above solution. Temperature of the mixture was slowly increased to 25 °C and kept at this temperature overnight. At the end of the reaction, it was quenched by the addition of cold aqueous sodium carbonate (10%). The organic layer was then washed sequentially with aqueous sodium carbonate (10%) and water and subsequently dried over sodium sulfate. After removal of the solvent, the crude product was purified by column chromatography (alumina TLC) using CHCl3–CH3OH (9.5:0.5, v/v) as the eluent to give a viscous light yellow liquid of 4-hydroxy-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)-butanamide C3N6C3-OH 5 in 60.0 % yield (1.32 g). Spectroscopic data of the compound 5: FT-IR (KBr) υmax 3304 (s, –O-H), 2957 (vs, –C-H stretching), 2933 (vs, –C-H stretching), 2872 (vs), 2807 (vs), 1650 (vs, amide carbonyl –C=O), 1555 (s), 1540 (s), 1459 (s), 1381 (m), 1189 (m, –C-C(=O)-NH– and –C(C=O)-NH-C– stretchings), 1077 (s, –C-O– stretching), and 749 (w) cm 1; 1H NMR (500 MHz, CDCl3, ppm) δ 3.66 (t, 2H, methylene protons of –CH2-OH), 3.27 (t, 2H, methylene protons of –CH2-NH2), 2.68–2.32 (br, 32H, amine N-attached –CH2– protons of ethyleneamino moiety), 1.83 (m, 2H, methylene protons of –CO-CH2-CH2-CH2-OH), 1.44 (m, 12H, CH3-CH2-CH2-N–), and 0.86 (m, 18H, CH3-CH2-CH2-N–). 13C NMR (500 MHz, CDCl3, ppm) δ 173.62 (amide –NH-C=O), 62.14, 57.45, 57.37, 57.00, 56.94, 56.93, 56.89, 56.84, 56.82, 56.77, 52.93, 52.63, 52.48, 52.43, 52.34, 51.74, 37.70, 34.19, 28.56, 20.44, 20.36, 20.34, 20.30, 20.20, 19.89, 12.12 (4C), and 12.05 (2C).
Synthesis of Bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[70]fullerenyl Malonate, Protonated Quaternary Ammonium Trifluoroacetate Salt, C70[>M(C3N6 +C3H)2] 7
The compound C70[>M(C3N6+C3H)2] 7 was synthesized by a transesterification reaction with a procedure as follows. To a solution of C70[>M(t-Bu)2] 4 (0.15 g, 0.14 mmol) in 1,2-dichloroethane was added C3N6C3-OH 5 (0.23 g, 0.40 mmol) and trifluoroacetic acid (1.0 g, 8.8 mmol). The solution was heated under reflux for a period of 3.0 d until no C70[>M(t-Bu)2] was detected on an analytical TLC plate (SiO2 with toluene as the eluent). Upon completion of the reaction, the mixture was washed by ice-cold aqueous potassium carbonate and water briefly, dried over sodium sulfate, and followed by solvent evaporation to give brown solids. The excessive C3N6C3-OH was removed by several ethyl acetate washings. The product was then quaternized by the addition of trifluoroacetic acid to afford bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19- hexaaza-tricosan-23-yl) [60]fullerenyl malonate, protonated quaternary ammonium trifluoroacetate salt, C70[>M(C3N6 +C3H)2] 7 in a yield of 71 % (0.32 g). Spectroscopic data of the compound 7: FT-IR (KBr) υmax 3441 (vs, water peak), 3006 (m), 2968 (s, –C-H stretching), 2929 (s, –C-H stretching), 2871 (m), 2852 (m), 1727 (m, malonyl ester –C=O), 1648 (vs, amide carbonyl –C=O), 1551 (w), 1453 (vs, anti-symmetric deformations of –CH3), 1433 (s), 1387 (m), 1372 (m, symmetric deformations of CH3), 1280 (w), 1252 (w), 1197 (w), 1155 (s, C(C60)-C-C(=O)– deformations and – C-O-C– stretching), 1095 (m), 1034 (m), 946 (s), 880 (w), 840 (w), 796 (s, C70), 750 (s), 708 (w), 658 (w), 625 (w), 582 (s, C70), 536 (s, C70), and 469 (br) cm−1; UV-vis (DMF, cutoff at 268 nm, 1.0 × 10−5 M) λmax 327–563 nm (a broad shoulder band); 1H NMR (500 MHz, CDCl3–CS2–DMF-d7, ppm) δ 4.0–4.15 (m, br, 4H, carboxylated methylene protons –C(=O)-O-CH2–), 3.32–4.0 (m, br, 56H, protonated quaternary ammonium ethylene protons –N+-CH2–), 3.11–3.20 (m, br, 8H, including –C(=O)-NH-CH2–), 2.28 (m, 4H, –CO-CH2-CH2-CH2-OH), 1.76 (m, 24H, CH3-CH2-CH2-N–), and 0.97 (m, 36H, CH3-CH2-CH2-N–); 13C NMR (500 MHz, CDCl3–CS2–DMSO-d6, ppm) 170.45 (amide –NH-C=O), 165.50 (ester –O-C=O), 155.81, 155.65, 155.51, 154.24, 153.64, 150.31, 150.28, 149.83, 149.65, 149.37, 149.11, 148.85, 148.84, 147.53, 147.49, 147.41, 147.36, 147.15, 146.83, 146.74, 146.72, 146.63, 146.61, 146.55, 146.54, 146.50, 145.88, 145.80, 145.62, 145.55, 145.18, 145.15, 144.59, 144.29, 144.18, 144.10, 144.04, 143.99, 143.44, 142.51, 142.25, 142.19, 142.08, 142.00, 141.89, 141.53, 141.45, 141.21, 140.82, 140.75, 140.31, 140.19, 139.73, 139.03, 138.67, 138.60, 137.83, 137.16, 136.02, 132.08, 131.86, 130.96, 130.92, 130.27, 129.05, 128.96 (2C), and 128.88 for the fullerenyl sp2 carbons, indicating a C1-symmetry of the fullerene cage. Signals of aliphatic carbons of the polycationic arm moiety were low due to high incompatibility of this moiety under the current solvent mixture used.
A similar procedure was applied for the preparation of bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl) [60]fullerenyl malonate, protonated quaternary ammonium trifluoroacetate salt, C60[>M(C3N6 +C3H)2] 6 in 65.2 % yield. Spectroscopic data: FT-IR (KBr) υmax 3436 (vs), 2957 (s, –C-H stretching), 2927 (s, –C-H stretching), 2868 (m), 2843 (m), 2809 (m), 1714 (m, malonyl ester –C=O), 1622 (vs, amide carbonyl –C=O), 1523 (w), 1456 (s, anti-symmetric deformations of –CH3), 1428 (m), 1361 (m, symmetric deformations of CH3), 1322 (w), 1184 (m), 1152 (m, C(C60)-C-C(=O)– deformations and –C-O-C– stretching), 1046 (m), 740 (s), 706 (s), 573 (m), and 526 (vs, a characteristic band of C60 monoadduct) cm−1; UV-vis (CHCl3, cutoff at 245 nm, 1.0 × 10−5 M) λmax 285 nm (shoulder peak); 1H NMR (500 MHz, CDCl3–CS2–DMF-d7, ppm) δ 4.0–4.25 (m, br, 4H, carboxylated methylene protons –C(=O)-O-CH2–), 3.32–4.10 (m, br, 56H, protonated quaternary ammonium ethylene protons –N+-CH2–), 3.11–3.20 (m, br, 8H, including –C(=O)-NH-CH2–), 2.34 (m, 4H, –CO-CH2-CH2-CH2-OH), 1.68 (m, 24H, CH3-CH2-CH2-N–), and 0.88 (m, 36H, CH3-CH2-CH2-N–); 13C NMR (500 MHz, CDCl3–CS2–DMF-d7, ppm) 147.99 (2C), 145.79 (2C), 144.82 (2C), 144.36 (2C), 144.18 (2C), 144.14 (2C), 144.10 (2C), 144.05, 143.74, 143.66 (2C), 143.64 (2C), 143.55 (2C), 143.46 (2C), 143.28 (2C), 142.96 (2C), 142.88, 142.73 (2C), 142.25 (2C), 142.12 (4C), 141.97 (2C), 141.93 (2C), 141.82 (2C), 141.55 (2C), 141.23 (2C), 141.13 (2C), 140.07, 139.80 (2C), 139.48 (2C), 136.74 (2C), and 135.46 (2C) for the fullerenyl sp2 carbons, indicating a C2-symmetry of the fullerene cage. Signals of aliphatic carbons of the polycationic arm moiety were low due to high incompatibility of this moiety under the current solvent mixture used.
Synthesis of Decacationic Bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaazatricosan- 23-yl)[70]fullerenyl Malonate, Methyl Quaternary Ammonium Iodide Salt, C70[>M(C3N6 +C3)2] 2
A suspension solution of 7 (0.20 g, 0.063 mmol) in chloroform (20 mL) was added potassium carbonate / DMF solution (20 mL) and subsequently stirred for a period of 1.0 h. To this brown solution was added iodomethane (12 mL, excess, addition in portions over the full reaction period). The reaction mixture was kept at 45 °C for 3.0 d. At the end of reaction, excessive iodomethane and solvent were removed via evaporation and the excess K2CO3 was removed by quick wash using water to give the product C70[>M(C3N6 +C3)2] 2 in a yield of 91.0 % (0.20 g). Spectroscopic data: MALDI–TOF–MS (sinapic acid as the matrix, rel. intensity) m/z 819 (40%), 841 (C70H+, 100%), 855 (C70-CH3 +, 15%), 1355, 1584, 1654, 1682, 1753, 2043, 2079, and 3340 (M – I−); m/z 841 (C70H+, 80%), 855 (C70-CH3 +, 100%), 913 (40%), 1036 (15%), 1124 (15%), 1344 (15%), 1369 (15%), 1440 (10%), 1577 (15%), and 1794 (15%); FT-IR (KBr) υmax 3437 (vs, water peak), 3006 (m), 2957 (m, –C-H stretching), 2923 (s, –C-H stretching), 2852 (m), 1735 (m, malonyl ester – C=O), 1642 (vs, amide carbonyl –C=O), 1465 (vs), 1427 (m), 1385 (m), 1317 (w), 1243 (w), 1209 (w), 1157 (w), 1111 (w), 1043 (m), 943 (m), 794 (m, C70), 757 (w), 723 (w), 671 (m, C70), 618 (m), 579 (s, C70), 530 (s, C70), 501 (m), and 459 (m) cm−1; UV-vis (DMF, cutoff at 268 nm, 1.0 × 10−5 M) λmax 327–563 nm (a broad shoulder band); 1H NMR (500 MHz, DMSO-d6, ppm) δ 3.82–4.43 (m, br, 36H, carboxylated methylene protons –C(=O)-O-CH2– and methylated quaternary ammonium ethylene protons –N+-CH2–), 3.10–3.69 (m, br, 32H, methylated quaternary ammonium ethylene protons –N+-CH2– and –C(=O)-NH-CH2–), 2.22 (m, 4H, –CO-CH2-CH2-CH2-OH), 1.58–1.90 (m, 24H, CH3-CH2-CH2-N–), and 0.97 (m, 36H, CH3-CH2-CH2-N–). Elemental analysis of 2 gave C, 43.90; H, 4.38; N, 5.09; I, 37.35 %.C, 54.08; H, 4.53; N, 4.24; I, 29.14%, indicating a measured I/Nq (quaternary amine) ratio of 0.97 as the same degree of quaternization on average.
A similar procedure was applied for the preparation of decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate, methyl quaternary ammonium iodide salt, C60[>M(C3N6 +C3)2] 1 in 92.0 % yield. Spectroscopic data: MALDI–TOF–MS (sinapic acid as the matrix, rel. intensity) m/z 703 (50%), 721 (C60H+, 100%), 759 (20%), 928 (40%), 946 (20%), 1595, 1648, 1682, 1839, 1989, 2039, 2152 (10%), 2359 (10%), 2468, 2626, 2794, 2838, 2852, and 3220 (M – I−); ESI–MS (rel. intensity) m/z 630, 720 (C60), 871 [C60-C(CO-O-CH=CH)2 +, 100%], 872, 885, 942, 949, 984, 985, 1043 (30%), 1045, 1067, 1114, 1208, 1210, 1282, 1570 [a group of 6 peaks, 8%, M+ – 10I – 8(CH2CH2CH3) – 4HCH3 – HN(CH2CH2CH3)2, consistent with further fragmentations from the peak of m/z 1760], 1587, 1591, 1608, 1623, 1625, 1741, 1742 (3%), and 1760 [a group of 7 peaks, 2%, MH+ – 10I – 6(CH2CH2CH3)– 4CH3, consistent with a mass of fully de-cationized 1 giving the evidence of a C60 malonate monoadduct with two 4-hydroxy-hexa(aminoethyl)butanamide main chains]; FT-IR (KBr) υmax 3427 (vs, water peak), 2964 (s, –C-H stretching), 2928 (s, –C-H stretching), 2874 (m), 2850 (m), 1752 (w, malonyl ester –C=O), 1711 (w), 1650 (vs, amide carbonyl –C=O), 1457 (vs), 1373 (m), 1320 (w), 1182 (m), 1102 (w), 1080 (m), 1032 (s), 947 (m), 756 (w), 727 (m), 708 (w), 693 (w), 578 (m), and 526 (vs, a characteristic band of C60 monoadduct) cm−1; UV-vis (DMF, cutoff at 268 nm, 1.0 × 10−5 M) λmax 323 nm (shoulder peak); 1H NMR (500 MHz, DMSO-d6, ppm) δ 3.82–4.43 (m, br, 36H, carboxylated methylene protons –C(=O)-O-CH2– and methylated quaternary ammonium ethylene protons –N+-CH2–), 3.10–3.69 (m, br, 32H, methylated quaternary ammonium ethylene protons –N+-CH2– and –C(=O)-NH-CH2–), 2.22 (m, 4H, –CO-CH2-CH2-CH2-OH), 1.58–1.90 (m, 24H, CH3-CH2-CH2-N–), and 0.97 (m, 36H, CH3-CH2-CH2-N–). Elemental analysis of 1 giving C, 54.08; H, 4.53; N, 4.24; I, 29.14% indicated a measured I/Nq (quaternary amine) ratio of 0.92 as the same degree of quaternization on average.
Synthesis of Decacationic Bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaazatricosan- 23-yl)[70]fullerenyl Malonate, Methyl Quaternary Ammonium Triflouroacetate Salt, C70[>M(C3N6 +C3)2] 2′
Ion-exchange reaction of the compound C70[>M(C3N6 +C3)2] 2 (30 mg, 0.0087 mmol) was carried out in DMF solution by the addition of an excess quantity of aqueous CF3COONa. The mixture was heated at 50 °C and sonicated for a period of 1.0 h. The product was precipitated from the solution upon the addition of methanol. The solids were washed by methanol and diethyl ether. The resulting solids were subjected to a repeated ion-exchange procedure and followed by drying under vacuum to afford the corresponding methyl quaternary ammonium salt C70[>M(C3N6 +C3)2] 2′with triflouroacetate anions in a nearly quantitative yield. Spectroscopic data: FT-IR (KBr) υmax 3436 (vs, water peak), 3006 (m), 2969 (s, –C-H stretching), 2932 (m, –C-H stretching), 2877 (m), 2757 (m), 1726 (m, malonyl ester –C=O), 1640 (s, amide carbonyl –C=O), 1457 (s), 1431 (m), 1332 (w), 1209 (m), 1156 (s), 1086 (m), 1018 (m), 945 (s), 880 (m), 794 (m, C70), 754 (w), 673 (m, C70), 576 (s, C70 ), 532 (s, C70), and 472 (m) cm−1; 1H NMR (500 MHz, CDCl3–CS2–DMSO-d6, ppm) δ 4.09–4.39 (m, br, 36H, carboxylated methylene protons –C(=O)-OCH2– and methylated quaternary ammonium ethylene protons –N+-CH2–), 2.86–3.50 (m, br, 32H, methylated quaternary ammonium ethylene protons –N+-CH2– and –C(=O)-NH-CH2–), 2.12 (m, 4H, –CO-CH2-CH2-CH2-OH), 1.45–1.95 (m, 24H, CH3-CH2-CH2-N–), and 0.74–1.11 (m, 36H, CH3- CH2-CH2-N–); 13C NMR (500 MHz, CDCl3–CS2–DMSO-d6, ppm) 170.45 (amide –NH-C=O), 155.81, 155.67, 155.50, 154.20, 153.10, 150.31, 149.83, 149.65, 149.37, 149.11, 148.87, 148.84, 148.82, 147.84, 147.64, 147.53, 147.49, 147.41, 147.36, 147.15, 146.83, 146.74, 146.72, 146.63, 146.61, 146.23, 146.21, 146.15, 146.10, 145.88, 145.80, 145.62, 145.55, 145.18, 145.15, 144.59, 144.38, 144.36, 144.34, 144.29, 144.10, 144.04, 143.99, 143.45, 142.51, 142.26, 142.22, 142.21, 142.19, 141.53, 141.45, 141.21, 141.15, 140.82, 140.31, 140.19, 139.73, 137.79, 137.77, 134.97, 134.15, 131.86, 130.96, 130.92, 129.05, 129.00, 128.94, and 128.88 for the fullerenyl sp2 carbons, indicating a C1-symmetry of the fullerene cage. Signals of aliphatic carbons of the polycationic arm moiety were low due to high incompatibility of this moiety under the current solvent mixture used.
A similar procedure was applied for the preparation of decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate, methyl quaternary ammonium triflouroacetate salt, C60[>M(C3N6 +C3)2] 1′. Spectroscopic data: FT-IR (KBr) υmax 3448 (vs, water peak), 2970 (s, –C-H stretching), 2924 (s, –C-H stretching), 2877 (m), 2846 (m), 1728 (s, malonyl ester –C=O), 1647 (vs, amide carbonyl –C=O), 1457 (vs), 1393 (w), 1368 (m), 1251 (w), 1213 (m), 1186 (m), 1154 (vs), 1111 (s), 1090 (s), 1019 (m), 944 (m), 752 (m), 613 (m), 576 (m), and 526 (vs, a characteristic band of C60 monoadduct) cm−1; 1H NMR (500 MHz, CDCl3–CS2–DMSO-d6, ppm) δ 4.09–4.39 (m, br, 36H, carboxylated methylene protons –C(=O)-O-CH2– and methylated quaternary ammonium ethylene protons –N+-CH2–), 2.86–3.50 (m, br, 32H, methylated quaternary ammonium ethylene protons –N+-CH2– and –C(=O)-NH-CH2–), 2.12 (m, 4H, –CO-CH2-CH2-CH2- OH), 1.45–1.95 (m, 24H, CH3-CH2-CH2-N–), and 0.74–1.11 (m, 36H, CH3-CH2-CH2-N–); 13C NMR (500 MHz, CDCl3–CS2–DMSO-d6, ppm) 147.09 (2C), 144.37 (2C), 143.85, 143.81 (2C), 143.69 (2C), 143.67, 143.61 (2C), 143.28 (2C), 143.23 (2C), 143.14 (2C), 143.03 (2C), 142.65, 142.35 (2C), 142.22 (2C), 141.89, 141.60 (4C), 141.48 (2C), 141.45 (2C), 140.70 (2C), 139.52 (2C), 139.38 (2C), 139.16 (2C), 138.77 (2C), 138.53 (2C), 138.23 (2C), 137.76 (2C), 137.33 (2C), 136.68 (2C), 136.67 (2C), and 136.58 (2C) for the fullerenyl sp2 carbons, indicating a C2-symmetry of the fullerene cage. Signals of aliphatic carbons of the polycationic arm moiety were low due to high incompatibility of this moiety under the current solvent mixture used.
Cell Culture
A human cervical cancer cell line HeLa37 was obtained from ATCC (Manassas, VA). The cells were cultured in RPMI medium with L-glutamine and NaHCO3 supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/mL) (Sigma, St. Louis, MO) at 37 °C in 5% CO2-humidified atmosphere in 75 cm2 flasks (Falcon, Invitrogen, Carlsbad, CA). When the cells reached 80% confluence, they were washed with phosphate-buffered saline (PBS) and harvested with 2.0 mL of 0.25% trypsin-EDTA solution (Sigma). Cells were then centrifuged and counted in trypan blue to ensure viability and plated at a density of 5000/well in flat-bottom 96-well plates (Fisher Scientific, Pittsburgh, PA).
Bacterial Culture
Staphylococcus aureus 8325-4 and Escherichia coli K12 (both wild type) were obtained from ATCC. Planktonic bacterial cells were cultured in brain-heart infusion (BHI) broth with aeration at 37 °C to stationary phase overnight and refreshed to mid-log growth phase for 2.0 h the next day. Cell numbers were estimated by measuring the optical density (OD) at 600 nm [OD of 0.5 = 10(8) cells/mL].
Light Source for Bio-experiments
We used a white (400–700 nm) broad-band light source (Lumacare, Newport Beach, CA) set to deliver a spot of 5.0 cm diameter at an irradiance of 100 mW/cm2. The power was measured with a power meter (Model DMM 199 with 201 Standard head, Coherent, Santa Clara, CA).
PDT of Bacteria
Mid-log phase cells were collected through centrifugation (1000 g) for five min then suspended in PBS. A cell suspension consisting of 10(8) cells/mL for bacteria was incubated with fullerene for 30 min at room temperature in the dark. 500 μL aliquots of cell suspension were transferred to a 48-well plate and fullerene derivatives, 1 or 2, were added (up to 10 μM for S. aureus and up to 80 μM for E. coli) from a DMA stock solution prepared at 2.0 mM. The highest concentration (for 80 μM fullerene) of DMA used was 4% and this did not cause any toxicity to the cells. Cells were illuminated at room temperature for 16.6 min to deliver 100 J/cm2. At the completion of the illumination period, 100 μL aliquots were removed from illuminated and non-illuminated wells (cells incubated with fullerene but kept in 48-well plates covered with aluminum foil at room temperature for the duration of the illumination were used as control) and serially diluted 10-fold in PBS to give dilutions of 10−1 to 10−6 times the original concentrations and 10 μl aliquots of each of the dilutions were streaked horizontally on square BHI plates by the method of Jett and colleagues38 Plates were streaked in triplicate and incubated for 24 h at 37 °C in the dark to allow colony formation. Controls groups included cells that were not treated with fullerene or light, and cells treated with light but not with fullerene. Survival fractions (SF) were routinely expressed as ratios of CFU of microbial cells treated with light and fullerene or treated with fullerene in the dark, to CFU of microbes treated with neither.
PDT of Cancer Cells
After cells had grown for 24 h, dilutions of fullerene derivatives were prepared in complete RPMI medium containing 10% serum, penicillin (100 U/mL), and streptomycin (100 μg/mL), and added to the cells at concentrations of 2.0 μM for an additional incubation of 24 h. The highest DMA concentration in the medium did not exceed 0.1%. The medium was replaced with fresh complete medium and white light (400–700 nm with the fluence of 0, 10, 20, 40 and 80 J/cm2) was delivered at an irradiance of 100 mW/cm2. The light spot covered 4 wells, which were considered as one experimental group illuminated at the same time. Control groups were as follows: no treatment, light alone, and fullerene alone (at the same dilution used for PDT experiments). Following PDT treatment the cells were returned to the incubator overnight and a 4.0-h MTT assay was carried out the next day and read at 562 nm using a microplate spectrophotometer (Spectra Max 340 PC, Molecular Devices, Sunnyvale, CA). Each experiment was repeated 3 times.
Fluorescence Probe Assay Procedures
96-well black-sided plates were used for fluorescence probe experiments. SOG (in methanol) or HPF (in DMF as provided by Invitrogen, Ltd. with a final concentration of 5.0 μM) was added to 5.0 μM C60[>M(C3N6 +C3)2] 1 or C70[>M(C3N6 +C3)2] 2 in phosphate buffered saline (PBS) solution (200 μL) per well. Fluorescence spectrometry (SpectraMax M5 plate reader, Molecular Devices, Sunnyvale, CA) used excitation and emission at 504 and 525 nm for SOG and 490 and 515 nm for HPF, respectively. Increasing fluences (J/cm2) were delivered using a blue LED light source (415 ± 15 nm, Clear-U, Photomedex, Montgomeryville, PA) at an irradiance of 20 mW/cm2. Each time after an incremental fluence was delivered, the fluorescence was measured.
Statistics
Values are means of three separate experiments, and bars presented in the graphs are standard errors of the means (SEM). Differences between means were statistically analyzed by one way ANOVA in Microsoft Excel, and p<0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank the financial support of National Institute of Health (NIH) under the grant number 1R01CA137108.
Footnotes
Supporting Information. Additional details of spectroscopic data for the structural characterization of all compounds given in Scheme 1. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nature Reviews Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 2.Hamblin MR, Hasan T. Photodynamic therapy: a new antimicrobial approach to infectious disease? Photochem Photobiol Sci. 2004;3:436–450. doi: 10.1039/b311900a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Riordan K, Akilov OE, Hasan T. The potential for photodynamic therapy in the treatment of localized infections. Photodiagnosis and Photodynamic Therapy. 2005;2:247–262. doi: 10.1016/S1572-1000(05)00099-2. [DOI] [PubMed] [Google Scholar]
- 4.Silva JN, Filipe P, Maziere P, Maziere JC, Freitas JP, Cine de Castro JP, Santus R. Photodynamic therapy: Dermatology and ophthalmology as main fields of current applications in clinic. Bio-Med Mater Eng. 2006;16:S147–S154. [PubMed] [Google Scholar]
- 5.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one--photosensitizers, photochemistry and cellular localization. Photodiagn Photodyn Ther. 2004;1:279–293. doi: 10.1016/S1572-1000(05)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part two-cellular signalling, cell metabolism and modes of cell death. Photodiagn Photodyn Ther. 2005;2:1–23. doi: 10.1016/S1572-1000(05)00030-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Livermore DM. The threat from the pink corner. Ann Med. 2003;35:226–234. doi: 10.1080/07853890310001609. [DOI] [PubMed] [Google Scholar]
- 8.Tang HM, Hamblin MR, Yow CM. A comparative in vitro photoinactivation study of clinical isolates of multidrug-resistant pathogens. J Infect Chemother. 2007;13:87–91. doi: 10.1007/s10156-006-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mroz P, Tegos GP, Gali H, Wharton T, Sarna T, Hamblin MR. Photodynamic therapy with fullerenes. Photochem Photobiol Sci. 2007;6:1139–1149. doi: 10.1039/b711141j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navarre WW, Schneewind O. Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott JR, Barnett TC. Surface proteins of Gram-positive bacteria and how they get there. Annu Rev Microbiol. 2006;60:397–423. doi: 10.1146/annurev.micro.60.080805.142256. [DOI] [PubMed] [Google Scholar]
- 12.Kell AJ, Stewart G, Ryan S, Peytavi R, Boissinot M, Huletsky A, Bergeron MG, Simard B. Vancomycin-modified nanoparticles for efficient targeting and preconcentration of Grampositive and Gram-negative bacteria. ACS Nano. 2008;2:1777–1788. doi: 10.1021/nn700183g. [DOI] [PubMed] [Google Scholar]
- 13.Demidova TN, Hamblin MR. Photodynamic therapy targeted to pathogens. Int J Immunopathol Pharmacol. 2004;17:245–254. doi: 10.1177/039463200401700304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bingel C. Cyclopropylation of fullerenes. Chem Ber. 1993;126:1957–1959. [Google Scholar]
- 15.Birkett PR, Avent AG, Darwish AD, Kroto HW, Taylor R, Walton DRM. Formation and characterisation of C70Cl10. J Chem Soc, Chem Commun. 1995:683–685. [Google Scholar]
- 16.Thilgen C, Hermann A, Diederich F. The covalent chemistry of higher fullerenes: C70 and beyond. Angew Chem Int Ed Engl. 1997;36:2268–2280. [Google Scholar]
- 17.Thilgen C, Diederich F. The higher fullerenes: covalent chemistry and chirality. Topics in Current Chemistry. 1999;199:136–171. [Google Scholar]
- 18.Sawamura M, Nagahama N, Toganoh M, Hackler UE, Isobe H, Nakamura E, Zhou S, Chu B. Chem Lett. 2000:1098–1099. doi: 10.1126/science.291.5510.1944. [DOI] [PubMed] [Google Scholar]
- 19.Brettreich M, Burghardi S, Bottcher C, Bayerl T, Bayerl S, Hirsch A. Angew Chem Int Ed. 2000;39:1845–1848. doi: 10.1002/(sici)1521-3773(20000515)39:10<1845::aid-anie1845>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 20.Zhou S, Burger C, Chu B, Sawamura M, Nagahama N, Toganoh M, Hackler UE, Isobe H, Nakamura E. Science. 2001;291:1944–1947. doi: 10.1126/science.291.5510.1944. [DOI] [PubMed] [Google Scholar]
- 21.Foley S, Berberan-Santos MN, Fedorov A, McGarvey DJ, Santos C, Gigante B. Photophysical properties of pseudo-dihydro derivatves of C70. J Phys Chem A. 1999;103:8173–8178. [Google Scholar]
- 22.Brites MJ, Santos C, Nascimento S, Gigante B, Luftmann H, Fedorov A, Berberan-Santos MN. Synthesis and fluorescence properties of [60] and [70]fullerene-coumarin dyads: Efficient dipole-dipole resonance energy trasfer from coumarin to fullerene. New J Chem. 2006;30:1036–1045. [Google Scholar]
- 23.Guldi DM, Prato M. Excited-state properties of C60 fullerene derivatives. Acc Chem Res. 2000;33:695–703. doi: 10.1021/ar990144m. [DOI] [PubMed] [Google Scholar]
- 24.Yamakoshi Y, Umezawa N, Ryu A, Arakane K, Miyata N, Goda Y, Masumizu T, Nagano T. Active oxygen species generated from photoexcited fullerene (C60) as potential medicines: O2 −· versus 1O2. J Am Chem Soc. 2003;125:12803–12809. doi: 10.1021/ja0355574. [DOI] [PubMed] [Google Scholar]
- 25.Kuznetsova NA, Gretsova NS, Yuzhakova OA, Negrimovskii VM, Kaliya OL, Luk’yanets EA. New reagents for determination of quantum efficiency of singlet oxygen generation in aquesou media. Russ J Gen Chem. 2001;71:36–41. [Google Scholar]
- 26.Raga’s X, Jimenez-Banzo A, Sanchez-Garcia D, Batllori X, Nonell S. Singlet oxygen photosensitisation by the fluorescent probe Singlet Oxygen Sensor Greens. Chem Commun. 2009:2920–2922. doi: 10.1039/b822776d. [DOI] [PubMed] [Google Scholar]
- 27.Setsukinai KI, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem. 2003;278:3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 28.Gupta AK, Rohatgi-Mukherjee KK. Solvent effect on photosensitized oxidation of iodide ion by anthracene sulphonates. Photochem Photobiol. 1978;27:539–543. [Google Scholar]
- 29.Huang L, Dai T, Hamblin MR. Antimicrobial photodynamic inactivation photodynamic therapy for infections. Methods Mol Biol. 2010;635:155–173. doi: 10.1007/978-1-60761-697-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang L, Xuan Y, Koide Y, Zhiyentayev T, Tanaka M, Hamblin MR. Type 1 and type 2 mechanisms of antimicrobial photodynamic therapy: An in vitro study on Gram-negative and Gram-positive bacteria. Laser Surg Med. 2011 doi: 10.1002/lsm.22045. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valduga G, Bertoloni G, Reddi E, Jori G. Effect of extracellularly generated singlet oxygen on gram-positive and gram-negative bacteria. J Photochem Photobiol B. 1993;21:81–86. doi: 10.1016/1011-1344(93)80168-9. [DOI] [PubMed] [Google Scholar]
- 32.Dahl TA, Midden WR, Hartman PE. Comparison of killing of gram-negative and gram-positive bacteria by pure singlet oxygen. J Bacteriol. 1989;171:2188–2194. doi: 10.1128/jb.171.4.2188-2194.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Z, Dai T, Huang L, Kurup DB, Tegos GP, Jahnke A, Wharton T, Hamblin MR. Photodynamic therapy with a cationic functionalized fullerene rescues mice from fatal wound infections. Nanomedicine (UK) 2010;5:1525–1533. doi: 10.2217/nnm.10.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mroz P, Xia Y, Asanuma D, Konopko A, Zhiyentayev T, Huang YY, Sharma SK, Dai T, Khan UJ, Wharton T, Hamblin MR. Intraperitoneal photodynamic therapy mediated by a fullerene in a mouse model of abdominal dissemination of colon adenocarcinoma. Nanomedicine. 2011;7:965–974. doi: 10.1016/j.nano.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma SK, Chiang LY, Hamblin MR. Photodynamic therapy with fullerenes in vivo: reality or a dream? Nanomedicine (UK) 2011;6:1813–1825. doi: 10.2217/nnm.11.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez GR, Garcia F, Catalani LH, Cadet J, Oliveira MCB, Ronsein GE, Miyamoto S, Medeiros MHG, Di Mascio P. Synthesis of a hydrophilic and non-ionic anthracene derivative, the N,N′-di(2,3-dihydroxypropyl)-9,10-anthracenedipropanamide as a chemical trap for singlet molecular oxygen detection in biological systems. Tetrahedron. 2006;62:10762–10770. [Google Scholar]
- 37.Perry VP. Cultivation of large cultures of HeLa cells in horse serum. Science. 1955;121:805. doi: 10.1126/science.121.3153.805. [DOI] [PubMed] [Google Scholar]
- 38.Jett BD, Hatter KL, Huycke MM, Gilmore MS. Simplified agar plate method for quantifying viable bacteria. Biotechniques. 1997;23:648–650. doi: 10.2144/97234bm22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.